

Abstract
The eukaryotic global genomic nucleotide excision repair (GG-NER) pathway is the major mechanism that removes most bulky and some nonbulky lesions from cellular DNA. There is growing evidence that certain DNA lesions are repaired slowly or are entirely resistant to repair in cells, tissues, and in cell extract model assay systems. It is well established that the eukaryotic DNA lesion-sensing proteins do not detect the damaged nucleotide, but recognize the distortions/destabilizations in the native DNA structure caused by the damaged nucleotides. In this article, the nature of the structural features of certain bulky DNA lesions that render them resistant to NER, or cause them to be repaired slowly, is compared to that of those that are good-to-excellent NER substrates. Understanding the structural features that distinguish NER-resistant DNA lesions from good NER substrates may be useful for interpreting the biological significance of biomarkers of exposure of human populations to genotoxic environmental chemicals. NER-resistant lesions can survive to replication and cause mutations that can initiate cancer and other diseases. Furthermore, NER diminishes the efficacy of certain chemotherapeutic drugs, and the design of more potent pharmaceuticals that resist repair can be advanced through a better understanding of the structural properties of DNA lesions that engender repair-resistance.
1. Introduction
The genome is continuously exposed to exogenous and endogenous genotoxic agents that generate different forms of DNA damage including DNA strand breaks, chemically modified nucleobases, and intrastrand and interstrand DNA cross-links that are collectively referred to as DNA lesions. Mammalian cells have developed repair pathways that remove the DNA damage and regenerate the intact DNA sequence. Among the major repair pathways are base excision repair1 (BER), global genomic nucleotide excision repair2,3 (GG-NER), and transcription coupled nucleotide excision repair (TC NER).2,4−6 The BER system removes specific and ubiquitous nonbulky DNA lesions such as 8-oxo-2′-deoxyguanosine (8-oxoG) and restores the original DNA by a series of exquisitely orchestrated steps that include the recognition of the damaged bases, their removal, and reinsertion of the correct nucleotides using the undamaged bases in the complementary strand as templates. By contrast, the nucleotide excision repair system can repair a large variety of different forms of DNA damage because it recognizes the local DNA distortions/destabilizations caused by the DNA lesion rather than the chemically modified base itself.7,8 The NER process involves ∼30 different proteins and a series of coordinated steps that excise a 24–32 nucleotide sequence that contains the damaged nucleotide, followed by the DNA polymerase-catalyzed resynthesis of the missing sequence using the unmodified complementary strand as the template.2,9
The two subpathways of NER, GG-NER and TC-NER, differ from one another in the sensing and recognition of the DNA lesions. In the case of mammalian GG-NER, the DNA distortions and thermodynamic destabilization caused by the damaged nucleotide(s) are sensed by the heterodimeric XPC-RAD23B protein (abbreviated as XPC below) that binds noncovalently to the damaged site5,10 and then recruits the subsequent NER factors that lead to the characteristic ladder of excised oligonucleotide sequences.11,12 In the case of TC-NER, the DNA damage in the transcribed strand is recognized by the stalling of the RNA polymerase RNAP II, an event that leads to the subsequent recruitment of the same NER factors as in the GG-NER XPC pathway.3,4 Thus, the same ladders of dual incision and excision fragments are observable after successful GG- and TC-NER that are the hallmarks of NER activity. The TC-NER mechanism occurs only on the transcribed strand of an active gene4 and thus affects only a small fraction of DNA lesions, while most of the lesions in genomic DNA are repaired by the GG-NER mechanism.
While it has been known for a long time that chemically and structurally different forms of nucleobase damage are repaired by the NER system with variable efficiencies,13 some nonbulky DNA lesions and certain bulky DNA lesions (traditionally called “DNA adducts”) are entirely resistant to NER. Interest in these phenomena has been growing because repair-resistant forms of DNA damage persist in human tissues, with detrimental consequences for the cells. Indeed, cellular DNA adduct levels have been correlated with levels of mutations in mammalian cells.14,15 The repair-resistant DNA lesions can result in error-prone translesion synthesis, genomic instability, and the initiation of cell proliferation and tumorigenesis. These processes may depend on base sequence context and the reactivity patterns of DNA adduct formation. Thus, human exposure to chemical carcinogens can give rise to characteristic signatures of mutations that can provide information about the nature of the carcinogen that caused the cancer.16−18
Understanding the structural features of repair-resistant DNA lesions can provide novel insights into the molecular basis of their recognition by damage-sensing proteins in a sea of normal, unmodified DNA bases. In this perspective, we focus on the eukaryotic NER pathway, while damage recognition in prokaryotes has been reviewed by Kisker et al.19 A resemblance in the relative efficiencies of dual incisions elicited by the same sets of DNA lesions by prokaryotic and eukaryotic NER systems in vitro has been noted and attributed to common, apparently conserved molecular mechanisms of recognition of DNA damage.20 Here we review our current knowledge of GG-NER-resistant bulky DNA lesions and the structural characteristics that render them poor substrates of NER, in contrast to those that are good-to-excellent human NER substrates. Interstrand cross-linked DNA lesions also pose challenges to cellular repair mechanisms but are not considered here since this topic has been reviewed elsewhere.21
2. Recognition of DNA Damage and Initiation of GG-NER
Exposure of human skin to UV irradiation generates the well-known cyclobutane thymine dimers (T^T CPDs) and the pyrimidine (6–4) pyrimidone photoproducts (Figure 1A and B, respectively). If not repaired, these DNA lesions cause mutations and skin cancers.22 While the (6–4) photoproduct is readily repaired via the human NER pathway, the CPD thymine dimer and other bipyrimidine CPDs are repaired much more slowly than the (6–4) lesions in human skin,23 in cell extracts,24,25 and in cells.26 These premutagenic lesions, if not repaired by NER mechanisms, are the primary causes of skin disorders and melanomas.27 While XPC recognizes and binds strongly to the (6–4) photoproduct, its binding affinity to CPD lesions is very weak, at best.10 Incubation of the (6–4) photoproduct in cell-free extracts yields the characteristic NER dual incision products, while CPD is resistant to NER under the same conditions.10,25 These latter observations are consistent with the XPC binding results. However, in intact cells, the binding of XPC to the CPD lesion is primarily mediated by the DNA damage binding protein DDB2 that is part of the UV-DDB1/2 complex,28−30 although the repair of CPD in intact cells is still significantly slower than the removal of the 6–4 lesions that are directly recognized by XPC.23,26,30 While the thymine dimer T^T CPD opposite its canonical adenine (AA) bases in the complementary strand is NER-resistant, it becomes an excellent substrate of NER when these AA bases are replaced by “mismatched” GG.10 The development of genome-wide methods of analysis of the formation and repair of these UV photolesions in human fibroblasts,31 human lymphocytes,32 and yeast30 genomes has provided novel insights into the base sequence dependence of mutational processes at the single nucleotide level of resolution.
Figure 1.

Structures of UV photodimers: (A) T^T cyclobutane pyrimidine dimer (CPD) and (B) 6–4 UV photoproduct. (C) Co-crystal structure of yeast Rad4-Rad23 with a T^T CPD (not resolved experimentally) opposite two thymines (blue) in the complementary strand (Min and Pavletich7; PDB ID: 2QSG).
2.1. Recognition of DNA Lesions by NER Damage-Sensing Proteins
Valuable insights into the mechanisms of recognition of chemically damaged DNA by the eukaryotic NER system have been obtained from the X-ray crystallographic structure of a truncated form of Rad4-Rad23 (abbreviated as Rad4), the S. cerevisiae homologue of the XPC-RAD23B heterodimer.7 The Rad4 dimer was complexed with an oligonucleotide containing a T^T CPD lesion opposite two mismatched thymine bases in the complementary strand (Figure 1C). One of the three β-hairpin domains, BHD2 (orange in Figure 1C), contacts the minor groove side without penetrating the DNA. A second hairpin, BHD3 (shown in green in Figure 1C), is inserted into the DNA helix from the major groove, thus separating the damaged and the complementary strands at the site of the lesion. The CPD lesion is positioned in a disordered region of the crystal and its coordinates could not be established, and no contacts were evident between CPD and the protein. However, the two mismatched thymines opposite the CPD in the complementary strand are flipped out of the duplex and interact with Rad4 amino acid residues in specific binding pockets. This structure represents the productive open complex that can stimulate the subsequent NER steps, and it reveals that the BHD3 β-hairpin insertion and the concomitant interaction with the protein of the two flipped-out thymine bases in the complementary strand (Figure 1C) are most likely important elements of lesion recognition in eukaryotic NER.7 The mechanistic aspects of the recognition of DNA lesions by Rad4 are of great current interest and are being investigated by temperature-jump perturbation spectroscopy (T-Jump) techniques in combination with fluorescence resonance energy transfer (FRET) methods.33,34 In these studies, a two stage binding mechanism for Rad4 was observed, a first fast step (∼100–500 μs) followed by a slow second step (5–10 ms). A “twist-open” mechanism was proposed to account for the binding of Rad4 to its DNA substrates.34 The first, rapid step involves DNA untwisting, while the slower, second step involves a local separation of the two strands in the DNA duplex and the full flipping of two nucleotides out of the duplex. It was proposed that the rates of these two steps depend on the DNA lesion, and the distortion and local destabilization caused by the lesion.
On the basis of the results of T-jump experiments, it was also suggested that lesion recognition is under kinetic control via a “kinetic gating” mechanism that would allow efficient recognition of DNA-destabilizing lesions.33 Single molecule fluorescence microscopy studies35 have provided further insights into the “kinetic gate”33 and “twist-open”34 lesion recognition steps. The molecular details and associated energetics and pathway of Rad4 binding to the CPD lesion have been investigated by molecular dynamics simulation methods, which provided atomic level views of the “twist-open” mechanism and the free energy profile along the binding path.36 While XPC is a much larger protein than Rad4, the homologies in the DNA binding domains suggest that the human protein adopts similar key elements of recognition of DNA damage as Rad4.33,34
Once the XPC-RAD23B–damaged DNA complex is formed, the ten-protein complex TFIIH11 and other NER factors12,24,37,38 are assembled at the site of the DNA lesion in a sequential manner.10,12,39 The assembly of the TFIIH complex leads to a subsequent, second level of DNA lesion recognition called the verification step. The TFIIH complex contains the helicases XPB and XPD that lead to an ATPase-driven enlargement of the six-base single stranded region, initially caused by XPC, to a 24–32 nucleotide single-stranded region.37 The detailed mechanisms of this verification step are not yet completely understood, but it is widely assumed that the presence of a true DNA lesion is signaled by the stalling of XPD at the site of the lesion.40,41 The recruitment of the endonucleases XPF and XPG to the double-single strand junctions leads to the excision of the 24–32-mer damaged oligonucleotides and the subsequent DNA synthesis step that regenerates the intact DNA sequence.2 Like other forms of DNA damage, DNA lesions, such as those derived from intrastrand cross-linked cisPt12,37,42 and bulky benzo[a]pyrene-derived adducts,43 cause the partial opening and strand separation of a six base-pair sequence containing the lesion site. The local weakening of the DNA duplex facilitates the extrusion of the nucleobases opposite the lesion in the complementary strand and favors the insertion of the BHD3 β-hairpin between the two strands from the major groove side of the DNA.
2.2. Nucleotide Excision versus BER Pathways and Resistance to NER
The general paradigm in the field of DNA repair is that small nonbulky DNA lesions derived from the oxidation of nucleobases in DNA are substrates of BER mechanisms, but not of NER. Well known BER substrates include 8-oxoG that is excised by the BER protein hOGG1 in humans. A very weak NER activity in human cell-free cell extracts associated with hOGG1 has been reported.44 Many other nonbulky DNA lesions are substrates of BER pathways but are resistant to NER. On the other hand, the CPD and the 6–4 UV photoproducts,22 as well as the intranucleotide, cross-linked and oxidatively generated 8,5′-cyclopurine adenine and guanine lesions, are substrates of NER only,45,46 but not of BER.47,48 Interestingly, the oxidatively generated spiroiminodihydantoin and guanidinohydantoin lesions,49 and the intrastrand cross-linked G[C8–N3]T lesions,50 are substrates of both BER and NER in human cell-free extracts. These results indicate that the susceptibility or resistance to NER of nonbulky DNA lesions is dependent on their unique structural features and the kind of distortions to the local B-DNA structure they engender.
The GG-NER mechanism is the most universal and versatile DNA repair mechanism because it recognizes the distortions/destabilizations caused by DNA damage rather than the lesion itself. Some DNA lesions like the CPD UV photoproduct, as well as bulky DNA adducts, are slowly repaired by NER, while some others are completely resistant to NER. Before discussing NER resistance, we discuss the physical characteristics and structural features of bulky forms of DNA damage that affect their response, including resistance, to the human NER apparatus. We first compare the characteristics of site-specifically modified DNA lesions in double-stranded DNA with their excision by the human NER system in human cell extracts. These in vitro systems are suitable for comparing the intrinsic NER efficiencies of different structurally defined bulky and nonbulky forms of DNA damage in defined base sequence contexts. In chromatin, remodeling of the latter is necessary before NER can occur.2 We conclude with a survey of known examples of NER-resistant DNA lesions in mammalian cellular environments.
3. Structural Features of Bulky PAH–DNA Adducts
3.1. Stereoselective Covalent DNA Adduct Formation
Polycyclic aromatic hydrocarbons (PAH) are products of fossil fuel combustion and are therefore ubiquitous, genotoxic contaminants in the environment. Two classes of PAH have been distinguished, the bay region PAH that includes the most extensively studied representative, benzo[a]pyrene,51 and the fjord PAH that includes the most tumorigenic compound known to date (dibenzo[a,l]pyrene)52−54 (Figure 2A).
Figure 2.

(A) Structures and carbon atom numbering systems of bay region benzo[a]pyrene (B[a]P), and fjord region benzo[c]phenanthrene (B[c]Ph), benzo[g]chrysene (B[g]C), and dibenzo[a,l]pyrene (DB[a,l]P). (B) Metabolic activation of PAH by P450 cytochrome and epoxy hydrolases that generate the enantiomeric diol epoxides is shown. The absolute configurations of substituents around the 7,8,9,10 (B[a]P); 4,3,2,1 (B[c]Ph); 11,12,13,14 (DB[a,l]P and B[g]C) of the metabolized aromatic rings, shaded in gray, are also shown.
In human cells, the PAH are metabolically activated by Cytochrome P450 (CYP) enzymes to epoxides that are converted by epoxide hydrolases (EH) to PAH dihydrodiol intermediates (Figure 2B). A second round of epoxidation by CYP leads to stereochemically distinct and tumorigenic55 PAH diol epoxides (Figure 2)56,57 that can react chemically with the exocyclic amino groups of guanine58−61or adenine62−64 in DNA to form stable, stereoisomeric covalent DNA adducts (Figure 3).
Figure 3.

Stereochemistry-dependent conformational motifs of DNA adducts that result from the reactions of B[a]PDE with (A, B) dG, and with (C) dA in double-stranded DNA (see the text for details).
The most genotoxic metabolite of B[a]P is the (+)-7R,8S-dihydrodiol,9S,10R-epoxy-benzo[a]pyrene enantiomer ((+)-7R,8S,9S,10R-anti-B[a]PDE) that is generated with a ∼6-fold greater yield in rat liver microsomes65 than its (−)-7S,8R,9R,10S mirror image enantiomer (−)-anti-B[a]PDE (Figure 2B). The (+) and (−) signs denote the signs of the optical rotatory dispersion signals beyond the UV absorption bands of these compounds, while the anti designation indicates that the 7-OH group and the 9,10-epoxy group are on opposite sides of the planar polycyclic aromatic ring system; since this is the only form of diol epoxides discussed in this article, the prefix “anti” is omitted throughout. These B[a]PDE enantiomers exhibit different mutagenic66 and tumorigenic properties.55
The stereochemical properties of (+)- and (−)-B[a]PDE and their adducts are summarized in Figure 3. The (+)-B[a]PDE and other PAH diol epoxides can react with the exocyclic amino groups of guanine by trans- or cis-addition mechanisms, as illustrated in Figure 3A and B. Similar reaction patterns are observed when the same B[a]PDE enantiomers react and bind with the exocyclic amino group of adenine, but only the major trans-N6–dA products will be discussed here (Figure 3C). The cis-N2-guanine adduct is formed when the exocyclic N2-amino group of dG approaches the C10 carbon atom from the same side as the epoxide group, while in the case of trans addition, the N2–dG and the epoxide groups are on opposite sides. The absolute R and S configurations of the N2–dG adducts at the C10 carbon atom are also indicated in Figures 3 and 4. In mammalian cells, the dominant benzo[a]pyrene-derived DNA product is the (+)-trans-B[a]PDE-N2–dG adduct.60,67,68 The reactions of racemic (±)-B[a]PDE with DNA in vitro in aqueous solutions yield predominantly N2–dG adducts with lesser amounts of N6–dA adducts, and some dC adducts as well.58,59 The (+)-7R,8S,9S,10R B[a]P diol epoxide forms the major proportion of guanine adducts in cellular DNA,67,68 while smaller proportions of DNA adducts derived from the (−)-SRRS-enantiomer are also observed. Direct reactions of (±)-B[a]PDE and other PAH diol epoxides with 2′-deoxyribooligonucleoides in aqueous solution69,70 produce cis- and trans-N2–dG and N6–dA adducts in yields sufficient for structural71 and biochemical studies using site-specifically modified DNA templates.72−76
Figure 4.
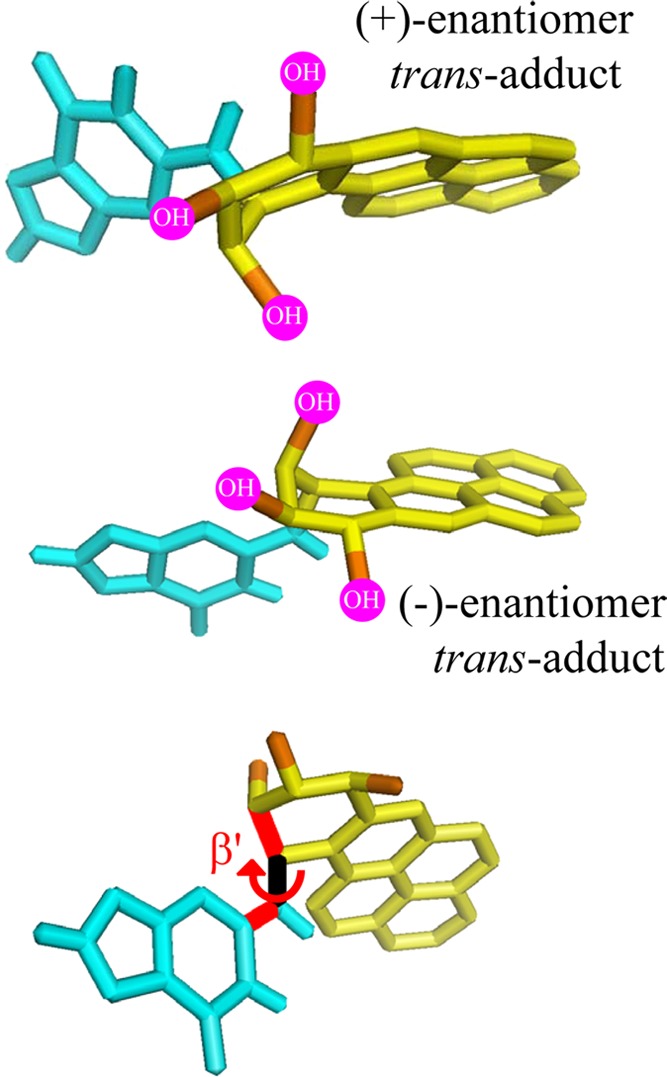
Steric hindrance due to bulky −OH groups limits the allowed values of the torsion angle β′ and thus the conformational space of the bulky polycyclic aromatic residues in the (+)-trans and (−)-trans-B[a]P-N2–dG adducts in double stranded DNA. The two top structures illustrate the principles of opposite orientations of the PAH residues relative to the planes of dG that result from the binding of enantiomeric PAH diol epoxides to the exocyclic amino groups of purines in DNA. The bottom structure designates the torsion angle β′.
The structural features of a variety of stereochemically and structurally distinct PAH diol epoxide-derived DNA adducts have been reviewed.71 Additional results have been published since then.77−80 Other significant contributions to our understanding of the structures of PAH-derived DNA adducts include those from the Stone laboratory81−89 as well as by Jerina and collaborators.90−94 Other types of bulky DNA adducts that have been structurally characterized include those derived from 2-aminofluorenes (AF) and N-acetylaminofluorene (AAF),95−103 2-aminopyrene,104 fluorene-labeled AF and AAF,105−108 heterocyclic aromatic amines,109−112 and other bulky and nonbulky DNA lesions described below.113,114 Collectively, these publications represent a rich source of information on the conformational features of structurally diverse forms of bulky DNA adducts for studying the relationships between their properties and their impact on biological phenomena.115
3.2. Conformational Motifs of Bulky DNA Adducts
The conformational and stereochemical features of polycyclic aromatic diol epoxide-derived DNA adducts can exert a significant impact on their excision by the human NER system.116 Therefore, the structural features of these forms of DNA damage and their relationships to DNA repair are of considerable interest. Examples of such structural studies are summarized in this section.
The effects of stereochemistry on the conformations of B[a]PDE-N2–dG adducts (G*) have been most extensively studied in the 11-mer duplex 5′-d(CCATCG*CTACC)·d(GGTAGCGATGG) (duplex I) by NMR methods.71 The different conformational themes can be divided into several broad classes.
3.2.1. Minor Groove Conformations with Minimal Perturbations of Base Pairing
The reactions of (+)- and (−)-B[a]PDE by trans-addition to the exocyclic amino group of guanine give rise to stereoisomeric 10S (+)-trans-B[a]PDE-N2–dG and 10R (−)-trans-B[a]PDE-N2–dG adducts, respectively, in oligonucleotide duplexes I. These 10R and 10S stereoisomeric adducts are characterized by opposite absolute configurations at the N2–dG linkage sites. The planar aromatic ring systems reside in the minor groove of these duplexes pointing either toward the 5′-(10S adduct) or the 3′-end (10R adduct) of the modified strands117,118 as shown in Figure 3A. These solution NMR structures illustrate the important principle that enantiomeric pairs of PAH diol epoxides form trans (or cis) adducts with purines in DNA that adopt opposite orientations relative to the modified bases. The conformations of these bulky adducts in the crowded DNA environment are governed by the absolute configurations of the hydroxyl groups119,120 and by the relative orientations of the planar bulky B[a]PDE aromatic residue relative to the guanine ring system. This orientation is characterized by the torsion angle β′121 shown in Figure 4. A survey of sterically allowed values of β′ and the sets of torsion angles χ and α′ (the glycosidic and the C2-N2 torsion angles, respectively) revealed four low potential energy domains, each with a limited range of allowed β′ values.71,120 The experimentally observed β′ values of the (+)-trans- and the (−)-trans-B[a]PDE-N2–dG adducts117,118 are found within the lowest potential energy domains identified by Xie et al.120 The preferred domains are the least crowded for each stereoisomeric adduct and explain the opposite orientation phenomenon (Figure 4), which has proved to be a principle that is generally followed based on many examples.71
3.2.2. Base-Displaced Intercalation
Examples are the guanine adducts derived from the reactions of (+)- and (−)-B[a]PDE to the exocyclic amino groups of guanine by cis-addition (Figure 3B).122,123 The planar aromatic ring systems of the (+)- and (−)-cis-B[a]PDE-N2–dG adducts are inserted between neighboring base pairs (intercalation) and displace both the modified and partner bases out of the double helix.
3.2.3. Intercalation of N2–dG Adducts from Minor Groove without Base Displacement
The diol epoxide (−)-11R,12S,13S,14R DB[a,l]PDE derived from the metabolic activation of DB[a,l]P) has the same absolute configurations as the (+)-7R,8S,9S,10R-B[a]PDE enantiomer. The reaction of this (−)-11R,12S,13S,14R DB[a,l]PDE enantiomer with N2–dG yields the 14S (−)-trans-DB[a,l]PDE-N2–dG adduct (dG*) that is positioned in the minor groove pointing toward the 5′-direction of the modified strand (Figure 5).124 This minor groove conformation is related to the one characterizing the stereochemically analogous 10S (+)-trans-B[a]PDE-N2–dG adduct (Figure 3A); however, it causes a much greater distortion of the minor groove and a rupture of the modified dG*:dC base pair because of the greater bulk of the aromatic DB[a,l]PDE residue.
Figure 5.
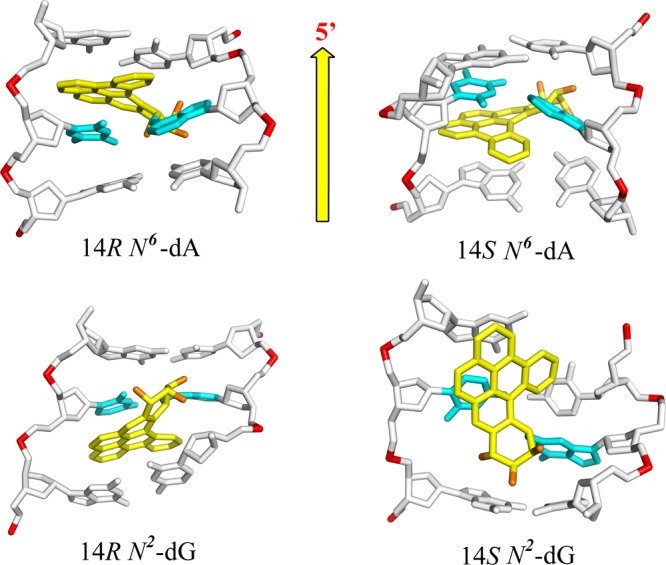
Conformations of DB[a,l]PDE-N6–dA (top) and DB[a,l]PDE-N2–dG (bottom) adducts. 14R-N6–dA: intercalated on the 5′-side of the intact dA*–dT base pair from the major groove without base displacement; 14S-N6–dA: same, but intercalated on the 3′-side of dA*–dT. 14R-N2–dG: intercalated from the minor groove on the 3′-side of the disrupted dG*–dC base pair. 14S-N2–dG: the DB[a,l]PDE residue is positioned in a distorted and widened minor groove on the 5′-side of the dG*–dC base pair.
The (+)-11S,12R,13R,14S DB[a,l]PDE enantiomer generates the 14R (+)-trans-DB[a,l]PDE-N2–dG adduct that has the same absolute configuration as the 10R (−)-trans-B[a]PDE-N2–dG adduct. While the latter assumes a minor groove conformation in double-stranded DNA, the 14R (+)-trans-DB[a,l]PDE-N2–dG adduct is intercalated from the minor groove between two adjacent base pairs (Figure 5); furthermore, the larger size and additional ring in the fjord region of the covalently attached DB[a,l]PDE residue, as well as its intercalation from the narrow minor groove, cause the disruption of the modified dG*–dC base pair; by contrast, in the less bulky minor groove 10R B[a]PDE-N2–dG adduct, the analogous base pair remains intact and the B[a]P ring system is positioned in the minor groove (Figure 3B). These examples illustrate how the size and shape of the polycyclic aromatic ring system can give rise to different conformations of DNA adducts and hence the degree of distortion/destabilization of the DNA duplexes. However, in all cases, regardless of conformation, the N2–dG adducts with S absolute configuration at the linkage sites are oriented on the 5′-side of the modified guanine dG*, while adducts with R-stereochemistry are oriented on the 3′-side of dG* (Figure 5).
3.2.4. Intercalation from Major Groove without Base Displacement
The exocyclic amino group of adenine (N6) is positioned on the major groove side of B DNA. All PAH diol epoxides that have been studied up until now bind covalently to N6–dA via the intercalative insertion of their aromatic ring systems between adjacent base pairs without base displacement. The 10S (+)- and 10R (−)-B[a]PDE-N6–dA adducts (A*)81,90,92−94,125−127 are examples of this intercalation motif (Figure 3C). However, the (+)-10S A* adduct is conformationally more flexible than the (−)-10R A* adduct, and the A–T Watson–Crick base pair at the lesion site is destabilized. Other examples of intercalation without base displacement include the fjord B[c]PhDE-N6–dA,128,129 B[g]CDE-N6–dA,130 and DB[a,l]PDE-N6–dA adducts in double-stranded DNA. Our molecular dynamics simulation studies131 suggest that the bulky aromatic residues of the 14R and 14S DB[a,l]PDE-N6–dA adducts are intercalated on the 5′- and 3′-sides of dA*, respectively, without disrupting adjacent base pairs and with significant, stabilizing π–π base stacking interactions (Figure 5).
3.2.5. Thermodynamic Destabilization of Modified DNA Duplexes
As discussed in section 2.1, the recognition of DNA lesions by XPC most likely involves the insertion of a β-hairpin into the duplex at the site of the lesion and the flipping out of two bases on the complementary, unmodified strand (Figure 1).7 These phenomena are facilitated when DNA damage destabilizes the duplex, and it is therefore of interest to examine the impact of different DNA lesions on the stabilities of the modified DNA duplexes. The destabilizing effects of DNA lesions are easily determined by measuring the thermal dissociation temperatures of relatively short duplexes, typically 11-mer duplexes under standard solution conditions.132 However, longer, 43-mer duplexes can also be employed.133 The experimentally measured duplex melting points, Tm, are defined as the temperatures at which 50% of the DNA strands are in the double-stranded form. The degree of destabilization caused by the DNA lesions is characterized by the difference in duplex melting points, ΔTm = Tm(modified duplex) – Tm(unmodified duplex).
4. Excision of Different Forms of DNA Damage by Human NER System Is Highly Variable
The relationships between the structural features of chemically modified nucleobases and their removal by the NER apparatus are best explored using structurally well-defined DNA modifications and a reliable and reproducible assay of NER activities. Analyses of structural features of many modified nucleobases by solution NMR methods71,82,98,108,124,125,134−137 have yielded a wealth of information on their conformations and the distortions of the local DNA structure that they cause.
There is a significant body of evidence that different forms of DNA damage are removed from mammalian cells and tissues with variable efficiencies.13 The critical recognition and dual incision steps can be determined by monitoring the relative yields of dual incision products. The subsequent steps of repair are the resynthesis of the missing nucleotides in the gap, which is no longer dependent on the nature of the lesion. Therefore, the relative NER efficiencies can be measured in cell extracts by determining the yields of dual incision products as a function of time.
4.1. Nucleotide Excision Repair Assays
The response of different DNA lesions to the human NER apparatus in vitro is assessed by incubating site-specifically modified and internally 32P-labeled oligo-2′-deoxyribonucleotide duplexes (typically 135–147 base pairs in length) that harbor a single chemically defined base in cell-free extracts from human cells.138 Such cell-free extracts are known to contain the complement of active NER proteins that can excise the 24–32-mer oligonucleotide sequences that contain the lesions. The yields of NER dual 24–32-mer incision products are evaluated by polyacrylamide gel electrophoresis and densitometric analysis of the gel autoradiographs. However, extracts from human cells prepared from different batches of cells at different times exhibit variable NER activities. To enhance reproducibility, we utilize the NER response of one particular adduct, normally the (+)-cis-B[a]PDE-N2–dG adduct that is 32P-labeled at the same time as the DNA adduct sample being studied, as a standard against which the relative yields of other NER substrates are measured in one and the same cell extract. This (+)-cis-B[a]PDE-N2–dG adduct exhibits approximately the same NER response as the 6–4-thymine dimer UV photoproduct, and the dG–C8-AAF adduct derived from the binding of N-acetylaminofluorene (AAF) to the C8-position of guanine.139,140 In this manner, good reproducibility is obtained that allows for comparisons of relative NER responses measured at different times and in different cell-free extracts.
4.2. NER Efficiencies Depend on Structural Features of DNA Lesions
A long-recognized example of the variability of NER efficiencies of DNA lesions is the difference in excision activities of the UV light-induced cyclobutane pyrimidine dimers (CPDs) that are very poor substrates of NER in cells,141 in cell extracts,10 and in human skin,23 while the (6−4) pyrimidine–pyrimidone photoproducts are efficiently excised under identical conditions. The effects of PAH diol epoxide–DNA adduct stereochemistry on NER dual incision efficiencies are striking manifestations of structure–function relationships. In this section, the structural features of bulky DNA adducts that favor efficient NER or that characterize NER-resistant adducts are described.
4.2.1. B[a]PDE-N2–dG Adducts in Sequence Context of Duplex I
The base-displaced intercalated (+)-cis- and (−)-cis-B[a]PDE-N2–dG adducts are among the most efficiently incised in human cell extracts (assigned NER efficiency of 100%) (Table 1), while the relative efficiencies of excision of the minor groove (+)-trans- and (−)-trans-B[a]PDE-N2–dG adducts are ∼5-times lower43,142 in the same sequence context (Figure 6); however, sequence context has a strong impact on relative efficiencies of excision in the case of the (+)-trans adduct, as detailed in section 4.2.2.
Table 1. Comparisons of NER Efficiencies in Human Cell Extracts of Different DNA Lesions Embedded in the Same 135-mer Duplexes with B[a]PDE- or DB[a,l]PDE-N2–dG/-N6–dA Adducts (G* or A*, Respectively)a.
| designation (text section) | sequence context in 135-mer duplexes | adduct (G*) B[a]PDE-N2–dG | adduct conformation | % NER efficiency |
|---|---|---|---|---|
| full duplex (4.2.1) | (CCATCG*CTACC)·(GGTAGC GATGG) (I-G*) | (+)/(−)-cis 10R/10S | base displaced intercalation | 100 |
| (+)/(−)-trans 10S/10R | minor groove | 20 ± 3 | ||
| deletion (Del) duplex (4.3.2) | (CCATCG*CTACC)·(GGTAG—GATGG) (G*:Del) | (+)-cis 10R | base displaced intercalation | ≤2 |
| (+)-trans 10S | base displaced intercalation | ≤2 | ||
| abasic (AB) duplex (4.3.2) | (CCATC G*CTACC)·(GGTAG[AB]GATGG) (G*:AB = THF) | (+)-cis 10R | base displaced intercalation | ≤3 |
| (+)-trans 10S | base displaced intercalation | ≤3 | ||
| mismatch (MM) duplex (4.3.3) | (CCATCG*CTACC)·(GGTAGXGATGG) | 10R (+)-cis X = G,A | base displaced intercalation | ≤3 |
| adduct (G*) DB[a,l]PDE-N2–dG | ||||
| full duplex (4.4.4) | (CCATCG*CTACC)·(GGTAGCGATGG) (I-G*) | (+)-trans 14R | intercalation, no base displacement | 65 ± 7 |
| (4.4.5) | (CCATCG*CTACC)·( GGTAGCGATGG) | (−)-trans 14S | minor groove | 19 ± 3 |
| adduct (A*) DB[a,l]PDE-N6–dA | ||||
| full duplex (4.4.6) | (CCATCA*CTACC)·(GGTAGTGATGG) | (+)-trans 14R | intercalation, no base displacement | <2 |
| full duplex (4.4.6) | (CCATCA*CTACC)·(GGTAGTGATGG) | (−)-trans 14S | intercalation, no base displacement | <2 |
Impact of deletion (Del), abasic (AB), or G*:X mismatches (MM) opposite G* positioned centrally in 135-mer duplexes.
Figure 6.

Relative NER efficiencies in Hela cell extracts of stereoisomeric (+)-trans- and (+)-cis-B[a]PDE-N2–dG adducts. (A) Typical autoradiograph of a gel depicting dual excision products in the 24–32 nucleotide range (size markers shown in lane M) after incubation of 135-mer control duplexes with (+)-trans- (lane 1) or (+)-cis-B[a]PDE-N2–dG adducts (lane 2) for 60 min. (B) Relative incision efficiencies after correcting for loading differences in each lane (data adapted from Mocquet et al.43). (C, D) Stereochemistry of the (+)-cis- and (+)-trans- B[a]P-N2–dG adducts, respectively.
The base-displaced intercalative (+) and (−) cis-adducts are considered to be excellent NER substrates that are repaired with the same efficiencies as the (6–4) UV photoproducts139 (as mentioned above), and the N-acetylaminofluorene-derived dG-C8-AAF adduct.140 These cis-adducts manifest full disruption of one Watson–Crick base pair, the extrusion of the modified base and its partner base out of the helix and, therefore, a loss of base–base stacking interactions. The π–π stacking interactions between the polycyclic aromatic ring system and the adjacent base pairs partly compensate for these distortions,143 as revealed by the overall modest impact of this lesion on the thermal melting of the modified DNA (see below). However, the key conformational property of this lesion appears to be the extruded partner cytosine base.144 It is likely that the initial conformational capture of this preflipped, extruded partner base C by the XPC NER recognition protein145 ultimately fosters its productive binding to the site of the lesion that can further stimulate the recruitment of the subsequent NER factors. Therefore, the strong NER response of the (+)-cis-B[a]PDE-N2–dG adduct is most probably due to its ability to efficiently capture the XPC protein.145
In the case of the stereoisomeric (+)- and (−)-trans-adducts that are positioned in the minor groove (Figure 3A), all base pairs remain intact, and there are no extruded bases. These minor groove conformations display slow repair in the same sequence context in which the cis adducts embedded in duplex I are efficiently repaired. The trans-B[a]PDE-N2–dG adducts that obstruct the minor groove may hinder the productive interaction of the BHD2 β-hairpin of XPC with the damaged DNA site.7,36,145 Furthermore, the minor structural distortions in base pairing caused by these trans-adducts may not be sufficient to elicit the same high NER response as in the case of the stereoisomeric cis-adducts.43
The 11-mer duplexes I with single (+)- and (−)-cis-B[a]PDE-N2–dG adducts are thermodynamically less destabilized (ΔTm = Tm (modified) – Tm (unmodified) = −4 to −5 °C, respectively) than the (+)- and (−)-trans-B[a]PDE-N2–dG adducts (ΔTm = −8 to −10 °C).71 Although the two cis-duplexes are structurally more distorted than the two trans-duplexes, the destabilization due to distortion in the cis cases is counterbalanced in part by stabilizing stacking interactions between the polycyclic aromatic ring systems and adjacent bases.143 Since the relative NER efficiencies are ∼5-times lower in the case of the trans than the cis-adducts, there is no correlation between the global destabilization of DNA duplexes and NER efficiencies in this case. However, in the case of other classes of DNA adducts, such qualitative correlations are indeed observable (section 4.3.1). A partial explanation of these observations is that the DNA lesions induce a local distortion/destabilization that is recognized by XPC, while the Tm experiments are performed with oligonucleotide duplexes of different lengths that contain the same lesions. The melting points reflect not only the local destabilization, but also the cooperative melting of the full duplexes that depends on the base sequence context and the lengths of the duplexes. The extent of local destabilization can be more accurately assessed by temperature-dependent NMR experiments, as was shown by Rodriquez et al.77
Furthermore, the binding of XPC may also depend on the structural features of the bulky DNA adducts. For example, as discussed above for the case of the cis adduct, partner base extrusion may foster initial conformational capture and efficient binding.145 In other cases, a specific structural feature might hinder productive binding of XPC to intercalated bulky lesions and impede the separation of the two DNA strands because of strong π–π base stacking interactions between the intercalated polycyclic aromatic residues and adjacent base pairs. Consequently, the insertion of the BHD3 hairpin between the two DNA strands would be hindered; this would prevent the proper alignment of the protein at the site of the lesion, thus leading to nonproductive binding complexes that are unable to stimulate the following NER steps.146 However, once the BHD3 hairpin is properly inserted, the XPC-damaged DNA complex would be stabilized and stimulate the subsequent NER steps. Hilton et al. studied the dynamics of XPC-Rad23B binding to AAF-modified DNA sequences by surface plasmon resonance methods and proposed that longer protein dissociation constants are correlated with enhanced NER efficiencies.147
In summary, it is well established that XPC binds to unmodified double-stranded DNA more weakly than to known DNA lesions.10,13,25,134,148−156Although the binding of XPC is essential for recruiting subsequent repair factors that lead to successful NER, it has been shown that XPC has high affinities for some DNA lesions that are nonetheless resistant to NER in cell-free extract experiments.146
4.2.2. NER Response of (+)-trans-B[a]PDE-N2–dG (G*) Adducts in Other Sequence Contexts
The 11-mer sequences shown in Figure 7 were embedded in otherwise fully identical 135-mer duplexes that were used as substrates in the usual NER assays in vitro. The CG*C–I sequence containing single (+)-trans-B[a]PDE-N2–dG adducts (G*) in the duplex I sequence context has a relative NER efficiency of ∼20% relative to the stereoisomeric (+)-cis-B[a]PDE-N2–dG adduct.43 Replacing the two cytosines flanking G* by thymines (TG*T sequence) enhances the efficiency by a factor of about two, which has been explained on the basis of the weaker hydrogen bonding and consequent greater dynamics when the base pairs adjacent to the G* are thymines instead of cytosines.133,157 Replacing two base pairs distant from G* (...ATCC on the 3′-side in CG*C–I by···ATAT in CG*C–II) diminishes the NER efficiency by a factor of ∼2 (Figure 7); these two different sequence contexts impose differences in duplex bending and flexibility that correlate with the differences in NER efficiencies observed.158
Figure 7.
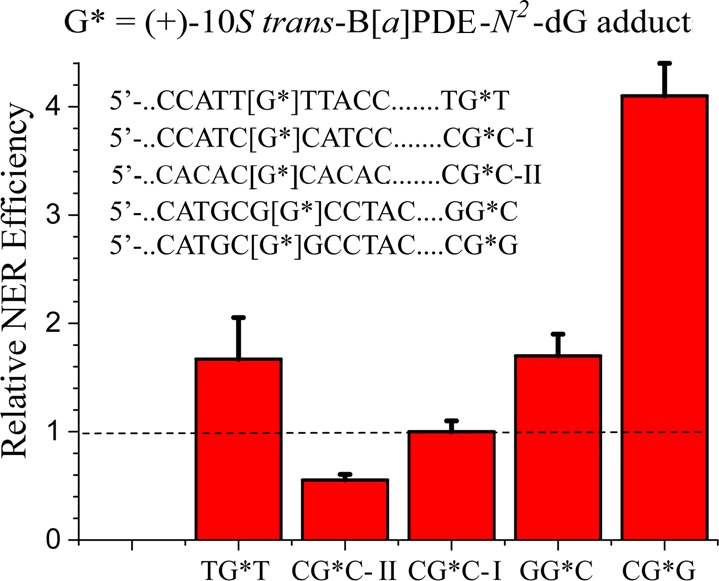
Effects of base sequence context on the NER efficiencies of the same 10S (+)-trans-B[a]PDE-N2–dG adduct in HeLa cell extracts.
Remarkably, different sequence effects have been observed with the (+)-trans-B[a]PDE-N2–dG (G*) adduct positioned in the identical sequence 5′-d(......CATGCG1G2CCTAC···) but with the modified guanine adduct positioned either on the 5′-guanine G1, or the 3′-guanine G2 in this same sequence context (labeled CG*G and GG*C in Figure 7). In the case of the adduct positioned on the 3′-side guanine in the GG*C sequence context, the NER efficiency is ∼35%. However, with the adduct positioned on the 5′-side guanine in the CG*G context, the NER efficiency is ∼80% (Figure 7). This strikingly enhanced efficiency has been attributed to steric crowding between the puckered, OH-containing aliphatic ring of the (+)-trans-B[a]PDE-N2–dG adduct and the minor groove positioned exocyclic amino group of the G flanking the G* on its 3′-side.139 As a result, the aromatic ring system is partially displaced within the minor groove and disrupts the Watson–Crick base pairing on its 5′-side, as demonstrated by NMR methods.77 The substantial differences in NER dual incision efficiencies associated with the same DNA adduct in special sequence contexts like CG*G/GG*C, indicates that the sequence context can play a very significant role in determining NER efficiencies.139,158 These few examples point to the importance of base sequence context effects on NER efficiencies assessed in human cell-free extracts using a limited set of site-specifically modified B[a]PDE-guanine adducts.
Recently, Li et al. developed a novel translesion excision repair-sequencing method (tXR-seq) to study genome-wide nucleotide excision repair maps of B[a]PDE–dG (G*) adducts derived from human lymphocyte cells treated with racemic (±)-B[a]PDE.32 This approach is based on the detection of DNA adducts that are substrates of NER in cellular environments. A sequencing library for tXR-seq analysis was generated by the immunoprecipitation of the intermediate NER dual excision products complexed with TFIIH, followed by the construction of a suitable library for next generation sequencing. The frequencies of nearest neighbor X and Y in the XG*Y sequence context (X,Y = A, C, or T) found in the NER dual excision products were analyzed. An enrichment by a factor of ∼2 of 5′-....CG*...over TG* and AG* sequences was found, and a somewhat greater preference for CG*C and CG*T sequences relative to other trinucleotide sequences was also documented.32 The groundbreaking results of Li et al. demonstrated the feasibility of the tXR-seq approach for studying genome-wide repair maps.
These (±)-B[a]PDE-derived adduct maps are probably not significantly affected by the diversity of stereoisomeric DNA adducts formed because a single adduct is dominant (see below). In general, however, DNA adduct heterogeneity and differences in susceptibilities to GG-NER, especially differences in repair resistance, will need to be considered. The treatment of cells and tissues with reactive mutagens generally yields a spectrum of structurally different DNA lesions, including some that are good substrates of NER, some that are slowly repaired, and some that are resistant to GG-NER. In the case of native DNA treated with racemic diol epoxide (±)-B[a]PDE, Cheng et al. have shown that the dominant adducts formed are the trans-B[a]PDE-N2–dG (∼79% trans- and 11% cis-B[a]PDE-N2–dG, and 10% trans-B[a]PDE-N6–dA adducts).61 It is therefore not surprising that the disappearance of DNA lesions in cellular environments associated with GG-NER as a function of time, may exhibit more than one kinetic decay phase (examples are cited in section 5, below). In human A549 lung epithelial carcinoma cells treated with (+)-B[a]PDE cells, the disappearance of the covalent B[a]PDE–DNA adducts formed was indeed biphasic, with about ∼40% of the adducts removed within ∼1 h (the stereochemistry of the adducts was not specified), while the remainder disappeared more slowly.159 Other workers showed that the treatment of mouse skin with benzo[a]pyrene gave rise to (+)-cis- and (+)-trans-B[a]PDE-N2–dG adducts that decayed at similar rates over a period of ∼100 h after the administration of B[a]P.160 In mammalian cells, the different DNA adducts formed by mutagens other than (±)-B[a]PDE are repaired at different rates, as suggested by the experimentally observed biphasic decays of DNA adducts (see section 5, below). Therefore, in general, differences in rates of repair of the different DNA adducts formed by a given mutagen need to be considered when interpreting the results of genome-wide sequencing experiments.
4.2.3. Intercalated B[a]PDE-N6–dA Adducts: NER Efficiencies and Thermodynamic Destabilization
The reaction of the (+)-7R,8S,9S,10R-B[a]P diol epoxide with the exocyclic amino group of adenine yields the 10S B[a]PDE-N6–dA adduct that is intercalated on the 3′-side of dA*,90,94 without any base displacement out of the double helical DNA. The (−)-7S,8R,9R,10S-B[a]PDE enantiomer yields the stereoisomeric 10R B[a]PDE-N6–dA adduct that is intercalated on the 5′-side of dA*in double-stranded DNA without base displacement.93,125 These B[a]PDE-N6–dA adducts are characterized by NER efficiencies in the range of ∼5–30% (relative to the 10R (+)-cis-B[a]PDE-N2–dG adduct), depending on the sequence context and adduct stereochemistry (Figure 8). The 10S adducts are better NER substrates than the stereoisomeric 10R adducts in several different sequence contexts. This pair of stereochemically related DNA lesions represent an excellent opportunity for correlating differences in their conformational features with their NER responses.
Figure 8.
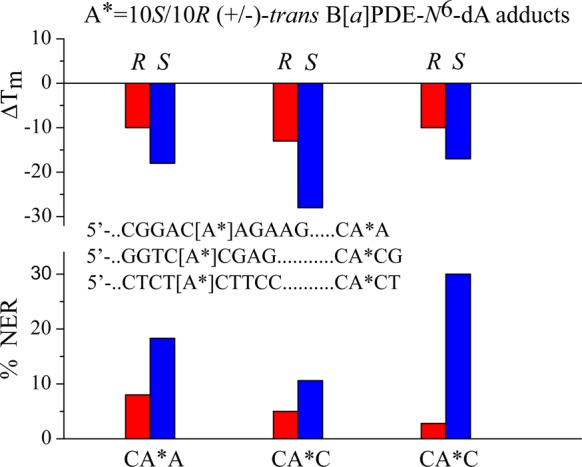
Dependence of NER efficiencies in HeLa cell extracts of 10R (−)-trans- and 10S (+)-trans-B[a]PDE-N6–dA adducts in different sequence contexts (NER data adapted from Buterin et al.165).
The NMR solution structures of these diastereomeric B[a]PDE-N6-adenine adducts (dA*) in double-stranded oligonucleotides have been evaluated in the CA*C90,91,93,161 and CAA*81,92,125,126,162 sequence contexts and are very different from those of the N2-guanine adducts. In the case of the 10R (−)-trans-B[a]PDE-N6–dA adduct, all Watson–Crick base pairs, including the modified dA*:dT base pair, are intact.81,125 The dA* residue is predominantly (95%) in the anti glycosidic bond conformation that is in equilibrium with a minor syn conformation. The NMR structure of the stereoisomeric 10S (+)-trans adduct in the same sequence context could not be resolved because of structural disorder at the binding site126 and the local destabilization of the duplex.93 However, its conformation was inferred from NMR studies of duplexes with a mismatched dA*:dG base pair,94 and a stereochemically related syn-B[a]PDE-N6–dA* adduct paired with dT in the complementary strand. Together with these experimental findings, molecular modeling and dynamic simulation studies131,163 indicate that the polycyclic aromatic ring system of the 10S B[a]PDE-N6–dA adduct is intercalated from the major groove on the 3′-side of the dA*:dT base pair. The 10S adduct is conformationally more heterogeneous and more disordered than the 10R adduct. In both cases, the duplex is stretched and partially unwound to accommodate the bulky intercalated aromatic ring system of the trans-B[a]PDE-N6–dA adducts.
The bulky aliphatic ring of the 10S (+)-trans adduct is situated on the 5′-side of the modified base and, because of the right-handed helical twist, there is steric crowding with the 5′-base adjacent to dA*. To partially relieve this crowding and to maintain stacking interactions between the aromatic ring system and neighboring base pairs, the glycosidic bond in the 10S (+)-trans-modified–dA* residue adopts a syn orientation for the major conformer that is in equilibrium with a minor anti orientation, thus disrupting the dA*:dT Watson–Crick hydrogen bonding.90,126
Because the aliphatic ring of the 10R adduct is positioned on the 3′-side of the modified base, the adjacent base pair is rotated away from this aliphatic residue because of the helical twist, thus avoiding the steric crowding that occurs in the 10S (+)-trans-adduct. Therefore, the normal anti glycosydic bond conformation and dA*:dT Watson–Crick hydrogen bonding remain essentially intact in the intercalated 10R (−)-trans-adduct.
Consistent with the greater structural disorder, the thermal melting points of the 10S adducts are significantly and consistently lower than the Tm values of the 10R B[a][P-N6–dA adduct in 9–11-mer duplexes.164 Buterin et al.165 investigated the NER efficiencies of the 10R (−)-trans- and the 10S (+)-trans-B[a]PDE-N6–dA adducts embedded in different sequence contexts in 135-mer duplexes and incubated in human cell extracts. The results of these NER assays are compared to the ΔTm values of shorter oligonucleotides (9–11 base pairs long) containing the same DNA adducts positioned in the same sequence contexts. The NER efficiency of the 10S adduct was ∼2–3-times greater than that of the stereoisomeric 10R adduct in ···CA*CG··· (ΔTm (10R) = −13 °C and ΔTm (10S) = −28 °C) and in the ···ACA*AG··· sequence contexts (ΔTm (10R) = −10 °C and ΔTm (10S) = −18 °C), and ∼10-times greater in the ···TCA*CT··· sequence contexts (ΔTm= −10 °C (10R) and −17 °C (10S)).164−167 The ΔTm values are negative in all three sequence contexts, and the B[a]PDE-N6–dA adducts with S adduct stereochemistry are consistently more destabilized than the stereoisomeric 10R adducts (Figure 8). Therefore, the differences in the magnitudes of the negative ΔTm values qualitatively parallel the higher NER efficiencies in the case of all three pairs of B[a]PDE-N6–dA adducts. These differences are consistent with the greater structural distortions of the duplexes caused by the 10S than the 10R B[a]PDE-N6–dA adducts that were observed by NMR methods (section 4.3.1) and suggested by molecular dynamics simulations.131,163
This positive correlation is in sharp contrast to the results obtained with the conformationally distinct pairs of cis- and trans-B[a]PDE-N2–dG adducts (section 4.2.1). These observations may be related to the similarities of the conformational intercalation motifs adopted by the 10S and 10R B[a]PDE-N6–dA adducts, whereas the conformations of the cis- and trans-B[a]PDE-N2–dG adducts are dramatically different from one another (base displaced intercalation and minor groove, respectively). We infer that thermodynamic destabilization is one, but not necessarily the only, factor that plays a role in the recognition steps of DNA lesions. Other factors including the structural properties and conformational features of specific bulky DNA adducts can also play an important role, as exemplified by the set of stereoisomeric B[a]PDE-N2–dG and – N6–dA adducts discussed in this section.
4.3. Unusual NER Resistance of Duplexes with Deleted or Noncanonical Bases Opposite B[a]PDE-N2–dG Adducts
If DNA replication occurs before the DNA lesions or adducts are removed by DNA repair mechanisms, translesion synthesis (TLS) catalyzed by various polymerases can be error-prone and thus induce mutations during the next round of replication.168 The TLS past B[a]PDE-N2–dG (G*) adducts in vitro(169,170) and in cellular environments171,172 have been documented. The insertion of nucleotides opposite G* other than dCTP gives rise to point mutations, while slipped frameshift intermediates can stimulate the bypass of G* without the insertion of any nucleotide, which can yield DNA duplexes with single nucleotide deletions.
Following DNA replication past a guanine adduct G*, the resulting duplexes may also contain mismatched partner bases designated as G*:A, G*:G, or G*:T duplexes, or G*:Del duplexes without a nucleotide opposite G*. Therefore, the repair of such mismatched and deletion duplexes is of significant interest.
4.3.1. G:Del* and G*:AB Duplexes Are NER-Resistant
A remarkable observation is that the absence of the single nucleotide dC opposite the trans- or cis-B[a]PDE-N2–dG adduct (G*:Del duplex) completely abrogates NER activity (Figure 9A). Removing just the cytosine, but leaving the phosphodiester backbone and attached deoxyribose residue intact, creates an abasic site opposite the trans- or cis-B[a]PDE-N2–dG adduct (G*:AB duplex), which also abolishes the NER activity that is normally observed in the full duplex.80 To gain mechanistic insights into the NER resistance exhibited by the G*:Del and G*:AB duplexes, the structural features of these duplexes were compared to those of the full 11-mer G*:C duplexes I with all complementary nucleotides intact that are good to-excellent NER substrates (section 4.2.1).
Figure 9.

Deleting the partner C from double-stranded DNA (135-del duplexes) abolishes the NER efficiency that is observed in full (135-mer) duplexes (Full) with C opposite G* ((+)-cis-B[a]PDE-N2–dG). (A) Gel autoradiograph of NER dual incision products incubated in HeLa cell extracts for 0, 10, 20, and 30 min (lanes 1, 2, 3, and 4, respectively; lane M, size markers). (B) Thermal melting curves of 11-mer Del duplexes with and without the adduct (data adapted from Reeves et al.143).
As discussed in section 3.2.2, the (+)-cis-B[a]PDE-N2–dG adduct in G*:C full duplexes assumes a base displaced intercalative conformation and is fully NER-active. The G*:Del and G*:AB 11-mer duplexes also assume intercalative conformations80,143,173 but are NER-inactive and differ from the full duplexes because there are no preflipped cytosines as in the full G*:C duplexes with (+)-cis-B[a]PDE-N2–dG adducts. The (+)-trans-B[a]PDE-N2–dG adduct in G*:C full duplexes assumes a minor groove conformation (Figure 3A), but like the stereosiomeric (+)-cis-adduct, it is also intercalated in the G*:Del and G*:AB duplexes143,174 and is NER-resistant.80,142,173 This kind of NER resistance of Del duplexes does not seem to be limited to B[a]PDE-N2–dG adducts144 since a similar resistance to NER was also observed in the case of the dG–C8-N-acetylaminofluorene adduct, G*(AAF):Del duplexes.144
4.3.2. Molecular Basis of NER Resistance in Deletion and Abasic Duplexes
The first insights into the origins of the NER resistance of G*:Del duplexes were obtained from experimental measurements of the thermal stabilities of deletion duplexes with a (+)-cis-B[a]PDE-N2–dG adduct (G*:Del) and the unmodified (G:Del) duplex. The difference in Tm values between the G*:Del and the G:Del 11-mer duplexes, ΔTm = Tm (G*:Del) – Tm (G:Del) = +19 °C (Figure 9B). This bulky adduct strongly stabilizes the deletion duplex by an astonishing 19 degrees. By contrast, the same lesion strongly destabilizes the full duplex with the canonical C opposite G* since ΔTm = Tm (G*:C) – Tm (G:C) = −12 degrees. This remarkable stabilization of G*:Del duplexes is attributed to the intercalation of the bulky polycyclic aromatic ring system between adjacent base pairs that maximizes the hydrophobic effect and the π–π base stacking interactions with the polycyclic aromatic ring system.143 On the other hand, the presence of the extruded cytosine residue in the base-displaced intercalated full G*:C duplex (Figure 3B) results in an overall modest thermal destabilization. These observations have led to the conclusion that the presence of a bulky DNA adduct produces a change in the local stability of the DNA adduct, which depends on the balance between stabilizing and destabilizing effects of the lesion that are modulated by sequence effects, lesion topology, and bulky adduct stereochemistry.131
These conclusions are supported by solution NMR structural studies: the (+)-cis-B[a]PDE-N2–dG adduct adopts a base-displaced intercalated conformation with a complete rupturing of the Watson–Crick G*:C base pair (Figure 3B). Further molecular modeling and dynamics simulation studies show that steric crowding due to some of the hydroxyl groups in the bulky benzylic ring also cause episodic propeller twisting and buckling of the two adjacent base pairs. This local destabilization and the displaced C partner base of G* should facilitate the insertion of the XPC BHD3 β-hairpin and the interaction of the flipped cytosine base with XPC, leading to a productive XPC protein–DNA complex that can successfully recruit the other NER factors that lead to the dual incisions. However, the absence of the partner nucleotide dC in the deletion duplexes causes a compression of the duplex on the complementary strand side (a wedge shape), which pushes the benzylic ring of the B[a]PDE residue into the minor groove, relieving some of the steric crowding and stabilizing the flanking base pairs.143,174 Notably, the compression or wedge shape causes a significant enhancement of the B[a]P aromatic ring-base stacking interactions that stabilize the site of the adduct; this would hinder insertion of the XPC BHD3 β-hairpin into the damaged DNA site and prevent formation of the strand-separated state that represents the productive open complex. Furthermore, the absence of the dC nucleotide in the dG*:Del duplexes corresponds to the loss of one of the flippable nucleobases144 that interact with amino acid pockets in the XPC protein.7 These considerations are consistent with the observed NER-resistance of the G*:Del duplexes.142−144 Similar explanations may also apply to the dG*:AB duplexes that are also NER resistant.80 However, loss of one partner base may still permit modest repair when an adduct is itself particularly destabilizing, as observed for the food mutagen-derived C8–dG-PHIP adduct that is base displaced-intercalated but possesses a disruptive mobile phenyl ring.143
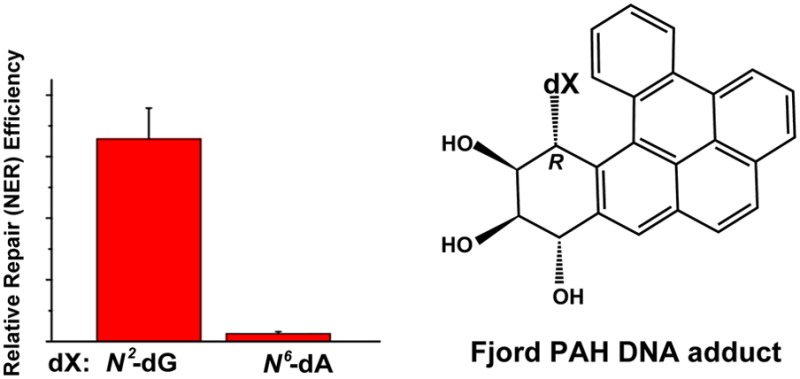
4.3.3. dG*:dX Duplexes with Mismatched Partner Bases dX = dA or dG Are Also NER-Resistant
Experimentally, in the case of the dG*:dC duplex with a canonical dC nucleotide opposite the modified guanine (+)- trans- or (+)-cis-B[a]PDE-N2–dG adduct, NER activity is observed. In the case of the cis-B[a]PDE-N2–dG adduct opposite dC in duplex I, the NER activity has an arbitrary value of 100% (section 4.2.1), and the duplex is moderately destabilized (ΔTm = −4 °C). When dC is replaced by dT, the NER activity is reduced by a factor of ∼3, while in the case of duplexes with dG*:dA142 and dG*:dG, the NER activity is completely abolished.175 The values of ΔTm in the case of dG*:dX* mismatches (ΔTm = −3 °C for X = A, or +2 °C for X = T) are similar to those observed in the case of the normal G*:C duplex I sequence (ΔTm = −4 °C).
Such base sequence context effects on NER activity involving different bases opposite DNA lesions are not limited to bulky PAH-derived DNA adducts. Similar phenomena have been reported in the case of some of the 8,5′-cyclopurine-2′-deoxyguanosine (cdG) and–deoxyadenosine (cdA) lesions by Pande et al.47 The 5′S cdA stereoisomers with a dC opposite the lesion (cdA:C) are excellent NER substrates, but the NER activities of cdA:dT or cdA:dA were 6–10-times smaller. However, entirely different results were observed in the case of dG*(AAF):dX) mismatched duplexes with dX = dT, dA, dG since the NER efficiencies were similar to those in the case of dG*:dC full duplexes with dG* = dG-C8-AAF.144 Such duplexes are strongly destabilized by the bulky AAF residue with ΔTm = −18 °C in the CG*C sequence context of the NarI 12-mer mutation hotspot sequence.140
4.3.4. Molecular Basis of NER Resistance of dG*:dX Mismatch Duplexes with dX = dA, dG, dT
The (+)-cis-B[a]PDE dG*:dT duplex is a weaker NER substrate than the (+)-cis-B[a]PDE–dG*:dC duplex by a factor of ∼3. On the basis of NMR studies of the conformation of the dG*:dC duplex with dG* = (+)-cis-B[a]PDE-N2–dG, it is known that this bulky aromatic B[a]PDE-derived adduct assumes a base-displaced intercalative conformation with the nonaromatic benzylic ring and guanine residue of dG* positioned in the minor groove, and the “orphaned” dC residue displaced into the major groove.122 However, the exact position of the external dC residue could not be ascertained by NMR. Molecular modeling and molecular dynamics simulations of this structure suggests that a stabilizing dynamic hydrogen bond can form between the cytosine amino group and a pendant phosphate oxygen on the 5′-side of dC,175 which helps to maintain the dC residue in the extruded position that is preflipped to interact with the XPC.
Recent studies of the Rad4–DNA binding mechanisms have revealed that the yeast ortholog of human XPC does initially capture the extruded partner C base.145 In the case of the dT duplex, the NER efficiency is reduced by a factor of ∼3 relative to the dG*:dC duplex. The molecular dynamics simulations show that the hydrophobic methyl group of thymine interacts with DNA residues in the major groove via van der Waals interactions and is thus more rarely fully extruded to a flipped out external position than the dC residue. The (+)-cis-B[a]PDE-N2–dG:dA and dG*:dG duplexes are fully resistant to NER. The larger purines, in contrast to the smaller pyrimidines, manifest strong van der Waals interactions with the DNA major groove where they are anchored or occasionally even intercalated into the duplex. Consequently, neither the dA nor the dG moieties are found in a flipped out conformation.175
Overall, these results suggest that the dG*:dG/dA purine mismatched duplexes might prevent the insertion of the XPC BHD3 β-hairpin between the two DNA strands from the major groove and that these purines do not readily assume a flipped out external conformation, thus accounting for the NER resistance of the dG*:dG/dA duplexes. By contrast, in normal dG*:dC duplexes with base-displaced conformations, the dC partner base is preflipped and can interact with XPC. In the case of the pyrimidine mismatched dG*:dT duplexes, molecular dynamics simulation studies indicate that the ensemble contains a small population of extruded dT in equilibrium with dT that interacts with the major groove, which could account for the ∼30% NER efficiency (section 4.4.4). Relative to the dG*:dG/dA mismatch duplex, the van der Waals interactions between the thymine methyl group and the DNA residues are much weaker. Therefore, some extrusion of the dT mismatch and β-hairpin insertion could occur that allows for productive XPC binding that, in turn, stimulates the subsequent NER steps.
4.4. NER-Resistance of Fjord PAH Diol Epoxide-N6-Adenine, but Not N2-Guanine Adducts in Full DNA Duplexes
4.4.1. Fjord and Bay Region PAH Diol Epoxide-DNA Adducts
The key structural feature that distinguishes fjord from bay region PAH compounds is the steric crowding in the fjord regions of the B[c]Ph, B[g]C, and DB[a,l]P molecules that results from the proximities of the pairs of hydrogen atoms at C1 and C12 (B[c]Ph), and C14 and C1 in B[g]C and DB[a,l]P (Figure 2). As a consequence, the 9,10,11,12 aromatic ring of B[c]Ph and the 1,2,3,4-aromatic rings of B[g]C and DB[a,l]P in the fjord region adopt nonplanar conformations.176 The out-of-plane twist is flexible and adopts a conformation that optimizes the stacking interactions between the fjord PAH and adjacent base pairs at the DNA intercalation sites.177 Furthermore, the more compact arrangement of aromatic rings in the fjord PAH compounds due to the closer proximity of the 1,2,3,4 aromatic ring to the sites of covalent attachment of purines in double-stranded DNA, also facilitates optimal stacking with adjacent base pairs in intercalative conformations. By contrast, all aromatic rings are rigid and coplanar in the sterically uncrowded bay region PAH aromatic compounds,178,179 the elongated bay region pyrenyl ring system cannot accommodate itself as well into the sterically crowded interior of the DNA duplex and thus tends to destabilize double-stranded DNA.
4.4.2. Fjord PAH Diol Epoxide–DNA Adducts Are Highly Genotoxic
The fjord PAH, especially dibenzo[a,l]pyrene, have attracted significant attention from the chemical carcinogenesis community52,53,180,181 because they are by far the most tumorigenic PAH ever tested.182 It has been estimated that the fjord PAH are up to ∼100-fold more tumorigenic than the bay region PAH benzo[a]pyrene.183 Both types of PAH are metabolically activated to reactive diol epoxide intermediates that react predominantly with guanine or adenine in cellular DNA to form premutagenic covalent adducts in mammalian cells and tissues.14,15,63,180,181,184−187 While the bay region B[a]PDE adducts react predominantly with N2–dG in DNA with only minor quantities of N6–dA adducts formed,61 the fjord PAH diol epoxides react with both N2–dG and N6–dA resulting in a greater proportion of adenine adducts in DNA in vitro.63,186 Consistent with this DNA adduct distribution, Yoon et al. found that treatment of BigBlue mouse cells with different stereoisomeric DB[a,l]PDE diol epoxides produced predominantly A to T transversions.188 Higher proportions of DB[a,l]PDE-N6–dA adducts were found in human MCF-7 cells189 and in rodent tissues.190,191 The genotoxic activities of fjord PAH diol epoxides are well correlated with DNA adduct levels in V79192 and other cells.193,194 Correlations between DNA adduct levels and tumorigenicity have also been reported in various mammalian systems.180,181,187,190 Since the stereochemical features are similar in the bay and fjord PAH diol epoxide-derived DNA adducts, the differences in their biological impact are related to the chemical reactivities of the respective diol epoxides with DNA and the biological response to the genotoxic DNA adducts formed.
4.4.3. NER and Other Characteristics of Fjord PAH-N6-Adenine and N2-Guanine Adducts
The NER efficiencies of the fjord DB[a,l]PDE-N2–dG and DB[a,l]PDE-N6–dA adducts are compared in Figure 10A and B. The NER experiments were conducted with the G* = 14R (+)-trans- and 14S (−)-trans-DB[a,l]PDE-N2–dG adducts (labeled G*-14R and G*-14S, respectively) embedded in the [5′-d(CCATCG*CTACC)]·[5′-d(GGTAGCGATGG)] 11-mer duplex (I-G*) sequence context that was also used to study the NER efficiencies of the stereoisomeric B[a]PDE-N2–dG adducts195 (Table 1). The A* = 14R (+)-trans- and 14S (−)-trans-DB[a,l]PDE-N6–dA adducts (labeled A*-14R and A*-14S, respectively) were embedded in the similar [5′-d(CCATCA*CTACC)]·[5′-d(GGTAGTGATGG)] 11-mer duplex (I-A*) sequence context, which allows for a direct comparison of the NER responses of the DB[a,l]PDE-derived adenine and guanine adducts in double-stranded DNA. The 11-mer duplexes I-G* and I-A* were used in the NMR structural studies79,124 and in the determinations of duplex thermal melting points. The NER studies were performed with the same I-G* or I-A* sequences embedded in fully complementary 135-mer duplexes.
Figure 10.

(A) Autoradiograph of dual excision products after incubation of stereoisomeric B[a]PDE-N2−dA (G*-10), DB[a,l]PDE-N6–dA (A*-14), or DB[a,l]PDE-N2–dG (G*-14) adducts embeded in identical 135-mer base sequence contexts in HeLa cell extracts. (B) Relative NER efficiencies and (C) impact of the same stereoisomeric DNA adducts on thermal stabilities (ΔTm); the adducts were embedded in the 11-mer duplexes [5′-d(CCATCX*CTACC)]·[5′-d(GGTAGYGATGG)] with X* = DB[a,l]PDE-N6–dA or DB[a,l]PDE-N2–dG, and Y = T or C, respectively; these same 11-mers were embedded in the 135-mer duplexes in the NER experiments depicted in panel A.
4.4.4. The 14R (+)-trans-DB[a,l]PDE–N2-Guanine Adduct
This adduct manifests a smaller NER response (∼65%) in HeLa cell-free extracts (G*-14R in Figure 10A,B) than the standard base-displaced intercalated 10R (+)-cis-B[a]PDE-N2–dG adduct (G*-10R). The strong NER response of this 14R (+)-trans-DB[a,l]PDE–dG adduct (Figure 10A,B) is in sharp contrast to the NER response of the minor groove 10R (−)-trans-B[a]PDE–dG adduct43 that is about three-times smaller and that has the identical absolute configurations of substituents of the nonaromatic ring as the intercalated G*-14R adduct (we recall that the + and–signs denote the optical rotatory dispersions of the PAH diol epoxides and not the absolute configurations). These differences are attributed to the structural features of the G*-14R DB[a,l]PDE-N2–dG adducts that were elucidated by NMR methods.79 The bulky aromatic ring system of the G*-14R adduct is intercalated from the minor groove and the G*:C Watson–Crick base pairing is ruptured although the bases remain partly stacked with the aromatic ring system of the DB[a,l]PDE residue. The bulky aliphatic ring in the crowded minor groove further destabilizes the duplex. These destabilizing structural properties are reflected in the rather large negative ΔTm = −10 °C value (Figure 10C).
The stereochemically identical minor groove 10R (+)-trans-B[a]PDE-N2–dG and the intercalated 14R (−)-trans-DB[a,l]PDE-N2–dG adduct differ from one another by the single additional 1,2,3,4-aromatic ring in the latter. However, because of this additional aromatic ring, the DB[a,l]PDE-derived DNA adduct adopts an intercalative conformation that favors hydrophobic interactions that are further enhanced by the compact fjord topology that positions the 1,2,3,4-aromatic ring close to the N2–dG binding site. However, this intercalative motif is accompanied by the unfavorable rupturing of the base pair and steric crowding associated with intercalation from the minor groove that destabilize this G*-14R adduct and thus favors efficient NER.
4.4.5. The 14S (−)-trans-DB[a,l]PDE–N2-Guanine Adduct
The relative NER efficiency of the minor groove 14S (−)-trans-DB[a,l]PDE-N2–dG adduct is ∼20% (Figure 10B), which is similar to the NER efficiencies of the minor groove 10S trans-B[a]PDE-N2–dG adduct (Figure 6B). Like the stereochemically identical 10S (+)-trans-B[a]PDE adduct, the 14S (−)-trans-DB[a,l]PDE-derived guanine adduct is also positioned in the minor groove and is oriented on the 5′-side of the modified guanine residue.124 However, because of the larger size of the aromatic ring system, the minor groove is much more structurally distorted in the G*-14S case, and the distortions of the double helix extend to the two adjacent base pairs flanking the adduct G* on its 5′-side. The Watson–Crick base pairing is ruptured at the G*-C base pair and also at the adjacent 5′-base pair. By contrast, in the case of the stereochemically analogous B[a]PDE-derived 10S (+)-trans-B[a]PDE-N2–dG adduct, all base pairs are intact, including the G*-C base pair. Surprisingly, the smaller-size of the 10S (+)-trans-B[a]PDE-N2–dG adduct is more destabilizing (ΔTm = −10 °C), while ΔTm is only −2 °C in the case of the more bulky fjord 14S (−)-trans-DB[a,l]PDE-N2–dG adduct. We proposed that the greater stability of this G*-14S adduct is due to enhanced van der Waals interactions of the more bulky aromatic ring system of the 14S (−)-trans-DB[a,l]PDE-N2–dG residue with the DNA residues in the minor groove; the latter is greatly widened and opened to accommodate these interactions. Intercalation of this bulky 14S stereoisomeric adduct from the minor groove is disfavored, although this intercalated conformation is observed in the case of the stereochemically identical N2–dG adduct derived from the smaller fjord region PAH fjord benzo[c]phenanthrene diol epoxide.130
Although the modified duplexes are destabilized to different extents, the differences in the overall NER efficiencies of the two trans-14S DB[a,l]PDE- and 10S B[a]PDE-N2–dG adducts are not well correlated with their impact on the absolute ΔTm values. A perhaps dominant and common feature of the bay region B[a]PDE- and fjord region DB[a,l]PDE-derived guanine adducts is the similar alignment of the bulky polycyclic aromatic ring systems in the minor grooves of the DNA duplexes. The presence of the bulky aromatic ring systems in the minor groove may interfere with the proper alignment of XPC protein, which may be the key factor that determines the relatively weak NER response of N2–dG adducts that occupy the minor groove. This notion is consistent with the NER resistance of DNA adducts derived from the binding of aromatic amines to the exocyclic amino groups of guanines in DNA described in section 6 that are also NER-resistant. Indeed, the BHD2 β-hairpin binds to the minor groove of the damaged DNA in the productive binding complex7 and a lesion in the minor groove would impede this binding.
4.4.6. The A*-14S (−)- and 14R (+)-trans-DB[a,l]PDE–N6-Adenine Adducts
The NER efficiencies of these two trans-DB[a,l]PDE-N6–dA adducts are ≤ 2% relative to the NER efficiency of the 10R (+)-cis-B[a]PDE-N2–dG adduct standard (Figure 10B) and are considered to be NER-resistant. The ΔTm value for the A*-14R adduct is +11 °C indicating that the 14R (+)-trans-DB[a,l]PDE-N6–dA adduct stabilizes double-stranded DNA (Figure 10C), in contrast to the stereochemically identical 10R (−)-trans-B[a]PDE-N6–dA adduct (ΔTm = −12 °C, section 4.3.2). On the other hand, the thermal melting of the stereoisomeric A*-14S adduct is very modestly destabilized (ΔTm = −3 °C) in the full duplex (Figure 10C). It is therefore of interest to examine the structural features of the stereoisomeric A*-14R and A*-14S adducts in DNA duplex I-A*.
The structures of the 14R (+)-trans- and 14S (−)-trans-DB[a,l]PDE-N6–dA (A*-14R and A*-14S adducts, respectively, in Figure 10) in sequence I-A* were investigated by molecular modeling and molecular dynamics simulations that were based on solution NMR studies of stereochemically and structurally related adducts derived from the reactions of the smaller fjord region trans-B[c]PhDE PAH diol epoxides (Figure 2) with N6-adenine in DNA duplexes.78,128,129 Like the analogous B[c]PhDE- and B[g]CDE-N6–dA adducts (55), the 14R (+)-trans-and 14S (−)-trans-DB[a,l]PDE-N6–dA adducts are intercalated from the major groove side on the 5′- and 3′-sides of the modified adenine residues, respectively (Figure 5). Molecular dynamics simulations indicate that all hydrogen bonds remain intact in both the 14R and 14S adducts, but the Propeller Twist and Buckle are perturbed at the 3′-flanking dC:dG base pair adjacent to the 14S dA*:dT base pair.177 In addition, the aromatic DB[a,l]PDE residue-base stacking interactions are weaker in the case of the 14S adduct, and its hydrophobic aromatic ring system is more exposed to the aqueous solvent environment than the aromatic ring system of the stereoisomeric 14R adduct.
These structural differences between the A*-14S and A*-14R adducts can account for the slightly negative ΔTm value of the A*-14S adduct, while the A*-14R adduct is significantly more stable as indicated by its large and positive ΔTm value (Figure 10C). Yet, both of these adducts are similarly resistant to NER with observed NER efficiencies close to the background levels of ∼2%. Our hypothesis is based on the Rad4-DNA structure (Figure 1) showing that the recognition of DNA lesions and adducts is a highly localized phenomenon that involves the interaction of the BHD2 hairpin with the minor groove and the insertion of the BHD3 β-hairpin between the two DNA strands at the site of the lesion from the major groove side. Since this intrusion of the BHD3 hairpin occurs from the major groove, the interaction of these bulky, major groove intercalated adenine adducts likely obstructs the hairpin insertion, which is also resisted by the stacking interactions of the DB[a,l]P aromatic ring system with adjacent base pairs.196 Together, these phenomena could be sufficient to inhibit productive binding by XPC. The overall binding affinities of the DNA lesion-sensing XPC-RAD23B factor to the 14R and 14S trans-DB[a,l]PDE-N6–dA are experimentally indistinguishable.146 As discussed elsewhere,146 productive and unproductive modes of binding may coexist, as shown for example by Sugusawa,41 who found that there are two modes of XPC binding and only one of these can lead to the subsequent steps that permit successful NER. This is an area of research that is in need of further development to reach a better understanding of the basic phenomena underlying the mechanisms of efficient NER, as well as resistance of DNA lesions to NER.
4.5. B[c]Ph and B[g]C Diol Epoxide Derived DNA Adducts
Other fjord PAH diol epoxide–DNA adducts depicted in Figure 11 are also resistant to repair. The 1S (−)-trans- and 1R (+)-trans-B[c]PhDE-N6–dA adducts are intercalated without base displacement on the 3′- and 5′-sides, respectively, of the dA*–dT base pairs128,129 and are fully NER-resistant in standard HeLa cell extracts.165 Both duplexes are characterized70 by ΔTm ≈ 0, which is not destabilizing. The lower van der Waals interactions between the 1R B[c]PhDE-N6–dA aromatic ring system with neighboring base pairs are, however, insufficient to stabilize the duplexes as observed in the case of the larger DB[a,l]PDE-N6–dA adducts (Figure 10C). However, with one additional aromatic ring relative to the analogous B[c]Ph diol epoxide-derived adenine adduct, the 14R (+)-trans-B[g]CDE-N6–dA adduct in the same duplex is also intercalated on the 5′-side of the dA*–dT base pair, but the adduct stabilizes the duplex with ΔTm = +9 °C,78 and is NER-resistant in human cells.197 In all cases, the adenine adducts are intercalated from the more spacious major groove with little destabilization that is further favored by the compact topology of the fjord region PAH adducts that places aromatic rings close to their site of attachment. Taken together, these factors enhance stacking interactions with adjacent base pairs at DNA intercalation sites, and this conclusion applies to all of the intercalated (without base-displacement) fjord PAHDE-N6–dA adducts studied up until now.
Figure 11.

Summary of stereochemical features of the fjord PAH diol epoxide -N6–dA and -N2–dG adducts.
4.6. Implications for Molecular Basis of NER Resistance
The extensive series of structural studies and NER efficiencies observed under standardized and reproducible conditions in human cell-free extracts in vitro, have provided insights into relationships between the properties of a variety of stereisomeric polycyclic aromatic diol epoxide-derived DNA adducts and their susceptibilities to NER. Several themes have emerged that can explain the high susceptibility of some substrates to NER as well as resistance to NER. A number of different DNA lesions such as the 6–4 UV photoproduct, other nonbulky DNA lesions that include spiroiminodihydantoins (Sp) and guanidinohydantoin,49 base-displaced intercalated cis-B[a]PDE-N2–dG, and N-acetyaminofluorene-derived C8–dG140 adducts manifest approximately equivalent susceptibilities to NER with relative efficiencies designated by us as ∼100%. The relative NER efficiencies of other DNA lesions and adducts are compared to this standardized value. In our experience, only cisplatin intrastrand G*TG*cross-linked DNA adducts are better NER substrates than the bulky PAH-diol epoxide–DNA adducts we have studied (∼160% on this scale139). Although Sp is a nonbulky lesion, its relative NER efficiency is ∼100% on this same scale.49 The propeller-like structure strongly destabilizes the duplexes (as shown by calorimetric methods) because of weakened interactions between Sp and flanking base pairs as shown by NMR methods.198 The NER efficiencies of benzo[a]pyrene diol epoxide-derived intercalated N6–dA adducts, as well as the minor groove-positioned B[a]PDE-N2–dG adducts, are in the range of ∼10–30%43,164,165 and are dependent on sequence contexts (Figures 7 and 8).
Some of the stereoisomeric fjord PAH diol epoxide N6–dA adducts are resistant to NER (efficiencies ≤2%) and result from intercalative insertions from the major groove of the compact fjord aromatic ring systems (Figure 3C) that maximize π–π base stacking interactions with neighboring base pairs. The stacking interactions stabilize the DNA duplexes by hindering the separation of the two DNA strands at or near the sites of the intercalated adducts, thus impeding the intrusion of the XPC BHD3 hairpin, which is also sterically obstructed by the intercalation from the major groove; these features are believed to inhibit lesion recognition by XPC and the subsequent NER steps.20,143 On the other hand, intercalation by the benzo[a]pyrene-derived dA adducts is much more distorting and destabilizing due to the extended, planar, and rigid nature of the bay region adduct, which prevents more optimal intercalation as in the more compact and twisted fjord adducts.131 Detailed analysis of these interactions by molecular modeling and molecular dynamic simulations indicates that the overall stability of different covalent fjord and bay region PAH diol epoxide DNA adducts depends on the size and shape of the intercalated fjord PAH aromatic ring systems and on the balance between stabilizing and destabilizing PAH residue–DNA interactions.131 Contact between XPC and the DNA damaging moiety itself is not essential for XPC binding, but bulky DNA adducts could play an indirect role by hindering the proper alignment of the XPC proteins at the perturbed DNA binding site, thus resulting in the formation of unproductive XPC–DNA adduct complexes that are not efficiently processed by subsequent NER factors.146 Such effects may, in part, account for the lower NER efficiencies of DNA adducts that occupy the minor groove of DNA and that may sterically hinder the interaction of hairpin BHD2 with the damaged DNA duplexes on the minor groove side.
In summary, the combination of steric effects and strong stacking and other van der Waals interactions associated with large PAH ring systems, as well as obstruction of the appropriate alignment of the XPC-RAD23B damage-sensing NER factor at the site of DNA lesions, could explain the observed NER resistance observed in human cell extracts. We now consider the available evidence that NER-resistant forms of DNA damage also exist in cellular and mammalian tissues.
5. Repair-Resistance of DNA Adducts in Cellular Environments
5.1. Fjord PAH–DNA Adducts
An early report of the persistence of DNA adducts derived from the reactions of fjord PAH benzo[g]chrysene diol epoxide with DNA in human fibroblasts in culture was presented by Lloyd and Hanawalt in 2002.197 The levels of (+) and (−)-B[g]CDE–dG and–dA adducts were determined by the sensitive 32P-postlabeling method and were found to persist at least until 3 days after treatment with 10 nM concentrations of B[g]CDE. Dreij et al.159 incubated A549 human epithelial lung carcinoma cells with the bay region (+)-B[a]PDE or the highly reactive fjord region (−)-DB[a,l]PDE diol epoxide stereoisomer and measured the persistence of the covalent DNA adducts formed. They found that the overall rate of removal of adducts derived from (−)-DB[a,l]PDE was slower relative to those derived from (+)-B[a]PDE. The dA/dG adduct ratios increased from 2.9 after a 20 min incubation time with (−)-anti-DB[a,l]DE to 4.0 after 6 h, which suggested that the adenine adducts were repaired more slowly than the guanine adducts. However, the ratio of dA/dG adducts remained constant during the same time interval upon incubation with the stereochemically identical (+)-anti-B[a]PDE. The slower repair of the DB[a,l]PDE-N6–dA adducts is consistent with the higher fraction of mutations at A:T than G:C sites in the Hprt gene in Chinese hamster V79 cells treated with (−)-DB[a,l]PDE.188 It was shown by Jankowiak et al.,888 that the exposure of mouse skin leads to a 2:1 ratio of covalent deoxyadenosine to deoxyguanosine adducts. By using a DNA adduct conformation-sensitive fluorescence method, it was also found that DNA-adducts that adopt intercalative conformations are more resistant to repair than adducts that adopt external conformations; these conclusions are consistent with the resistance of the intercalated fjord DB[a,l]PDE-N6–dA adducts to NER in cell extracts (Figure 10). Lagerqvist et al.199 reported that DNA adducts in DNA repair-proficient Chinese Hamster Ovary cell lines treated with racemic (±)-DB[a,l]PDE generated adducts that were repaired ∼5-times more slowly than those produced by (+)-B[a]PDE; however, adenine adducts were not distinguished from guanine adducts in this study. A 32P-postlabeling method for monitoring DNA adduct levels in NER-deficient and NER-proficient SV40-transformed human skin fibroblasts and MCF7 cells with racemic (±)-DB[a,l]PDE was employed by Spencer et al.;200 the major DNA reaction products detected were guanine adducts that exhibited biphasic repair kinetics and the DNA adducts persisted for at least 34 h after treatment.
More recently, the effects of the bay region B[a]PDE-N6–dA and the fjord B[c]PhDE-N6–dA adducts on transcription and transcription-coupled repair (TCR) were assessed based on a pCI-neo-G-less-T7 plasmid that incorporated either of these adducts downstream from a CMV immediate-early promoter/enhancer element. These plasmids were transfected into primary human fibroblasts of different genetic backgrounds.201 Both adducts were found to be substrates of TCR; the B[a]PDE-N6–dA adduct was also repaired by the GG-NER pathway, while the fjord B[c]PhDE-N6–dA adduct was resistant to GG-NER. Previous NER assays conducted in cell-free extracts165 are thus consistent with these findings in intact human cells. While more studies using these cellular assays201 are needed, these preliminary results suggest that the cell-free extract NER assays may be relevant to understanding NER in cellular environments.
Zhang and El-Bayoumy181,187 developed an oral cancer model based on the treatment of the oral cavities of mice with DB[a,l]P and DB[a,l]PDE. The genotoxic impact of DB[a,l]P on oral tissues is predominantly due to its metabolic activation to the (−)-anti-DB[a,l]PDE enantiomer. The levels of (−)-trans-DB[a,l]PDE-N6–dA adducts were greater by a factor of 2 than the levels of the stereoisomeric (−)-cis-, (+)-trans-, and the (+)-cis-DB[a,l]PDE-N2–dG adducts. However, all adducts persisted in the oral tissues of mice for at least 28 days after the last dose of DB[a,l]P administration. While the development of mutations and tumorigenesis was attributed to both DB[a,l]PDE-adenine and -guanine adducts, it was concluded that the levels and persistence of the (−)-trans-DB[a,l]PDE-N6–dA adducts in oral tissue may play a significant role in accounting for the carcinogenic activity of DB[a,l]P in the oral tissues of mice.
5.2. Aflatoxin B1–Derived Guanine Adducts
The major DNA adduct derived from Aflatoxin B1 in human cells stems from its metabolic activation to AFB1-exo-8,9-epoxide202,203 and its reaction with N7-guanine in DNA to yield the cationic adduct trans-8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 adduct (AFB1-N7–dG).204 The subsequent base-catalyzed hydrolysis cleaves the imidazole ring of guanine to yield the AFB1 FAPY adduct trans-8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxy aflatoxin B1 (Figure 12).205 The AFB1-N7–dG adduct is rapidly repaired, while the AFB1 FAPY adduct is long-lived in the livers of rats treated with AFB1.206 Analogous results were obtained in human fibroblasts treated with rat liver microsome-activated AFB1.207 Insights into the molecular basis of the repair-resistance of the AFB1 FAPY adduct were obtained from studies of their conformations in double-stranded DNA.208,209 The solution NMR structures showed that in a 10-mer duplex, the cationic AFB1 N7–dG and the FAPY adducts assume intercalated conformations on the 5′-side of the modified G*:C base pair; all Watson–Crick base pairs were normal except at the G*:C and 5′-flanking base pair (G:C). None of these two AFB1 adducts destabilized the 10-mer 2′-deoxyoligonucleotide duplexes in which it was embedded. Instead, the cationic duplex was moderately stabilizing with ΔTm = +2–3 °C, while the AFB1 FAPY adduct yielded a remarkably large increase in the thermal stability of the duplex with ΔTm = +15 °C. These phenomena were attributed to the intercalation of AFB1 between adjacent base pairs and significant residue–base stacking interactions that have been correlated with the repair-resistance of these adducts.209
Figure 12.

Structures of Aflatoxin B1-derived guanine adducts.
5.3. DNA Adducts Derived from Nitroaromatic Compounds and Aromatic Amines
The genotoxic properties of nitroaromatic compounds and aromatic amines have been studied extensively.210,211 The metabolic activation of these compounds generates electrophilic intermediates that react predominantly with purines in DNA to yield adducts that can, if not repaired by cellular defense systems, lead to mutations, genomic instability, and cancers.211 A number of these DNA adducts have been identified that are resistant to repair or are slowly repaired by the NER mechanism as described below.
5.3.1. 2-Nitrofluorene-Derived 2-Aminofluorene and 2-Acetylaminofluorene-Guanine Adducts
2-Nitrofluorene is an environmental pollutant that is metabolically activated in mammalian cells to the widely studied mutagenic and carcinogenic aromatic amines 2-aminofluorene (AF) and 2-acetylaminofluorene (AAF).212 These electrophilic derivatives react with guanine in DNA to form predominantly dG-C8-AF and dG-C8-AAF (N-deoxyguanosin-8-yl)-2-AF and -AAF, respectively) and, to a lesser extent, the dG-N2-AAF (3-(deoxyguanosin-N2-yl)-2-AAF) DNA adducts (Figure 13).210 Treatment of primary cultures of rat hepatocytes with N-hydroxy-AAF generates three adducts that are removed with a half-life of ∼10 h (dG-C8-AAF), or more slowly in a biphasic manner (dG-C8-AF and dG-N2-AAF); the dG-N2-AAF adduct was found to be particularly persistent.213 Similar results were reported in animal experiments based on the dietary administration of AAF to rats and the subsequent analysis of the DNA in the liver; the dG-C8-AAF adduct was present in ∼10-times higher amounts than the dG-C8-AF and dG-N2-AAF adducts, and the latter were the most persistent adducts in these tissues.214 Cui et al.215 examined the formation and persistence of four DNA adducts formed in different tissues of rats after long-term dietary administration of AF. Two of these adducts were not identified but were found to be persistent in liver tissues. Two other DNA adducts found in the liver were identified as dG-C8-AF and dG-N2-AAF. The latter adduct persisted for at least 10 months in the livers of these rodents after the cessation of feeding 2-nitrofluorene to these animals. By contrast, the levels of dG-C8-AF adducts decreased by a factor of more than ∼100 during the same period of time.215
Figure 13.

Structures of 2-aminofluorene (AF) and 2-acetylaminofluorene (AAF)-derived guanine adducts.
The structural features of the dG-N2-AAF adduct were investigated by Zaliznyak et al. by NMR spectroscopy and restrained molecular dynamics.98 The AAF residue was positioned in the minor groove pointing to the 5′-end of the modified strand, and all Watson–Crick base pairs remained intact; a minor groove position for this adduct was previously predicted by Grad et al.216 This structure is reminiscent of the (+)-trans-B[a]PDE-N2–dG adduct positioned in the minor groove with the bulky aromatic residue pointing toward the 5′-direction of the modified strand.117 The latter PAH diol epoxide adduct destabilizes double-stranded DNA,71 and its NER efficiency varies from weak to strong, depending on the sequence context.43,142,158 However, the dG-N2-AAF adduct stabilizes double-stranded DNA and is repair-resistant in rodent tissues. The difference between these two N2–dG adducts stems from the planarity and curved topology of the fluorenyl ring system in the dG-N2–AAF adduct, which fits tightly into the B-DNA minor groove. On the other hand, the (+)-trans-B[a]PDE-N2–dG adduct is larger, more extended, and has a nonplanar benzylic ring; it therefore significantly widens the minor groove that tends to destabilize the duplex and can account for its moderate NER activity.43
5.3.2. 3-Nitrobenzanthrone
DNA adducts derived from the metabolic activation of the nitroaromatic compound 3-nitrobenzanthrone (3NBA) are known to be mutagenic.217 Like other nitro-PAH derivatives, 3NBA undergoes nitroreduction to 3-aminobenzanthrone (ABA) that is subsequently metabolically activated to the electrophilic arylnitrenium ion. This intermediate reacts with purine bases to yield the three DNA adducts N-(deoxyguanosin-8-yl)-3-aminobenzanthrone (C8–dG-ABA), 2-(deoxyguanosin-N2-yl)-3-aminobenzanthrone (N2–dG-ABA), and 2-(deoxyadenosin-N6-yl)-3-aminobenzanthrone (N6–dA-ABA) (Figure 14). After treatment of rats by intratracheal instillation of 3NBA, persistent DNA adducts were found in various organs but were not identified.218 Treatment of human hepatoma HepG2 cells with 3NBA gives rise to two major adducts, C8–dG-ABA and N6–dA-ABA, while the N2–dG-ABA was found to be a minor adduct. The C8- dG-ABA and N6–dA-ABA adducts were rapidly repaired, but the N2–dG-ABA adduct was significantly more persistent.217 Interestingly, the N2–dG-ABA adduct assumes a minor groove conformation pointing toward the 5′-end of the modified strand and its thermodynamic stability is characterized by a positive ΔTm value (+7.5 °C),219 while the C8–dG adduct assumes a base-displaced intercalative conformation and strongly destabilizes the DNA duplex (ΔTm= −11 °C).220 These features of the ABA adducts are similar to those of the dG-C8-AAF and dG-N2-AAF adducts (section 5.3.1).
Figure 14.

Structures of 3-nitrobenzanthrone (ABA)-derived guanine and adenine adducts.
5.3.3. 6-Aminochrysene Reduction Products of 6-Nitrochrysene
It has been shown that 6-nitrochrysene (6NC) is metabolically activated in rodents and in vitro systems by the nitroreduction of 6NC to N-hydroxy-6-aminochrysene that yields three major adducts that include N-(dG-8-yl)-6AC (N-(deoxyguanosin-8-yl)-6-aminochrysene), 5-(dG-N2-yl)-6AC (5-(deoxyguanosin-N2-yl)-6-aminochrysene), and N-(dA-8-yl)-6AC (N-(deoxyadenosine-8-yl)-6-aminochrysene) (Figure 15).221 This pattern of adduct formation is analogous to the pattern of purine adduct formation associated with 2-nitrofluorene and 3-aminobenzanthrone-derived DNA adducts. The relative NER efficiencies of these adducts in Hela cell extracts222,223 showed that the 5-(dG-8-yl)-6AC and the N-(dA-8-yl)-6AC adducts were repaired with similar efficiencies, while the 5-(dG-N2-yl)-6AC adduct was almost one order of magnitude less well incised in human cell extracts than the dG-C8–6AC adducts. However, the conformations of these 6-aminochrysene-derived guanine and adenine adducts have not been investigated. It has been hypothesized that the slow repair of the various lesions derived from 6NC/6AC and their potential persistence in mammalian tissue could in part account for the powerful carcinogenicity of 6NC.223
Figure 15.

Structures of 6-nitrochrysene (6AC)-derived guanine and adenine adducts.
5.3.4. Aristolochic Acid-Derived Adenine Adduct, ALII–dA
The nephrotoxin aristolochic acids AAI (8-methoxy-6-nitrophenanthro[3,4-d][1,3]dioxole-5-carboxylic acid) and AAII (same as AAI except that the 8-methoxy group is replaced by a hydrogen atom) is present in plants of the Aristolochiaceae family and was first linked to Balkan endemic nephropathy, a disease that was concentrated among communities living along the Danube river in the Balkans and adjacent East European countries. It was eventually discovered that chronic exposure to aristolochic acids in the human diet leads not only to terminal kidney failure, but also to transitional cell carcinoma of the urinary tract.224,225 Aristolochic acid is a complex derivative of the PAH phenanthrene that has a carboxylic acid and a nearby nitro group that play key roles in its metabolic activation to aristolactam derivatives (ALI and ALII).226,227 Both ALI and ALII react with the exocyclic amino groups of guanine and adenine in cellular DNA228 to form genotoxic and persistent DNA adducts (Figure 16) in human kidney tissues of cancer patients that had been exposed to AA.229
Figure 16.

Structures of aristolochic acid-derived adenine adducts. X = COCH3 for ALI, and X = H for ALII.
Most of the somatic mutations found in human urethelial carcinomas attributed to AA have a distinct preponderance of A:T → T:A transversions. This unusual mutational signature was discovered by genome-wide sequencing of DNA extracted from tumor tissues of upper urothelial cell carcinoma patients. These results clearly established a link between exposure to an environmental carcinogen and the etiology of these urothelial cancers.17,18
Sidorenko et al. showed that ALII–dG adducts are repaired by NER mechanisms while ALII-N6–dA adducts are resistant to NER in human fibroblasts exposed to AA. The ALII adducts site-specifically incorporated into plasmids were also NER-resistant in human cell extracts.230 Therefore, there has been considerable interest in elucidating the conformational and biological properties of the DNA adducts formed by the reactions of these aristolactams with DNA.
The structural features of ALII–dA (A*) lesions have been investigated by Lukin et al.136 With the normal partner base dT in the complementary strand, this aristolactam AL–DNA adduct adopts a novel intercalative conformation with the aromatic ring system of the adduct, including the modified adenine, intercalated between adjacent base pairs. The Watson–Crick hydrogen bonding with the dT base is fully disrupted and the T is displaced into the major groove.136 The 11-mer duplexes containing this adduct are moderately destabilized as is evident from the relatively small lowering of the Tm of these duplexes relative to the unmodified controls in different sequence contexts (ΔTm = −3.0, −3.6, and −6.4 °C in duplexes with ..CA*G.., TA*G.., and ..CA*C.. sequence contexts, respectively). It was proposed that the loss of one Watson–Crick base pair is compensated by stabilizing π–π stacking interactions between the aromatic ring system of the ALII residue and the flanking guanine residues in the CA*C sequence context.136 This effect is reminiscent of the weak destabilizing effects of the NER-resistant 14S DB[a,l]PDE-N6–dA adduct (Figure 10C).231 The ALII–dA lesion is resistant to GG-NER in human fibroblasts treated with aristolochic acid, which is accounted for by a lack of specific binding of XPC-RAD23B to this DNA adduct; however, when the ALII–dA adduct is mismatched with CCC, XPC-RAD23B binding is observed.230 These latter observations with mismatches are analogous to similar effects observed with the cross-linked T^T CPD lesion (see section 2). The resistance of the ALII–dA lesions to NER is also paralleled by the bulky DB[a,l]PDE-N6–dA and B[c]PhDE-N6–dA lesions in cell-free extracts,165,195 although XPC-RAD23B binds robustly to these fjord adducts,146 in contrast to the ALII–dA adducts.230 The ALII–dA adducts persist in rat genomic DNA and are repaired only by TC-NER in human fibroblasts, while the ALII–dG adducts are substrates of both TC-NER and GG-NER.230
The structural and dynamic characteristics of these two adducts were analyzed by molecular dynamics simulations.232 The basic difference suggested by the simulations is in the preferred relative orientations of the planar bases and the planar aromatic ALII ring systems. In the case of the ALII–dA adducts, these moieties are approximately coplanar, while in the case of the ALII–dG adducts, they are not. This leads to a greater structural distortion at the lesion site, enhanced dynamics in the case of the guanine adduct, and thus diminished carcinogen-base stacking interactions. Similar differences in local structural distortions in the case of fjord PAH-N2–dG and N6–dA adducts were associated with the NER resistance of the adenine, but not guanine adducts.195
5.3.5. Heterocyclic Aromatic Amines
Heterocyclic aromatic amines (HAA) are present in cooked meats and fish and are metabolized by CYP P450 1A2 to N-hydroxylamines that are highly mutagenic and carcinogenic.233 The N-hydroxylamines are acetylated by the N-acetyl transferases NAT2. The resulting highly reactive nitrenium ions are generated by solvolysis and react mostly with C8–dG, and to a minor extent with N2–dG in DNA to form stable adducts.
The reactions of the HAA 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) with DNA lead to the formation of the major guanine adduct C8-[(3-methyl-3H-imidazo[4,5-f]quinolin-2-yl)amino]–dG (dG-C8-IQ) and, to a lesser extent, to the N2-(2-amino-3-methyl-3H-imidazo[4,5-f]quinol-5yl)–dG (dG-N2-IQ) adduct (Figure 17).234 The formation and rates of removal of these DNA adducts were investigated in rats after administration of an oral dose of IQ; after cessation of treatment, the dG-C8-IQ adduct was cleared from the liver and kidney more quickly than the dG-N2-IQ adduct, which persisted for at least 4 weeks after treatment.234 It is therefore interesting to compare the physical and structural difference between the dG-C8-IQ and the dG-N2-IQ adducts.
Figure 17.

Structures of 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) derived guanine adducts.
Stone and co-workers demonstrated that the dG-C8-IQ adduct adopts a base-displaced intercalative conformation when positioned at G3, and minor groove conformations when positioned at G1 and G2 in the 12-mer NarI sequence context 5′-d(CTCG1G2CG3CCATC) in a duplex hybridized with its fully complementary 12-mer strand.111,112 All three 12-mer duplexes are destabilized by these adducts with ΔTm in the range of −4 to −7 °C.110
The dG-N2-IQ adducts positioned at G1 and G3 also adopt base-displaced conformations,109,110 but the thermal destabilization is negligible. In the case of all three duplexes with the adducts at G1, G2, or G3, the ΔTm is within the −1 to +1 range, close to the experimental error of these measurements. Thus, the duplex melting points Tm of the dG-N2-IQ duplexes are practically the same as the Tm of the unmodified 12-mer duplex, while the dG-C8-IQ duplexes are destabilized. In this case, the differences in reported NER repair resistance of these adducts appear to be better correlated with the thermal destabilization of these DNA adducts110 than their conformations in double-stranded DNA. However, the relative NER efficiencies in standardized NER assays using the six site-specifically modified dG-C8-IQ/dG–N2–dG would need to be established to determine the impact of the adduct conformations on NER in this set of related DNA adducts. Nevertheless, the dG-N2-IQ involves the linkage of a bulky adduct to the exocyclic amino group of guanine which protrudes into the minor groove. Therefore, this is yet another example of an N2–dG DNA adduct that is partially or fully resistant to NER.
The heterocyclic aromatic amine DNA adduct dG-C8-PhIP, a guanine adduct derived from 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, has been found in prostate tumors.235 The relative dual incision efficiency of site-specifically modified oligonucleotide duplexes with dG-C8-PhIP adducts, is about 90% of the cis-B[a]PDE-N2–dG adduct value.143 This adduct adopts a base-displaced intercalated conformation with its adducted G and partner C both extruded into the major groove,236 and the heterocyclic aromatic ring system that is intercalated has a characteristic destabilizing mobile phenyl ring.143 Thermal melting data revealed a destabilization of −10.6 °C of PhiP-modified relative to unmodified 11-mer duplexes. The dG-C8-PhIP adduct thus fits the pattern established by other base-displaced intercalated bulky C8–dG adducts that are thermally destabilized, and are very good NER substrates.
6. Summary and Conclusions
In this perspective, some of the known classes of bulky NER-resistant DNA lesions have been discussed (Table 2). There is little doubt that other GG-NER-resistant DNA lesions remain to be discovered, or have already been documented but not characterized structurally. An example of the latter are the DNA adducts derived from the alkylating agent acylfulvene237 and their natural product precursor and antitumor agent illudine S.238 An important motivation for understanding the molecular and structural origins of repair-resistance of DNA lesions is the potential for designing improved drugs for chemotherapeutic applications. In this respect, the interstrand cross-linked lesions are particularly important,21 and include the well-known cisplatin intrastrand cross-linked DNA lesions, and other forms of DNA damaging agents.113 Among the best NER substrates besides the cisplatin intrastrand cross-linked DNA lesions are the 6–4 UV photoproducts and other forms of DNA damage reviewed by Gillet and Schärer,9 certain oxidatively generated DNA lesions,49,239 and base-displaced intercalated bulky DNA adducts (section 4.2.1).
Table 2. Summary of Some NER-Resistant/Slowly Repaired DNA Lesions in Human/Mammalian Cells and Mammalian Tissues.
| parent compound or reactive metabolite | DNA lesion and stereochemistry | DNA repair environment | reference | remarks |
|---|---|---|---|---|
| I. Polycyclic Aromatic Epoxides | ||||
| B[c]PhDE (fjord) | 1R trans-dA | human fibroblasts | (201) | plasmid, site-specific |
| B[g]CDE (fjord) | (±)-trans-dA/dG | human fibroblasts | (197) | 32P postlabeling adduct recovery |
| DB[a,l]PDE (fjord) | 14R and S trans and cis-dA/dG | mouse oral tissues | (181) | mass spectrometry |
| (187) | ||||
| DB[a,l]PDE (fjord) | 14R and S trans- and cis-dA/dG | A549 human lung carcinoma cells | (159) | identified by HPLC biphasic kinetics |
| DB[a,l]PDE (fjord) | unspecified dG adducts | human cell lines | (200) | 32P post labeling |
| aflatoxin B1 | AFB1 Fapy | rat liver | (206) | radioactively labeled AFB1 |
| human fibroblasts | (207) | |||
| II. Aromatic Amines | ||||
| AF and AAF | dG-N2-AAF | rat hepatocytes | (213) | |
| dietary administration | ||||
| dG-C8-AF | other rat tissues | (214) | ||
| 3-aminobenzanthrone (3-ABA) | dG-N2-ABA | human hepatoma HepG2 cells | (217) | treatment of cells with 3NBA |
| aristolochic acid | ALII–dA | human fibroblasts | (230) | plasmid, site-specific lesion |
| III. Heterocyclic Aromatic Amines | ||||
| IQ | dG-N2-IQ | rat liver and kidney | (234) | oral administration of IQ |
What are the features of DNA lesions that are associated with good substrates of GG-NER and of NER resistant lesions? Some typical examples of strong and weak NER substrates and NER-resistant DNA lesions, including their structural characteristics and impact on the thermodynamic properties of the modified DNA duplexes, are compared in Table 3. Our overall conclusion is that the NER response in standardized human cell-free extracts depends on (1) the bulk and the specific chemical structure of the individual lesion, (2) the modified nucleotide and the site of attachment of the lesion to this nucleotide, (3) the sequence context that the lesion is embedded in, (4) the DNA conformation adopted by the lesion which depends on factors 1–3, and (5) the impact of the lesions on the local thermodynamic stability of double-stranded DNA.
Table 3. Some Examples of DNA Lesions That Are NER-Resistant, Weak or Strong NER Substrates, and Their Characteristics.

Abbreviations are those used in the original references and the main text.
See the relevant section of the main text for additional details.
The effect of DNA sequence context can be particularly dominant as shown by the examples in Table 1. A DNA lesion can be an excellent substrate in the normal full duplex but becomes NER-resistant when the partner base in the complementary strand is absent (the Del duplexes).
The impact of the lesions on the overall thermodynamic characteristics of DNA duplexes, as measured by thermal melting experiments, can be stabilizing or destabilizing, depending on the lesion. The concept has emerged that bulky adducts, because of their enhanced potential for stabilizing van der Waals interactions with DNA residues, can locally stabilize DNA duplexes especially via intercalation and thus impart NER resistance to the DNA adducts.131,143
While accurate a priori predictions of the effects of DNA lesion structure on susceptibilities to GG-NER are difficult if not impossible, certain trends have nevertheless been identified. DNA repair resistance is most clearly associated with DNA adducts that are intercalated between adjacent base pairs without expulsion of either the modified or partner base (Table 3). All of the identified NER-resistant bulky lesions either thermodynamically stabilize the local DNA sites (presumable by van der Waals adduct-base stacking interactions) or have little or no negative impact. Weaker NER substrates can be thermodynamically destabilizing but are characterized by conformations that do not strongly affect the stacking of base pairs within the duplex. Thus, minor groove DNA adducts involving covalent linkages with the exocyclic N2-amino groups of guanine that thermally destabilize DNA tend to be among the weaker substrates of NER.
Specific examples of the known NER-resistant DNA substrates can be categorized according to the following three groups:
-
(I)
Certain bulky polycyclic aromatic diol epoxide-N6-adenine (sections 4.4,5.1) and aristolochic acid-derived (5.3.4) N6-adenine adducts. The former are intercalated between adjacent base pairs without base displacement and all Watson–Crick pairs are intact, while the aristolochic acid-derived adduct has a partner T base that is displaced into the major groove; both manifest strong π–π stacking interactions between the aromatic ring system and adjacent bases.
-
(II)
Bulky polycyclic aromatic N2-guanine adducts that are excellent substrates of NER in full double-stranded DNA, but are fully resistant when the canonical cytosine nucleotides are deleted, or replaced by noncanonical purine nucleotides (section 4.3). Such duplexes with “deletion” or mismatches are biologically significant because they can arise during DNA replication due to error-prone bypass of lesions by replicative or by translesion DNA polymerases.
-
(III)
DNA lesions that do not greatly distort the structure of DNA include, for example, most of the nonbulky DNA lesions that are substrates of BER, and the UV-induced CPD thymine dimers that are GG-NER-resistant in cell-free extracts. However, the CPD lesions are repaired by NER in vivo with the assistance of the proteins DDB1 in the DDB1/2 complex.29,240
-
(IV)
Adducts that are bound to DNA via the exocyclic amino groups of guanine are often repaired slowly by NER mechanisms in human cell extracts. Such adducts are also known to persist in cells and tissues for significant periods of time. Since the amino group of guanine protrudes into the minor groove, such bulky PAH N2–dG adducts are often positioned in the minor groove of DNA. It is interesting to note that some DNA repair-resistant heterocyclic aromatic amine-N2–dG adducts have indeed been found in cells and tissues of animals exposed to various heterocyclic aromatic amines (Table 2). Some bulky minor groove adducts are readily recognized by the damage-sensing NER factor XPC-RAD23B146 (sections 4.2.1 and 4.2.2), but they may interfere with the proper NER-productive alignment of XPC, thus limiting the efficiency of NER.
7. Perspectives
In this article, we have implicitly assumed that the appearance of the characteristic 24–32 nucleotide-long NER dual incision products reflect the early stages of the complex NER multiple step mechanism, which involves the recognition and excision of the 24–32 nucleotide-long oligonucleotides that contains the damaged base. The subsequent gap-filling steps are no longer dependent on the nature of the excised lesion, and it is therefore assumed that the relative NER efficiencies in human cell-free extracts are critical to understanding these early recognition and excision steps that must occur for the successful completion of all of the other subsequent NER steps. The successful GG-NER mechanisms involve the recognition and the binding of XPC-RAD23B to the site of the damage that is followed by a verification mechanism that involves TFIIH. The initial binding of XPC-RAD23B may be either productive or unproductive and thus not necessarily lead to the dual incisions.146 The subsequent recruitment of TFIIH to the XPC-RAD23B-damaged DNA complex leads to the verification step that involves the local unwinding of the DNA duplex on both sides of the lesion and the verification of the presence of a genuine DNA lesion. The details of the verification mechanism are still not well understood. Since XPC-RAD23B binds well to at least some DNA lesions that are nevertheless resistant to NER,146 it remains to be determined whether and how the verification mechanism plays a role in the DNA lesion recognition phenomenon in NER and somehow fails to recognize such DNA lesions.
DNA lesions that are resistant to excision by the NER mechanism can still be recognized and removed by TC–NER, thus ensuring the accurate transcription of the genome in spite of the presence of NER-resistant DNA lesions. However, error-prone bypass of these lesions by DNA polymerases adds to the mutagenic burden of the cells and ultimately to transcription errors, and it is therefore important to distinguish the forms of DNA damage that are resistant to DNA repair by GG-NER mechanisms.
In chromatin in intact cells and tissues, the access of repair proteins to DNA lesions is strongly hindered.241,242 Therefore, NER activity is significantly slower in chromatinized DNA than in cell extracts. In principle, it may be more difficult to distinguish NER-resistant from good NER substrates in cellular and tissue environments. On the other hand, there are several examples that show that the same genotoxic substances can give rise to different adducts, some persistent and others repaired more rapidly (Table 2). In such cases, differences in DNA lesion-specific repair capacities are discernible even at the cellular level.
An important general question remains whether the hierarchies of GG-NER efficiencies observed in cell-free extracts in vitro, especially NER resistance, are relevant to cellular DNA repair capacities, that is, the removal of similar forms of DNA damage in cellular environments and tissues. These questions should be addressed using the same well-defined site-specifically positioned DNA lesions in cell-free extracts and in intact cells. At this time, there are only two explicit comparisons of this type summarized in Table 2, both involving adenine adducts: (1) a site-specific fjord B[c]Ph-N6–dA adduct is NER resistant in cell extracts as well as in intact fibroblasts,165,201 and (2) the aristolochic acid derived ALII–dA adduct is repair resistant in fibroblasts and in Hela cell-free extracts.230 The NER efficiencies of the other bulky DNA lesions listed in Table 2 that have been found to be DNA repair-resistant in mammalian cellular environments have not yet been studied in mammalian cell extracts (e.g., the dG-N2-AAF aromatic amines). The in vitro cell extract experiments are more convenient than cell- or tissue-based methods for establishing GG-NER resistance. The potential biological impact of previously uncharacterized forms of DNA damage that are discoverable by mass spectrometric adductomics methods243 could be assessed by cell-free NER experiments to distinguish NER-resistant from easily repaired DNA adducts. Such GG-NER-resistant DNA adducts may turn out to be particularly relevant and useful as biomarkers of exposure of human populations to toxic environmental chemicals and the prevention of human diseases that they engender.
Acknowledgments
We thank Dr. Yuqin Cai for outstanding help with the figures and manuscript preparation.
Glossary
Abbreviations
- NER
nucleotide excision repair
- BER
base excision repair
- GG-NER
global genomic nucleotide excision repair
- TC-NER
transcription coupled nucleotide excision repair
- hRNAPII
human RNA polymerase II
- 8-oxoG
8-oxo-2′-deoxyguanosine
- T^T CPD
cyclobutane pyrimidine dimer
- 6–4 photoproduct
pyrimidine–pyrimidone photoproduct
- PAH
polycylic aromatic hydrocarbons
- Del
deletion duplex
- TLS
translesion synthesis
- CYP
cytochrome P450
- EH
epoxide hydrolase
- G*, A*
guanine or adenine adduct
- THF
tetrahydrofuran
- XPC
XPC-RAD23B
Biographies
Nicholas E. Geacintov is Professor of Chemistry at New York University in New York and the Vice Dean of Science at NYU Shanghai. He served as the Chair of the NYU Department of Chemistry (2000–2009) and is currently the Chair of the ACS Division of Chemical Toxicology (2017–2018). He was trained as a polymer physical chemist and received his PhD from the Environmental Sciences and Forestry College at Syracuse University. He gradually switched fields to areas at the interface of chemistry and biology and is currently interested in unraveling the mechanisms of repair of DNA lesions formed by reactions of endogenous and environmental genotoxic chemicals with DNA.
Suse Broyde is Professor of Biology and Affiliate Professor of Chemistry at New York University. She received her PhD in Physical Chemistry from the Polytechnic Institute of Brooklyn, now NYU Tandon School of Engineering. Her research has focused for many years on molecular modeling of DNA damaged by environmental and endogenous mutagens and carcinogens, in settings that include DNA and RNA polymerases, DNA repair proteins, and with the DNA packaged in nucleosomes. The goal is to gain understanding on a molecular level of the mutagenic properties of the damaged DNA and its susceptibility to nucleotide excision repair using state-of-the-art computational chemistry approaches.
The support of the National Institute of Environmental Health Sciences (NIEHS) Grant No. ES024050 (N.E.G.) and National Cancer Institute (NCI) Grant No. CA168469 (N.E.G.), and NIEHS Grant No. ES025987 (S.B.), NCI Grant Nos. CA75449 (S.B.), and CA 28038 (S.B.), are gratefully acknowledged. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (NSF) Grant No. MCB060037 (S.B.), and the high performance computing resources of New York University (NYU-ITS).
The authors declare no competing financial interest.
References
- David S. S.; O’Shea V. L.; Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950. 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schärer O. D. (2013) Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspect. Biol. 5, a012609. 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak G. (2015) Nucleotide excision repair in humans. DNA Repair 36, 13–18. 10.1016/j.dnarep.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt P. C.; Spivak G. (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970. 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Reardon J. T.; Mu D.; Sancar A. (1996) Overproduction, purification, and characterization of the XPC subunit of the human DNA repair excision nuclease. J. Biol. Chem. 271, 19451–19456. 10.1074/jbc.271.32.19451. [DOI] [PubMed] [Google Scholar]
- Marteijn J. A.; Lans H.; Vermeulen W.; Hoeijmakers J. H. (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465–481. 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- Min J. H.; Pavletich N. P. (2007) Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 449, 570–575. 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- Schärer O. D. (2007) Achieving broad substrate specificity in damage recognition by binding accessible nondamaged DNA. Mol. Cell 28, 184–186. 10.1016/j.molcel.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Scharer O. D. (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 106, 253–276. 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- Sugasawa K.; Okamoto T.; Shimizu Y.; Masutani C.; Iwai S.; Hanaoka F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 15, 507–521. 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi M.; Masutani C.; Maekawa T.; Sugasawa K.; Ohkuma Y.; Hanaoka F. (2000) The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J. Biol. Chem. 275, 9870–9875. 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- Riedl T.; Hanaoka F.; Egly J. M. (2003) The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 22, 5293–5303. 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood R. D. (1999) DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie 81, 39–44. 10.1016/S0300-9084(99)80036-4. [DOI] [PubMed] [Google Scholar]
- Nesnow S.; Ross J. A.; Mass M. J.; Stoner G. D. (1998) Mechanistic relationships between DNA adducts, oncogene mutations, and lung tumorigenesis in strain A mice. Exp. Lung Res. 24, 395–405. 10.3109/01902149809087376. [DOI] [PubMed] [Google Scholar]
- Ross J. A.; Nesnow S. (1999) Polycyclic aromatic hydrocarbons: correlations between DNA adducts and ras oncogene mutations. Mutat. Res., Fundam. Mol. Mech. Mutagen. 424, 155–166. 10.1016/S0027-5107(99)00016-0. [DOI] [PubMed] [Google Scholar]
- Alexandrov L. B.; Nik-Zainal S.; Wedge D. C.; Aparicio S. A.; Behjati S.; Biankin A. V.; Bignell G. R.; Bolli N.; Borg A.; Borresen-Dale A. L.; Boyault S.; Burkhardt B.; Butler A. P.; Caldas C.; Davies H. R.; Desmedt C.; Eils R.; Eyfjord J. E.; Foekens J. A.; Greaves M.; Hosoda F.; Hutter B.; Ilicic T.; Imbeaud S.; Imielinsk M.; Jager N.; Jones D. T.; Jones D.; Knappskog S.; Kool M.; Lakhani S. R.; Lopez-Otin C.; Martin S.; Munshi N. C.; Nakamura H.; Northcott P. A.; Pajic M.; Papaemmanuil E.; Paradiso A.; Pearson J. V.; Puente X. S.; Raine K.; Ramakrishna M.; Richardson A. L.; Richter J.; Rosenstiel P.; Schlesner M.; Schumacher T. N.; Span P. N.; Teague J. W.; Totoki Y.; Tutt A. N.; Valdes-Mas R.; van Buuren M. M.; van ’t Veer L.; Vincent-Salomon A.; Waddell N.; Yates L. R.; Australian Pancreatic Cancer Genome I.; Consortium I. B. C.; Consortium I. M.-S.; PedBrain I.; Zucman-Rossi J.; Futreal P. A.; McDermott U.; Lichter P.; Meyerson M.; Grimmond S. M.; Siebert R.; Campo E.; Shibata T.; Pfister S. M.; Campbell P. J.; Stratton M. R. (2013) Signatures of mutational processes in human cancer. Nature 500, 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang M. L.; Chen C. H.; Sidorenko V. S.; He J.; Dickman K. G.; Yun B. H.; Moriya M.; Niknafs N.; Douville C.; Karchin R.; Turesky R. J.; Pu Y. S.; Vogelstein B.; Papadopoulos N.; Grollman A. P.; Kinzler K. W.; Rosenquist T. A. (2013) Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. 5, 197ra102. 10.1126/scitranslmed.3006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon S. L.; Pang S. T.; McPherson J. R.; Yu W.; Huang K. K.; Guan P.; Weng W. H.; Siew E. Y.; Liu Y.; Heng H. L.; Chong S. C.; Gan A.; Tay S. T.; Lim W. K.; Cutcutache I.; Huang D.; Ler L. D.; Nairismagi M. L.; Lee M. H.; Chang Y. H.; Yu K. J.; Chan-On W.; Li B. K.; Yuan Y. F.; Qian C. N.; Ng K. F.; Wu C. F.; Hsu C. L.; Bunte R. M.; Stratton M. R.; Futreal P. A.; Sung W. K.; Chuang C. K.; Ong C. K.; Rozen S. G.; Tan P.; Teh B. T. (2013) Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci. Transl. Med. 5, 197ra101. 10.1126/scitranslmed.3006086. [DOI] [PubMed] [Google Scholar]
- Kisker C.; Kuper J.; Van Houten B. (2013) Prokaryotic nucleotide excision repair. Cold Spring Harbor Perspect. Biol. 5, a012591. 10.1101/cshperspect.a012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Reeves D.; Kropachev K.; Cai Y.; Ding S.; Kolbanovskiy M.; Kolbanovskiy A.; Bolton J. L.; Broyde S.; Van Houten B.; Geacintov N. E. (2011) Probing for DNA damage with beta-hairpins: similarities in incision efficiencies of bulky DNA adducts by prokaryotic and human nucleotide excision repair systems in vitro. DNA Repair 10, 684–696. 10.1016/j.dnarep.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauson C.; Scharer O. D.; Niedernhofer L. (2013) Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harbor Perspect. Biol. 5, a012732. 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E. C., Graham W. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2006) DNA Repair and Mutagenesis, 2nd ed., ASM Press, Washington, D.C. [Google Scholar]
- Mouret S.; Baudouin C.; Charveron M.; Favier A.; Cadet J.; Douki T. (2006) Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. U. S. A. 103, 13765–13770. 10.1073/pnas.0604213103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumoto R.; Masutani C.; Sugasawa K.; Iwai S.; Araki M.; Uchida A.; Mizukoshi T.; Hanaoka F. (2001) Diversity of the damage recognition step in the global genomic nucleotide excision repair in vitro. Mutat. Res., DNA Repair 485, 219–227. 10.1016/S0921-8777(00)00082-3. [DOI] [PubMed] [Google Scholar]
- Reardon J. T.; Sancar A. (2003) Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 17, 2539–2551. 10.1101/gad.1131003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H.; Kim S. Y.; Kim S. K.; Kemp M. G.; Sancar A. (2015) An integrated approach for analysis of the DNA damage response in mammalian cells: Nucleotide excision repair, DNA damage checkpoint, and apoptosis. J. Biol. Chem. 290, 28812–28821. 10.1074/jbc.M115.690354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A. (2016) Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew. Chem., Int. Ed. 55, 8502–8527. 10.1002/anie.201601524. [DOI] [PubMed] [Google Scholar]
- Fitch M. E.; Nakajima S.; Yasui A.; Ford J. M. (2003) In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem. 278, 46906–46910. 10.1074/jbc.M307254200. [DOI] [PubMed] [Google Scholar]
- Scrima A.; Konickova R.; Czyzewski B. K.; Kawasaki Y.; Jeffrey P. D.; Groisman R.; Nakatani Y.; Iwai S.; Pavletich N. P.; Thoma N. H. (2008) Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 135, 1213–1223. 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P.; Smerdon M. J.; Roberts S. A.; Wyrick J. J. (2016) Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. U. S. A. 113, 9057–9062. 10.1073/pnas.1606667113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Adebali O.; Adar S.; Sancar A. (2017) Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl. Acad. Sci. U. S. A. 114, 6758–6763. 10.1073/pnas.1706522114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Hu J.; Adebali O.; Adar S.; Yang Y.; Chiou Y. Y.; Sancar A. (2017) Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene. Proc. Natl. Acad. Sci. U. S. A. 114, 6752–6757. 10.1073/pnas.1706021114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Velmurugu Y.; Zheng G.; Park B.; Shim Y.; Kim Y.; Liu L.; Van Houten B.; He C.; Ansari A.; Min J. H. (2015) Kinetic gating mechanism of DNA damage recognition by Rad4/XPC. Nat. Commun. 6, 5849. 10.1038/ncomms6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmurugu Y.; Chen X.; Slogoff Sevilla P.; Min J. H.; Ansari A. (2016) Twist-open mechanism of DNA damage recognition by the Rad4/XPC nucleotide excision repair complex. Proc. Natl. Acad. Sci. U. S. A. 113, E2296–2305. 10.1073/pnas.1514666113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kad N. M.; Wang H.; Kennedy G. G.; Warshaw D. M.; Van Houten B. (2010) Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single- molecule imaging of quantum-dot-labeled proteins. Mol. Cell 37, 702–713. 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu H.; Geacintov N. E.; Zhang Y.; Broyde S. (2015) Recognition of Damaged DNA for Nucleotide Excision Repair: A Correlated Motion Mechanism with a Mismatched cis-syn Thymine Dimer Lesion. Biochemistry 54, 5263–5267. 10.1021/acs.biochem.5b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E.; Moggs J. G.; Hwang J. R.; Egly J. M.; Wood R. D. (1997) Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO. J. 16, 6559–6573. 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias A.; Auriol J.; Forget D.; Enzlin J. H.; Scharer O. D.; Coin F.; Coulombe B.; Egly J. M. (2004) Ordered conformational changes in damaged DNA induced by nucleotide excision repair factors. J. Biol. Chem. 279, 19074–19083. 10.1074/jbc.M312611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M. S.; von Bornstaedt G.; Gourdin A. M.; Politi A. Z.; Mone M. J.; Warmerdam D. O.; Goedhart J.; Vermeulen W.; van Driel R.; Hofer T. (2010) Stochastic and reversible assembly of a multiprotein DNA repair complex ensures accurate target site recognition and efficient repair. J. Cell Biol. 189, 445–463. 10.1083/jcb.200909175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu N.; Kaczmarek N.; Naegeli H. (2010) Strand- and site-specific DNA lesion demarcation by the xeroderma pigmentosum group D helicase. Proc. Natl. Acad. Sci. U. S. A. 107, 17545–17550. 10.1073/pnas.1004339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K.; Akagi J.; Nishi R.; Iwai S.; Hanaoka F. (2009) Two-step recognition of DNA damage for mammalian nucleotide excision repair: Directional binding of the XPC complex and DNA strand scanning. Mol. Cell 36, 642–653. 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
- Evans E.; Fellows J.; Coffer A.; Wood R. D. (1997) Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO. J. 16, 625–638. 10.1093/emboj/16.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocquet V.; Kropachev K.; Kolbanovskiy M.; Kolbanovskiy A.; Tapias A.; Cai Y.; Broyde S.; Geacintov N. E.; Egly J. M. (2007) The human DNA repair factor XPC-HR23B distinguishes stereoisomeric benzo[a]pyrenyl-DNA lesions. EMBO J. 26, 2923–2932. 10.1038/sj.emboj.7601730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon J. T.; Bessho T.; Kung H. C.; Bolton P. H.; Sancar A. (1997) In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. U. S. A. 94, 9463–9468. 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P. J.; Wise D. S.; Berry D. A.; Kosmoski J. V.; Smerdon M. J.; Somers R. L.; Mackie H.; Spoonde A. Y.; Ackerman E. J.; Coleman K.; Tarone R. E.; Robbins J. H. (2000) The oxidative DNA lesion 8,5′-(S)-cyclo-2′-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 275, 22355–22362. 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- Kuraoka I.; Bender C.; Romieu A.; Cadet J.; Wood R. D.; Lindahl T. (2000) Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc. Natl. Acad. Sci. U. S. A. 97, 3832–3837. 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande P.; Das R. S.; Sheppard C.; Kow Y. W.; Basu A. K. (2012) Repair efficiency of (5 S)-8,5 -cyclo-2 -deoxyguanosine and (5′S)-8,5′-cyclo-2′-deoxyadenosine depends on the complementary base. DNA Repair 11, 926. 10.1016/j.dnarep.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropachev K.; Ding S.; Terzidis M. A.; Masi A.; Liu Z.; Cai Y.; Kolbanovskiy M.; Chatgilialoglu C.; Broyde S.; Geacintov N. E.; Shafirovich V. (2014) Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucleic Acids Res. 42, 5020–5032. 10.1093/nar/gku162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafirovich V.; Kropachev K.; Anderson T.; Liu Z.; Kolbanovskiy M.; Martin B. D.; Sugden K.; Shim Y.; Chen X.; Min J. H.; Geacintov N. E. (2016) Base and Nucleotide Excision Repair of Oxidatively Generated Guanine Lesions in DNA. J. Biol. Chem. 291, 5309–5319. 10.1074/jbc.M115.693218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S.; Kropachev K.; Cai Y.; Kolbanovskiy M.; Durandina S. A.; Liu Z.; Shafirovich V.; Broyde S.; Geacintov N. E. (2012) Structural, energetic and dynamic properties of guanine(C8)-thymine(N3) cross-links in DNA provide insights on susceptibility to nucleotide excision repair. Nucleic Acids Res. 40, 2506–2517. 10.1093/nar/gkr1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. H. (1983) Fifty years of benzo(a)pyrene. Nature 303, 468–472. 10.1038/303468a0. [DOI] [PubMed] [Google Scholar]
- Amin S.; Desai D.; Dai W.; Harvey R. G.; Hecht S. S. (1995) Tumorigenicity in newborn mice of fjord region and other sterically hindered diol epoxides of benzo[g]chrysene, dibenzo[a,l]pyrene (dibenzo[def,p]chrysene), 4H-cyclopenta[def]chrysene and fluoranthene. Carcinogenesis 16, 2813–2817. 10.1093/carcin/16.11.2813. [DOI] [PubMed] [Google Scholar]
- Amin S.; Krzeminski J.; Rivenson A.; Kurtzke C.; Hecht S. S.; El-Bayoumy K. (1995) Mammary carcinogenicity in female CD rats of fjord region diol epoxides of benzo[c]phenanthrene, benzo[g]chrysene and dibenzo[a,l]pyrene. Carcinogenesis 16, 1971–1974. 10.1093/carcin/16.8.1971. [DOI] [PubMed] [Google Scholar]
- Higginbotham S.; RamaKrishna N. V.; Johansson S. L.; Rogan E. G.; Cavalieri E. L. (1993) Tumor-initiating activity and carcinogenicity of dibenzo[a,l]pyrene versus 7,12-dimethylbenz[a]anthracene and benzo[a]pyrene at low doses in mouse skin. Carcinogenesis 14, 875–878. 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- Buening M. K.; Wislocki P. G.; Levin W.; Yagi H.; Thakker D. R.; Akagi H.; Koreeda M.; Jerina D. M.; Conney A. H. (1978) Tumorigenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides in newborn mice: exceptional activity of (+)-7beta,8alpha-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Proc. Natl. Acad. Sci. U. S. A. 75, 5358–5361. 10.1073/pnas.75.11.5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey R. G. (1981) Activated Metabolites of Carcinogenic Hydrocarbons. Acc. Chem. Res. 14, 218–226. 10.1021/ar00067a004. [DOI] [Google Scholar]
- Thakker D. R.; Yagi H.; Akagi H.; Koreeda M.; Lu A. H.; Levin W.; Wood A. W.; Conney A. H.; Jerina D. M. (1977) Metabolism of benzo[a]pyrene. VI. Stereoselective metabolism of benzo[a]pyrene and benzo[a]pyrene 7,8-dihydrodiol to diol epoxides. Chem.-Biol. Interact. 16, 281–300. 10.1016/0009-2797(77)90108-9. [DOI] [PubMed] [Google Scholar]
- Meehan T.; Straub K. (1979) Double-stranded DNA steroselectively binds benzo(a)pyrene diol epoxides. Nature 277, 410–412. 10.1038/277410a0. [DOI] [PubMed] [Google Scholar]
- Meehan T.; Straub K.; Calvin M. (1977) Benzo[alpha]pyrene diol epoxide covalently binds to deoxyguanosine and deoxyadenosine in DNA. Nature 269, 725–727. 10.1038/269725a0. [DOI] [PubMed] [Google Scholar]
- Jeffrey A. M.; Jennette K. W.; Blobstein S. H.; Weinstein I. B.; Beland F. A.; Harvey R. G.; Kasai H.; Miura I.; Nakanishi K. (1976) Letter: Benzo[a]pyrene-nucleic acid derivative found in vivo: structure of a benzo[a]pyrenetetrahydrodiol epoxide-guanosine adduct. J. Am. Chem. Soc. 98, 5714–5715. 10.1021/ja00434a060. [DOI] [PubMed] [Google Scholar]
- Cheng S. C.; Hilton B. D.; Roman J. M.; Dipple A. (1989) DNA adducts from carcinogenic and noncarcinogenic enantiomers of benzo[a]pyrene dihydrodiol epoxide. Chem. Res. Toxicol. 2, 334–340. 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- Pruess-Schwartz D.; Baird W. M.; Yagi H.; Jerina D. M.; Pigott M. A.; Dipple A. (1987) Stereochemical specificity in the metabolic activation of benzo(c)phenanthrene to metabolites that covalently bind to DNA in rodent embryo cell cultures. Cancer Res. 47, 4032–4037. [PubMed] [Google Scholar]
- Dipple A.; Pigott M. A.; Agarwal S. K.; Yagi H.; Sayer J. M.; Jerina D. M. (1987) Optically active benzo[c]phenanthrene diol epoxides bind extensively to adenine in DNA. Nature 327, 535–536. 10.1038/327535a0. [DOI] [PubMed] [Google Scholar]
- Jerina D. M.; Sayer J. M.; Agarwal S. K.; Yagi H.; Levin W.; Wood A. W.; Conney A. H.; Pruess-Schwartz D.; Baird W. M.; Pigott M. A.; et al. (1986) Reactivity and tumorigenicity of bay-region diol epoxides derived from polycyclic aromatic hydrocarbons. Adv. Exp. Med. Biol. 197, 11–30. 10.1007/978-1-4684-5134-4_2. [DOI] [PubMed] [Google Scholar]
- Conney A. H. (1982) Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 42, 4875–4917. [PubMed] [Google Scholar]
- Wood A. W.; Chang R. L.; Levin W.; Yagi H.; Thakker D. R.; Jerina D. M.; Conney A. H. (1977) Differences in mutagenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides. Biochem. Biophys. Res. Commun. 77, 1389–1396. 10.1016/S0006-291X(77)80133-2. [DOI] [PubMed] [Google Scholar]
- Koreeda M.; Moore P. D.; Wislocki P. G.; Levin W.; Yagi H.; Jerina D. M. (1978) Binding of benzo[a]pyrene 7,8-diol-9,10-epoxides to DNA, RNA, and protein of mouse skin occurs with high stereoselectivity. Science 199, 778–781. 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- Weinstein I. B.; Jeffrey A. M.; Jennette K. W.; Blobstein S. H.; Harvey R. G.; Harris C.; Autrup H.; Kasai H.; Nakanishi K. (1976) Benzo(a)pyrene diol epoxides as intermediates in nucleic acid binding in vitro and in vivo. Science 193, 592–595. 10.1126/science.959820. [DOI] [PubMed] [Google Scholar]
- Cosman M.; Ibanez V.; Geacintov N. E.; Harvey R. G. (1990) Preparation and isolation of adducts in high yield derived from the binding of two benzo[a]pyrene-7,8-dihydroxy-9,10-oxide stereoisomers to the oligonucleotide d(ATATGTATA). Carcinogenesis 11, 1667–1672. 10.1093/carcin/11.9.1667. [DOI] [PubMed] [Google Scholar]
- Laryea A.; Cosman M.; Lin J. M.; Liu T.; Agarwal R.; Smirnov S.; Amin S.; Harvey R. G.; Dipple A.; Geacintov N. E. (1995) Direct synthesis and characterization of site-specific adenosyl adducts derived from the binding of a 3,4-dihydroxy-1,2- epoxybenzo[c]phenanthrene stereoisomer to an 11-mer oligodeoxyribonucleotide. Chem. Res. Toxicol. 8, 444–454. 10.1021/tx00045a017. [DOI] [PubMed] [Google Scholar]
- Geacintov N. E.; Cosman M.; Hingerty B. E.; Amin S.; Broyde S.; Patel D. J. (1997) NMR solution structures of stereoisometric covalent polycyclic aromatic carcinogen-DNA adduct: principles, patterns, and diversity. Chem. Res. Toxicol. 10, 111–146. 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- Fernandes A.; Liu T.; Amin S.; Geacintov N. E.; Grollman A. P.; Moriya M. (1998) Mutagenic potential of stereoisomeric bay region (+)- and (−)-cis-anti- benzo[a]pyrene diol epoxide-N2–2′-deoxyguanosine adducts in Escherichia coli and simian kidney cells. Biochemistry 37, 10164–10172. 10.1021/bi980401f. [DOI] [PubMed] [Google Scholar]
- Shukla R.; Jelinsky S.; Liu T.; Geacintov N. E.; Loechler E. L. (1997) How stereochemistry affects mutagenesis by N2-deoxyguanosine adducts of 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene: configuration of the adduct bond is more important than those of the hydroxyl groups. Biochemistry 36, 13263–13269. 10.1021/bi971195z. [DOI] [PubMed] [Google Scholar]
- Avkin S.; Goldsmith M.; Velasco-Miguel S.; Geacintov N.; Friedberg E. C.; Livneh Z. (2004) Quantitative analysis of translesion DNA synthesis across a benzo[a]pyrene-guanine adduct in mammalian cells: the role of DNA polymerase kappa. J. Biol. Chem. 279, 53298–53305. 10.1074/jbc.M409155200. [DOI] [PubMed] [Google Scholar]
- Cohen I. S.; Bar C.; Paz-Elizur T.; Ainbinder E.; Leopold K.; de Wind N.; Geacintov N.; Livneh Z. (2015) DNA lesion identity drives choice of damage tolerance pathway in murine cell chromosomes. Nucleic Acids Res. 43 (3), 1637–1645. 10.1093/nar/gku1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Wu X.; Guo D.; Rechkoblit O.; Wang Z. (2002) Activities of human DNA polymerase kappa in response to the major benzo[a]pyrene DNA adduct: error-free lesion bypass and extension synthesis from opposite the lesion. DNA Repair 1, 559–569. 10.1016/S1568-7864(02)00055-1. [DOI] [PubMed] [Google Scholar]
- Rodriguez F. A.; Cai Y.; Lin C.; Tang Y.; Kolbanovskiy A.; Amin S.; Patel D. J.; Broyde S.; Geacintov N. E. (2007) Exocyclic amino groups of flanking guanines govern sequence-dependent adduct conformations and local structural distortions for minor groove-aligned benzo[a]pyrenyl-guanine lesions in a GG mutation hotspot context. Nucleic Acids Res. 35, 1555–1568. 10.1093/nar/gkm022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri A. K.; Mao B.; Amin S.; Geacintov N. E.; Patel D. J. (1999) Solution conformation of the (+)-trans-anti-benzo[g]chrysene-dA adduct opposite dT in a DNA duplex. J. Mol. Biol. 292, 289–307. 10.1006/jmbi.1999.2974. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Liu Z.; Ding S.; Lin C. H.; Cai Y.; Rodriguez F. A.; Sayer J. M.; Jerina D. M.; Amin S.; Broyde S.; Geacintov N. E. (2012) Nuclear magnetic resonance solution structure of an N(2)-guanine DNA adduct derived from the potent tumorigen dibenzo[a,l]pyrene: intercalation from the minor groove with ruptured Watson-Crick base pairing. Biochemistry 51, 9751–9762. 10.1021/bi3013577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Ding S.; Kropachev K.; Lei J.; Amin S.; Broyde S.; Geacintov N. E. (2015) Resistance to nucleotide excision repair of bulky guanine adducts opposite abasic sites in DNA duplexes and relationships between structure and function. PLoS One 10, e0137124. 10.1371/journal.pone.0137124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegar I. S.; Chary P.; Jabil R. J.; Tamura P. J.; Johansen T. N.; Lloyd R. S.; Harris C. M.; Harris T. M.; Stone M. P. (1998) Multiple conformations of an intercalated (−)-(7S,8R,9S, 10R)-N6-[10- (7,8,9,10-tetrahydrobenzo[a]pyrenyl)]-2′-deoxyadenosyl adduct in the N- ras codon 61 sequence. Biochemistry 37, 16516–16528. 10.1021/bi9817616. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Schnetz-Boutaud N. C.; Kroth H.; Yagi H.; Sayer J. M.; Kumar S.; Jerina D. M.; Stone M. P. (2008) 3′-Intercalation of a N2-dG 1R-trans-anti-benzo[c]phenanthrene DNA adduct in an iterated (CG)3 repeat. Chem. Res. Toxicol. 21, 1348–1358. 10.1021/tx7004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennard C.; Finneman J.; Harris C. M.; Harris T. M.; Stone M. P. (2001) The nonmutagenic (R)- and (S)-beta-(N(6)-adenyl)styrene oxide adducts are oriented in the major groove and show little perturbation to DNA structure. Biochemistry 40, 9780–9791. 10.1021/bi010564v. [DOI] [PubMed] [Google Scholar]
- Li Z.; Kim H. Y.; Tamura P. J.; Harris C. M.; Harris T. M.; Stone M. P. (1999) Intercalation of the (1S,2R,3S,4R)-N6-[1-(1,2,3,4-tetrahydro-2,3, 4-trihydroxybenz[a]anthracenyl)]-2′-deoxyadenosyl adduct in an oligodeoxynucleotide containing the human N-ras codon 61 sequence. Biochemistry 38, 16045–16057. 10.1021/bi9903650. [DOI] [PubMed] [Google Scholar]
- Li Z.; Kim H. Y.; Tamura P. J.; Harris C. M.; Harris T. M.; Stone M. P. (1999) Role of a polycyclic aromatic hydrocarbon bay region ring in modulating DNA adduct structure: the non-bay region (8S,9R,10S, 11R)-N(6)-[11-(8,9,10,11-tetrahydro-8,9, 10-trihydroxybenz[a]anthracenyl)]-2′-deoxyadenosyl adduct in codon 61 of the human N-ras protooncogene. Biochemistry 38, 14820–14832. 10.1021/bi991607z. [DOI] [PubMed] [Google Scholar]
- Li Z.; Mao H.; Kim H. Y.; Tamura P. J.; Harris C. M.; Harris T. M.; Stone M. P. (1999) Intercalation of the (−)-(1R,2S,3R, 4S)-N6-[1-benz[a]anthracenyl]-2′-deoxyadenosyl adduct in an oligodeoxynucleotide containing the human N-ras codon 61 sequence. Biochemistry 38, 2969–2981. 10.1021/bi982072x. [DOI] [PubMed] [Google Scholar]
- Li Z.; Tamura P. J.; Wilkinson A. S.; Harris C. M.; Harris T. M.; Stone M. P. (2001) Intercalation of the (1R,2S,3R,4S)-N6-[1-(1,2,3,4-tetrahydro-2,3,4-trihydroxybenz[a]anthracenyl)]-2′-d eoxyadenosyl adduct in the N-ras codon 61 sequence: DNA sequence effects. Biochemistry 40, 6743–6755. 10.1021/bi002785r. [DOI] [PubMed] [Google Scholar]
- Painter S. L.; Zegar I. S.; Tamura P. J.; Bluhm S.; Harris C. M.; Harris T. M.; Stone M. P. (1999) Influence of the R(61,2)- and S(61,2)-alpha-(N6-adenyl)styrene oxide adducts on the A.C mismatched base pair in an oligodeoxynucleotide containing the human N-ras codon 61. Biochemistry 38, 8635–8646. 10.1021/bi9900323. [DOI] [PubMed] [Google Scholar]
- Setayesh F. R.; DeCorte B. L.; Horton P.; Harris C. M.; Harris T. M.; Stone M. P. (1998) Styrene oxide adducts in an oligodeoxynucleotide containing the human N-ras codon 12: minor groove structures of the R(12,1)- and S(12,1)-alpha-(N2-guanyl) stereoisomers determined by 1H nuclear magnetic resonance. Chem. Res. Toxicol. 11, 766–777. 10.1021/tx9800147. [DOI] [PubMed] [Google Scholar]
- Pradhan P.; Tirumala S.; Liu X.; Sayer J. M.; Jerina D. M.; Yeh H. J. (2001) Solution structure of a trans-opened (10S)-dA adduct of (+)- (7S,8R,9S,10R)-7,8-dihydroxy-9,10-epoxy-7,8,9,10- tetrahydrobenzo[a]pyrene in a fully complementary DNA duplex: evidence for a major syn conformation. Biochemistry 40, 5870–5881. 10.1021/bi002896q. [DOI] [PubMed] [Google Scholar]
- Schwartz J. L.; Rice J. S.; Luxon B. A.; Sayer J. M.; Xie G.; Yeh H. J.; Liu X.; Jerina D. M.; Gorenstein D. G. (1997) Solution structure of the minor conformer of a DNA duplex containing a dG mismatch opposite a benzo[a]pyrene diol epoxide/dA adduct: glycosidic rotation from syn to anti at the modified deoxyadenosine. Biochemistry 36, 11069–11076. 10.1021/bi971306u. [DOI] [PubMed] [Google Scholar]
- Volk D. E.; Thiviyanathan V.; Rice J. S.; Luxon B. A.; Shah J. H.; Yagi H.; Sayer J. M.; Yeh H. J.; Jerina D. M.; Gorenstein D. G. (2003) Solution structure of a cis-opened (10R)-N6-deoxyadenosine adduct of (9S,10R)-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene in a DNA duplex. Biochemistry 42, 1410–1420. 10.1021/bi026745u. [DOI] [PubMed] [Google Scholar]
- Schurter E. J.; Yeh H. J.; Sayer J. M.; Lakshman M. K.; Yagi H.; Jerina D. M.; Gorenstein D. G. (1995) NMR solution structure of a nonanucleotide duplex with a dG mismatch opposite a 10R adduct derived from trans addition of a deoxyadenosine N6-amino group to (−)-(7S,8R,9R,10S)-7,8-dihydroxy-9,10-epoxy-7,8,9,10- tetrahydrobenzo[a]pyrene. Biochemistry 34, 1364–1375. 10.1021/bi00004a031. [DOI] [PubMed] [Google Scholar]
- Yeh H. J.; Sayer J. M.; Liu X.; Altieri A. S.; Byrd R. A.; Lakshman M. K.; Yagi H.; Schurter E. J.; Gorenstein D. G.; Jerina D. M. (1995) NMR solution structure of a nonanucleotide duplex with a dG mismatch opposite a 10S adduct N6-amino derived from trans addition of a deoxyadenosine group to (+)-(7R,8S,9S,10R)-7,8-dihydroxy-9,10-epoxy-7,8,9,10- tetrahydrobenzo[a]pyrene: an unusual syn glycosidic torsion angle at the modified dA. Biochemistry 34, 13570–13581. 10.1021/bi00041a037. [DOI] [PubMed] [Google Scholar]
- O’Handley S. F.; Sanford D. G.; Xu R.; Lester C. C.; Hingerty B. E.; Broyde S.; Krugh T. R. (1993) Structural characterization of an N-acetyl-2-aminofluorene (AAF) modified DNA oligomer by NMR, energy minimization, and molecular dynamics. Biochemistry 32, 2481–2497. 10.1021/bi00061a005. [DOI] [PubMed] [Google Scholar]
- Eckel L. M.; Krugh T. R. (1994) 2-Aminofluorene modified DNA duplex exists in two interchangeable conformations. Nat. Struct. Mol. Biol. 1, 89–94. 10.1038/nsb0294-89. [DOI] [PubMed] [Google Scholar]
- Patel D. J.; Mao B.; Gu Z.; Hingerty B. E.; Gorin A.; Basu A. K.; Broyde S. (1998) Nuclear magnetic resonance solution structures of covalent aromatic amine-DNA adducts and their mutagenic relevance. Chem. Res. Toxicol. 11, 391–407. 10.1021/tx9702143. [DOI] [PubMed] [Google Scholar]
- Zaliznyak T.; Bonala R.; Johnson F.; de Los Santos C. (2006) Structure and stability of duplex DNA containing the 3-(deoxyguanosin-N2-yl)-2-acetylaminofluorene (dG(N2)-AAF) lesion: a bulky adduct that persists in cellular DNA. Chem. Res. Toxicol. 19, 745–752. 10.1021/tx060002i. [DOI] [PubMed] [Google Scholar]
- Cho B. P.; Beland F. A.; Marques M. M. (1994) NMR structural studies of a 15-mer DNA duplex from a ras protooncogene modified with the carcinogen 2-aminofluorene: conformational heterogeneity. Biochemistry 33, 1373–1384. 10.1021/bi00172a013. [DOI] [PubMed] [Google Scholar]
- Gu Z.; Gorin A.; Hingerty B. E.; Broyde S.; Patel D. J. (1999) Solution structures of aminofluorene [AF]-stacked conformers of the syn [AF]-C8-dG adduct positioned opposite dC or dA at a template-primer junction. Biochemistry 38, 10855–10870. 10.1021/bi991266p. [DOI] [PubMed] [Google Scholar]
- Mao B.; Cosman M.; Hingerty B. E.; Broyde S.; Patel D. J. (1995) Solution conformation of [AF]dG opposite a −1 deletion site in a DNA duplex: intercalation of the covalently attached aminofluorene ring into the helix with base displacement of the C8-modified Syn guanine into the major groove. Biochemistry 34, 6226–6238. 10.1021/bi00018a027. [DOI] [PubMed] [Google Scholar]
- Mao B.; Hingerty B. E.; Broyde S.; Patel D. J. (1998) Solution structure of the aminofluorene [AF]-external conformer of the anti-[AF]-C8-dG adduct opposite dC in a DNA duplex. Biochemistry 37, 95–106. 10.1021/bi972258g. [DOI] [PubMed] [Google Scholar]
- Mao B.; Hingerty B. E.; Broyde S.; Patel D. J. (1998) Solution structure of the aminofluorene [AF]-intercalated conformer of the syn-[AF]-C8-dG adduct opposite dC in a DNA duplex. Biochemistry 37, 81–94. 10.1021/bi972257o. [DOI] [PubMed] [Google Scholar]
- Gu Z.; Gorin A.; Krishnasamy R.; Hingerty B. E.; Basu A. K.; Broyde S.; Patel D. J. (1999) Solution structure of the N-(deoxyguanosin-8-yl)-1-aminopyrene ([AP]dG) adduct opposite dA in a DNA duplex. Biochemistry 38, 10843–10854. 10.1021/bi9912138. [DOI] [PubMed] [Google Scholar]
- Patnaik S.; Cho B. P. (2010) Structures of 2-acetylaminofluorene modified DNA revisited: insight into conformational heterogeneity. Chem. Res. Toxicol. 23, 1650–1652. 10.1021/tx100341u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F.; Cho B. P. (2010) Enthalpy-entropy contribution to carcinogen-induced DNA conformational heterogeneity. Biochemistry 49, 259–266. 10.1021/bi901629p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V.; Hilton B.; Lin B.; Patnaik S.; Liang F.; Darian E.; Zou Y.; Mackerell A. D. Jr.; Cho B. P. (2013) Unusual sequence effects on nucleotide excision repair of arylamine lesions: DNA bending/distortion as a primary recognition factor. Nucleic Acids Res. 41, 869–880. 10.1093/nar/gks1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V.; Hilton B.; Patnaik S.; Zou Y.; Chiarelli M. P.; Cho B. P. (2012) Conformational and thermodynamic properties modulate the nucleotide excision repair of 2-aminofluorene and 2-acetylaminofluorene dG adducts in the NarI sequence. Nucleic Acids Res. 40, 3939–3951. 10.1093/nar/gkr1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavros K. M.; Hawkins E. K.; Rizzo C. J.; Stone M. P. (2014) Base-displaced intercalation of the 2-amino-3-methylimidazo[4,5-f]quinolone N2-dG adduct in the NarI DNA recognition sequence. Nucleic Acids Res. 42, 3450–3463. 10.1093/nar/gkt1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavros K. M.; Hawkins E. K.; Rizzo C. J.; Stone M. P. (2015) Base-Displaced Intercalated Conformation of the 2-Amino-3-methylimidazo[4,5-f]quinoline N(2)-dG DNA Adduct Positioned at the Nonreiterated G(1) in the NarI Restriction Site. Chem. Res. Toxicol. 28, 1455–1468. 10.1021/acs.chemrestox.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; DeMuro N. E.; Elmquist C. E.; Stover J. S.; Rizzo C. J.; Stone M. P. (2006) Base-displaced intercalated structure of the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in the recognition sequence of the NarI restriction enzyme, a hotspot for −2 bp deletions. J. Am. Chem. Soc. 128, 10085–10095. 10.1021/ja062004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Elmquist C. E.; Stover J. S.; Rizzo C. J.; Stone M. P. (2007) DNA sequence modulates the conformation of the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in the recognition sequence of the NarI restriction enzyme. Biochemistry 46, 8498–8516. 10.1021/bi700361u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukin M.; de Los Santos C. (2006) NMR structures of damaged DNA. Chem. Rev. 106, 607–686. 10.1021/cr0404646. [DOI] [PubMed] [Google Scholar]
- Stone M. P.; Huang H.; Brown K. L.; Shanmugam G. (2011) Chemistry and structural biology of DNA damage and biological consequences. Chem. Biodiversity 8, 1571–1615. 10.1002/cbdv.201100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2010) The Chemical Biology of DNA Damage (Geacintov N. E., and Broyde S., Eds.) Wiley-VCH Verlag GMBH & Co., Weinheim, Germany. [Google Scholar]
- Cai Y., Kropachev K., Kolbanovskiy M., and Kolbanovskiy A. (2010) Recognition and Removal of Bulky DNA Lesions by the Nucleotide Excision Repair System, in Chemical Biology of DNA Damage (Geacintov N. E., and Broyde S., Eds.) pp 261–298, Wiley-VCH, Weinheim, Germany. [Google Scholar]
- Cosman M.; de los Santos C.; Fiala R.; Hingerty B. E.; Singh S. B.; Ibanez V.; Margulis L. A.; Live D.; Geacintov N. E.; Broyde S.; et al. (1992) Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc. Natl. Acad. Sci. U. S. A. 89, 1914–1918. 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Santos C.; Cosman M.; Hingerty B. E.; Ibanez V.; Margulis L. A.; Geacintov N. E.; Broyde S.; Patel D. J. (1992) Influence of benzo[a]pyrene diol epoxide chirality on solution conformations of DNA covalent adducts: the (−)-trans-anti-[BP]G.C adduct structure and comparison with the (+)-trans-anti-[BP]G.C enantiomer. Biochemistry 31, 5245–5252. 10.1021/bi00138a002. [DOI] [PubMed] [Google Scholar]
- Xie X. M.; Geacintov N. E.; Broyde S. (1999) Origins of conformational differences between cis and trans DNA adducts derived from enantiomeric anti-benzo[a]pyrene diol epoxides. Chem. Res. Toxicol. 12, 597–609. 10.1021/tx990021a. [DOI] [PubMed] [Google Scholar]
- Xie X. M.; Geacintov N. E.; Broyde S. (1999) Stereochemical origin of opposite orientations in DNA adducts derived from enantiomeric anti-benzo[a]pyrene diol epoxides with different tumorigenic potentials. Biochemistry 38, 2956–2968. 10.1021/bi9825605. [DOI] [PubMed] [Google Scholar]
- Geacintov N. E.; Shafirovich V. Y.; B L.; Mao B.; Ya N. (1997) Photoinduced electron transfer, fluorescence, and intrastrand migration of reactive intermediates in pyrenyl sensitizer - modified DNA duplexes. Spectrum 10, 2–9. [Google Scholar]
- Cosman M.; de los Santos C.; Fiala R.; Hingerty B. E.; Ibanez V.; Luna E.; Harvey R.; Geacintov N. E.; Broyde S.; Patel D. J. (1993) Solution conformation of the (+)-cis-anti-[BP]dG adduct in a DNA duplex: intercalation of the covalently attached benzo[a]pyrenyl ring into the helix and displacement of the modified deoxyguanosine. Biochemistry 32, 4145–4155. 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- Cosman M.; Hingerty B. E.; Luneva N.; Amin S.; Geacintov N. E.; Broyde S.; Patel D. J. (1996) Solution conformation of the (−)-cis-anti-benzo[a]pyrenyl-dG adduct opposite dC in a DNA duplex: intercalation of the covalently attached BP ring into the helix with base displacement of the modified deoxyguanosine into the major groove. Biochemistry 35, 9850–9863. 10.1021/bi9605346. [DOI] [PubMed] [Google Scholar]
- Rodriguez F. A.; Liu Z.; Lin C. H.; Ding S.; Cai Y.; Kolbanovskiy A.; Kolbanovskiy M.; Amin S.; Broyde S.; Geacintov N. E. (2014) Nuclear magnetic resonance studies of an N2-guanine adduct derived from the tumorigen dibenzo[a,l]pyrene in DNA: impact of adduct stereochemistry, size, and local DNA sequence on solution conformations. Biochemistry 53, 1827–1841. 10.1021/bi4017044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegar I. S.; Kim S. J.; Johansen T. N.; Horton P. J.; Harris C. M.; Harris T. M.; Stone M. P. (1996) Adduction of the human N-ras codon 61 sequence with (−)-(7S,8R,9R,10S)- 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a] pyrene: structural refinement of the intercalated SRSR(61,2) (−)-(7S,8R,9S,10R)-N6-[10- (7,8,9,10- tetrahydrobenzo[a]pyrenyl)]-2′-deoxyadenosyl adduct from 1H NMR. Biochemistry 35, 6212–6224. 10.1021/bi9524732. [DOI] [PubMed] [Google Scholar]
- Volk D. E.; Rice J. S.; Luxon B. A.; Yeh H. J.; Liang C.; Xie G.; Sayer J. M.; Jerina D. M.; Gorenstein D. G. (2000) NMR evidence for syn-anti interconversion of a trans opened (10R)-dA adduct of benzo[a]pyrene (7S,8R)-diol (9R,10S)-epoxide in a DNA duplex. Biochemistry 39, 14040–14053. 10.1021/bi001669l. [DOI] [PubMed] [Google Scholar]
- Mao B.; Gu Z.; Gorin A.; Chen J.; Hingerty B. E.; Amin S.; Broyde S.; Geacintov N. E.; Patel D. J. (1999) Solution structure of the (+)-cis-anti-benzo[a]pyrene-dA ([BP]dA) adduct opposite dT in a DNA duplex. Biochemistry 38, 10831–10842. 10.1021/bi991212f. [DOI] [PubMed] [Google Scholar]
- Cosman M.; Fiala R.; Hingerty B. E.; Laryea A.; Lee H.; Harvey R. G.; Amin S.; Geacintov N. E.; Broyde S.; Patel D. (1993) Solution conformation of the (+)-trans-anti-[BPh]dA adduct opposite dT in a DNA duplex: intercalation of the covalently attached benzo[c]phenanthrene to the 5′-side of the adduct site without disruption of the modified base pair. Biochemistry 32, 12488–12497. 10.1021/bi00097a029. [DOI] [PubMed] [Google Scholar]
- Cosman M.; Laryea A.; Fiala R.; Hingerty B. E.; Amin S.; Geacintov N. E.; Broyde S.; Patel D. J. (1995) Solution conformation of the (−)-trans-anti-benzo[c]phenanthrene-dA ([BPh]dA) adduct opposite dT in a DNA duplex: intercalation of the covalently attached benzo[c]phenanthrenyl ring to the 3′-side of the adduct site and comparison with the (+)-trans-anti-[BPh]dA opposite dT stereoisomer. Biochemistry 34, 1295–1307. 10.1021/bi00004a024. [DOI] [PubMed] [Google Scholar]
- Lin C. H.; Huang X.; Kolbanovskii A.; Hingerty B. E.; Amin S.; Broyde S.; Geacintov N. E.; Patel D. J. (2001) Molecular topology of polycyclic aromatic carcinogens determines DNA adduct conformation: a link to tumorigenic activity. J. Mol. Biol. 306, 1059–1080. 10.1006/jmbi.2001.4425. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Geacintov N. E.; Broyde S. (2012) Nucleotide excision repair efficiencies of bulky carcinogen-DNA adducts are governed by a balance between stabilizing and destabilizing interactions. Biochemistry 51, 1486–1499. 10.1021/bi201794x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaLucia J. Jr.; Allawi H. T.; Seneviratne P. A. (1996) Improved nearest-neighbor parameters for predicting DNA duplex stability. Biochemistry 35, 3555–3562. 10.1021/bi951907q. [DOI] [PubMed] [Google Scholar]
- Ruan Q.; Liu T.; Kolbanovskiy A.; Liu Y.; Ren J.; Skorvaga M.; Zou Y.; Lader J.; Malkani B.; Amin S.; Van Houten B.; Geacintov N. E. (2007) Sequence context- and temperature-dependent nucleotide excision repair of a benzo[a]pyrene diol epoxide-guanine DNA adduct catalyzed by thermophilic UvrABC proteins. Biochemistry 46, 7006–7015. 10.1021/bi700294k. [DOI] [PubMed] [Google Scholar]
- Brown K. L.; Roginskaya M.; Zou Y.; Altamirano A.; Basu A. K.; Stone M. P. (2010) Binding of the human nucleotide excision repair proteins XPA and XPC/HR23B to the 5R-thymine glycol lesion and structure of the cis-(5R,6S) thymine glycol epimer in the 5′-GTgG-3′ sequence: destabilization of two base pairs at the lesion site. Nucleic Acids Res. 38, 428–440. 10.1093/nar/gkp844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Das R. S.; Basu A. K.; Stone M. P. (2011) Structure of (5′S)-8,5′-cyclo-2′-deoxyguanosine in DNA. J. Am. Chem. Soc. 133, 20357–20368. 10.1021/ja207407n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukin M.; Zaliznyak T.; Johnson F.; de los Santos C. (2012) Structure and stability of DNA containing an aristolactam II-dA lesion: implications for the NER recognition of bulky adducts. Nucleic Acids Res. 40, 2759–2770. 10.1093/nar/gkr1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaliznyak T.; Lukin M.; de los Santos C. (2012) Structure and stability of duplex DNA containing (5′S)-5′,8-cyclo-2′-deoxyadenosine: an oxidatively generated lesion repaired by NER. Chem. Res. Toxicol. 25, 2103–2111. 10.1021/tx300193k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji M. K.; Moggs J. G.; Kuraoka I.; Wood R. D. (1999) Dual-incision assays for nucleotide excision repair using DNA with a lesion at a specific site. Methods Mol. Biol. 113, 373–392. 10.1007/978-1-4612-1608-7_30. [DOI] [PubMed] [Google Scholar]
- Kropachev K.; Kolbanovskii M.; Cai Y.; Rodriguez F.; Kolbanovskii A.; Liu Y.; Zhang L.; Amin S.; Patel D.; Broyde S.; Geacintov N. E. (2009) The sequence dependence of human nucleotide excision repair efficiencies of benzo[a]pyrene-derived DNA lesions: insights into the structural factors that favor dual incisions. J. Mol. Biol. 386, 1193–1203. 10.1016/j.jmb.2008.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu H.; Kropachev K.; Wang L.; Zhang L.; Kolbanovskiy A.; Kolbanovskiy M.; Geacintov N. E.; Broyde S. (2012) Nucleotide excision repair of 2-acetylaminofluorene- and 2-aminofluorene-(C8)-guanine adducts: molecular dynamics simulations elucidate how lesion structure and base sequence context impact repair efficiencies. Nucleic Acids Res. 40, 9675–9690. 10.1093/nar/gks788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H.; Gaddameedhi S.; Kim S. Y.; Hu J.; Kemp M. G.; Sancar A. (2014) Highly specific and sensitive method for measuring nucleotide excision repair kinetics of ultraviolet photoproducts in human cells. Nucleic Acids Res. 42, e29. 10.1093/nar/gkt1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess M. T.; Gunz D.; Luneva N.; Geacintov N. E.; Naegeli H. (1997) Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol. Cell. Biol. 17, 7069–7076. 10.1128/MCB.17.12.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves D. A.; Mu H.; Kropachev K.; Cai Y.; Ding S.; Kolbanovskiy A.; Kolbanovskiy M.; Chen Y.; Krzeminski J.; Amin S.; Patel D. J.; Broyde S.; Geacintov N. E. (2011) Resistance of bulky DNA lesions to nucleotide excision repair can result from extensive aromatic lesion-base stacking interactions. Nucleic Acids Res. 39, 8752–8764. 10.1093/nar/gkr537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buterin T.; Meyer C.; Giese B.; Naegeli H. (2005) DNA quality control by conformational readout on the undamaged strand of the double helix. Chem. Biol. 12, 913–922. 10.1016/j.chembiol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Mu H.; Geacintov N. E.; Min J. H.; Zhang Y.; Broyde S. (2017) The nucleotide excision repair lesion-recognition protein Rad4 captures a pre-flipped partner base in a benzo[a]pyrene-derived DNA lesion: how structure impacts the binding pathway. Chem. Res. Toxicol. 30, 1344. 10.1021/acs.chemrestox.7b00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. C.; Cai Y.; Mu H.; Broyde S.; Amin S.; Chen X.; Min J. H.; Geacintov N. E. (2014) The relationships between XPC binding to conformationally diverse DNA adducts and their excision by the human NER system: Is there a correlation?. DNA Repair 19, 55–63. 10.1016/j.dnarep.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton B.; Gopal S.; Xu L.; Mazumder S.; Musich P. R.; Cho B. P.; Zou Y. (2016) Dissociation Dynamics of XPC-RAD23B from Damaged DNA Is a Determining Factor of NER Efficiency. PLoS One 11, e0157784. 10.1371/journal.pone.0157784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty D.; Rapic’-Otrin V.; Levine A. S.; Wood R. D. (2000) Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J. Mol. Biol. 300, 275–290. 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- Clement F. C.; Camenisch U.; Fei J.; Kaczmarek N.; Mathieu N.; Naegeli H. (2010) Dynamic two-stage mechanism of versatile DNA damage recognition by xeroderma pigmentosum group C protein. Mutat. Res., Fundam. Mol. Mech. Mutagen. 685, 21–28. 10.1016/j.mrfmmm.2009.08.005. [DOI] [PubMed] [Google Scholar]
- Hey T.; Lipps G.; Sugasawa K.; Iwai S.; Hanaoka F.; Krauss G. (2002) The XPC-HR23B complex displays high affinity and specificity for damaged DNA in a true-equilibrium fluorescence assay. Biochemistry 41, 6583–6587. 10.1021/bi012202t. [DOI] [PubMed] [Google Scholar]
- Shell S. M.; Hawkins E. K.; Tsai M. S.; Hlaing A. S.; Rizzo C. J.; Chazin W. J. (2013) Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA Repair 12, 947–953. 10.1016/j.dnarep.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K. (2010) Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat. Res., Fundam. Mol. Mech. Mutagen. 685, 29–37. 10.1016/j.mrfmmm.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Sugasawa K.; Masutani C.; Uchida A.; Maekawa T.; van der Spek P. J.; Bootsma D.; Hoeijmakers J. H.; Hanaoka F. (1996) HHR23B, a human Rad23 homolog, stimulates XPC protein in nucleotide excision repair in vitro. Mol. Cell. Biol. 16, 4852–4861. 10.1128/MCB.16.9.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K.; Shimizu Y.; Iwai S.; Hanaoka F. (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair 1, 95–107. 10.1016/S1568-7864(01)00008-8. [DOI] [PubMed] [Google Scholar]
- Yeo J. E.; Khoo A.; Fagbemi A. F.; Scharer O. D. (2012) The efficiencies of damage recognition and excision correlate with duplex destabilization induced by acetylaminofluorene adducts in human nucleotide excision repair. Chem. Res. Toxicol. 25, 2462–2468. 10.1021/tx3003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon J. T.; Nichols A. F.; Keeney S.; Smith C. A.; Taylor J. S.; Linn S.; Sancar A. (1993) Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6–4]T, and T[Dewar]T. J. Biol. Chem. 268, 21301–21308. [PubMed] [Google Scholar]
- Cai Y.; Patel D. J.; Geacintov N. E.; Broyde S. (2007) Dynamics of a benzo[a]pyrene-derived guanine DNA lesion in TGT and CGC sequence contexts: enhanced mobility in TGT explains conformational heterogeneity, flexible bending, and greater susceptibility to nucleotide excision repair. J. Mol. Biol. 374, 292–305. 10.1016/j.jmb.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Patel D. J.; Broyde S.; Geacintov N. E. (2010) Base sequence context effects on nucleotide excision repair. J. Nucleic Acids 2010, 1. 10.4061/2010/174252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreij K.; Seidel A.; Jernstrom B. (2005) Differential removal of DNA adducts derived from anti-diol epoxides of dibenzo[a,l]pyrene and benzo[a]pyrene in human cells. Chem. Res. Toxicol. 18, 655–664. 10.1021/tx0497090. [DOI] [PubMed] [Google Scholar]
- Suh M.; Ariese F.; Small G. J.; Jankowiak R.; Hewer A.; Phillips D. H. (1995) Formation and persistence of benzo[a]pyrene-DNA adducts in mouse epidermis in vivo: importance of adduct conformation. Carcinogenesis 16, 2561–2569. 10.1093/carcin/16.10.2561. [DOI] [PubMed] [Google Scholar]
- Schurter E. J.; Sayer J. M.; Oh-hara T.; Yeh H. J.; Yagi H.; Luxon B. A.; Jerina D. M.; Gorenstein D. G. (1995) Nuclear magnetic resonance solution structure of an undecanucleotide duplex with a complementary thymidine base opposite a 10R adduct derived from trans addition of a deoxyadenosine N6-amino group to (−)-(7R,8S,9R,10S)-7,8-dihydroxy- 9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Biochemistry 34, 9009–9020. 10.1021/bi00028a009. [DOI] [PubMed] [Google Scholar]
- Zegar I. S.; Stone M. P. (1996) Solution structure of an oligodeoxynucleotide containing the human N-ras codon 12 sequence refined from 1H NMR using molecular dynamics restrained by nuclear Overhauser effects. Chem. Res. Toxicol. 9, 114–125. 10.1021/tx9500787. [DOI] [PubMed] [Google Scholar]
- Yan S.; Shapiro R.; Geacintov N. E.; Broyde S. (2001) Stereochemical, structural, and thermodynamic origins of stability differences between stereoisomeric benzo[a]pyrene diol epoxide deoxyadenosine adducts in a DNA mutational hot spot sequence. J. Am. Chem. Soc. 123, 7054–7066. 10.1021/ja0043035. [DOI] [PubMed] [Google Scholar]
- Yan S.; Wu M.; Buterin T.; Naegeli H.; Geacintov N. E.; Broyde S. (2003) Role of base sequence context in conformational equilibria and nucleotide excision repair of benzo[a]pyrene diol epoxide-adenine adducts. Biochemistry 42, 2339–2354. 10.1021/bi0270081. [DOI] [PubMed] [Google Scholar]
- Buterin T.; Hess M. T.; Luneva N.; Geacintov N. E.; Amin S.; Kroth H.; Seidel A.; Naegeli H. (2000) Unrepaired fjord region polycyclic aromatic hydrocarbon-DNA adducts in ras codon 61 mutational hot spots. Cancer Res. 60, 1849–1856. [PubMed] [Google Scholar]
- Geacintov N. E.; Broyde S.; Buterin T.; Naegeli H.; Wu M.; Yan S.; Patel D. J. (2002) Thermodynamic and structural factors in the removal of bulky DNA adducts by the nucleotide excision repair machinery. Biopolymers 65, 202–210. 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- Geacintov N. E., Naegeli H., Patel D. J., and Broyde S. (2006) Structural aspects of polycyclic aromatic carcinogen-damaged DNA and its recognition by NER proteins, in DNA Damage and Recognition (Siede W., Kow Y. W., and Doetsch P. W., Eds.) Taylor & Francis, London. [Google Scholar]
- Moriya M.; Spiegel S.; Fernandes A.; Amin S.; Liu T.; Geacintov N.; Grollman A. P. (1996) Fidelity of translesional synthesis past benzo[a]pyrene diol epoxide-2′-deoxyguanosine DNA adducts: marked effects of host cell, sequence context, and chirality. Biochemistry 35, 16646–16651. 10.1021/bi9608875. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Ohashi E.; Kolbanovskiy A.; Geacintov N. E.; Grollman A. P.; Ohmori H.; Shibutani S. (2002) Translesion synthesis by human DNA polymerase kappa on a DNA template containing a single stereoisomer of dG-(+)- or dG-(−)-anti-N(2)-BPDE (7,8-dihydroxy-anti-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene). Biochemistry 41, 6100–6106. 10.1021/bi020049c. [DOI] [PubMed] [Google Scholar]
- Xu P.; Oum L.; Lee Y. C.; Geacintov N. E.; Broyde S. (2009) Visualizing sequence-governed nucleotide selectivities and mutagenic consequences through a replicative cycle: processing of a bulky carcinogen N2-dG lesion in a Y-family DNA polymerase. Biochemistry 48, 4677–4690. 10.1021/bi802363f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adar S.; Izhar L.; Hendel A.; Geacintov N.; Livneh Z. (2009) Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res. 37, 5737–5748. 10.1093/nar/gkp632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan C. J.; Bacolod M. D.; Vyas R. R.; Liu T.; Geacintov N. E.; Loechler E.C L.; Basu A. K. (1997) Sequence specific mutagenesis of the major (+)-anti-benzo[a]pyrene diol epoxide-DNA adduct at a mutational hot spot in vitro and in Escherichia coli cells. Chem. Res. Toxicol. 10, 369–377. 10.1021/tx9601925. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Ding S.; Kropachev K.; Jia L.; Amin S.; Broyde S.; Geacintov N. E. (2015) Correction: Resistance to nucleotide excision repair of bulky guanine adducts opposite abasic sites in DNA duplexes and relationships between structure and function. PLoS One 10, e0142068. 10.1371/journal.pone.0142068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman M.; Fiala R.; Hingerty B. E.; Amin S.; Geacintov N. E.; Broyde S.; Patel D. J. (1994) Solution conformation of the (+)-cis-anti-[BP]dG adduct opposite a deletion site in a DNA duplex: intercalation of the covalently attached benzo[a]pyrene into the helix with base displacement of the modified deoxyguanosine into the minor groove. Biochemistry 33, 11518–11527. 10.1021/bi00204a014. [DOI] [PubMed] [Google Scholar]
- Mu H.; Kropachev K.; Chen Y.; Zhang H.; Cai Y.; Geacintov N. E.; Broyde S. (2013) Role of structural and energetic factors in regulating repair of a bulky DNA lesion with different opposite partner bases. Biochemistry 52, 5517–5521. 10.1021/bi4009177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz A. K.; Carrell H. L.; Glusker J. P. (1998) Dibenzo[a,l]pyrene (dibenzo[def,p]chrysene): fjord-region distortions. Carcinogenesis 19, 1641–1648. 10.1093/carcin/19.9.1641. [DOI] [PubMed] [Google Scholar]
- Cai Y.; Ding S.; Geacintov N. E.; Broyde S. (2011) Intercalative Conformations of the 14R (+)- and 14S (−)-trans-anti-DB[a,l]P-N(6)-dA Adducts: Molecular Modeling and MD Simulations. Chem. Res. Toxicol. 24, 522. 10.1021/tx1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell C. J.; Carrell H. L.; Glusker J. P.; Abu-Shaqara E.; Cortez C.; Harvey R. G. (1994) Bay- and fjord-region distortions in dibenz[a,j]anthracene and tetrabenzo[de,hi,mn,qr]naphthacene. Carcinogenesis 15, 2931–2936. 10.1093/carcin/15.12.2931. [DOI] [PubMed] [Google Scholar]
- Karle I. L.; Yagi H.; Sayer J. M.; Jerina D. M. (2004) Crystal and molecular structure of a benzo[a]pyrene 7,8-diol 9,10-epoxide N2-deoxyguanosine adduct: absolute configuration and conformation. Proc. Natl. Acad. Sci. U. S. A. 101, 1433–1438. 10.1073/pnas.0307305101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahalad A. K.; Ross J. A.; Nelson G. B.; Roop B. C.; King L. C.; Nesnow S.; Mass M. J. (1997) Dibenzo[a,l]pyrene-induced DNA adduction, tumorigenicity, and Ki-ras oncogene mutations in strain A/J mouse lung. Carcinogenesis 18, 1955–1963. 10.1093/carcin/18.10.1955. [DOI] [PubMed] [Google Scholar]
- Zhang S. M.; Chen K. M.; Aliaga C.; Sun Y. W.; Lin J. M.; Sharma A. K.; Amin S.; El-Bayoumy K. (2011) Identification and quantification of DNA adducts in the oral tissues of mice treated with the environmental carcinogen dibenzo[a,l]pyrene by HPLC-MS/MS. Chem. Res. Toxicol. 24, 1297–1303. 10.1021/tx200188j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri E. L.; Higginbotham S.; RamaKrishna N. V.; Devanesan P. D.; Todorovic R.; Rogan E. G.; Salmasi S. (1991) Comparative dose-response tumorigenicity studies of dibenzo[alpha,l]pyrene versus 7,12-dimethylbenz[alpha]anthracene, benzo[alpha]pyrene and two dibenzo[alpha,l]pyrene dihydrodiols in mouse skin and rat mammary gland. Carcinogenesis 12, 1939–1944. 10.1093/carcin/12.10.1939. [DOI] [PubMed] [Google Scholar]
- (2009) On the Impact of the Molecule Structure in Chemical Carcinogenesis (Luch A., Ed.) Vol. 1, Molecular Toxicology, Birkhauser Verlag, Switzerland. [DOI] [PubMed] [Google Scholar]
- Nesnow S.; Davis C.; Nelson G.; Ross J. A.; Allison J.; Adams L.; King L. C. (1997) Comparison of the morphological transforming activities of dibenzo[a,l]pyrene and benzo[a]pyrene in C3H10T1/2CL8 cells and characterization of the dibenzo[a,l]pyrene-DNA adducts. Carcinogenesis 18, 1973–1978. 10.1093/carcin/18.10.1973. [DOI] [PubMed] [Google Scholar]
- Ross J. A.; Nelson G. B.; Wilson K. H.; Rabinowitz J. R.; Galati A.; Stoner G. D.; Nesnow S.; Mass M. J. (1995) Adenomas induced by polycyclic aromatic hydrocarbons in strain A/J mouse lung correlate with time-integrated DNA adduct levels. Cancer Res. 55, 1039–1044. [PubMed] [Google Scholar]
- Szeliga J.; Dipple A. (1998) DNA adduct formation by polycyclic aromatic hydrocarbon dihydrodiol epoxides. Chem. Res. Toxicol. 11, 1–11. 10.1021/tx970142f. [DOI] [PubMed] [Google Scholar]
- Zhang S. M.; Chen K. M.; Sun Y. W.; Aliaga C.; Lin J. M.; Sharma A. K.; Amin S.; El-Bayoumy K. (2014) Simultaneous detection of deoxyadenosine and deoxyguanosine adducts in the tongue and other oral tissues of mice treated with Dibenzo[a,l]pyrene. Chem. Res. Toxicol. 27, 1199–1206. 10.1021/tx5001078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. H.; Besaratinia A.; Feng Z.; Tang M. S.; Amin S.; Luch A.; Pfeifer G. P. (2004) DNA damage, repair, and mutation induction by (+)-Syn and (−)-anti-dibenzo[a,l]pyrene-11,12-diol-13,14-epoxides in mouse cells. Cancer Res. 64, 7321–7328. 10.1158/0008-5472.CAN-04-1094. [DOI] [PubMed] [Google Scholar]
- Ralston S. L.; Seidel A.; Luch A.; Platt K. L.; Baird W. M. (1995) Stereoselective activation of dibenzo[a,l]pyrene to (−)-anti (11R,12S,13S,14R)- and (+)-syn(11S,12R,13S,14R)-11,12-diol-13,14- epoxides which bind extensively to deoxyadenosine residues of DNA in the human mammary carcinoma cell line MCF-7. Carcinogenesis 16, 2899–2907. 10.1093/carcin/16.12.2899. [DOI] [PubMed] [Google Scholar]
- Arif J. M.; Gupta R. C. (1997) Microsome-mediated bioactivation of dibenzo[a,l]pyrene and identification of DNA adducts by 32P-postlabeling. Carcinogenesis 18, 1999–2007. 10.1093/carcin/18.10.1999. [DOI] [PubMed] [Google Scholar]
- Mahadevan B.; Dashwood W. M.; Luch A.; Pecaj A.; Doehmer J.; Seidel A.; Pereira C.; Baird W. M. (2003) Mutations induced by (−)-anti-11R,12S-dihydrodiol 13S,14R-epoxide of dibenzo[a,l]pyrene in the coding region of the hypoxanthine phosphoribosyltransferase (Hprt) gene in Chinese hamster V79 cells. Environ. Mol. Mutagen. 41, 131–139. 10.1002/em.10136. [DOI] [PubMed] [Google Scholar]
- Phillips D. H.; Hewer A.; Seidel A.; Steinbrecher T.; Schrode R.; Oesch F.; Glatt H. (1991) Relationship between mutagenicity and DNA adduct formation in mammalian cells for fjord- and bay-region diol-epoxides of polycyclic aromatic hydrocarbons. Chem.-Biol. Interact. 80, 177–186. 10.1016/0009-2797(91)90023-Z. [DOI] [PubMed] [Google Scholar]
- Leavitt S. A.; George M. H.; Moore T.; Ross J. A. (2008) Mutations induced by benzo[a]pyrene and dibenzo[a,l]pyrene in lacI transgenic B6C3F1 mouse lung result from stable DNA adducts. Mutagenesis 23, 445–450. 10.1093/mutage/gen033. [DOI] [PubMed] [Google Scholar]
- Topinka J.; Marvanova S.; Vondracek J.; Sevastyanova O.; Novakova Z.; Krcmar P.; Pencikova K.; Machala M. (2008) DNA adducts formation and induction of apoptosis in rat liver epithelial ’stem-like’ cells exposed to carcinogenic polycyclic aromatic hydrocarbons. Mutat. Res., Fundam. Mol. Mech. Mutagen. 638, 122–132. 10.1016/j.mrfmmm.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Kropachev K.; Kolbanovskiy M.; Liu Z.; Cai Y.; Zhang L.; Schwaid A. G.; Kolbanovskiy A.; Ding S.; Amin S.; Broyde S.; Geacintov N.E. (2013) Adenine-DNA adducts derived from the highly tumorigenic dibenzo[a,l]pyrene are resistant to nucleotide excision repair while guanine adducts are not. Chem. Res. Toxicol. 26, 783–793. 10.1021/tx400080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Zheng H.; Ding S.; Kropachev K.; Schwaid A. G.; Tang Y.; Mu H.; Wang S.; Geacintov N. E.; Zhang Y.; Broyde S. (2013) Free energy profiles of base flipping in intercalative polycyclic aromatic hydrocarbon-damaged DNA duplexes: energetic and structural relationships to nucleotide excision repair susceptibility. Chem. Res. Toxicol. 26, 1115–1125. 10.1021/tx400156a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd D. R.; Hanawalt P. C. (2002) p53 controls global nucleotide excision repair of low levels of structurally diverse benzo(g)chrysene-DNA adducts in human fibroblasts. Cancer Res. 62, 5288–5294. [PubMed] [Google Scholar]
- Khutsishvili I.; Zhang N.; Marky L. A.; Crean C.; Patel D. J.; Geacintov N. E.; Shafirovich V. (2013) Thermodynamic profiles and nuclear magnetic resonance studies of oligonucleotide duplexes containing single diastereomeric spiroiminodihydantoin lesions. Biochemistry 52, 1354–1363. 10.1021/bi301566v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowiak R.; Ariese F.; Hewer A.; Luch A.; Zamzow D.; Hughes N. C.; Phillips D.; Seidel A.; Platt K.-L.; Oesch F.; Small G. J. (1998) Structure, Conformations, and Repair of DNA Adducts from Dibenzo[a,l]pyrene: 32P-Postlabeling and Fluorescence Studies. Chem. Res. Toxicol. 11, 674–685. 10.1021/tx970232k. [DOI] [PubMed] [Google Scholar]
- Lagerqvist A.; Hakansson D.; Prochazka G.; Lundin C.; Dreij K.; Segerback D.; Jernstrom B.; Tornqvist M.; Seidel A.; Erixon K.; Jenssen D. (2008) Both replication bypass fidelity and repair efficiency influence the yield of mutations per target dose in intact mammalian cells induced by benzo[a]pyrene-diol-epoxide and dibenzo[a,l]pyrene-diol-epoxide. DNA Repair 7, 1202–1212. 10.1016/j.dnarep.2008.03.022. [DOI] [PubMed] [Google Scholar]
- Spencer W. A.; Singh J.; Orren D. K. (2009) Formation and differential repair of covalent DNA adducts generated by treatment of human cells with (±)-anti-dibenzo[a,l]pyrene-11,12-diol-13,14-epoxide. Chem. Res. Toxicol. 22, 81–89. 10.1021/tx8001675. [DOI] [PubMed] [Google Scholar]
- Nadkarni A.; Burns J. A.; Gandolfi A.; Chowdhury M. A.; Cartularo L.; Berens C.; Geacintov N. E.; Scicchitano D. A. (2016) Nucleotide Excision Repair and Transcription-coupled DNA Repair Abrogate the Impact of DNA Damage on Transcription. J. Biol. Chem. 291, 848–861. 10.1074/jbc.M115.685271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson W. W.; Harris T. M.; Guengerich F. P. (1996) Kinetics and Mechanism of Hydrolysis of Aflatoxin B1 exo-8,9-Epoxide and Rearrangement of the Dihydrodiol. J. Am. Chem. Soc. 118, 8213–8220. 10.1021/ja960525k. [DOI] [Google Scholar]
- Baertschi S. W.; Raney K. D.; Stone M. P.; Harris T. M. (1988) Preparation of the 8, 9-epoxide of the mycotoxin aflatoxin B1: the ultimate carcinogenic species. J. Am. Chem. Soc. 110, 7929–7931. 10.1021/ja00231a083. [DOI] [Google Scholar]
- Essigmann J. M.; Croy R. G.; Nadzan A. M.; Busby W. F. Jr.; Reinhold V. N.; Buchi G.; Wogan G. N. (1977) Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc. Natl. Acad. Sci. U. S. A. 74, 1870–1874. 10.1073/pnas.74.5.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzog P. J.; Smith J. R.; Garner R. C. (1982) Characterisation of the imidazole ring-opened forms of trans-8,9-dihydro-8,9-dihydro-8-(7-guanyl)9-hydroxy aflatoxin B1. Carcinogenesis 3, 723–725. 10.1093/carcin/3.6.723. [DOI] [PubMed] [Google Scholar]
- Croy R. G.; Wogan G. N. (1981) Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1. Cancer Res. 41, 197–203. [PubMed] [Google Scholar]
- Leadon S. A.; Tyrrell R. M.; Cerutti P. A. (1981) Excision repair of aflatoxin B1-DNA adducts in human fibroblasts. Cancer Res. 41, 5125–5129. [PubMed] [Google Scholar]
- Giri I.; Jenkins M. D.; Schnetz-Boutaud N. C.; Stone M. P. (2002) Structural refinement of the 8,9-dihydro-8-(N7-guanyl)-9-hydroxy-aflatoxin B(1) adduct in a 5′-Cp(AFB)G-3′ sequence. Chem. Res. Toxicol. 15, 638–647. 10.1021/tx010187n. [DOI] [PubMed] [Google Scholar]
- Giri I.; Stone M. P. (2002) Thermal stabilization of the DNA duplex by adducts of aflatoxin B1. Biopolymers 65, 190–201. 10.1002/bip.10222. [DOI] [PubMed] [Google Scholar]
- Heflich R. H.; Neft R. E. (1994) Genetic toxicity of 2-acetylaminofluorene, 2-aminofluorene and some of their metabolites and model metabolites. Mutat. Res., Rev. Genet. Toxicol. 318, 73–114. 10.1016/0165-1110(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Purohit V.; Basu A. K. (2000) Mutagenicity of nitroaromatic compounds. Chem. Res. Toxicol. 13, 673–692. 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]
- Beije B.; Moeller L. (1988) 2-Nitrofluorene and related compounds: prevalence and biological effects. Mutat. Res., Rev. Genet. Toxicol. 196, 177–209. 10.1016/0165-1110(88)90019-X. [DOI] [PubMed] [Google Scholar]
- Howard P. C.; Casciano D. A.; Beland F. A.; Shaddock J. G. (1981) The binding of N-hydroxy-2-acetylaminofluorene to DNA and repair of the adducts in primary rat hepatocyte cultures. Carcinogenesis 2, 97–102. 10.1093/carcin/2.2.97. [DOI] [PubMed] [Google Scholar]
- Culp S. J.; Poirier M. C.; Beland F. A. (1993) Biphasic removal of DNA adducts in a repetitive DNA sequence after dietary administration of 2-acetylaminofluorene. Environ. Health Perspect 99, 273–275. 10.1289/ehp.9399273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X. S.; Eriksson L. C.; Moeller L. (1999) Formation and persistence of DNA adducts during and after a long-term administration of 2-nitrofluorene. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 442, 9–18. 10.1016/S1383-5718(99)00053-4. [DOI] [PubMed] [Google Scholar]
- Grad R.; Shapiro R.; Hingerty B. E.; Broyde S. (1997) A molecular mechanics and dynamics study of the minor adduct between DNA and the carcinogen 2-(acetylamino)fluorene (dG-N2-AAF). Chem. Res. Toxicol. 10, 1123–1132. 10.1021/tx970092e. [DOI] [PubMed] [Google Scholar]
- Kawanishi M.; Fujikawa Y.; Ishii H.; Nishida H.; Higashigaki Y.; Kanno T.; Matsuda T.; Takamura-Enya T.; Yagi T. (2013) Adduct formation and repair, and translesion DNA synthesis across the adducts in human cells exposed to 3-nitrobenzanthrone. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 753, 93–100. 10.1016/j.mrgentox.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Bieler C. A.; Cornelius M. G.; Stiborova M.; Arlt V. M.; Wiessler M.; Phillips D. H.; Schmeiser H. H. (2007) Formation and persistence of DNA adducts formed by the carcinogenic air pollutant 3-nitrobenzanthrone in target and non-target organs after intratracheal instillation in rats. Carcinogenesis 28, 1117–1121. 10.1093/carcin/bgl219. [DOI] [PubMed] [Google Scholar]
- Lukin M.; Zaliznyak T.; Johnson F.; de Los Santos C. R. (2011) Incorporation of 3-aminobenzanthrone into 2′-deoxyoligonucleotides and its impact on duplex stability. J. Nucleic Acids 2011, 521035. 10.4061/2011/521035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politica D. A.; Malik C. K.; Basu A. K.; Stone M. P. (2015) Base-Displaced Intercalated Structure of the N-(2′-Deoxyguanosin-8-yl)-3-aminobenzanthrone DNA Adduct. Chem. Res. Toxicol. 28, 2253–2266. 10.1021/acs.chemrestox.5b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae Y. H.; Delclos K. B.; Blaydes B.; el-Bayoumy K. (1996) Metabolism and DNA binding of the environmental colon carcinogen 6-nitrochrysene in rats. Cancer Res. 56, 2052–2058. [PubMed] [Google Scholar]
- Krzeminski J.; Kropachev K.; Kolbanovskiy M.; Reeves D.; Kolbanovskiy A.; Yun B. H.; Geacintov N. E.; Amin S.; El-Bayoumy K. (2011) Inefficient nucleotide excision repair in human cell extracts of the N-(deoxyguanosin-8-yl)-6-aminochrysene and 5-(deoxyguanosin-N(2)-yl)-6-aminochrysene adducts derived from 6-nitrochrysene. Chem. Res. Toxicol. 24, 65–72. 10.1021/tx100284h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzeminski J.; Kropachev K.; Reeves D.; Kolbanovskiy A.; Kolbanovskiy M.; Chen K. M.; Sharma A. K.; Geacintov N.; Amin S.; El-Bayoumy K. (2013) Adenine-DNA adduct derived from the nitroreduction of 6-nitrochrysene is more resistant to nucleotide excision repair than guanine-DNA adducts. Chem. Res. Toxicol. 26, 1746–1754. 10.1021/tx400296x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollman A. P.; Shibutani S.; Moriya M.; Miller F.; Wu L.; Moll U.; Suzuki N.; Fernandes A.; Rosenquist T.; Medverec Z.; Jakovina K.; Brdar B.; Slade N.; Turesky R. J.; Goodenough A. K.; Rieger R.; Vukelic M.; Jelakovic B. (2007) Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. U. S. A. 104, 12129–12134. 10.1073/pnas.0701248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist T. A.; Grollman A. P. (2016) Mutational signature of aristolochic acid: Clue to the recognition of a global disease. DNA Repair 44, 205–211. 10.1016/j.dnarep.2016.05.027. [DOI] [PubMed] [Google Scholar]
- Schmeiser H. H.; Frei E.; Wiessler M.; Stiborova M. (1997) Comparison of DNA adduct formation by aristolochic acids in various in vitro activation systems by 32P-post-labelling: evidence for reductive activation by peroxidases. Carcinogenesis 18, 1055–1062. 10.1093/carcin/18.5.1055. [DOI] [PubMed] [Google Scholar]
- Stiborova M.; Mareis J.; Frei E.; Arlt V. M.; Martinek V.; Schmeiser H. H. (2011) The human carcinogen aristolochic acid i is activated to form DNA adducts by human NAD(P)H:quinone oxidoreductase without the contribution of acetyltransferases or sulfotransferases. Environ. Mol. Mutagen 52, 448–459. 10.1002/em.20642. [DOI] [PubMed] [Google Scholar]
- Arlt V. M.; Stiborova M.; Schmeiser H. H. (2002) Aristolochic acid as a probable human cancer hazard in herbal remedies: a review. Mutagenesis 17, 265–277. 10.1093/mutage/17.4.265. [DOI] [PubMed] [Google Scholar]
- Nortier J. L.; Martinez M. C.; Schmeiser H. H.; Arlt V. M.; Bieler C. A.; Petein M.; Depierreux M. F.; De Pauw L.; Abramowicz D.; Vereerstraeten P.; Vanherweghem J. L. (2000) Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med. 342, 1686–1692. 10.1056/NEJM200006083422301. [DOI] [PubMed] [Google Scholar]
- Sidorenko V. S.; Yeo J. E.; Bonala R. R.; Johnson F.; Scharer O. D.; Grollman A. P. (2012) Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of aristolactam-DNA adducts. Nucleic Acids Res. 40, 2494–2505. 10.1093/nar/gkr1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q.; Kolbanovskiy A.; Zhuang P.; Chen J.; Krzeminski J.; Amin S.; Geacintov N. E. (2002) Synthesis and characterization of site-specific and stereoisomeric fjord dibenzo[a,l]pyrene diol epoxide-N(6)-adenine adducts: unusual thermal stabilization of modified DNA duplexes. Chem. Res. Toxicol. 15, 249–261. 10.1021/tx010157k. [DOI] [PubMed] [Google Scholar]
- Kathuria P.; Sharma P.; Wetmore S. D. (2015) Adenine versus guanine DNA adducts of aristolochic acids: role of the carcinogen-purine linkage in the differential global genomic repair propensity. Nucleic Acids Res. 43, 7388–7397. 10.1093/nar/gkv701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky R. J.; Le Marchand L. (2011) Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: lessons learned from aromatic amines. Chem. Res. Toxicol. 24, 1169–1214. 10.1021/tx200135s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turesky R. J.; Markovic J.; Aeschlimann J. M. (1996) Formation and differential removal of C-8 and N2-guanine adducts of the food carcinogen 2-amino-3-methylimidazo[4,5-f]quinoline in the liver, kidney, and colorectum of the rat. Chem. Res. Toxicol. 9, 397–402. 10.1021/tx950131r. [DOI] [PubMed] [Google Scholar]
- Xiao S.; Guo J.; Yun B. H.; Villalta P. W.; Krishna S.; Tejpaul R.; Murugan P.; Weight C. J.; Turesky R. J. (2016) Biomonitoring DNA Adducts of Cooked Meat Carcinogens in Human Prostate by Nano Liquid Chromatography-High Resolution Tandem Mass Spectrometry: Identification of 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine DNA Adduct. Anal. Chem. 88, 12508–12515. 10.1021/acs.analchem.6b04157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.; Hingerty B. E.; Guenther E. A.; Krishnan V. V.; Broyde S.; Turteltaub K. W.; Cosman M. (2001) Solution structure of the 2-amino-1- methyl-6-phenylimidazo[4,5-b]pyridine C8-deoxyguanosine adduct in duplex DNA. Proc. Natl. Acad. Sci. U. S. A. 98, 8507–8512. 10.1073/pnas.151251898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto C.; Spivak G.; Aloisi C. M.; Menigatti M.; Naegeli H.; Hanawalt P. C.; Tanasova M.; Sturla S. J. (2017) Modulation of cytotoxicity by transcription-coupled nucleotide excision repair Is independent of the requirement for bioactivation of acylfulvene. Chem. Res. Toxicol. 30, 769–776. 10.1021/acs.chemrestox.6b00240. [DOI] [PubMed] [Google Scholar]
- Jaspers N. G.; Raams A.; Kelner M. J.; Ng J. M.; Yamashita Y. M.; Takeda S.; McMorris T. C.; Hoeijmakers J. H. (2002) Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair 1, 1027–1038. 10.1016/S1568-7864(02)00166-0. [DOI] [PubMed] [Google Scholar]
- Brooks P. J. (2008) The 8,5′-cyclopurine-2′-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair 7, 1168–1179. 10.1016/j.dnarep.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Wang Q. E.; Zhu Q.; El-Mahdy M. A.; Wani G.; Praetorius-Ibba M.; Wani A. A. (2006) DNA damage binding protein component DDB1 participates in nucleotide excision repair through DDB2 DNA-binding and cullin 4A ubiquitin ligase activity. Cancer Res. 66, 8590–8597. 10.1158/0008-5472.CAN-06-1115. [DOI] [PubMed] [Google Scholar]
- Smerdon M. J.; Lieberman M. W. (1978) Nucleosome rearrangement in human chromatin during UV-induced DNA- reapir synthesis. Proc. Natl. Acad. Sci. U. S. A. 75, 4238–4241. 10.1073/pnas.75.9.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara R.; Mo J.; Sancar A. (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 20, 9173–9181. 10.1128/MCB.20.24.9173-9181.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbo S.; Turesky R. J.; Villalta P. W. (2014) DNA adductomics. Chem. Res. Toxicol. 27, 356–366. 10.1021/tx4004352. [DOI] [PMC free article] [PubMed] [Google Scholar]