

Abstract
Objective(s):
The use of antisense oligonucleotides (AOs) to restore normal splicing by blocking the recognition of aberrant splice sites by the spliceosome represents an innovative means of potentially controlling certain inherited disorders affected by aberrant splicing. Selection of the appropriate target site is essential in the success of an AO therapy. In this study, in search for a splice model system to facilitate the evaluation of AOs to redirect defective splicing of IVSI-110 β-globin intron, an EGFP-based IVSI-110 specific cellular reporter assay system has been developed and a number of AOs were tested in this cellular splicing assay.
Materials and Methods:
A recombinant plasmid (pEGFP/I-110) carrying the EGFP gene interrupted by a mutated human β-globin intron 1 (IVSI-110) was developed and transfected into K562 cells. A number of AOs with a 2’-O-methyl oligoribonucleotide (2’-O-Me) backbone system were systematically tested in this cellular splicing assay.
Results:
The mutation in the intron causes aberrant splicing of EGFP pre-mRNA, preventing translation of EGFP; however, treatment of the cells with specific concentration of a sequence specific 2’-O-Me AO targeted to the aberrant splice site induced correct splicing and resulted in restoring of EGFP activity.
Conclusion:
This cellular splicing assay provides a novel functional assay system in assessing the cellular delivery efficiency of AOs and therapeutic effect of AOs in restoration of aberrant splicing.
Keywords: Antisense, Beta-thalassemia, Gene therapy, Oligonucleotides, Splicing
Introduction
The use of antisense oligonucleotides (AOs) to restore normal splicing by blocking the recognition of aberrant splice sites by the spliceosome represents an innovative means of potentially controlling certain inherited disorders affected by aberrant splicing (1, 2). AOs are short single-stranded sequences (8–50 bp), which hybridize with the target mRNA via Watson–Crick base pairing. After an AO binds with the mRNA, either the target complex will be degraded by endogenous cellular ribonuclease H (RNase H) or a functional blockade of mRNA occurs due to steric hindrance. RNase H-independent AO can be used to block cryptic splice sites arising from intronic or exonic mutations, to induce exon skipping to overcome pre-mature stop codon or frame-shift mutations, or to change exon selection to create specific isoforms (3, 4). AOs are short synthetic strand of deoxyribonucleotide analogue that hybridize with the complementary mRNA via Watson–Crick base pairing. The mRNA in RNA-DNA duplex is a substrate for cellular RNase H, an enzyme that destroys the RNA.
Selection of the appropriate target site is essential to the success of an antisense therapy. The majority of active AOs have been chosen either empirically, or by consideration of the thermodynamic or structural properties of the target mRNA. Conventional approa-ches, such as selection by ‘sequence-walking’ require the screening of up to 30–50 AO compounds in a cellular environment for the discovery of a few active AO sequences. The success of such intensive AO walking experiments is reliant on an efficient and readily available readout system.
An in vitro AO-based strategy was first utilized to redirect splicing by blocking the aberrant splice site of β-globin pre-mRNA containing common β-thalassemia splicing mutations (2). This in vitro system was not suitable for testing large numbers of AO. In order to identify and evaluate optimal AO target sequences, which can block aberrant splicing of IVS2-654 huβ-globin mRNA, an EGFP-based cellular assay system was developed (5). This IVS2-654 huβ-globin intron was inserted into the coding sequence of EGFP preventing EGFP expression in both stably transfected HeLa cell lines and in transgenic mice ubiquitously expressing the EGFP-654 construct (6). Increased EGFP expression in HeLa cells was accomplished by AO directed to the aberrant 5’ splice site at position 654 in the intron. In addition, systemic delivery of AO in the EGFP-654 transgenic mice was able to restore EGFP expression in various tissues. This animal model provided strong evidence that AO can be delivered in vivo to restore splicing specificity in a large variety of tissues. Increase in the EGFP production was proportional to antisense activity of a given AO. Using this EGFP systems, RNase H-inactive AO with novel or established chemistry were compared in both the cellular and the animal model, on a standard platform with a standardized sequence. Therefore, any variability in sequence and output associated with different assays could be eliminated.
In search for an alternative splice model system to facilitate the evaluation of AOs to redirect defective splicing of IVSI-110β-globin intron I, the Cell and Gene Therapy Group created a ‘humanized’ mouse model containing the β-thalassemia IVSI-110 (G→A) mutation in the context of the huβ-globin locus (7).
The IVSI-110 β-thalassemia mutation is one of the most common β-globin splicing mutations found in β-thalassemia patients with Mediterranean origins. The G to A substitution at position 110 in the first intron of the β-globin gene generates a new donor-like splice site, leading to 90% aberrant splicing. In homozygous state, the β-globin chain synthesis is markedly reduced and severe β-thalassemia phenotype occurs. Severe cases of β-thalassemia result in pronounced anemia, bone deformities, hepatosplenomegaly and, if left untreated, death (7, 8).
The heterozygous mouse model carrying the IVSI-110β-globin locus and the normal mouse locus was found to display many of the classical clinical features associated with β-thalassemia intermedia, including extramedullary erythropoiesis, splenomegaly, anemia, and enhanced erythropoiesis. The heterozygous IVSI-110 β-thalassemic mice represent one of the first BAC transgenic animal models of any disease resulting from a known human splicing mutation.
While the heterozygous IVSI-110 β-thalassemic mice represents an excellent animal model to test the therapeutic potential of AO, unfortunately, the breeding of IVSI-110 β-thalassemic mice yielded unexpectedly low number of progeny containing the desired IVSI-110> //KO+/- genotype. This severely restricted identification and evaluation of various AO sequences, in a time- and dose-dependent manner. The simplest explanation for the low number of progeny is low rate of fertility due to hypogonatotrophic hypogonadism, since this occurs in thalassemia patients. This inherent problem with the IVSI-110 β-thalassemic mouse line led us to investigate an alternative splicing assay system, which could be used to evaluate the efficiency of various AOs in a low to high-throughput manner.
In this study, the development and evaluation of an EGFP-based IVSI-110-specific cellular reporter assay system (EGFP-110) is described. The EGFP gene was interrupted by the insertion of the huβ-globin intron I containing the IVSI-110 (G→A) substitution. In this investigation, sequence-specific evaluation of AO was used to restore splicing as measured by RT-PCR and flowcytometry.
Materials and Methods
The phosphorothioate 2’-O-methyl-oligoribonucleo tides used in the present study were kindly provided by Professor Steve Wilton (Head of the Experimental Molecular Medicine Group, Australian Neuromus-cular Research Institute, Nedlands, Western Australia) (Table 1). The 2’-O-methyl RNA modification combined with a full length phosphorothioate backbone is the most used chemistry for splicing modulation. The 2’-O-methyl modification renders the AO RNase H resistance and the phosphorothioate backbone greatly improves transmembrane uptake in cultured cells and increases serum half-life in vivo (9). The major advantages of 2’-O-methyl modifications in addition to the resistancy to RNase H cleavage is displaying lower toxicity and slightly increased hybridization affinities (10-12).
Table 1.
Sequence of antisense oligonucleotides
| Name | Antisense oligonucleotide sequence |
|---|---|
| CR 1 | 5’-GAG AGA GTC AGT GCC TAT CAG AAA C 3’ |
| Mt 2 | 5’-TGG GAA AAT AGA CTA ATA GGC A-3’ |
| H36A | 5’ CTG GTA TTC CTT AAT TGT ACA GAG A 3’ |
AOs were targeted to respective sequences in the intron 1 of human β-globin gene. As an additional control, a randomized oligonucleotide pool (H36A), that is, a preparation synthesized as a library of all possible 18-mers[A GA2][A GA3], was used. K562 cell line, EGFP C1 plasmid, and pEGFP C1 were purchased from Clontech Laboratories, Inc. (Palo Alto, CA).
Plasmid and cell line construction
The thalassemic β-globin intron IVSI-110 or its wild-type equivalent IVSI was inserted into the EGFP cDNA sequence of the pEGFP C1 plasmid, between nucleotides 274 and 275 of the EGFP cassette using “gene splicing by overlap extension PCR” method resulting in an 854 bp PCR product [EGFP/I1-110 or pEGFP/ Intron wild type (Iwt]. (The PCR product was cloned in pEGFP resulting in plasmids pEGFP/I1-110 or pEGFP/Iwt, respectively (Figure 1). Sequence analysis of the pEGFP C1/I 1-110 using primer Fwd EGFP or Rev EGFP was used to confirm the presence of the IVSI-110 intron I in the expected position without any random mutation or deletion. The details of the construction are available on request. K562 cells were transfected with the recombinant plasmids via electroporation using the Gene Pulser II (Bio-Rad, Hercules, California, USA) at the following conditions; 1.8 kV, 25 μF, 200 Ω. Cells were plated on selective LB agar plates and incubated overnight at 37 °C.
Figure 1.
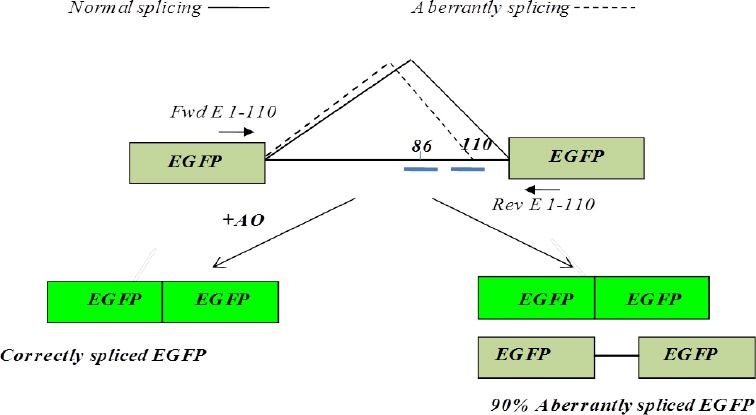
Splicing of EGFP-IVSI-110 mRNA. Schematic diagram illustrating EGFP pre-mRNA containing intron I of huβ-globin carrying the G>A splicing mutation at nucleotide 110. The mutation activates an aberrant splice site. Short bars at position 86 and 110 of intron I represent targeted 2’-O-Me phosphorothioate antisense oligonucleotide (AOs), which have been targeted to redirect aberrant splicing. Primers Fwd E 1-110 and Rev E 1-110 were used in RT-PCR
Oligonucleotide treatment
K562 cells (5 x 106 cells/0.5 ml) were transiently co-transfected by electroporation with pEGFP C1+ I1-110 plasmid and 2’-O-Me phosphorothioate AOs targeted to the branch point and the IVSI-110 mutant splice. The electroporation was performed as follows. A 0.1-ml aliquot of an oligonucleotide at a given concentration in Opti-MEM media was mixed with 0.4 ml of Opti-MEM containing 5 μg of pEGFP C1/I1-110. After being briefly mixed, the preparation was electroporated at the following conditions; 1.8 kV, 25 μF, 200 Ω. The cells were incubated in DMEM containing 10% foetal calf serum (FCS), penicillin (100 U/ml) and streptomycin (100 μg/ml) and following 24 , 48 hr and 72 hr, the cells were analysed using flow cytometry for EGFP and RNA was collected for RT-PCR assays. Forward and reverse primers (5’-TTTAGTGAACCGTCAGATCC-3’ and 5’-ATGT-GGTATGGCTGATTATTG-3’, respectively) hybridized to the EGFP sequences flanking the intron.
Reverse transcriptase–PCR (RT-PCR) analysis
Total RNA was prepared using the Tri-Reagent BD system (Molecular Research Center, Cincinnati, OH) as described by the manufacturer. First-strand cDNA was synthesised by using a Superscript II reverse transcriptase kit (Invitrogen, Carlsbad, Calif). Primers used to amplify the sequence between exon 1 and exon 2 were: forward primer -5’- GGCGTGCAGTGCTTCAGCCGC-3’ and reverse primer 5’-CCCTCGAACTTCACCTCGGCGC-3’. The PCR was per-formed using the following conditions: denaturation at 94 °C for 30 min, annealing at 60 °C for 30 sec, and extension at 72 °C for 30 sec. After 30 cycles, the PCR products were resolved on a 3% Nusieve GTG-agarose gel. The above primers amplified a 140-bp and a 159-bp products from construct, which correspond to the normal and aberrantly spliced mRNA, respectively.
Experion chip capillary electrophoresis
RT-PCR products were analysed by chip capillary electrophoresis using Experion DNA 1K Analysis Kit (Experion-Bio-Rad Life Sciences city state) according to the manufacturer’s instructions. In brief, following preparation of the gel-stain mix, it was applied to the gel priming well and primed using the Experion priming station. Next, 9 μl of the gel-stain was applied to three other designated wells labelled “gel-stain”. Supplied DNA loading buffer (5 μl) was loaded into each sample and ladder wells. Next, each RT-PCR product (1 μl) and DNA 1K ladder molecular weight standard were loaded into corresponding wells, followed by mixing using vortex station. The loaded chip was run in the electrophoresis station for 40 min. Finally, Experion Software was used for data processing. All experiments were repeated at least three times in this study and quantitative variables were expressed as mean±standard deviation (SD) by descriptive statistics.
Flow cytometry analysis of EGFP positive cells- K562 cells transfected with i) pEGFP C1, ii) pEGFP C1+IWT or iii) pEGFP C1+I1- 110 were analysed using flow cytometry analysis. Dead and abnormal cells were excluded using 7AAD staining. The gating was set based on untransfected cells so that the background fluorescence was less than 0.5% EGFP-positive cells. Transfected cells were analysed in terms of fluorescence index (FI). This number was derived by multiplying percentage of EGFP-positive Cells in the subpopulation by median peak fluorescence.
For each electroporation, 0.5 ml of final cell suspension (5 x 106 cells) was transferred to 0.4 cm cuvettes and mixed with 10 μg of DNA. Electro-poration was performed on the Gene Pulser (Bio-Rad, Hercules, California) using the following conditions 250 V, 950 μF, ∞ Ω. Electroporated cells were resuspended in 10 ml DMEM supplemented with 10% foetal calf serum (FCS), penicillin (100 U/ml) and streptomycin (100 μg/ml) and incubated at 37 °C for 24 or 48 hr.
Results
Assessment of splicing in EGFP assay system
Following 24 hr to 72 hr at 37°C, electroporated cells were analysed using flow cytometry and RT-PCR analysis.
Flow cytometric analysis
Transfected cells were analysed in terms of the FI. The FI is derived by multiplying the percentage of EGFP positive cells by the median peak fluorescence. K562 cells transiently transfected with the pEGFP C1+ Iwt yielded an equivalent FI of 302 ± 47 equivalent to that of the control pEGFP C1 (330 ± 60 FI). This result indicated that wild-type β-globin intron I can be spliced out even though it is located in an unnatural sequence context. K562 cells transiently transfected with pEGFP C1+ I1-110 exhibited a much-reduced FI (72 ± 35 FI) relative to pEGFP C1 and pEGFP C1+ Iwt suggesting that the IVSI-110 mutation adversely interfered with the splicing of intron I (Figure 2 and Figure 3).
Figure 2.

Representative flowcytometric analysis of EGFP expression in splicing assays system. Panel A corresponds to K562 cells transfected with pEGFP C1, panel B corresponds to K562 cells transfected with pEGFP C1+ Iwt and panel C corresponds to K562 cells transfected with pEGFP C1+ I1-110
Figure 3.
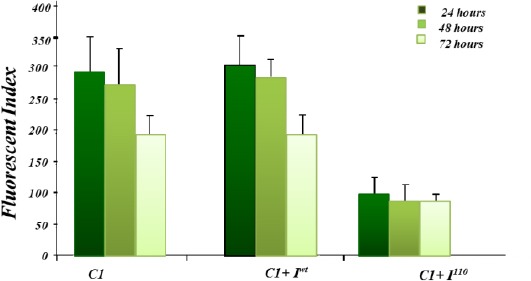
Comparison of FI values following transient transfection of EGFP plasmids. pEGFP C1, pEGFP C1+ Iwt and pEGFP C1+ I 1-110in K562 cells. Multiplying the percentage of EGFP positive cells with median peak fluorescence was used to derive FI. The FI values following transfection were calculated at 24 hrs, 48 hrs and 72 hr (Results represent the average of three independent experiments ± SD)
RT-PCR analysis
To obtain direct evidence that the reduction in EGFP fluorescence was due to aberrant splicing, total cellular RNA was extracted and treated with DNase I enzyme prior to RT-PCR analysis. RNA was extracted from K562 cells transfected with pEGFP C1, pEGFP C1+ Iwt or pEGFP C1+ I1-110 after 24 hr incubation. RT-PCR analysis was performed on 1 μg of total RNA using PCR primers (Fwd E 1-110 and Rev E 1-110), which amplified the correctly and aberrantly spliced EGFP mRNA spanning the 3’end of EGFP pseudo exon 1 to the 5’end of EGFP pseudo exon 2 regions (Figure 1). The RT-PCR products were analysed by agarose gel (Figure 4) and capillary electrophoresis (Figure 4 and Figure 5).
Figure 4.

RT-PCR analysis of the EGFP assay constructs. Lane 1, 2 and 3 correspond to RT-PCR analysis on pEGFP C1, pEGFP C1+ Iwt and pEGFP C1+ I1-110, respectively. Lane M corresponds to molecular marker VIII (Roche). Lower panel illustrates the capillary electrophoresis of the corresponding lanes
Figure 5.

Analysis of RT-PCR products by capillary electro-phoresis following transient transfection of EGFP plasmids into K562 cells. A) pEGFP C1. B) pEGFP C1 + Iwt, C) pEGFP C1+ I1-110. Peaks “a” and “f” correspond to lower (15 bp) and upper (1500 bp) internal markers in capillary electrophoresis. Peak “b” corresponds to β-actin. Peak “c” corresponds the correctly spliced product (140 bp). Peak “d” in panel C corresponds to the aberrantly spliced product (159 bp). Percentages (19.6% and 80.4%) in panel C correspond to correctly and aberrantly product, respectively. Peak “e” in panel B and C indicate to pre mRNA in pEGFP C1 + Iwt and pEGFP C1+ I1-110 (270 bp)
In cells transfected with pEGFP C1 and pEGFP C1+ Iwt, one peak of 140 bp in size was detected, which indicates that intron I was removed from the EGFP cassette through splicing process (Figure 5-A and B). In K562 cells transfected with pEGFP C1+ I1-110, two RT-PCR amplification products were detected; a low peak of 140 bp in size, corresponding to correctly spliced mRNA and a larger peak of 159 bp corresponding to aberrantly spliced transcript (as expected +19 nt large than wild type). Interestingly, the correctly spliced product represented 20% of the total EGFP (Figure 5-C). This result further demonstrated that the mutated IVSI-110 intron is aberrantly spliced to a similar extent observed in its natural counterpart.
Using agarose gel electrophoresis and capillary electrophoresis, another peak with the size of 270 bp that was not present in pEGFP C1 transfected cells, was identified in pEGFP C1 + Iwt and pEGFP C1+ I1-110 (Figure 4 and Figure 7). The 270 bp peak corresponded to pre-mRNA containing intron I of β-globin, which was not evident in pEGFP C1 transfected cells, since the EGFP cassette in pEGFP C1 does not contain an intron.
Figure 6.
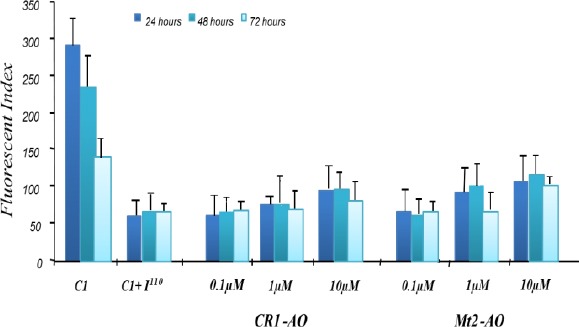
Time and dose-dependent effect of antisense oligonucleotide (AO) treatment on fluorescent index (FI) with pEGFP C1 +I1-110 transfected cells. EGFP activity in K562 cells co-transiently transfected with AOs and pEGFP C1 + IIVSI-110 plasmid (n=2). The activity of EGFP was normalized to total cellular EGFP fluorescence and is presented as FI
Figure 7.

Time dependent effect of antisense oligonucleotide (AO) treatment on fluorescent index (FI) in pEGFP C1 +Iwt transfected cells. EGFP activity in K562 cells co-transiently transfected with AOs and pEGFP + Iwt plasmid. The activity of EGFP was normalized to total cellular EGFP fluorescence and is presented as FI. This figure shows that CR1-AO and Mt2-AO did not influence EGFP expression in cells co-transfected with pEGFP C1 + Iwt plasmid
AO therapy in pEGFP C1+ I1-110
In order to validate the EGPF-based cellular splicing assay system, K562 cells were transiently co-transfected by electroporation with pEGFP C1+ I1-110 plasmid and 2’-O-Me phosphorothioate AOs targeted to the branch point and the IVSI-110 mutant splice.
In this study, two AOs, Mt2-AO and CR1-AO, were tested. Mt2-AO is directed to the mutant 3’ splice site and was found previously (unpublished data) to restore correct splicing in erythroid progenitor cells derived from the IVSI-110 β-thalassemia mouse model. CR1-AO was designed based on the AO target sequence, which was previously reported to inhibited aberrant splicing of IVSI-110 pre-mRNA (13). Flow cytometry analysis of cells co-transfected with pEGFP C1+ I1-110 and Mt2-AO showed an increase in EGFP expression in an AO dose-dependent manner. Mt2-AO, at 0.1, 1 and 10 μM was found to increase EGFP expression to 1.1, 1.5 and 1.8-fold, respectively relative to untreated cells transfected with pEGFP C1+ I1-110 (Figure 6).
Flow cytometry analysis of cells co-transfected with pEGFP C1+ I1-110 and CR1-AO showed an increase in EGFP expression in an AO dose-dependent manner. CR1-AO concentrations at 0.1, 1 and 10 μM resulted in a 1, 1.2 and 1.86 fold increase in EGFP expression, respectively relative to untreated cells (Figure 6). While co-transfection of pEGFP C1+ Iwt and CR1-AO showed no increase in the EGFP level at 10 μM.
When Mt2-AO or CR1-AO were co-transfected with pEGFP C1+ Iwt, no increase in EGFP expression was observed indicating that the increase in EGFP expression was due to a sequence-specific restoration of aberrant splicing and increase in correctly spliced EGFP mRNA (Figure 7). These results indicate that the effect of CR1-AO on the pEGFP C1+ I1-110 appeared to be a sequence-specific interaction resulting in an increase in normal splicing as measured by increase of EGFP.
However, the RT-PCR results following co- transfection with pEGFP C1+ I1-110 and AOs did not correlate completely with the flowcytometric analysis (Figure 6 and Figure 8). It was found that in cells co-transfected with pEGFP C1+ I1-110 and Mt2-AO at 1 μM, the total amount of the normally spliced EGFP mRNA increased from 18% to 30% of total, while at 10 μM Mt2-AO concentration the total amount of correctly spliced product appeared to decrease relative to untreated cells (Figure 8). Co-transfection of CR1-AO also showed an increase of approximately 6% in correctly spliced product compared to untreated cells. Higher concentration of both AOs was not effective in the restoration of normal splicing. Mt2-AO at 10 μM showed decrease in relative levels of the normal splice product, while CR2-AO at this concentration had no effect (Figure 8).
Figure 8.

Dose-dependent effects of antisense oligonucleotide (AO) treatment on pEGFP C1 +I1-110 transfected cells. (A) RT-PCR analysis on K562 cells co-transiently transfected with AOs and pEGFP C1 + IIVSI-110 plasmid. Lane 1-3 correspond to untreated and pEGFP C1 and pEGFP C1 + Iwt, pEGFP C1 + IIVSI-110, respectively. Lanes 4-5 correspond to cells co-transfected with pEGFP C1 + IIVSI-110 and Mt2-AOs at 0.1, 1 and 10 μM, respectively. Lanes 6-9 correspond to cells pEGFP C1 + IIVSI-110 co-transfected with CR1-AOs at 0.1, 1 and 10 μM, respectively. Lane 10 and 11 correspond to pEGFP C1 + Iwt co-transfected with Mt2-AO and CR1-AO at 10 μM concentration, respectively. (B) Relative amounts between normal (blue) and aberrant (red) splice RT-PCR products following AO therapy. Using Mt2-AO and CR2-AO at 1 μM resulted in 10% and 6% increase in normal splicing, respectively
Discussion
The development of effective means to evaluate AOs effect on the intended intracellular target has been hampered in part by the lack of a simple, rapid, and reliable way to evaluate AO efficiency. An assay system that provides clear, sequence-specific and sensitive read-out system would be very useful in identification and evaluation of AOs intended to redirect splicing (5, 6). In this study, an EGFP IVSI-110-specific cellular reporter assay system (EGFP-110) was developed to facilitate the in vitro selection of AOs. This assay was devised and exploited in this laboratory in order to analyse the potential antisense effects of several AOs in cell culture. The assay utilizes the coding sequence of the EGFP reporter, which is interrupted by a wild type, or mutated intron I derived from huβ-globin gene. The mutated intron contains the common IVSI-110 β-thalassemic mutation that creates an aberrant 3’ splice site 19 nt upstream from the intact natural acceptor site (8). In this splicing assay system, K562 cells were transiently transfected with an EGFP expression vector, which contains the IVSI-110 sequence that induces aberrant splicing. The resultant aberrant splicing significantly reduced the production of correct EGFP mRNA and protein as measured by RT-PCR and flow cytometry. In the absence of AO treatment, 17-30% of EGFP protein is produced as measured by the FI value. However, upon delivery of the appropriate AO, functional EGFP is then produced presumably because aberrant splicing is corrected in a dose-dependent manner and functional EGFP is then produced (Figure 6). The AO treatment did not affect the expression of EGFP when they co-transfected with pEGFP C1 + Iwt and pEGFP C1. The attraction of this procedure is that it scores positive events, rather than simple inhibition of gene expression, and thus should be less susceptible to non-AO artifacts.
This system tests sequence-specific antisense activity with the generated fluorescence being proportional to the effectiveness of the antisense molecules in splice shifting. A positive readout confirms that the antisense molecule was able to cross the plasma and nuclear membranes, bind to its correct target pre-mRNA in the nucleus and interfere with the normal functioning of the spliceosome in a sequence specific manner. The results can be quantified at the mRNA or protein level by fluorescence-activated cell sorting (FACS) analysis of cultured cells. The assay allows comparison of effectiveness of AO with different sequence and can be used for optimization of AO delivery. In theory, this approach should allow separate assessment of the relative contributions of uptake versus hybridization to the target sequence.
Previously, cellular splicing assay had been used for IVS2-705 beta-globin mutation in which a recombinant plasmid (pLuc/705) carrying the luciferase gene interrupted by a mutated huβ-globin gene intron II (IVS2-705). This HeLa pLuc/705 cell culture model was used to investigate putative activity of 2’-O-methyl-oligoribonucleotide to treat β-thalassemia (14). The investigators showed that their system would be ideal in assessing the cellular delivery efficiency of AOs. This research group in 2001 used an EGFP-based cellular assay system to identify and evaluate AO targets for blocking aberrant splicing of IVS2-654 huβ-globin mRNA (5). Evaluation of EGFP expression from this construct in both stably transfected HeLa cell lines and in transgenic mice (ubiquitously expressing the EGFP-654 construct) (6) showed increased EGFP expression by AO directed to the aberrant 5’ splice site at position 654 in the intron.
Using this assay system, three concentrations of each AO was tested for their effect on splicing in the ex vivo murine erythroid culture system. Using RT-PCR analysis, it was found that 0.1 μM of AO was not sufficient to restore normal splicing, while 1 μM of AO was effective in restoring normal splicing. In 2008, peripheral blood mononuclear cells derived from 10 Egyptian thalassemic patients with IVSI-110 mutation were treated with 20 micromol/ml morpholino AO targeted against the 3’ aberrant splice site. In cultured erythroid cells derived from 5 patients, they showed correction following AO treatment. The sequence of used AO was similar to the Mt2 AO(15).
However, when using higher concentrations of AO (10 μM CR1-AO and Mt2-AO), a decrease in normal splice was observed. This was in contrast to the increased level of EGFP expression measured by flow cytometry. At this stage, we can only hypothesize that the decrease in normal spliced product may be attributed to non-specific effects as has previously been reported by Kole et al (14). However, the increased level of EGFP expression in spite of reduced level of correctly splice mRNA represents an unexpected result, which will require further investigation.
Using this assay, the target sequence for restoration of IVSI-110 mutation with 2’-O-Me AOs was further investigated. Mt2-AO, which was found to restore normal splicing in vitro using erythroid progenitor cells derived from the IVSI-110 mouse model, was demonstrated to restore normal splicing in the EGFP-110 cellular assay to a similar extent. Moreover, this assay system showed that targeting the cryptic splice with CR2-AO was also able to restore normal splicing, which was found to be ineffective at restoring normal splicing using the erythroid culture system (16, 17).
Conclusion
These results indicate that CR1-AO and Mt2-AO were able to restore correct splicing of EGFP containing the IVSI-110 intron; however, we could not exclude the possibility of non-specific activation of EGFP gene expression due to other mechanisms. While these differences were not further investigated due to time constraints, it suggests that the artificial EGFP-110 assay system may not parallel the native splicing process and clearly will be the subject of further investigations.
Acknowledgment
This study was funded by Ministry of Health and Medical Education of Iran. The results described in this paper were part of Ph.D. student thesis.
Conflict of interest
The authors declare that they have no conflict of interests in this paper.
References
- 1.Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol Ther J Am Soc Gene Ther. 2007;15:1288–1296. doi: 10.1038/sj.mt.6300095. [DOI] [PubMed] [Google Scholar]
- 2.Sazani P, Astriab-Fischer A, Kole R. Effects of base modifications on antisense properties of 2’-O-methoxyethyl and PNA oligonucleotides. Antisense Nucleic Acid Drug Dev. 2003c;13:119–128. doi: 10.1089/108729003768247583. [DOI] [PubMed] [Google Scholar]
- 3.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleo-tides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andronescu M, Zhang ZC, Condon A. Secondary structure prediction of interacting RNA molecules. J Mol Biol. 2005;345:987–1001. doi: 10.1016/j.jmb.2004.10.082. [DOI] [PubMed] [Google Scholar]
- 5.Sazani P, Kang SH, Maier MA, Wei C, Dillman J, Summerton J, et al. Nuclear antisense effects of neutral, anionic and cationic oligonucleotide analogs. Nucleic Acids Res. 2001;29:3965–3974. doi: 10.1093/nar/29.19.3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sazani P, Gemignani F, Kang SH, Maier MA, Manoharan M, Persmark M, et al. Systemically delivered antisense oligomers upregulate gene expression in mouse tissues. Nat Biotechnol. 2002;20:1228–1233. doi: 10.1038/nbt759. [DOI] [PubMed] [Google Scholar]
- 7.Vadolas J, Nefedov M, Wardan H, Mansoori-derakshan S, Voullaire L, Jamsai D, et al. Humanized beta-thalassemia mouse model containing the common IVSI-110 splicing mutation. J Biol Chem. 2006;281:7399–7405. doi: 10.1074/jbc.M512931200. [DOI] [PubMed] [Google Scholar]
- 8.Spritz RA, Jagadeeswaran P, Choudary PV, Biro PA, Elder JT, deRiel JK, et al. Base substitution in an intervening sequence of a beta+-thalassemic human globin gene. Proc Natl Acad Sci U S A. 1981;78:2455–249. doi: 10.1073/pnas.78.4.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popplewell LJ, Trollet C, Dickson G, Graham IR. Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol Ther. 2009;17:554–561. doi: 10.1038/mt.2008.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majlessi M, Nelson NC, Becker MM. Advantages of 2’-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998;26:2224–2229. doi: 10.1093/nar/26.9.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamm GM, Blencowe BJ, Sproat BS, Iribarren AM, Ryder U, Lamond AI. Antisense probes containing 2-aminoadenosine allow efficient depletion of U5 snRNP from HeLa splicing extracts. Nucleic Acids Res. 1991;19:3193–3198. doi: 10.1093/nar/19.12.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bharti Bhandari DC, Neeta Wardhan. Antisense oligonucleotide: basic concept and its therapeutic application. J Res Pharm Sci. 2014;2:01–13. [Google Scholar]
- 13.Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleo-tides. Proc Natl Acad Sci U S A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang SH, Cho MJ, Kole R. Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry. 1998;37:6235–6239. doi: 10.1021/bi980300h. [DOI] [PubMed] [Google Scholar]
- 15.El-Beshlawy A, Mostafa A, Youssry I, Gabr H, Mansour IM, El-Tablawy M, et al. Correction of aberrant pre-mRNA splicing by antisense oligonucleotides in beta-thalassemia Egyptian patients with IVSI-110 mutation. J Pediatr Hematol Oncol. 2008;30:281–284. doi: 10.1097/MPH.0b013e3181639afe. [DOI] [PubMed] [Google Scholar]
- 16.Suwanmanee T, Sierakowska H, Lacerra G, Svasti S, Kirby S, Walsh CE, et al. Restoration of human beta-globin gene expression in murine and human IVS2-654 thalassemic erythroid cells by free uptake of antisense oligonucleotides. Mol Pharmacol. 2002;62:545–553. doi: 10.1124/mol.62.3.545. [DOI] [PubMed] [Google Scholar]
- 17.Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, Weller D, et al. Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Proc Natl Acad Sci U S A. 2000;97:9591–9596. doi: 10.1073/pnas.97.17.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]