

Abstract
Purpose
Somatic mutations and copy number variation in the ERBB family are frequent in urothelial carcinoma (UC) and may represent viable therapeutic targets. We studied whether afatinib (an oral, irreversible inhibitor of the ErbB family) has activity in UC and if specific ERBB molecular alterations are associated with clinical response.
Patients and Methods
In this phase II trial, patients with metastatic platinum-refractory UC received afatinib 40 mg/day continuously until progression or intolerance. The primary end point was 3-month progression-free survival (PFS3). Prespecified tumor analysis for alterations in EGFR, HER2, ERBB3, and ERBB4 was conducted.
Results
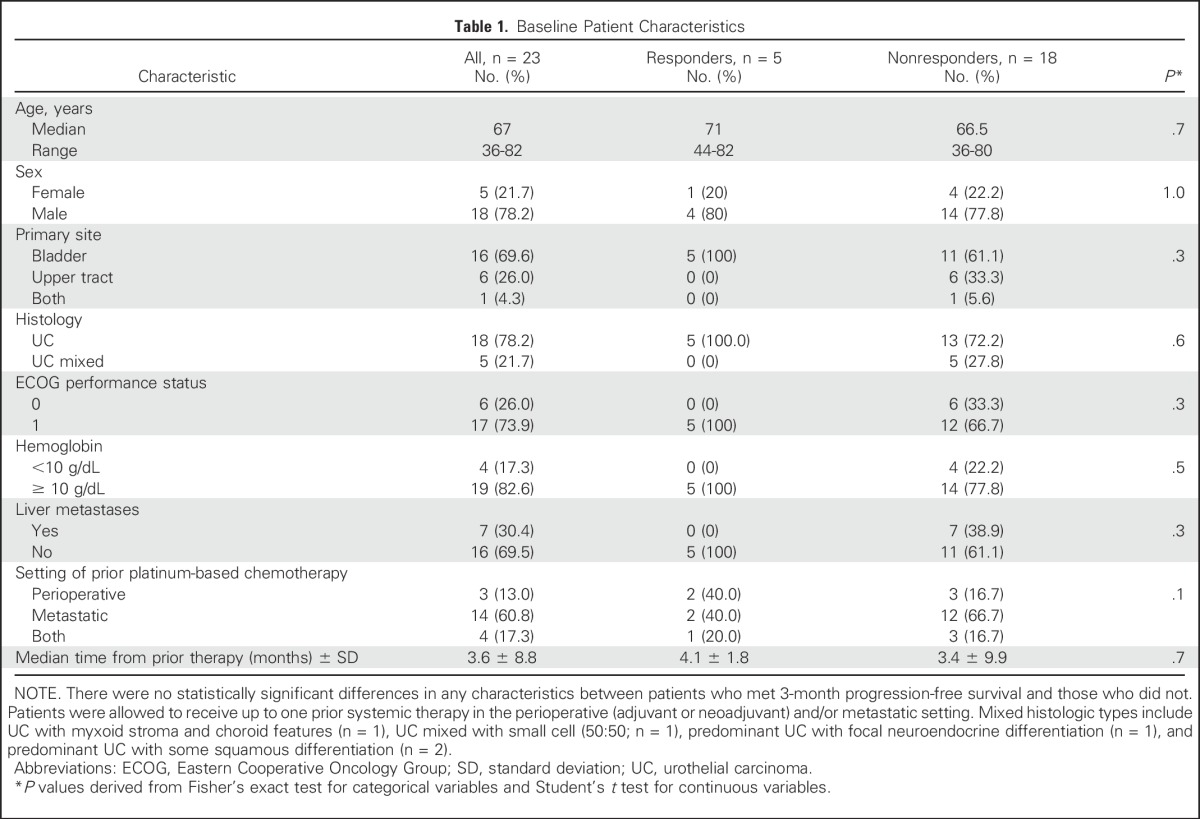
The first-stage enrollment goal of 23 patients was met. Patient demographic data included: 78% male, median age 67 years (range, 36 to 82 years), hemoglobin < 10 g/dL in 17%, liver metastases in 30%, median time from prior chemotherapy of 3.6 months, and Eastern Cooperative Oncology Group performance status ≤ 1 in 100%. No unexpected toxicities were observed; two patients required dose reduction for grade 3 fatigue and rash. Overall, five of 23 patients (21.7%) met PFS3 (two partial response, three stable disease). Notably, among the 21 tumors analyzed, five of six patients (83.3%) with HER2 and/or ERBB3 alterations achieved PFS3 (PFS = 10.3, 7.0, 6.9, 6.3, and 5.0 months, respectively) versus none of 15 patients without alterations (P < .001). Three of four patients with HER2 amplification and three of three patients with ERBB3 somatic mutations (G284R, V104M, and R103G) met PFS3. One patient with both HER2 amplification and ERBB3 mutation never progressed on therapy, but treatment was discontinued after 10.3 months as a result of depressed ejection fraction. The median time to progression/discontinuation was 6.6 months in patients with HER2/ERBB3 alterations versus 1.4 months in patients without alterations (P < .001).
Conclusion
Afatinib demonstrated significant activity in patients with platinum-refractory UC with HER2 or ERBB3 alterations. The potential contribution of ERBB3 to afatinib sensitivity is novel. Afatinib deserves further investigation in molecularly selected UC.
INTRODUCTION
Urothelial carcinoma (UC) remains the fourth most common cancer among males and the eighth leading cause of cancer death in the United States; 16,000 deaths were expected in 2015.1 Despite the significant prevalence and mortality of metastatic disease, there has been relatively little progress in therapeutic strategies for UC in the last 25 years, although immune checkpoint blockade has generated notable promise.2 Platinum-based therapy remains the only standard of care,3 with no approved second-line therapies. There is therefore significant interest in identifying new therapies.
The ErbB family, consisting of EGFR, HER2, ErbB3, and ErbB4, is a class of receptor tyrosine kinases that has been extensively investigated as potentially important in the pathogenesis of UC.4-6 Upon ligand binding for EGFR, ErbB3, and ErbB4, receptor homo- or heterodimerization activates downstream growth-signaling pathways.7 HER2, in contrast, has no known ligand and is constitutively active. EGFR overexpression in UC is correlated with higher tumor grade and muscle invasiveness,8 tumor recurrence,9,10 and overall survival.10,11 Similarly, HER2 overexpression in UC is associated with recurrence and metastasis.5,12
Recently, comprehensive molecular analysis demonstrated that EGFR amplifications (11%), HER2 amplifications (7%), and ERBB3 somatic mutations (11%) are relatively frequent in UC.13,14 Earlier clinical data on EGFR and HER2 inhibition in UC has been mixed, with one promising result of erlotinib in the neoadjuvant setting15 and two negative trials for gefitinib in chemotherapy-resistant UC.16,17 A phase II trial testing trastuzumab in a combination regimen in HER2-positive UC had a 70% response rate but higher than expected rates of cardiotoxicity.18 Separately, patients with chemotherapy-refractory UC whose tumors had 2+ or 3+ expression levels of EGFR or HER2 had prolonged survival when treated with lapatinib compared with those with 0/1+ expression,19 suggesting a possible role for dual inhibition in patients with HER2/EGFR overexpression.
Afatinib is a novel, oral, irreversible tyrosine kinase inhibitor of the ErbB receptor family. Afatinib is approved for treatment of metastatic non–small-cell lung carcinoma bearing EGFR exon 19 deletions or exon 21 (L858R) substitutions.20 A recent phase III trial (LUX-H&N 1) of platinum-refractory metastatic squamous-cell carcinoma of the head and neck also found that afatinib significantly improved progression-free survival (PFS) compared with methotrexate.21 Given the frequency and potential importance of ErbB family alterations in UC, we hypothesized that afatinib would demonstrate activity in this disease.
PATIENTS AND METHODS
Patients
Between November 2013 and May 2015, adults with a histologic diagnosis of UC of the bladder, upper tract, or urethra who had progressed despite receiving prior platinum-based combination chemotherapy in the perioperative or metastatic setting were enrolled. Patients who had received perioperative chemotherapy within 1 year were eligible. Inclusion criteria were age of 18 years or older; presence of measurable, unresectable/metastatic disease; Eastern Cooperative Oncology Group performance status ≤ 1, absolute neutrophil count ≥ 1,000/μL, platelets > 100,000/μL, hemoglobin ≥ 8.5 g/dL, total bilirubin ≤ 1.5 × the institutional normal upper limit, AST/ALT ≤ 2.5 × the normal upper limit, creatinine clearance ≥ 30 mL/min, and the ability to provide informed consent. Patients were only eligible if they had received no more than one prior systemic therapy in the metastatic setting. Patients were excluded if they received prior afatinib, were breastfeeding and/or pregnant, had uncontrolled intercurrent illness, had brain metastases, were concurrently receiving other investigational agents, had uncontrolled HIV or HIV currently treated with antiretroviral agents, had interstitial lung disease, or were unable to take oral medications. ErbB overexpression or alteration were not required for trial enrollment.
Study Design and Treatment
This was an open-label, single-arm phase II clinical trial in which patients received continuous therapy with afatinib 40 mg/day until disease progression or intolerability. Study drug was provided by Boehringer Ingelheim Pharmaceuticals. All patients were monitored for toxicity by physical examination, complete blood counts, and serum chemistry analysis every 2 weeks. Radiologic disease evaluation (computed tomography/magnetic resonance imaging) was performed every 6 weeks with disease response (stable disease, partial response, complete response, or progressive disease) characterized using RECIST version 1.1.
All adverse events from the initiation of treatment to 28 days after the last administration were graded according to the Common Terminology Criteria for Adverse Events, version 4.0. The Appendix (online only) details supportive treatment of frequently occurring adverse events.
Statistical Analyses
The primary end point was 3-month PFS (PFS3) using a Simon two-stage design. For 85% power at alpha = .10, seven of the first 23 patients (30%) would need to meet PFS3 for the study to proceed to the second stage, during which an additional 10 patients would be enrolled. The null hypothesis was a PFS3 rate of < 30%, which would be considered representative of lack of efficacy in the tested population. The alternate hypothesis was that ≥ 50% of patients reaching PFS3 would be indicative of activity, and ≥ 14 of 33 patients (42%) reaching PFS3 would be considered promising. To set the null and alternative hypotheses, we calculated the composite weighted-average PFS3 rate of 14 historical studies that evaluated second-line therapies in refractory UC. The median PFS of the cohort was < 3 months, consistent with previously reported median PFS.22 A random effects model, which took into account the heterogeneity among the studies, estimated the pooled PFS3 rate of these studies to be 28% (95% CI, 22% to 33%). In contrast, the pooled PFS3 rate among five studies of second-line agents that reported considerable activity in the refractory setting (including vinflunine and taxanes) was 45% (95% CI, 38.4% to 51.5%). The alternative hypothesis of 50% of patients meeting PFS3 as the criterion for activity was therefore considered sufficient to justify further testing. Secondary end points were overall response rate, overall survival, and median PFS estimated by the Kaplan-Meier method.
Biomarker Analyses
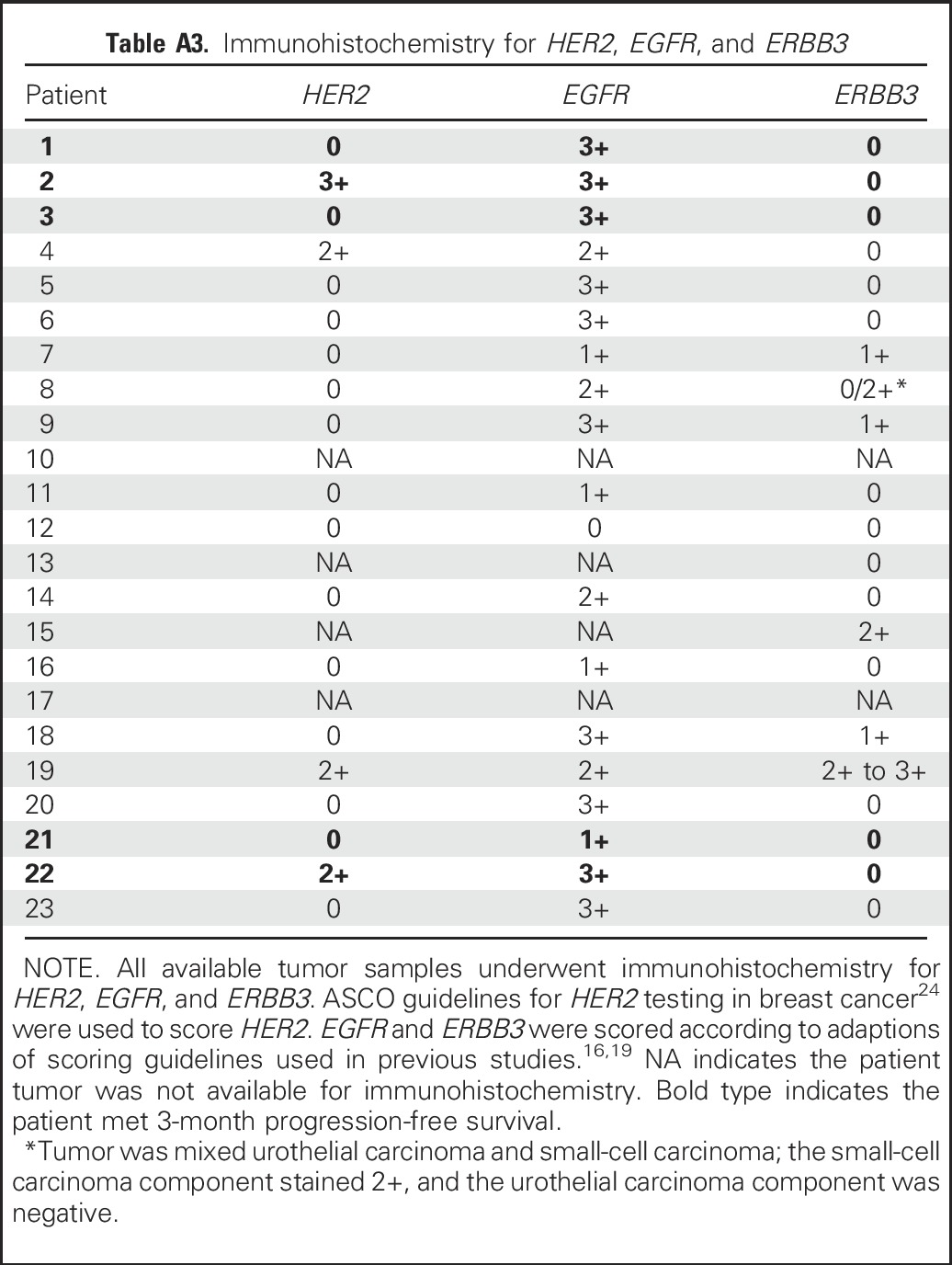
The salient exploratory end point for this trial was whether genomic alterations in EGFR, HER2, ERBB3, and ERBB4, including somatic mutations of all four genes plus copy number analysis of EGFR and HER2, were associated with PFS3 and/or response. These analyses were conducted using available archival formalin-fixed, paraffin-embedded sections from surgical specimens (Appendix Table A1, online only). Before analyses, tissue slides were stained with hematoxylin and eosin and assessed by a genitourinary pathologist (T.A.) so that selected sections had ≥ 60% tumor nuclei, lacked extensive necrosis, and excluded adjacent normal tissue. Peripheral blood served as the germline DNA control in all but one patient. DNA was extracted using the QIAamp DNA FFPE Tissue Kit (QIAgen, Valencia, CA). Targeted next-generation sequencing (NGS) was performed using libraries prepared with the Ion AmpliSeq Library Kit and Comprehensive Cancer Panel (ThermoFisher Scientific, Waltham, MA) on the Ion Personal Genome Machine (ThermoFisher) using 200-bp sequencing chemistry with the Ion 314 Chip. Single-nucleotide variations were called and annotated using IonReporter software. Only nonsynonymous somatic mutations in EGFR, HER2, ERBB3, and ERBB4 with read frequency > 10% were selected. Additional filtering parameters were followed according to previously published methods.23 All identified mutations were verified as somatic by comparing tumor and normal DNA sequencing results using Sanger sequencing on the 3500 Genetic Analyzer system (ThermoFisher).
Available samples were subjected to EGFR and HER2 copy number analysis using TaqMan Copy Number Assays (ThermoFisher) on the ViiA 7 Real-Time PCR System (Applied Biosystems, Foster City, CA) using RNase P as the control gene (EGFR: Hs02925916_cn; HER2: Hs00817646_cn). At least 3.5 copies were considered amplified. Fluorescent in situ hybridization (FISH) was performed using standard methods to confirm copy number assessment of HER2 and EGFR in select specimens (Abbott, PathVysion, HER2 DNA Probe Kit). Two independent reviewers (K.L.Y. and C.A.F.) scored all FISH samples according to the guidelines set by ASCO for HER2 testing in breast cancer.24
Finally, immunohistochemistry (IHC) staining for HER2 (HercepTest, Dako, Carpinteria, CA), EGFR (clone 31G7, ThermoFisher), and ERBB3 (C-17, Santa Cruz Biotechnology, Dallas, TX) was performed. ASCO guidelines were used for HER2 scoring,24 and standards used to score EGFR in earlier trials16,19 were adapted for EGFR and ERBB3 scoring (because there are no accepted guidelines for these proteins), using the following scale: 0 = no staining, 1+ = weak or focal staining, 2+ = moderate staining, and 3+ = strong staining. Performers of each analysis were blinded to clinical outcomes and to the results of concurrent analyses.
RESULTS
Patients
The first-stage enrollment goal of 23 patients was met. Baseline patient characteristics are summarized in Table 1. Of variables known to be associated with prognosis in UC,22 four patients (17.3%) had initial hemoglobin levels < 10 g/dL and seven (30.4%) had liver metastases. The median time from prior chemotherapy to the initiation of afatinib22 was 3.6 months (range, 0.2 to 44.3 months).
Table 1.
Baseline Patient Characteristics
Adverse Events
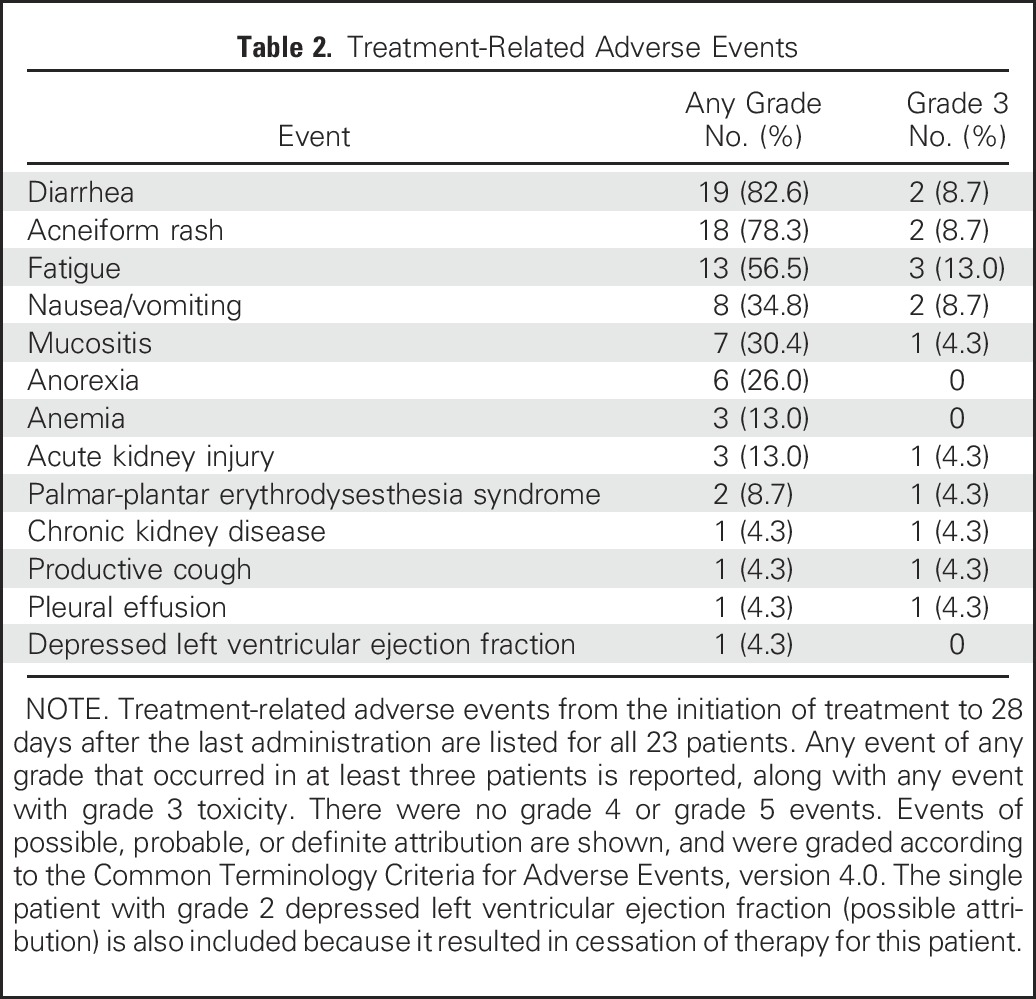
The safety profile of afatinib was similar to that in a previous report,21 and patients who experienced drug toxicity were successfully managed with supportive care. The most common treatment-related toxicities of any grade were diarrhea (82.6%), acneiform rash (78.3%), and fatigue (56.5%; Table 2). Three patients underwent dose reductions (grade 3 fatigue, grade 3 rash, grade 2 cardiotoxicity). Afatinib was discontinued for patient 2 as a result of asymptomatic grade 2 reduction in ejection fraction (an on-treatment decrease from 46% to 33% after 10.3 months of treatment was considered possibly related to the drug). There were no treatment-related deaths.
Table 2.
Treatment-Related Adverse Events
Treatment Response
Five of 23 patients (21.7%) achieved PFS3, the primary end point. The study did not meet the criterion of seven or more patients reaching PFS3 that was necessary to proceed to the second stage of enrollment. The median PFS for the entire cohort was 1.4 months. The overall response rate was 8.6%. The best overall responses were partial response observed in two patients (8.7%), stable disease in seven patients (30.4%), and progressive disease in 14 patients (60.9%). The median overall survival for all patients was 5.3 months via Kaplan-Meier survival analysis. Two patients remained alive at the time of submission.
Genomic Alterations as Predictors of Afatinib Sensitivity
Targeted NGS was performed on 21 available tumor samples. Overall, the rates of somatic nonsynonymous mutations and copy number amplifications found in our patient cohort were similar to previous reporting,13 as summarized in Table 3.
Table 3.
ERBB Family Molecular Analysis
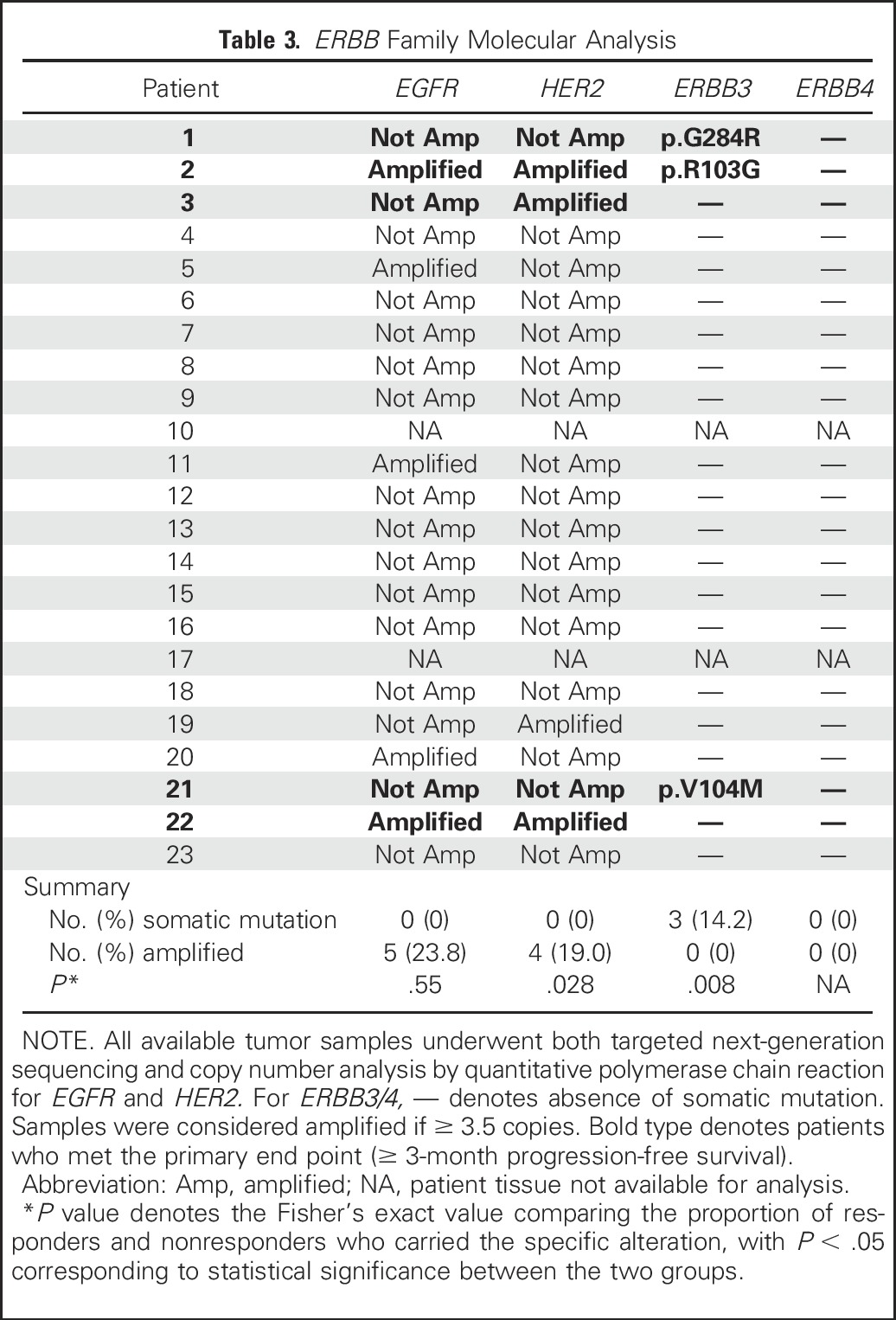
Importantly, molecular alterations of two specific genes—HER2 and ERBB3—were found to be significantly predictive of afatinib efficacy. Specifically, five of six patients (83%) with ERBB molecular alterations consisting of HER2 copy number amplification and/or ERBB3 somatic mutations achieved PFS3, whereas none of 15 patients (0%) without alterations reached PFS3 (P < .001, Fisher’s exact test; Fig 1). The median PFS in the six patients with HER2/ERBB3 alterations was 6.6 months versus 1.4 months in patients without alterations (Fig 2; P < .001, log-rank test). The findings for each molecular target are described below in detail.
Fig 1.

Treatment response: Swimmer’s plot of the 23 enrolled patients, with time on therapy (months) shown for each patient. Blue color indicates that the patient met 3-month progression-free survival (PFS3, the primary end point). Specific ERBB molecular alterations are noted for each patient. The asterisk (*) indicates that patient 2 carried two alterations (ERBB3 somatic mutation and HER2 amplification, as shown adjacent to the asterisk).
Fig 2.

Progression-free survival: Kaplan-Meier curve of progression-free survival. The six patients (1, 2, 3, 19, 21, and 22) with ERBB molecular alterations had a median progression-free survival of 6.6 months, compared with 1.4 months for the 15 patients without alterations (P < .001, log-rank test).
HER2.
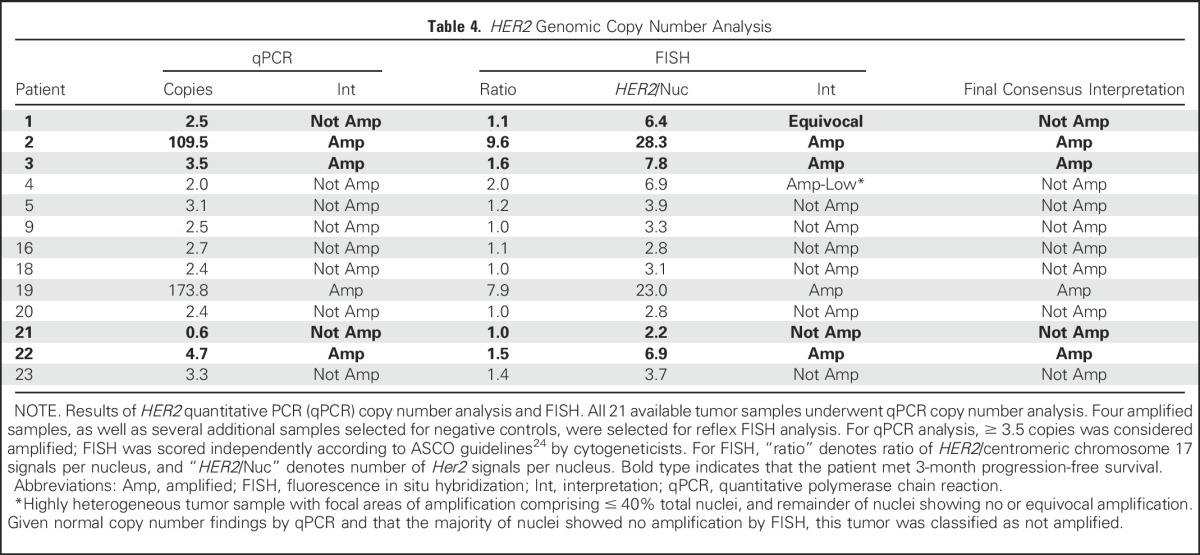
Genomic copy number analysis for HER2 amplification was performed, results of which are shown in Table 3. To corroborate identification of truly amplified samples, FISH was performed on all samples with HER2 amplification by quantitative polymerase chain reaction (qPCR) as well as select nonamplified tumors that served as internal controls. The four samples with ≥ 3.5 copies of HER2 by qPCR were the only samples to show amplification by FISH (Table 4). The tumor of patient 4 demonstrated low-level amplification by FISH and no amplification by qPCR; it was noted to be heterogeneous, with focal areas (comprising approximately 40% of nuclei) of HER2 amplification by FISH. Given that the majority of the tumor was unamplified by FISH and that qPCR confirmed no amplification, patient 4 was designated unamplified in the final consensus interpretation. A representative FISH image for patient 2, who had the longest PFS and whose tumor had high molecular amplification (copy number > 50), is shown in Appendix Figure A1 (online only). No patients were found to have HER2 mutations.
Table 4.
HER2 Genomic Copy Number Analysis
ERBB3.
NGS and Sanger sequencing confirmation identified somatic nonsynonymous mutations in ERBB3 in three patients—patient 1 (exon 7 p.G284R), patient 2 (exon 3 p.R103G), and patient 21 (exon 3 p.V104M)—all of which are in the extracellular domain of the receptor protein. Importantly, all three patients with ERBB3 somatic mutations met PFS3 (P < .001 v those without mutations). Whereas p.V104M25-28 (patient 21) and G284R29,30 (patient 1) have been reported in multiple cancer types (although not in UC), ERBB3 R103G has not been previously described in cancer.
EGFR.
No somatic nonsynonymous mutations in EGFR were detected, consistent with previously reported findings that EGFR somatic mutations are rare in UC.13,31 Because EGFR qPCR and FISH copy number results did not correlate well, definitive amplification status was not assigned (Appendix Table A2, online only). However, using the results of either assay, there were no patients meeting PFS3 who demonstrated EGFR amplification as their sole molecular alteration.
Finally, for ERBB3, HER2, and EGFR, IHC was performed on the 21 available tumor specimens. We found no correlations between IHC for any of the three targets and clinical response to afatinib (Appendix Table A3, online only). For HER2, although FISH and qPCR copy number results had high concordance, these genomic assays had weak correlation with IHC results. Representative images are shown in Appendix Figure A2 (online only).
DISCUSSION
Despite an overall PFS3 rate below the prespecified cutoff for the full cohort of this trial, we observed significant and clinically meaningful activity for afatinib in the predefined subpopulation of patients with platinum-refractory UC with somatic ERBB family alterations. The median PFS on afatinib for patients with alterations was 6.6 months, which is nearly three-fold longer than historical median PFS times in this disease setting.22 The median PFS for vinflunine, the only approved second-line agent in Europe, was 3.0 months,32 and the recently reported median PFS time for pembrolizumab in its phase Ib trial was 2.2 months.33 In our study, five of six patients (83.3%) with identified HER2 or ERBB3 alterations exceeded the primary PFS3 end point of this study, compared with 0% of patients without these alterations.
Our study reveals several important possibilities regarding the mechanism of afatinib sensitivity in UC. First, despite earlier evidence suggesting that EGFR alterations may identify patients with UC who might benefit from EGFR inhibitors, we were unable to demonstrate that EGFR amplification or protein overexpression identified patients benefitting from afatinib. This may be in part because the EGFR exon 19 and 21 alterations, for which afatinib is approved in non–small-cell lung cancer, are absent in UC.34 In contrast, HER2 amplification and ERBB3 somatic mutation were strongly associated with clinical response. In fact, the only patient (patient 2) with both HER2 and ERBB3 alterations had the longest PFS. Patient 2 never progressed on therapy, but afatinib was discontinued after 10.3 months per protocol rules as a result of depressed ejection fraction. This patient shortly thereafter resumed afatinib (off protocol) for an additional 5.7 months before progressing.
Our data also suggest a potential role for the ErbB3-HER2 interaction in mediating afatinib sensitivity. ErbB3, although lacking intrinsic tyrosine kinase activity, has notable oncogenic activity through its potent ability to form heterodimers with HER2, an interaction that induces activity of the phosphoinositide 3-kinase–protein kinase B signaling pathway.35 In HER2-amplified breast cancer, for example, the ErbB3-HER2 dimer is critical for tumor formation and maintenance.36 Clinically, the combination of docetaxel, trastuzumab (a HER2-targeted antibody), and pertuzumab (which blocks the HER2-ErbB3 interaction) significantly improved PFS in patients with breast cancer compared with trastuzumab and docetaxel alone.37
Moreover, transphosphorylation of ErbB3 is thought to be a potential mechanism of resistance to EGFR/HER2 kinase inhibitors by negative feedback.38-40 In fact, the only patient with HER2 amplification who did not achieve PFS3 (patient 19, with a HER2 copy number > 50, progressed at 1.4 months) had the only tumor to stain 3+ for ErbB3 protein expression. Because the three ERBB3 mutations found in our patients were in the extracellular domain, which is responsible for ligand binding and receptor dimerization,7 these mutations may preferentially induce ERBB3-HER2 dimerization, with a phenotype similar to HER2 amplification. Further investigation with in vitro functional assays is warranted to conclude whether the ErbB3-HER2 interaction is indeed responsible for mediating sensitivity to afatinib.
The potentially critical role of HER2 amplification in clinical response is partially consistent with a previous trial that found a high response rate (70%) in patients with UC with HER2-overexpressing tumors who were given trastuzumab with chemotherapy.18 However, the optimal method for identifying HER2 overexpression or amplification in UC has been controversial. HER2 gene amplification does not correlate well with protein overexpression by IHC in UC,41-44 and there have been conflicting results on which method may have greater prognostic significance.12,44,45
In earlier clinical trials of ERBB-targeting drugs, IHC has been more commonly used for patient selection.16,18,19 Our results indicate that HER2 amplification detected by qPCR or FISH, rather than protein overexpression detected by IHC, is a more sensitive predictive biomarker for afatinib in UC, with 75% of patients with amplification reaching PFS3 compared with only 25% of patients with 2+ or 3+ staining. Given that afatinib is a tyrosine kinase inhibitor (compared with trastuzumab, which is a monoclonal antibody), it is perhaps not surprising that specific ERBB genomic amplifications and mutations, rather than protein overexpression, appear more relevant.
Because this was a single-arm trial, we were not able to conclusively determine whether HER2 amplification and/or ERBB3 mutation, rather than being predictive biomarkers, are themselves simply associated with improved prognosis in this disease. Previous data, however, argue against this possibility,44 and in fact suggest that HER2 and ERBB3 alterations are associated with worse prognosis.26,46-48 Tumor specimens analyzed were from the primary site of disease. Although genomic concordance between primary and metastatic sites can vary,49-51 it is likely that these are conserved driver alterations that are also present in the metastases.
Finally, it is acknowledged that the sample size in this trial is relatively small, and therefore a small number of patients with molecular alterations were treated. It is nonetheless striking that we were able to detect significant outcomes differences with only a handful of such patients, raising the possibility that these molecular alterations are indeed highly correlated with afatinib responsiveness. Given this, afatinib deserves examination in a larger number of patients with molecularly altered UC, including evaluation in those with negative prognostic variables such as liver metastases and histologic variants, to characterize the range of alterations that are predictive of benefit. We would then likely proceed to randomized examination to formally quantify changes in disease outcomes. An important area of future investigation will also be improving understanding of mechanisms of resistance; all patients in our trial eventually had progressive disease, a problem that is seen across multiple EGFR/HER2 inhibitors.52
Molecular characterization of tumors is becoming increasingly used and more feasible to perform. In the era of personalized medicine, a nuanced understanding of molecular studies is vital for identifying patients most likely to benefit from selected therapies. With this in mind, to our knowledge, this report is the first to show that afatinib has significant activity in patients with platinum-refractory UC with somatic ERBB3 and HER2 genomic alterations.
ACKNOWLEDGMENT
We thank the Human Tissue Resource Center and the Cancer Center Support Grant at the University of Chicago, and specifically Dr Shihong Li, for their support in performing the immunohistochemistry studies, and Heather Mashek for her assistance in scoring the fluorescence in situ hybridization assays.
Appendix
Supportive Treatment Methods
Supportive treatment of frequently occurring adverse events was as follows: loperamide was used for diarrhea; topical hydrocortisone and/or topical clindamycin for grade 1 rash, with doxycycline and oral diphenhydramine added for grade 2 rash and oral corticosteroids added at investigator’s discretion; and antiemetics and as-needed intravenous hydration for persistent or grade ≥ 2 nausea and vomiting. Routine multigated acquisition scan was performed every 12 weeks to monitor development of left ventricular (LV) dysfunction, given concern for cardiotoxicity with other HER2-targeting agents.18 The occurrence of grade 2 LV dysfunction (new resting ejection fraction of 40% to 50% or a 10% to 19% drop from baseline) required therapy cessation for 14 days before repeating multigated acquisition scan and permanent discontinuation of therapy if ejection fraction did not resolve to grade 1 by that time. Toxicities that required prespecified 10 mg/day incremental dose reductions included presence of grade ≥ 2 diarrhea persisting for 2 or more consecutive days despite adequate antidiarrheal medication/hydration; grade ≥ 2 nausea and/or vomiting persisting for 3 or more consecutive days despite antiemetic treatment and hydration; grade 2 or grade 3 worsening of renal function measured by serum creatinine or newly developed decrease in glomerular filtration rate of more than 50% from baseline; and any other drug-related adverse events grade ≥ 3. Toxicities that required permanent discontinuation of therapy were grade 4 rash, grade ≥ 3 interstitial lung disease, grade 4 hepatic impairment, grade ≥ 3 keratitis, any symptomatic (≥ grade 3) LV dysfunction, and grade 4 worsening of renal function.
Fig A1.
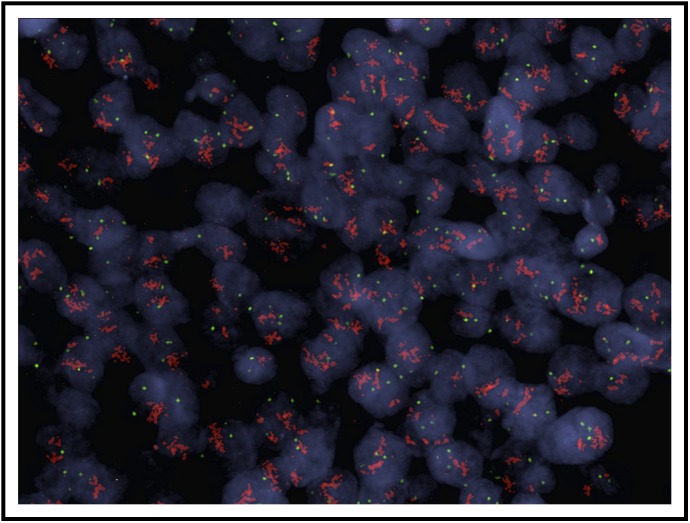
HER2 copy number assessment in a patient with robust HER2 amplification: A representative image of fluorescence in situ hybridization for patient 2 is shown. Within each nucleus, red color denotes HER2 signal and green color denotes the centromeric probe (D17Z1) for chromosome 17 used as the control. HER2/Cep17 ratio 9.6, average HER2/nucleus 28.3.
Fig A2.

Immunohistochemistry images for ErbB family proteins: Representative images from (A) HER2, (B) EGFR, and (C) ERBB3 are shown, with individual patients labeled. For HER2 and EGFR, the cases shown were selected to show the observed range across the cohort, which included samples with completely negative staining to some with marked overexpression. Patient 12, for example, was selected to demonstrate negative EGFR staining. Two images for patient 4 (HER2 staining) are shown side by side to demonstrate focality of staining. For patient 8, the histology of the tumor was mixed urothelial carcinoma (50%) and small-cell carcinoma (50%). Whereas the urothelial carcinoma component stained negative for ERBB3, the small-cell carcinoma component stained 2+ to 3+.
Table A1.
Archival Tissue Samples
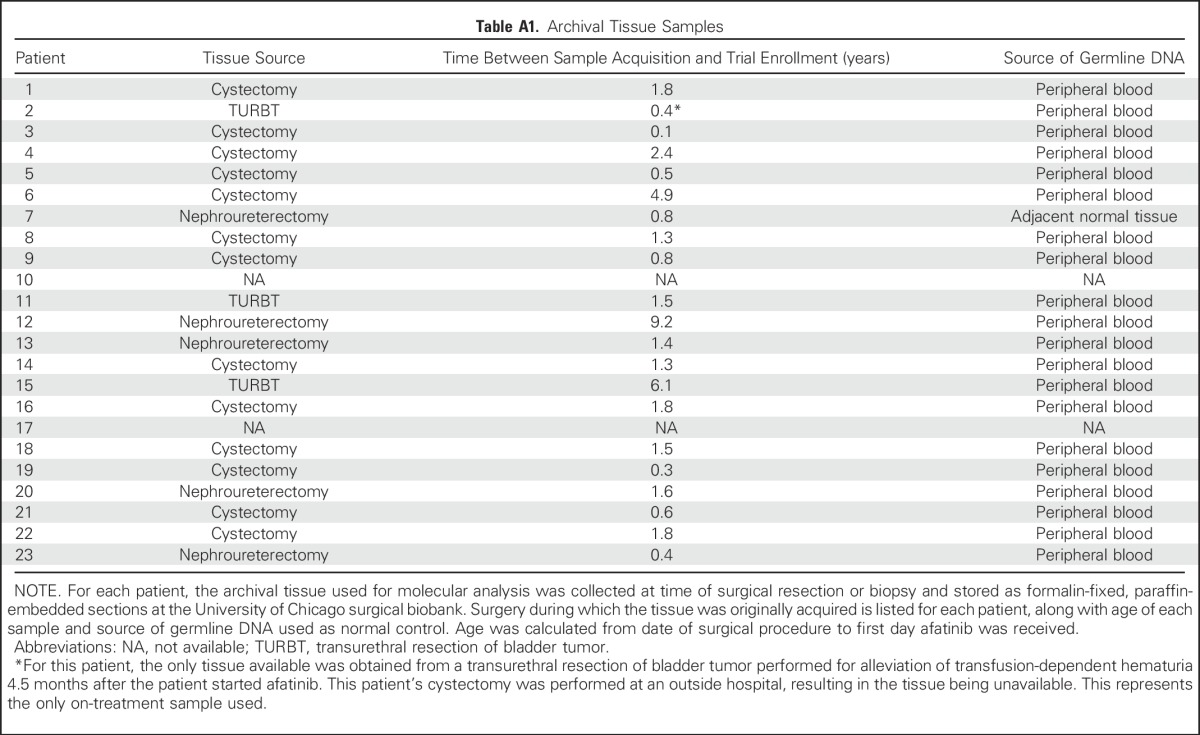
Table A2.
EGFR Genomic Copy Number Analysis
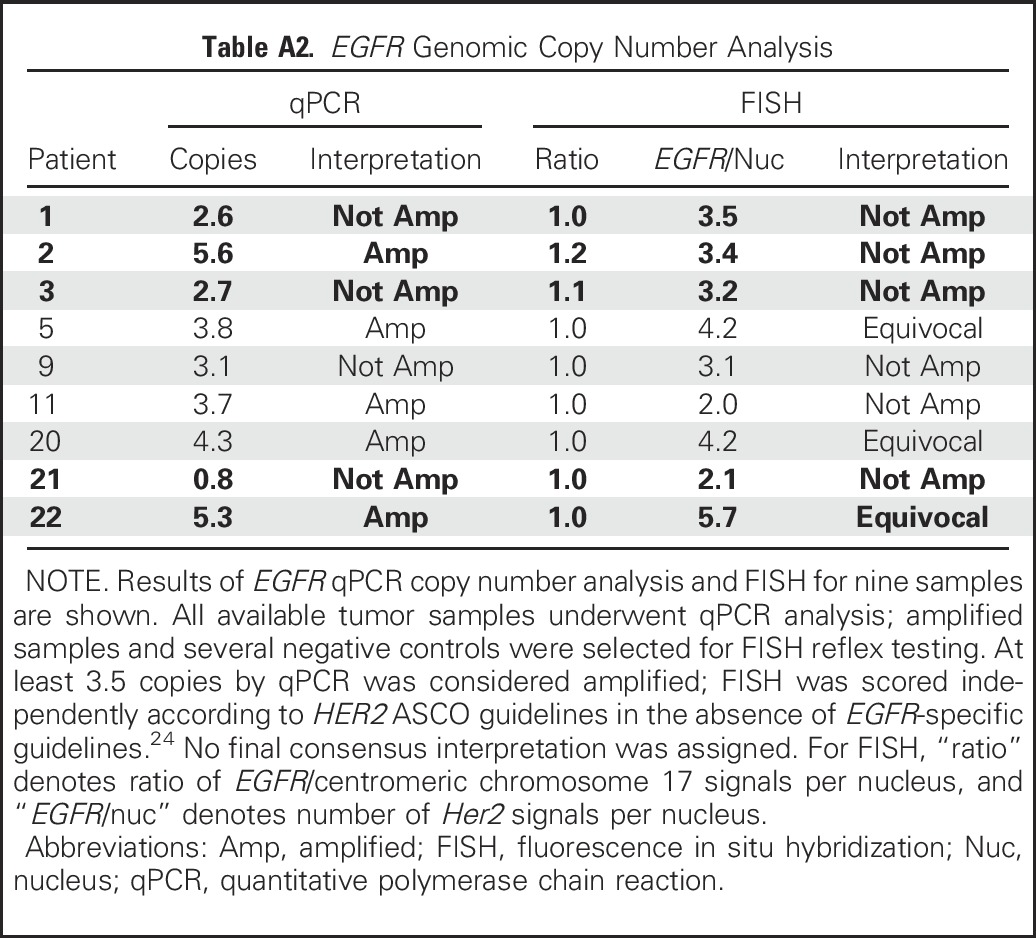
Table A3.
Immunohistochemistry for HER2, EGFR, and ERBB3
Footnotes
Supported by Boehringer Ingelheim Pharmaceuticals, Inc (BIPI). Additional support for correlative studies was provided by University of Chicago Cancer Center Auxiliary Board.
This study was supported by BIPI. BIPI had no role in the design, analysis, or interpretation of the results in this study; BIPI was given the opportunity to review the manuscript for medical and scientific accuracy as it relates to BIPI substances, as well as intellectual property considerations.
Presented in abstract form at the American Society of Clinical Oncology Genitourinary Cancers Symposium, Orlando, FL, February 26-28, 2015.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
See accompanying article on page 2088
AUTHOR CONTRIBUTIONS
Conception and design: Walter M. Stadler, Peter H. O'Donnell
Financial support: Peter H. O'Donnell
Administrative support: Alexa Campanile, Peter H. O'Donnell
Provision of study materials or patients: James L. Wade III, Peter H. O'Donnell
Collection and assembly of data: Noura J. Choudhury, Alexa Campanile, James L. Wade III, Peter H. O'Donnell
Data analysis and interpretation: Noura J. Choudhury, Tatjana Antic, Kai Lee Yap, Carrie A. Fitzpatrick, Theodore Karrison, Yusuke Nakamura, Peter H. O'Donnell
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Noura J. Choudhury
No relationship to disclose
Alexa Campanile
No relationship to disclose
Tatjana Antic
No relationship to disclose
Kai Lee Yap
No relationship to disclose
Carrie A. Fitzpatrick
No relationship to disclose
James L. Wade III
Employment: Johnson & Johnson (I)
Stock or Other Ownership: Seattle Genetics, Celgene
Theodore Karrison
No relationship to disclose
Walter M. Stadler
Honoraria: Bayer, CVS Caremark, Sotio, Astra-Zeneca, Genentech, Pfizer
Consulting or Advisory Role: CVS Caremark, Sotio, AstraZeneca (I), Genentech, Pfizer
Research Funding: Active Biotech (Inst), Bayer (Inst), Bristol-Myers Squibb (Inst), Boehringer Ingelheim (Inst), Dendreon (Inst), Exelixis (Inst), Novartis (Inst), Genentech (Inst), Medivation (Inst), Pfizer, (Inst), Merck (Inst), Janssen Pharmaceuticals (Inst), Johnson & Johnson (Inst), Medivation/Astellas (Inst)
Other Relationship: UpToDate
Yusuke Nakamura
No relationship to disclose
Peter H. O'Donnell
Stock or Other Ownership: Davita
Honoraria: Dendreon, OptumHealth, Algeta, Genentech, Novartis, American Medical Forum
Research Funding: Boehringer Ingelheim (Inst)
Patents, Royalties, Other Intellectual Property: Named as co-inventor on a pending patent for a genomic prescribing system for medication prescribing.
Travel, Accommodations, Expenses: Sequenom
Other Relationship: Advance Medical
REFERENCES
- 1.Society AC. Cancer Facts and Figures 2015. Atlanta, GA: American Cancer Society; 2015. [Google Scholar]
- 2.Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 3.Hussain MH, Wood DP, Bajorin DF, et al. Bladder cancer: Narrowing the gap between evidence and practice. J Clin Oncol. 2009;27:5680–5684. doi: 10.1200/JCO.2009.23.6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mooso BA, Vinall RL, Mudryj M, et al. The role of EGFR family inhibitors in muscle invasive bladder cancer: A review of clinical data and molecular evidence. J Urol. 2015;193:19–29. doi: 10.1016/j.juro.2014.07.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chow NH, Chan SH, Tzai TS, et al. Expression profiles of ErbB family receptors and prognosis in primary transitional cell carcinoma of the urinary bladder. Clin Cancer Res. 2001;7:1957–1962. [PubMed] [Google Scholar]
- 6.van Kessel KE, Zuiverloon TC, Alberts AR, et al. Targeted therapies in bladder cancer: An overview of in vivo research. Nat Rev Urol. 2015;12:681–694. doi: 10.1038/nrurol.2015.231. [DOI] [PubMed] [Google Scholar]
- 7.Baselga J, Swain SM. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 8.Neal DE, Marsh C, Bennett MK, et al. Epidermal-growth-factor receptors in human bladder cancer: Comparison of invasive and superficial tumours. Lancet. 1985;325:366–368. doi: 10.1016/s0140-6736(85)91386-8. [DOI] [PubMed] [Google Scholar]
- 9.Chow NH, Liu HS, Lee EI, et al. Significance of urinary epidermal growth factor and its receptor expression in human bladder cancer. Anticancer Res. 1997;17:1293–1296. [PubMed] [Google Scholar]
- 10.Neal DE, Sharples L, Smith K, et al. The epidermal growth factor receptor and the prognosis of bladder cancer. Cancer. 1990;65:1619–1625. doi: 10.1002/1097-0142(19900401)65:7<1619::aid-cncr2820650728>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen PL, Swanson PE, Jaszcz W, et al. Expression of epidermal growth factor receptor in invasive transitional cell carcinoma of the urinary bladder. A multivariate survival analysis. Am J Clin Pathol. 1994;101:166–176. doi: 10.1093/ajcp/101.2.166. [DOI] [PubMed] [Google Scholar]
- 12.Fleischmann A, Rotzer D, Seiler R, et al. Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur Urol. 2011;60:350–357. doi: 10.1016/j.eururo.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research Network Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer G, Al-Ahmadie H, Schultz N, et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J Clin Oncol. 2013;31:3133–3140. doi: 10.1200/JCO.2012.46.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pruthi RS, Nielsen M, Heathcote S, et al. A phase II trial of neoadjuvant erlotinib in patients with muscle-invasive bladder cancer undergoing radical cystectomy: Clinical and pathological results. BJU Int. 2010;106:349–354. doi: 10.1111/j.1464-410X.2009.09101.x. [DOI] [PubMed] [Google Scholar]
- 16.Petrylak DP, Tangen CM, Van Veldhuizen PJ, Jr, et al. Results of the Southwest Oncology Group phase II evaluation (study S0031) of ZD1839 for advanced transitional cell carcinoma of the urothelium. BJU Int. 2010;105:317–321. doi: 10.1111/j.1464-410X.2009.08799.x. [DOI] [PubMed] [Google Scholar]
- 17.Philips GK, Halabi S, Sanford BL, et al. A phase II trial of cisplatin (C), gemcitabine (G) and gefitinib for advanced urothelial tract carcinoma: Results of Cancer and Leukemia Group B (CALGB) 90102. Ann Oncol. 2009;20:1074–1079. doi: 10.1093/annonc/mdn749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hussain MH, MacVicar GR, Petrylak DP, et al. Trastuzumab, paclitaxel, carboplatin, and gemcitabine in advanced human epidermal growth factor receptor-2/neu-positive urothelial carcinoma: Results of a multicenter phase II National Cancer Institute trial. J Clin Oncol. 2007;25:2218–2224. doi: 10.1200/JCO.2006.08.0994. [DOI] [PubMed] [Google Scholar]
- 19.Wülfing C, Machiels JP, Richel DJ, et al. A single-arm, multicenter, open-label phase 2 study of lapatinib as the second-line treatment of patients with locally advanced or metastatic transitional cell carcinoma. Cancer. 2009;115:2881–2890. doi: 10.1002/cncr.24337. [DOI] [PubMed] [Google Scholar]
- 20.Afatinib. Silver Spring, MD: 2013. U.S. Food and Drug Administration. [Google Scholar]
- 21.Machiels JP, Haddad RI, Fayette J, et al. Afatinib versus methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): An open-label, randomised phase 3 trial. Lancet Oncol. 2015;16:583–594. doi: 10.1016/S1470-2045(15)70124-5. [DOI] [PubMed] [Google Scholar]
- 22.Sonpavde G, Pond GR, Fougeray R, et al. Time from prior chemotherapy enhances prognostic risk grouping in the second-line setting of advanced urothelial carcinoma: A retrospective analysis of pooled, prospective phase 2 trials. Eur Urol. 2013;63:717–723. doi: 10.1016/j.eururo.2012.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yap KL, Kiyotani K, Tamura K, et al. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res. 2014;20:6605–6617. doi: 10.1158/1078-0432.CCR-14-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 25.Lee YS, Cho YS, Lee GK, et al. Genomic profile analysis of diffuse-type gastric cancers. Genome Biol. 2014;15:R55. doi: 10.1186/gb-2014-15-4-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M, Zhang Z, Li X, et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat Genet. 2014;46:872–876. doi: 10.1038/ng.3030. [DOI] [PubMed] [Google Scholar]
- 27.Mouradov D, Sloggett C, Jorissen RN, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74:3238–3247. doi: 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- 28.Bentivegna S, Zheng J, Namsaraev E, et al. Rapid identification of somatic mutations in colorectal and breast cancer tissues using mismatch repair detection (MRD). Hum Mutat. 2008;29:441–450. doi: 10.1002/humu.20672. [DOI] [PubMed] [Google Scholar]
- 29.Wang K, Yuen ST, Xu J, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 30.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blehm KN, Spiess PE, Bondaruk JE, et al. Mutations within the kinase domain and truncations of the epidermal growth factor receptor are rare events in bladder cancer: Implications for therapy. Clin Cancer Res. 2006;12:4671–4677. doi: 10.1158/1078-0432.CCR-06-0407. [DOI] [PubMed] [Google Scholar]
- 32.Bellmunt J, Théodore C, Demkov T, et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J Clin Oncol. 2009;27:4454–4461. doi: 10.1200/JCO.2008.20.5534. [DOI] [PubMed] [Google Scholar]
- 33. Plimack E, Gupta S, Bellmunt J, et al: LBA23—A phase 1b study of pembrolizumab (Pembro; MK-3475) in patients (Pts) with advanced urothelial tract cancer. Ann Oncol 25: 2014 (suppl 4; abstr 1093)
- 34.Chaux A, Cohen JS, Schultz L, et al. High epidermal growth factor receptor immunohistochemical expression in urothelial carcinoma of the bladder is not associated with EGFR mutations in exons 19 and 21: A study using formalin-fixed, paraffin-embedded archival tissues. Hum Pathol. 2012;43:1590–1595. doi: 10.1016/j.humpath.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res. 2014;20:1410–1416. doi: 10.1158/1078-0432.CCR-13-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee-Hoeflich ST, Crocker L, Yao E, et al. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 37.Baselga J, Cortés J, Kim SB, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sergina NV, Rausch M, Wang D, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruser TJ, Wheeler DL. Mechanisms of resistance to HER family targeting antibodies. Exp Cell Res. 2010;316:1083–1100. doi: 10.1016/j.yexcr.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 40.Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15:429–440. doi: 10.1016/j.ccr.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 41.Krüger S, Weitsch G, Büttner H, et al. Overexpression of c-erbB-2 oncoprotein in muscle-invasive bladder carcinoma: Relationship with gene amplification, clinicopathological parameters and prognostic outcome. Int J Oncol. 2002;21:981–987. [PubMed] [Google Scholar]
- 42.Coombs LM, Pigott DA, Sweeney E, et al. Amplification and over-expression of c-erbB-2 in transitional cell carcinoma of the urinary bladder. Br J Cancer. 1991;63:601–608. doi: 10.1038/bjc.1991.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caner V, Turk NS, Duzcan F, et al. No strong association between HER-2/neu protein overexpression and gene amplification in high-grade invasive urothelial carcinomas. Pathol Oncol Res. 2008;14:261–266. doi: 10.1007/s12253-008-9027-y. [DOI] [PubMed] [Google Scholar]
- 44.Bellmunt J, Werner L, Bamias A, et al. HER2 as a target in invasive urothelial carcinoma. Cancer Med. 2015;4:844–852. doi: 10.1002/cam4.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jimenez RE, Hussain M, Bianco FJ, Jr, et al. Her-2/neu overexpression in muscle-invasive urothelial carcinoma of the bladder: Prognostic significance and comparative analysis in primary and metastatic tumors. Clin Cancer Res. 2001;7:2440–2447. [PubMed] [Google Scholar]
- 46.Krüger S, Weitsch G, Büttner H, et al. HER2 overexpression in muscle-invasive urothelial carcinoma of the bladder: Prognostic implications. Int J Cancer. 2002;102:514–518. doi: 10.1002/ijc.10731. [DOI] [PubMed] [Google Scholar]
- 47.Grivas PD, Day M, Hussain M. Urothelial carcinomas: A focus on human epidermal receptors signaling. Am J Transl Res. 2011;3:362–373. [PMC free article] [PubMed] [Google Scholar]
- 48.Ocana A, Vera-Badillo F, Seruga B, et al. HER3 overexpression and survival in solid tumors: A meta-analysis. J Natl Cancer Inst. 2013;105:266–273. doi: 10.1093/jnci/djs501. [DOI] [PubMed] [Google Scholar]
- 49.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xue R, Li R, Guo H, et al: Variable intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gastroenterology doi: S0016-5085(15)01861-2. [DOI] [PubMed]
- 51.Guancial EA, Werner L, Bellmunt J, et al. FGFR3 expression in primary and metastatic urothelial carcinoma of the bladder. Cancer Med. 2014;3:835–844. doi: 10.1002/cam4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31:3987–3996. doi: 10.1200/JCO.2012.45.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]