

Abstract
Objective
In RA, autoreactive B cells are pathogenic drivers and sources of anti-citrullinated protein antibodies (ACPA) that serve as a diagnostic biomarker and predictor of worse long-term prognosis. Yet the immunobiologic significance of persistent ACPA production at a cellular level is poorly understood.
Methods
In a cross-sectional study of RA patients, we investigated for the presence of continued defects in immune homeostasis as a function of disease activity. Using an ELISA and a sensitive multiplex bead-based immunoassay, we characterized fine-binding antibody-specificities in sera, synovial fluid (SF) and B-cell culture supernatants. In this manner, we determined the frequency and epitope reactivity patterns of ACPA produced by SF B cells and switched-memory blood B cells, and compared the latter to serum ACPA levels and disease activity scores.
Results
Cultured B cells from SF were shown to spontaneously secrete ACPA, while constitutive IgG-autoantibody production by PBMC was substantially less frequent. After in vitro stimulation, PBMC secreted IgG ACPA that was overwhelmingly from switched-memory B-cells, across all patient groups treated with MTX and/or a TNF-inhibitor. Intriguingly, frequencies of ACPA-producing switched-memory B cells significantly correlated with serum IgG anti-CCP3 (r=0.57, p=0.003). Moreover, treatment-induced clinical remission had little or no effect on the circulating burden of switched-memory ACPA-producing B cells, in part explaining the continued dysregulation of humoral immunity.
Conclusions
Our findings rationalize why therapeutic cessation most often results in disease reactivation and clinical flare. Hence, a clinical disease activity score is not a reliable indicator of the resolution of pathologic recirculating B-cell autoimmunity.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease, affecting ~1–2% of Western populations, that can lead to joint destruction, disability and early mortality (1, 2). Disease is postulated to be initiated by an inflammatory response in periodontal tissue, pulmonary airways, and/or the intestine (3–6). Progression to tissue injury is linked to a specific breach in immunologic tolerance to autoantigens generated by a post-translational enzymatic process, citrullination, that modifies the side-chains of arginine residues. In fact, circulating anti-citrullinated protein antibodies (ACPA), which recognize a progressively expanding number of citrullinated protein epitopes, may arise years before symptomatic joint involvement (7, 8). ACPA themselves and ACPA-containing immune complexes have been postulated to be directly pathogenic (9). Moreover, B lymphocytes, which infiltrate inflamed synovial membranes of affected joints, locally produce ACPA and rheumatoid factors (RFs), also contribute to pathogenesis through antigen presentation, co-stimulation of CD4+ T cells, and secretion of chemokines and cytokines such as IL-6, TNFα and RANKL (10–13).
The presence of serum ACPA, detected by cyclic citrullinated peptide (CCP) assay, serves both as a diagnostic biomarker as well as predicts a worse prognosis for progressive joint damage (14). At clinical onset RA patients also have expansions of memory B-cell subset(s) that correlate with worse long-term clinical outcomes (15). Importantly, even though treat-to-target strategies can more dependably induce low disease activity and clinical remission, serum ACPA nonetheless generally persist. The immunobiologic basis for this persistence remains obscure as ACPA production in ectopic synovial lymphoid tissue is presumably diminished with disease remission. It can be postulated that ACPA continue to be spontaneously produced by long-lived plasma cells in the bone marrow or other sites, which have become disconnected from a previous pathogenic synovial process. We decided to test this model by determining whether disease-associated autoreactive peripheral B cells disappeared or persisted in patients in treatment induced clinical remission.
Herein, we have compared the binding specificities of serum antibodies and peripheral blood B cells; with special focus on the representation of ACPA-expressing switched-memory B cells, which require additional stimuli to secrete Ig/Abs. We have also characterized the fine binding specificities of these memory B cells, and enumerated their frequencies in the peripheral blood. Importantly, to better understand the effect of treatment on the underlying autoimmune process, we assessed relationships with clinical disease activity (16). Our findings show that recirculating ACPA memory B cells generally persist despite conventional DMARD or TNF inhibition. Thus, the dysregulation of systemic autoimmunity in RA persists, even with remission of synovial disease, which likely accounts for the failure of most treatment regimens to induce drug-free remission.
Patients and Methods
Study design, human subjects, and sample procurement
All patients fulfilled the 2010 ACR/EULAR criteria (17) (Supplementary Table 1). For initial classification, patients were defined as “seropositive” or “seronegative“ based on the CCP2 clinical test (Axis-Shield PLC)(18). Patients were recruited at NYU and as part of the RACER trial at University of Pittsburgh. All patients and healthy donors provided written informed consent in accordance with protocols approved by the Human Subjects Institutional Review Board of the NYU School of Medicine and the University of Pittsburgh.
ELISA and multiplex bead-based assays
In a central laboratory, sera from all donors were tested for IgM rheumatoid factor (RF) and IgG CCP3 (Inova Diagnostics, San Diego)(19)(Supplementary Table 1). Samples with high activity were further evaluated at multiple dilutions for accurate activity assignment. Reactivity patterns for auto cyclizing biotinylated peptides (Supplementary Table 2) (20–28) were then defined (Supplementary Figure 1). Using a panel of avidin-coated microspheres and the same peptides/proteins, we also refined a multiplex bead-based array (Luminex Corp. Austin TX), which has enhancements compared to methods in a recent report (29). Signal strength was quantified by MFI from >35 beads for each analyte per well. During assay development, an additive (Surmodics Inc.) was included to limit the potential influence of RF in a sample. A recombinant human IgG1 antibody to citrullinated fibrinogen (clone 1F11, Modiquest, Oss Netherlands) was used as a positive control.
Synovial fluid mononuclear cell culture
Briefly, after joint aspiration an aliquot of synovial fluid was cleared by centrifugation then treated with hyaluronidase (20 U/mL). Cells were recovered by Ficoll separation, washed, assessed for viability, and cultured in RPMI1640 with 10%FBS, 10mM HEPES, 0.1%2-ME, penicillin/streptomycin/glutamine without or with CpG 2006, IL-21 and the CD40L-expressing feeder line, MS40L (30).
Detection of antigen-specific or IgG-secreting cells (ISC)
To determine the frequencies of total IgG and antigen-specific IgG-secreting B cells, we adapted methodology for ELISpot detection (31, 32). PBMCs, cultured without/with stimulants, were added at 2×105/well to 96-well PVDF-covered wells (Millipore) coated with anti-human IgG Fc-specific F(ab)’2 (Jackson Immunoresearch) to capture ISC products. To detect specificity, we added biotinylated-CCP3 or -CQP3 or -Tetanus Toxoid, or anti-human IgG antibody. To develop, we used streptavidin-HRP80 (RDI, Fitzgerald Industries) and AEC substrate. The enumeration of antigen-specific IgG-secreting cells (ISC) was normalized per 106 PBMC.
PBMC cultures
To assess the capacity for in vitro Ig/Ab secretion, PBMC were isolated and cryopreserved, and later cultured at 106 cells per well for 6 days in 1mL of RPMI complete media with 10% FBS, without or with CpG2006 (6μg/mL), IL-21 (50 ng/mL) and sCD40L (500 ng/mL), adapting reported conditions (31). We used flow cytometry to sort isolate four B-cell sub-populations; CD27+IgD−, CD27+IgD+, CD27−IgD−, and CD27−IgD+ (described in Supplementary Figure 2).
Statistical analysis
We assessed non-parametric paired-sample comparisons by Spearman correlations with Prism v6.0e (GraphPad, La Jolla, CA). P<0.05 was considered significant.
Results
Patient cohorts and the development of citrulline-specific immunoassays
Towards our goal of investigating the contributions of circulating ACPA-specific B lymphocytes to rheumatoid arthritis pathogenesis, we first assembled cohorts of well-characterized patients. A cross-sectional cohort was established at NYU that included new onset and established RA (see Supplementary Table 1). Assays for IgG CCP3 demonstrated near complete concordance with IgG CCP2 results obtained at enrollment (data not shown).
Development of a bead-based multiplex ACPA assay
To evaluate patterns of antibody reactivity with citrulline-containing proteins and autocyclizing synthetic peptides, we first independently evaluated reactivity by ELISA (Supplementary Figure 1). These same peptides and proteins were then adapted to a multiplex bead-based array using a panel of avidin-coated microspheres, and we found significant correlations between results obtained with ELISA and our custom bead-based multiplex assays (Supplementary Figure 3).
To better assess the limits of detection, we evaluated the binding reactivity pattern of a recombinant human antibody to citrullinated fibrinogen, 1F11. This IgG1 antibody demonstrated a dose-dependent hierarchy of reactivity with a range of citrulline-containing antigenic ligands; with the most active antigenic ligands, reactivity was detectable at 0.1 ng/ml (not shown).
With this multiplex assay, we characterized the circulating autoantibody fine-specificity of sera from seropositive and seronegative RA patients (Figure 1A). The strongest IgG binding reactivity was generally with the CCP3 peptide, while only infrequent and at best weak reactivity was observed for the CQP3 peptide (that contains the neutral side chain of glutamine residues instead of charged arginine residues). Otherwise, RA patients displayed great heterogeneity in the fine specificities of the ACPA in their sera when tested with a range of citrulline-containing antigens (Figures 1A&B).
Figure 1. Serum IgG in seropositive RA patients react with citrullinated but not native cyclized peptide epitopes in a multiplex bead-based array.
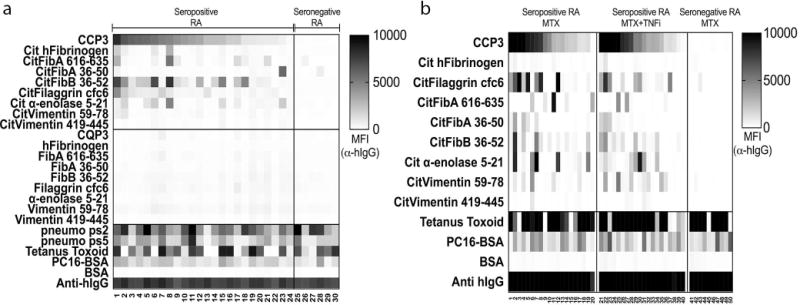
(A) Serum samples from seropositive RA (n = 24) and seronegative RA (n = 6) were diluted 1:1000 and assessed for IgG ACPA reactivity by multiplex bead-based array. Data are presented as Median Fluorescence Intensity (MFI). (B) A custom multiplex bead-based antigen array was used to assess ACPA in serum after different treatments. Herein, RA samples (n = 20 RA CCP3+ MTX treated, n = 20 RA CCP3+ MTX/TNF inhibitor treated, and n = 10 RA CCP3- MTX treated) were analyzed for IgG ACPA fine-specificities. Similar epitope-specificity patterns were detected in the different treatment groups. Data are presented as Median Fluorescence Intensity (MFI).
We also evaluated ACPA responses in the sera of RA patients who had received different treatment regimens (i.e., patient groups on oral methotrexate and/or TNF-inhibitors). We observed no major differences between these groups with respect to ACPA fine binding specificity, qualitatively or quantitatively, although we again found that RA seropositive patients were highly heterogeneous with respect to their IgG ACPA epitope specificity profiles (Figure 1B). Seronegative RA patients, as defined by CCP2 and CCP3 assays, were generally non-reactive with other citrulline-containing antigens, as our multiplex assay detected infrequent (0–4%) recognition by IgG binding of Cit-peptides/proteins, and then with only weak reactivity (Figure 1), which reiterates prior findings (33, 34).
To further evaluate the clinical specificity of our assay, we tested sera from healthy subjects, and patients with other rheumatic diseases; osteoarthritis, systemic lupus erythematosus, psoriatic arthritis and Sjogren’s syndrome (Supplementary Table 3). Our findings confirmed that the detection of IgG anti-citrulline antigen reactivity with our assay was highly specific for patients with RA, as amongst these 87 disease-specific controls we found that IgG reactivity with the CCP3 peptide or other citrullinated antigens was distinctly uncommon.
Specificity and frequency of disease-associated autoreactive B cells at site of disease
To evaluate ACPA produced at the site of disease, we assessed the capacity of our multiplex assay to detect ACPAs in synovial fluid (SF) samples from two seropositive RA patients. Substantial levels of CCP3+ IgG were present while no reactivity with the CQP3 control ligand was seen (Figure 2). Despite similar levels of CCP3 reactivity in these two SF, there were notable differences in the detection of IgG ACPA fine binding specificities with the other citrulline-containing peptides in the assay.
Figure 2. ACPA-reactive B cells are common in RA synovial fluid, and SFMC culture supernatant epitope-reactivity patterns mirror those in autologous synovial fluid.

Synovial fluid (SF) was obtained from RA patients (n = 2), and synovial fluid mononuclear cells (SFMC) isolated. We cultured 1,000 or 10,000 SFMC per well for 12 days +/− 3000 MS40L cells/well and CpG2006/IL-21. Culture supernatants were analyzed with an ELISA to detect IgG anti-CCP3, followed by multiplex assay for epitope specificity of all positive and some negative wells. SF was tested at 1:100 and 1:1000 dilution by ELISA and multiplex assay. Directly ex vivo cultured cells are referred to as “fresh”, while previously cryopreserved/thawed are designated “cryo”.
These SF mononuclear cell (SFMC) samples contained a substantial representation of lymphocytes with the surface phenotype of switched memory B cells (data not shown). SFMC were cultured in media supplemented with IL-21/CpG2006, in the presence of the MS40L feeder cell line that enables in vitro clonal expansion of preactivated B cells (30), which also induced 20-fold higher total IgG levels compared to non-stimulated cultures (data not shown). IgG ACPA were commonly detected in wells with 10,000 SFMC, but were infrequent at 1000 SFMC/well, and never detected at 100 SFMC/well. In each well, IgG ACPA fine specificity was heterogeneous, with the most common secreted ACPA epitope reactivities reiterating the same immunodominant ACPA specificities found in synovial fluid (Figure 2). While these studies document the high frequency of ACPA B cells at site of disease, unfortunately blood samples from these particular donors for comparisons were not available.
Detection of circulating antigen-specific peripheral B cells by ELISpot
To investigate the representation of circulating autoreactive B cells, we adapted ELISpot methods to evaluate the representation of ACPA B cells capable of constitutive ex vivo secretion (i.e., plasma cells/blasts) and B cells that required in vitro stimulation for IgG ACPA production (i.e., memory B cells) (31, 32). Without in vitro stimulation, we detected only a very low frequency of spontaneous total IgG-secreting cells (ISC) (range 200–321/106 PBMC), and by this method we detected neither ACPA reactivity nor other TT-specific antibodies (data not shown). From stimulated PBMC from seropositive RA patients, we found CCP3 reactive IgG-spots at 41.3+/−0.5 ISC/106 PBMC (mean +/−SE, range 0–149), representing mean 0.13% of all ISC (Figure 3A). Only background levels were detected with the control peptide, CQP3, (1.9+/−0.6 ISC/106 PBMC). In contrast, for seronegative RA and healthy adults, CCP3 and CQP3 ISC were below the limit of detection. These results confirmed that only RA patients have disease-specific circulating B cells capable of secreting IgG ACPA.
Figure 3. ACPA-secreting B cells are present in the circulation of seropositive RA patients, and PBMC cultures from seropositive RA secrete IgG ACPA with epitope-reactivity patterns that recapitulate ACPA patterns in serum.
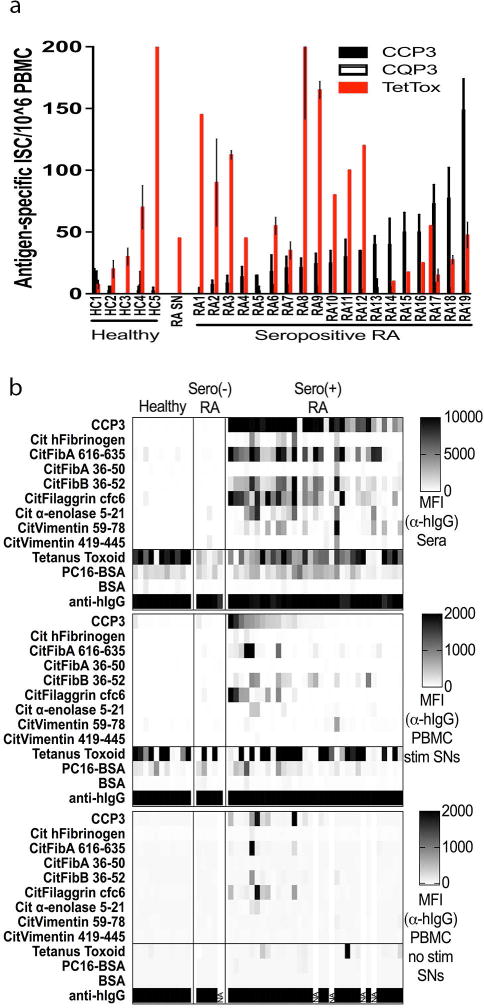
(A) Seropositive RA (n = 19), seronegative RA (n=1) and healthy (n = 5) day-6 PBMC cultures (+/− CpG2006/IL-21/CD40L) were assessed by ELISpot for antigen-specific B cells using 200,000 cells per well after overnight culture. We found IgG anti-TT ISC at 71.1+/−2.6 (range 0–145) and 65.4+/−1.2 (range 0–220) anti-TT ISC/106 PBMC in seropositive RA and healthy controls, respectively. (B) Seropositive RA (n = 33), seronegative RA (n = 5) and healthy control (n = 11) PBMCs were isolated and cultured without and with stimulation, adapting reported conditions (31). Culture supernatants were assayed for Ig/antibody content by multiplex antigen bead assays using anti-human IgG (Fc specific) R-phycoerythrin (Ebioscience), with values reported as MFI.
As an internal control, we found that almost all RA and healthy subjects had detectable circulating IgG-expressing memory B cells reactive the control bacterial antigen, Tetanus Toxoid (TT) from the preventative vaccine that has been given to almost every adult, as RA patients had IgG anti-TT ISC at 71.1+/−2.6 (range 0–145) and healthy adults had 65.4+/−1.2 (range 0–220) anti-TT ISC/106 PBMC (Figure 3A), which were not significantly different between these groups. Notably, these studies showed that the frequencies of recirculating anti-TT memory B cells overlap with the frequencies of ACPA-specific B cells in seropositive RA patients. Although these ELISpot assays were informative, we sought to develop a more efficient means for illuminating the broader range of fine binding auto-specificities expressed by peripheral B cells in a donor.
ACPA production by peripheral blood mononuclear cells from RA patients
To investigate the fine binding specificity of circulating citrulline-antigen reactive B cells, these above-described PBMC culture conditions were used to induce B-cell differentiation and antibody production, which we evaluated by multiplex bead array. Indeed, without stimulation we detected only low levels of IgG (mean 0.16 μg/mL, range 0.01–0.56 μg/mL), while with 6 days of stimulation IgG levels were greatly increased (mean 7.65 μg/mL, range 1.85–20.95 μg/mL). Based on IgG CCP3 reactivity, ACPA were found in 15/33 (45.5%) supernatants from stimulated seropositive RA PBMCs (Figure 3B). In general, wells that contained IgG anti-CCP3 antibodies also displayed a range of reactivities for other citrulline-containing epitopes (but not arginine-containing analogues) that greatly varied between different RA donors. Taken together with the above-described ELISpot results, these findings confirmed that seropositive RA patients commonly have circulating citrulline-epitope specific B cells.
Overall, IgG ACPA were only detected in cultures of PBMC from seropositive RA, and in most cases only after 4–6 days of in vitro stimulation – ACPA were infrequently detected in wells with unstimulated cells (i.e., 7/29 (24.1%) of seropositive RA patients) (Figure 3B). Furthermore, the autoreactive blood B-cell clones in different donors appeared to vary broadly by fine specificity for citrulline-containing self-antigen variants. These findings support the notion that RA patients have quiescent switched-memory B cells that can express ACPA in their blood, while circulating constitutively IgG-secreting cells (i.e., plasma cells/plasmablasts) are much less commonly detected.
The relationship between the fine specificity of serum ACPA and of the stimulated circulating ACPA-producing memory B cells within the PBMC was evaluated. Intriguingly, we found significant correlations between the serum IgG reactivities and in vitro secreted autoantibodies to CCP3 (p<0.0001), Cit-filaggrin cfc6 (p=0.0001), as well as seven other citrulinated-antigens that were tested (Supplementary Figure 4). Correlations for serum and in vitro produced anti-TT antibody responses were also observed (p=0.03). These results strongly support the notion that there is a connection between epitope-specific ACPA in serum and the fine specificities of circulating autoreactive B cells. However, clinical disease activity, based on DAS28 scores, correlated neither with the level nor the ACPA specificity in serum or supernatant (data not shown).
Evaluation of sorted peripheral blood B cells by multiplex assay
To directly determine which B-cell subsets in the peripheral blood are the source of RA disease-associated ACPA, we used flow cytometry to isolate CD3− CD14− CD19+ B cells from PBMC into four sub-populations: CD27+IgD−, CD27+IgD+, CD27−IgD−, and CD27−IgD+. The MS40L cell line was then used for the in vitro expansion of each sorted B cell subset of defined surface phenotype (30). Sorted B-cell subsets from 10 RA and 5 healthy individuals (see Supplementary Figure 2 for strategy) were separately cultured at 50 viable B cells per well. From the multiplex assay and confirmed by ELISA, only low or undetectable IgG levels were present after 0–2 days of in vitro culture, and ACPA reactivity was not detectable. Following culture of switched-memory B cells with stimulants for 12 days, we detected 10-fold greater levels of total IgG (mean 3.2+/-2.4 μg/mL) than was associated with any other defined B-cell subset.
Cultured switched-memory B cells (CD27+/IgD−) most consistently produced high levels of IgG ACPA. Otherwise ACPA were only infrequently detected in wells from “double-negative” B cells (CD27−IgD−). Other sorted subsets had no detectable ACPA activity (not shown), as IgG ACPA were never detected among pre-switched memory (CD27+IgD+) or naïve/transitional B cells (CD27−IgD+).
Overall, these data document that IgG ACPA primarily derive from switched memory B cells (i.e., ACPA MBC) in the circulation of RA patients (Table 1). Moreover, ACPA memory B cells were detected in the circulation of seropositive RA patients in both treatment groups (MTX or TNFi) tested, with detection most frequently based on reactivity with the CCP3 ligand, while there was much greater heterogeneity with other individual citrulline-containing peptides, but neither had detectable reactivity with the B cells of seronegative RA nor with unaffected control subjects. We also surveyed for IgM-autoantibodies but every case of IgM reactivity with citrulline-containing ligands was associated with high polyreactivity with other antigens as well (not shown), and therefore no truly monospecific IgM ACPA were detected.
Table 1.
RA patient data for memory B cell studies
| Group | All RA | Methotrexate | TNFi +/− Methotrexate |
Healthy Control |
|---|---|---|---|---|
| Number of subjects (n) | 24 | 12 | 12 | 5 |
| Treatment | – | MTX | TNFi+MTX (n = 11) TNFi (n = 1) |
– |
| Age (years) | 53.5 +/− 11.5 (27 – 82) |
51.1 +/− 13.5 (27 – 82) |
56.0 +/− 9.1 (42 – 72) |
36.6 +/− 12.6 (25 – 58) |
| DAS28 | 3.0 +/− 1.8 (1.1–7.2) |
3.1 +/− 2.0 (1.3–7.2) |
3.0 +/− 1.7 (1.1–6.8) |
– |
| Sex (% female) | 20F / 4M (83.3%) | 10F / 2M (83.3%) | 10F / 2M (83.3%) | 2F / 3M (40%) |
| CCP3 IgG (RU) | 1675.3 +/− 1906.7 (49.2–7725.9) |
1192.9 +/− 1105.4 (104.8 – 3424.3) |
2157.6 +/− 2423.2 (49.2 – 7725.9) |
2.9 +/− 1.1 (2.1–4.8) |
| ACPA sw-MBC (% wells) | 1.4% +/− 2.3 (0 – 7.7) |
0.86% +/− 2.2 (0 – 7.7) |
1.9% +/− 2.4 (0 – 7.7) |
0% |
| ACPA sw-MBC / 1 × 104 sw-MBC | 2.8 +/− 4.6 (0 – 15.4) |
1.7 +/− 4.4 (0 – 15.4) |
3.8 +/− 4.8 (0 – 15.4) |
0 |
| Disease duration (days) | 4200.5 +/− 4243.9 (226 – 16342) |
2379.2 +/− 1830.5 (239 – 6022) |
6187.5 +/− 5254.9 (315 – 16342) |
– |
| Methotrexate duration (days) | 1711.3 +/− 2854.1 (119 – 11204) |
1061.9 +/− 1490.5 (119 – 3878) |
2360.7 +/− 3760.2 (148 – 11204) |
– |
| TNFi duration (days) | – | – | 866.1 +/− 1301.2 (148 – 3584) |
– |
Data are mean +/− SD (range), unless otherwise stated.
sw-MBC = switched memory B cells, TNFi = TNF-α inhibitor
IgG anti-CCP3 IgG (INOVA) performed with sera diluted 1:1616
Burden of circulating ACPA-secreting B cells correlates with levels of serum ACPA
Based on evidence that seropositive RA patients commonly have circulating B cells that can secrete IgG ACPA, we wondered whether there was a relationship between levels of these autoantibody-secreting memory B cells and their serum IgG ACPA levels. We therefore performed surveys on samples from a cross-sectional study of RA patients.
Overall, we found ACPA secretion in a mean 1.3% (range 0–7.7%) of wells containing 50 viable switched-memory B cells (CD19+/CD27+/IgD−), while IgG ACPA-secreting B cells were never detected in healthy donors (Table 1). Considering the number of B cells that were tested, we estimated that ACPA reactivity was expressed by 0.03%+/−0.04 (range 0–0.15%) of switched-memory B cells in a seropositive RA patient. Notably, we found a significant direct correlation between the proportion of wells with detectable IgG ACPA and the serum levels of IgG anti-CCP3 (Spearman correlation R=0.58, p=0.003)(Figure 4A). In general, ACPA B cells in individual wells exhibited great heterogeneity in their recognition of multiple autoantigenic determinants (Figure 4B). Taken together, the data indicate that there is a biologic link between the frequencies of recirculating anti-citrulline memory B cells and serum ACPA levels.
Figure 4. A high burden of ACPA switched-memory B cells correlates with a high serum ACPA level, and epitope specificities largely reiterate those in circulating serum ACPA in the same subject.
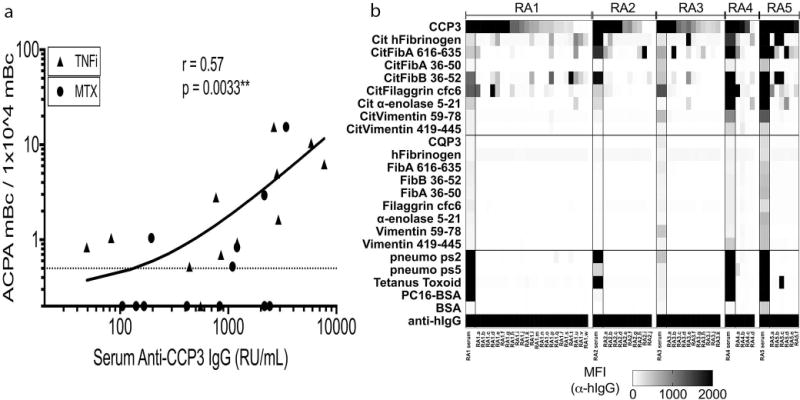
(A) Seropositive RA patient PBMC were stained and switched-memory (CD19+ IgD− CD27+) B cells sorted by FACS and cultured at 50 cells/well with MS40L cell line + CpG2006/IL-21 for 12 days. CCP3-positive wells by ELISA prescreening were subjected to multiplex analysis for citrullinated epitope reactivity. Multiplex wells were counted as positive if reactivity of a citrullinated peptide (based on MFI) was >50 MFI and 2-fold or greater than native peptide. (A) For seropositve RA on a TNF inhibitor (n = 12) and methotrexate (MTX, n = 12), ACPA-positive wells were counted, as these were assumed to contain one or more ACPA memory B cells per well. Data were converted to switched-memory B cells per 10,000 memory B cells cultured, and plotted against serum IgG anti-CCP3 (with RU/mL determined by IgG CCP3 clinical test, with additional serum dilutions performed when required). (B) Representative multiplex data from 5 seropositive RA patients, showing serum (diluted 1:1000, column on left for each patient) and supernatant (diluted 1:2) IgG ACPA reactivity.
Levels of circulating ACPA-secreting B cells do not correlate with disease activity
Over the past two decades there have been remarkable advances in the efficacy of treatment regimens, and in our capacity to accurately gauge overall disease activity in individual RA patients. Neither total blood levels of memory B cells nor IgG CCP3 antibody levels correlated with disease activity (not shown). We therefore next asked whether we could find relationships between RA clinical disease activity and levels of recirculating citrulline antigen-reactive memory B cells. Our cross-sectional studies were performed on patients that displayed a wide range of serum IgG anti-CCP3 levels, and based on the DAS28 scale (35) different individuals had from high disease activity (>5.1) to clinical remission (<2.6). ACPA switched-memory B cells were found in the majority (16/24, 66.7%) of seropositive RA patients (Figures 4&5). Yet from quantitation of the frequency of wells with detectable ACPA, we found only a non-significant trend towards a higher frequency of ACPA blood memory B cells in patients with higher DAS28 scores (Figure 5). Further analyses in subsets of patients that received either treatment with methotrexate, or with a biologic agent (TNF inhibitor), also failed to show a significant correlation. Most importantly, ACPA secreting memory B cells were detected in most (8/14) seropositive RA patients in clinical remission, based on DAS28 scores of less than 2.6 (Figure 5).
Figure 5. High burdens of circulating ACPA switched-memory B cells can be detected in the circulation of treated RA patients, including those in clinical remission.
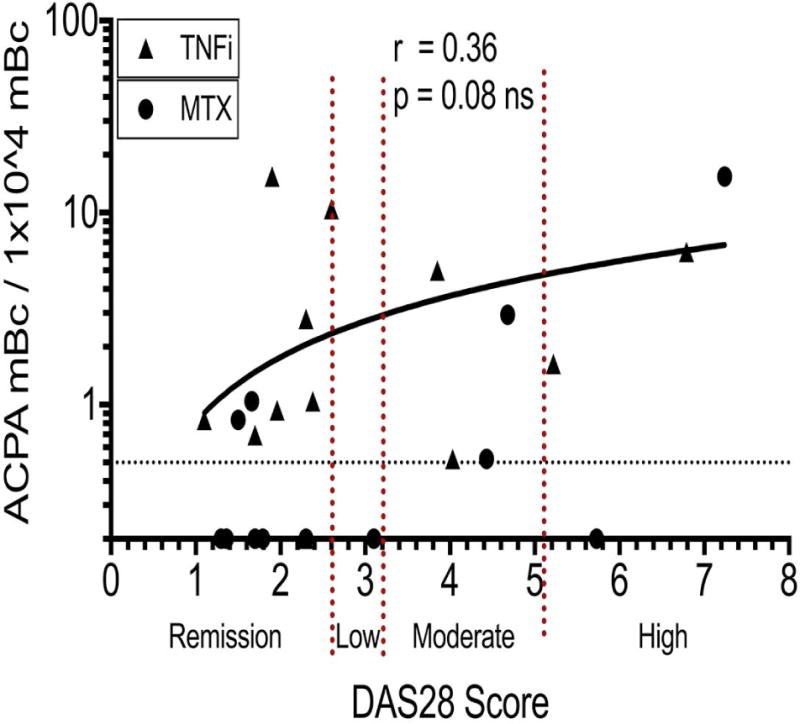
Levels of ACPA switched-memory B cells do not correlate significantly with DAS28 scores. For seropositive RA on TNF inhibitor therapy (TNFi, n = 12) and methotrexate (MTX, n = 12), ACPA-positive switched memory B cells (represented per 10,000 switched-memory B cells cultured) were quantified as described in Figure 5, and then plotted with matched DAS28 scores, and analyzed by Spearman correlation. DAS28-CRP (n = 21) and DAS28-ESR (n = 3) in cases wherein CRP data was not available. Results for correlation analyses for DAS28-CRP and DAS28-ESR were equivalent.
In summary, treatment with standard of care regimens, while sufficient to reduce disease activity and in some patients to attain clinical remission, was not sufficient to eliminate ACPA memory B cells from peripheral circulation, regardless of DAS28 score (Figure 5). A subset of RA patients had very high frequencies of circulating ACPA memory-B cells with a variety of ACPA epitope reactivity patterns in individual patients (Figure 4B). Serum IgG CCP3 was also detected in the patient subsets with high, moderate, and low disease activity, as well as those in DAS remission, although this association was not significant (not shown). Taken together, our results suggest that the presence of a high burden of circulating ACPA memory B cells may persist despite seemingly effective therapeutic interventions and are not reflected by standard measures of disease activity.
Discussion
We have found that seropositive RA patients, but not seronegative RA or healthy adults, have substantial levels of circulating anti-citrullinated epitope-specific B cells, at frequencies akin to those for an antigen-specific protective B-cell response. Circulating RA-associated ACPA B cells were predominantly switched memory (i.e., CD27+IgD−) B cells (36–38) that do not spontaneously secrete ACPA without in vitro activation. These B-cells displayed a range of binding auto-specificities for different citrullinated epitopes, and were also cross-reactive with the synthetic CCP3 peptide widely used to confirm the clinical diagnosis of RA, while they did not recognize native arginine-containing self –protein/peptide analogues.
Our surveys of a cross-sectional cohort of adult RA patients also discovered that the frequency of activatable citrullinated antigen-reactive B cells in a subject was directly proportional to serum IgG ACPA levels. While the range of expressed IgG ACPA specificities varied greatly between individual patients, for an individual RA patient the serum ACPA binding reactivity pattern was very similar to that induced by in vitro stimulation of disease-associated memory B cells. Whereas it has been argued that serum autoantibodies may be primarily produced by antibody-secreting B lineage cells that infiltrate affected synovial linings (20, 39) and/or in the bone marrow (40), our findings suggest that the same clonal sets may be represented among the memory B cells in the bloodstream.
Progressive advances in our understanding of RA pathogenesis have led to the introduction of therapeutic agents that often provide high hurdle clinical benefits, and biopsy/rebiopsy studies have shown this is generally paralleled by resolution of the pathogenic lymphocytic and myeloid synovial infiltrates in affected joints (41–43). Yet despite these therapeutic advancements, such treatments are generally associated with only modest decreases in circulating IgM RF or IgG ACPA/CCP and very rarely do these serologic disease markers become undetectable.
In this study, we demonstrated that therapeutic interventions (methotrexate and/or TNF inhibitors) did not dependably abolish the recirculating autoantigen-targeted memory B cells that reflect the disease-associated defect in B cell immunologic tolerance, even in patients who attained clinical remission. Our findings therefore confirm that, despite the capacity of TNF inhibition to disengage autoreactive B cells from the inflammatory destructive effector pathways in the rheumatoid synovial disease process, these standards of care do not appear to substantially impact the underlying pathogenic autoimmunity (as recently discussed (15)).
Our integrated technical approach provided insights that complement other cutting edge methods for the isolation and characterization of individual B cells in the RA autoimmune response. Recombinant monoclonal antibodies have been recovered and expressed from single synovium-infiltrating B cells, and RA BCR repertoire studies have also characterized the molecular characterization of ACPA binding specificity, B-cell clonal relatedness and immunogenetic origins (39, 44). Tetramer technology for flow cytometric sorting has also been adapted for the study of autoantigen-specific B cells from RA patients (38). While both approaches provide important perspectives, single B-cell analyses enable detailed examinations of individual antigen-reactive lymphocytes (akin to the in-depth study of “individual trees”), while the latter approaches provide broad surveys of disease-associated B-cell lineage “forests.” Our approach provided an informative middle ground as it was designed to quantitate memory B cells and also elucidate autoantibody fine specificity and cross-reactivity in groups of patients. Our approach may require larger blood samples to derive more accurate frequencies when in vivo levels of ACPA B cells amongst peripheral B cells are low. In future studies we would like to apply our now validated methods to longitudinal studies of individuals before and after starting of a new agent.
Our investigations have provided direct evidence that memory B cells are not eradicated in peripheral blood following therapeutic attainment of clinical remission – therefore, the regimens that we examined are insufficient for induction of long-term immune remission. In fact, patients with high serum IgG anti-CCP3 levels were significantly more likely to have high frequencies of ACPA-producing switched-memory B cells, suggesting there are likely ongoing roles for these cells in the production of serum ACPA. Our findings also suggest that serum ACPA levels are closely intertwined with the persistence of circulating ACPA memory B cells. We therefore wonder about the relationship between memory B cells and long-lived plasma cells, as our findings are consistent with a model in which memory B cells are required to replenish ACPA-producing plasma cells in the bone marrow and potentially at other sites.
The contributions of B cells to RA pathogenesis have been best documented by the clinical efficacy of rituximab, the prototypic anti-CD20 antibody, which induces efficient eradication of B cells from the blood. However, the impact at other sites may not be accurately reflected in the bloodstream levels. In treated RA patients with clinical benefits, when in vivo therapeutic anti-CD20 levels later wane, the return of detectable levels of blood B cells is often associated with an overall reduction in the levels of different CD27+ B-cell subsets that can persist for years (45). Yet, to the best of our knowledge the proportions of these post-treatment recovery memory B cells linked to host immune defenses vs. disease-associated autoimmunity have not been investigated.
In conclusion, our results highlight that commonly used treatment regimens are deficient in affecting the underlying pathologic autoimmune disease process. These findings therefore help to rationalize the outcome of two large trials in which cessation of therapy in RA patients in clinical remission led to relapse in the great majority of RA patients within the first year (46–48). Indeed, these regimens fail to provide an effective cure for RA because they do not have the capacity to target the reservoirs of pathogenic autoreactive B cells (as well as T cells).
We therefore propose that the next phase in the development of RA therapy should have a goal of deleting pathologic memory cells and reversing the defect in immune tolerance. Progress towards this goal will require the adoption of a new standard for differentiating clinical remission from true immune remission, which includes an assessment of cellular autoimmune players that will otherwise be responsible for disease reactivation and clinical relapse. Our technical approach for assessing the autoimmune B cell burden in RA patients may therefore provide this essential tool for assessing the capacity of treatment regimens to target and eliminate disease-specific autoimmunity, as well as for making better and more informed treatment decisions in daily clinical practice.
Supplementary Material
Supplementary Figure 1. Seropositive RA patient sera samples exhibit varied IgG ACPA fine binding specificity patterns by ELISA. These assays were performed on serum samples from seropositive RA (n = 29), seronegative RA (n = 8), and healthy controls (n = 2). Anti-citrullinated peptide reactivity patterns are detectable only in seropositive RA patients, with an increasing number of ACPA epitope reactivities as the values from the IgG CCP3 diagnostic assay (INOVA) increased but are not detected in seronegative RA or healthy individuals. ACPA reactivity patterns were first evaluated with neutravidin coated wells of EIA plates after incubation with individual ligands from a panel that included biotinylated CCP3 and the control CQP3 peptide (gift of INOVA) and 7 paired sets of biotinylated arginine- and citrulline-containing autocyclicizing peptides and control antigens/ligands (see Supplementary Table 2). Anti-human IgG HRP was used for detection. Values were interpolated from a standard curve using a pool of RA sera.
Supplementary Figure 2. B-cell sorting and stimulation strategy. PBMC were stained with mouse anti-human anti-IgD FITC (clone IA6-2), anti-CD19 APC-Cy7 (clone SJ25C1), anti-CD27 APC (clone 0323), anti-CD3 PerCP-Cy5.5 (clone HIT3a), and anti-CD14 PerCP-Cy5.5 (clone M5E2) in PBS with 1% BSA supplemented with mouse serum, followed by live/dead staining. B-cell subsets were sorted on a FACSAria II (Becton Dickinson) into FBS on ice. Sorted B cells of defined phenotype were assessed for viability, then plated at 5 or 50 cells in complete RPMI in individual wells in a 96-well plate that were seeded with a CD40-L expressing cell line, MS40L at 3,000 cells/well without or with the TLR9 agonist, CpG2006 (1 μg/mL) + IL-21 for 1–12 days. PBMC from seropositive RA patients and healthy control B cells were sorted into CD27+/IgD− (switched memory), CD27+IgD+ (unswitched memory), CD27− IgD− (double negative), and CD27− IgD+ (antigen-inexperienced transitional/naïve) populations. Supernatants were prescreened by ELISA for IgG CCP3 reactivity, followed by multiplex analysis of all positive and select negative wells from each patient. Supernatants were diluted at 1:10, 1:100, and 1:1000 and assayed for total IgG, and 1:2 for multiplex analyses.
Supplementary Figure 3. In seropositive RA serum, IgG ACPA reactivity by ELISA correlated with results from the multiplex bead-based assay for CCP3 and other cyclic citrullinated peptide epitopes. Using a biotinylated version of the CCP3 peptide (INOVA), we compared IgG ACPA levels assessed by CCP3 peptide ELISA and compared to values for CCP3 reactivity from the multiplex bead-based assay (MFI) in sera at a 1:1000 dilution. Similarly, we tested for binding to other Cit/non-Cit ligands by both methods (see Supplementary Table 2 for list of citrullinated and native peptides/proteins). Significant correlations were determined by Spearman correlation.
Supplementary Figure 4. Patterns of epitope-reactive ACPA from stimulated PBMC correlated with serum IgG ACPA reactivity. 1 × 106 PBMC were stimulated with CpG2006/IL-21/sCD40L for 6 days, and culture supernatants were tested for epitope-specific ACPA by multiplex bead-based array. For testing, sera were diluted 1:1000. For each peptide tested, Spearman correlations were performed. Assays included testing for tetanus toxoid (TT) and phosphorylcholine conjugated to albumin (PC-BSA).
Acknowledgments
This work was supported by National Institutes of Health Grants; R01AI090118 (GJS), R01AI068063 (GJS), R01-AR42455 (GJS), N01-AR-4-2271, an American Recovery and Reinvestment Act supplement (GJS), and the NIAID/NIH: NYU School of Medicine-Immunology & Inflammation Training Grant (T32 AI100853); the American College of Rheumatology Research Education Foundation (Disease Targeted Innovative Research Grant)(GJS); the P. Robert Majumder Charitable Trust (GJS) and the Stewart and Judith Colton Autoimmunity Center (GJS). We are grateful to Rufus Burlingame and INOVA Diagnostics for providing the CCP3 and CQP3 peptides. We acknowledge the assistance of the NYU Immune Monitoring Core and the NYU Flow Cytometry Core Facility, supported by NYU-HHC CTSI Grant UL1 TR000038, and the NYU the Laura and Isaac Perlmutter Cancer Center support grant, P30CA016087 from the National Center for Advancing Translational Sciences (NCATS).
Abbreviations
- CCP3
Cyclic citrullinated peptide 3
- CQP3
Cyclic glutamine-containing peptide 3
- Fib
Fibrinogen
Footnotes
Competing Interests
The authors declare that they have no competing interests.
Authors’ contributions
AJP and GJS designed studies and wrote paper. CG, AJP, and GJS developed ELISA assays. AJP performed most other studies. PR, JDG, MM, LM and WCR contributed to experimental design and to the writing and revision of the manuscript.
References
- 1.Sokka T, Abelson B, Pincus T. Mortality in rheumatoid arthritis: 2008 update. Clin Exp Rheumatol. 2008;26:S35–61. [PubMed] [Google Scholar]
- 2.Dadoun S, Zeboulon-Ktorza N, Combescure C, Elhai M, Rozenberg S, Gossec L, Fautrel B. Mortality in rheumatoid arthritis over the last fifty years: systematic review and meta-analysis. Joint Bone Spine. 2013;80:29–33. doi: 10.1016/j.jbspin.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Catrina AI, Joshua V, Klareskog L, Malmstrom V. Mechanisms involved in triggering rheumatoid arthritis. Immunol Rev. 2016;269:162–174. doi: 10.1111/imr.12379. [DOI] [PubMed] [Google Scholar]
- 4.de Smit MJ, Brouwer E, Vissink A, van Winkelhoff AJ. Rheumatoid arthritis and periodontitis; a possible link via citrullination. Anaerobe. 2011;17:196–200. doi: 10.1016/j.anaerobe.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Demoruelle MK, Weisman MH, Simonian PL, Lynch DA, Sachs PB, Pedraza IF, Harrington AR, Kolfenbach JR, Striebich CC, Pham QN, Strickland CD, Petersen BD, Parish MC, Derber LA, Norris JM, Holers VM, Deane KD. Brief report: airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: early injury or initiating site of autoimmunity? Arthritis Rheum. 2012;64:1756–1761. doi: 10.1002/art.34344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, Huttenhower C, Littman DR. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 8.van der Woude D, Rantapaa-Dahlqvist S, Ioan-Facsinay A, Onnekink C, Schwarte CM, Verpoort KN, Drijfhout JW, Huizinga TW, Toes RE, Pruijn GJ. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis. 2010;69:1554–1561. doi: 10.1136/ard.2009.124537. [DOI] [PubMed] [Google Scholar]
- 9.Holers VM. Antibodies to citrullinated proteins: pathogenic and diagnostic significance. Curr Rheumatol Rep. 2007;9:396–400. doi: 10.1007/s11926-007-0063-5. [DOI] [PubMed] [Google Scholar]
- 10.Silverman GJ, Carson DA. Roles of B cells in rheumatoid arthritis. Arthritis Res Ther. 2003;5(Suppl 4):S1–6. doi: 10.1186/ar1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorner T. Crossroads of B cell activation in autoimmunity: rationale of targeting B cells. J Rheumatol Suppl. 2006;77:3–11. [PubMed] [Google Scholar]
- 12.Marston B, Palanichamy A, Anolik JH. B cells in the pathogenesis and treatment of rheumatoid arthritis. Curr Opin Rheumatol. 2010;22:307–315. doi: 10.1097/BOR.0b013e3283369cb8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meednu N, Zhang H, Owen T, Sun W, Wang V, Cistrone C, Rangel-Moreno J, Xing L, Anolik JH. Production of RANKL by Memory B Cells: A Link Between B Cells and Bone Erosion in Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68:805–816. doi: 10.1002/art.39489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, Moreland LW, O’Dell J, Winthrop KL, Beukelman T, Bridges SL, Jr, Chatham WW, Paulus HE, Suarez-Almazor M, Bombardier C, Dougados M, Khanna D, King CM, Leong AL, Matteson EL, Schousboe JT, Moynihan E, Kolba KS, Jain A, Volkmann ER, Agrawal H, Bae S, Mudano AS, Patkar NM, Saag KG. 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2012;64:625–639. doi: 10.1002/acr.21641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silverman GJ, Pelzek A. Rheumatoid arthritis clinical benefits from abatacept, cytokine blockers, and rituximab are all linked to modulation of memory B cell responses. J Rheumatol. 2014;41:825–828. doi: 10.3899/jrheum.140022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fransen J, van Riel PL. The Disease Activity Score and the EULAR response criteria. Rheum Dis Clin North Am. 2009;35:745–757. vii–viii. doi: 10.1016/j.rdc.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2010;69:1580–1588. doi: 10.1136/ard.2010.138461. [DOI] [PubMed] [Google Scholar]
- 18.van Venrooij WJ, Zendman AJ. Anti-CCP2 antibodies: an overview and perspective of the diagnostic abilities of this serological marker for early rheumatoid arthritis. Clin Rev Allergy Immunol. 2008;34:36–39. doi: 10.1007/s12016-007-8029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debaugnies F, Servais G, Badot V, Noubouossie D, Willems D, Corazza F. Anti-cyclic citrullinated peptide antibodies: a comparison of different assays for the diagnosis of rheumatoid arthritis. Scand J Rheumatol. 2013;42:108–114. doi: 10.3109/03009742.2012.723746. [DOI] [PubMed] [Google Scholar]
- 20.Snir O, Widhe M, Hermansson M, Spee Cvon, Lindberg J, Hensen S, Lundberg K, Engstrom A, Venables PJ, Toes RE, Holmdahl R, Klareskog L, Malmstrom V. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010;62:44–52. doi: 10.1002/art.25036. [DOI] [PubMed] [Google Scholar]
- 21.Hill JA, Bell DA, Brintnell W, Yue D, Wehrli B, Jevnikar AM, Lee DM, Hueber W, Robinson WH, Cairns E. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J Exp Med. 2008;205:967–979. doi: 10.1084/jem.20072051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandra PE, Sokolove J, Hipp BG, Lindstrom TM, Elder JT, Reveille JD, Eberl H, Klause U, Robinson WH. Novel multiplex technology for diagnostic characterization of rheumatoid arthritis. Arthritis Res Ther. 2011;13:R102. doi: 10.1186/ar3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebbag M, Moinard N, Auger I, Clavel C, Arnaud J, Nogueira L, Roudier J, Serre G. Epitopes of human fibrin recognized by the rheumatoid arthritis-specific autoantibodies to citrullinated proteins. Eur J Immunol. 2006;36:2250–2263. doi: 10.1002/eji.200535790. [DOI] [PubMed] [Google Scholar]
- 24.Verpoort KN, Cheung K, Ioan-Facsinay A, van der Helm-van Mil AH, de Vries-Bouwstra JK, Allaart CF, Drijfhout JW, de Vries RR, Breedveld FC, Huizinga TW, Pruijn GJ, Toes RE. Fine specificity of the anti-citrullinated protein antibody response is influenced by the shared epitope alleles. Arthritis Rheum. 2007;56:3949–3952. doi: 10.1002/art.23127. [DOI] [PubMed] [Google Scholar]
- 25.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43:155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Dubucquoi S, Solau-Gervais E, Lefranc D, Marguerie L, Sibilia J, Goetz J, Dutoit V, Fauchais AL, Hachulla E, Flipo RM, Prin L. Evaluation of anti-citrullinated filaggrin antibodies as hallmarks for the diagnosis of rheumatic diseases. Ann Rheum Dis. 2004;63:415–419. doi: 10.1136/ard.2003.008623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, Mikuls TR, Venables PJ. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008;58:3009–3019. doi: 10.1002/art.23936. [DOI] [PubMed] [Google Scholar]
- 29.Wagner CA, Sokolove J, Lahey LJ, Bengtsson C, Saevarsdottir S, Alfredsson L, Delanoy M, Lindstrom TM, Walker RP, Bromberg R, Chandra PE, Binder SR, Klareskog L, Robinson WH. Identification of anticitrullinated protein antibody reactivities in a subset of anti-CCP-negative rheumatoid arthritis: association with cigarette smoking and HLA-DRB1 ‘shared epitope’ alleles. Ann Rheum Dis. 2015;74:579–586. doi: 10.1136/annrheumdis-2013-203915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo XM, Maarschalk E, O’Connell RM, Wang P, Yang L, Baltimore D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood. 2009;113:1422–1431. doi: 10.1182/blood-2008-09-177139. [DOI] [PubMed] [Google Scholar]
- 31.Cao Y, Gordic M, Kobold S, Lajmi N, Meyer S, Bartels K, Hildebrandt Y, Luetkens T, Ihloff AS, Kroger N, Bokemeyer C, Atanackovic D. An optimized assay for the enumeration of antigen-specific memory B cells in different compartments of the human body. J Immunol Methods. 2010;358:56–65. doi: 10.1016/j.jim.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Bonsignori M, Moody MA, Parks RJ, Holl TM, Kelsoe G, Hicks CB, Vandergrift N, Tomaras GD, Haynes BF. HIV-1 envelope induces memory B cell responses that correlate with plasma antibody levels after envelope gp120 protein vaccination or HIV-1 infection. J Immunol. 2009;183:2708–2717. doi: 10.4049/jimmunol.0901068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibodies: the past, the present and the future. Nat Rev Rheumatol. 2011;7:391–398. doi: 10.1038/nrrheum.2011.76. [DOI] [PubMed] [Google Scholar]
- 34.Sakkas LI, Bogdanos DP, Katsiari C, Platsoucas CD. Anti-citrullinated peptides as autoantigens in rheumatoid arthritis-relevance to treatment. Autoimmun Rev. 2014;13:1114–1120. doi: 10.1016/j.autrev.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Anderson J, Caplan L, Yazdany J, Robbins ML, Neogi T, Michaud K, Saag KG, O’Dell JR, Kazi S. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res (Hoboken) 2012;64:640–647. doi: 10.1002/acr.21649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bellatin MF, Han M, Fallena M, Fan L, Xia D, Olsen N, Branch V, Karp D, Stastny P. Production of autoantibodies against citrullinated antigens/peptides by human B cells. J Immunol. 2012;188:3542–3550. doi: 10.4049/jimmunol.1100577. [DOI] [PubMed] [Google Scholar]
- 37.Szarka E, Babos F, Magyar A, Huber K, Szittner Z, Papp K, Prechl J, Pozsgay J, Neer Z, Adori M, Nagy G, Rojkovich B, Gati T, Kelemen J, Baka Z, Brozik M, Pazar B, Poor G, Hudecz F, Sarmay G. Recognition of new citrulline-containing peptide epitopes by autoantibodies produced in vivo and in vitro by B cells of rheumatoid arthritis patients. Immunology. 2014;141:181–191. doi: 10.1111/imm.12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerkman PF, Fabre E, van der Voort EI, Zaldumbide A, Rombouts Y, Rispens T, Wolbink G, Hoeben RC, Spits H, Baeten DL, Huizinga TW, Toes RE, Scherer HU. Identification and characterisation of citrullinated antigen-specific B cells in peripheral blood of patients with rheumatoid arthritis. Ann Rheum Dis. 2016;75:1170–1176. doi: 10.1136/annrheumdis-2014-207182. [DOI] [PubMed] [Google Scholar]
- 39.Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, Engstrom M, Snir O, Israelsson L, Catrina AI, Wardemann H, Corti D, Meffre E, Klareskog L, Malmstrom V. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med. 2013;210:445–455. doi: 10.1084/jem.20121486. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. 2011;7:170–178. doi: 10.1038/nrrheum.2011.1. [DOI] [PubMed] [Google Scholar]
- 41.Kavanaugh A, Rosengren S, Lee SJ, Hammaker D, Firestein GS, Kalunian K, Wei N, Boyle DL. Assessment of rituximab’s immunomodulatory synovial effects (ARISE trial). 1: clinical and synovial biomarker results. Ann Rheum Dis. 2008;67:402–408. doi: 10.1136/ard.2007.074229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buch MH, Boyle DL, Rosengren S, Saleem B, Reece RJ, Rhodes LA, Radjenovic A, English A, Tang H, Vratsanos G, O’Connor P, Firestein GS, Emery P. Mode of action of abatacept in rheumatoid arthritis patients having failed tumour necrosis factor blockade: a histological, gene expression and dynamic magnetic resonance imaging pilot study. Ann Rheum Dis. 2009;68:1220–1227. doi: 10.1136/ard.2008.091876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klaasen R, Thurlings RM, Wijbrandts CA, van Kuijk AW, Baeten D, Gerlag DM, Tak PP. The relationship between synovial lymphocyte aggregates and the clinical response to infliximab in rheumatoid arthritis: a prospective study. Arthritis Rheum. 2009;60:3217–3224. doi: 10.1002/art.24913. [DOI] [PubMed] [Google Scholar]
- 44.Tan YC, Kongpachith S, Blum LK, Ju CH, Lahey LJ, Lu DR, Cai X, Wagner CA, Lindstrom TM, Sokolove J, Robinson WH. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:2706–2715. doi: 10.1002/art.38754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roll P, Mahmood Z, Muhammad K, Feuchtenberger M, Dorner T, Tony HP. Long-term repopulation of peripheral B-cell subsets after single and repeated rituximab infusions in patients with rheumatoid arthritis. Clin Exp Rheumatol. 2015;33:347–353. [PubMed] [Google Scholar]
- 46.Smolen JS, Nash P, Durez P, Hall S, Ilivanova E, Irazoque-Palazuelos F, Miranda P, Park MC, Pavelka K, Pedersen R, Szumski A, Hammond C, Koenig AS, Vlahos B. Maintenance, reduction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis (PRESERVE): a randomised controlled trial. Lancet. 2013;381:918–929. doi: 10.1016/S0140-6736(12)61811-X. [DOI] [PubMed] [Google Scholar]
- 47.Nagy G, van Vollenhoven RF. Sustained biologic-free and drug-free remission in rheumatoid arthritis, where are we now? Arthritis Res Ther. 2015;17:181. doi: 10.1186/s13075-015-0707-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moghadam MG, Vonkeman HE, Ten Klooster PM, Tekstra J, van Schaardenburg D, Starmans-Kool M, Brouwer E, Bos R, Lems WF, Colin EM, Allaart CF, Meek IL, Landewe R, Bernelot Moens HJ, van Riel PL, van de Laar MA, Jansen TL, P.C. Dutch National Stopping Tumor Necrosis Factor-inhibitors in Patients with Established Rheumatoid Arthritis in Remission or Stable Low Disease Activity: A Pragmatic Randomized Multicenter Open-Label Controlled Trial. Arthritis Rheumatol. 2016 doi: 10.1002/art.39626. [DOI] [PubMed] [Google Scholar]
- 49.Gronwall C, Akhter E, Oh C, Burlingame RW, Petri M, Silverman GJ. IgM autoantibodies to distinct apoptosis-associated antigens correlate with protection from cardiovascular events and renal disease in patients with SLE. Clin Immunol. 2012;142:390–398. doi: 10.1016/j.clim.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Seropositive RA patient sera samples exhibit varied IgG ACPA fine binding specificity patterns by ELISA. These assays were performed on serum samples from seropositive RA (n = 29), seronegative RA (n = 8), and healthy controls (n = 2). Anti-citrullinated peptide reactivity patterns are detectable only in seropositive RA patients, with an increasing number of ACPA epitope reactivities as the values from the IgG CCP3 diagnostic assay (INOVA) increased but are not detected in seronegative RA or healthy individuals. ACPA reactivity patterns were first evaluated with neutravidin coated wells of EIA plates after incubation with individual ligands from a panel that included biotinylated CCP3 and the control CQP3 peptide (gift of INOVA) and 7 paired sets of biotinylated arginine- and citrulline-containing autocyclicizing peptides and control antigens/ligands (see Supplementary Table 2). Anti-human IgG HRP was used for detection. Values were interpolated from a standard curve using a pool of RA sera.
Supplementary Figure 2. B-cell sorting and stimulation strategy. PBMC were stained with mouse anti-human anti-IgD FITC (clone IA6-2), anti-CD19 APC-Cy7 (clone SJ25C1), anti-CD27 APC (clone 0323), anti-CD3 PerCP-Cy5.5 (clone HIT3a), and anti-CD14 PerCP-Cy5.5 (clone M5E2) in PBS with 1% BSA supplemented with mouse serum, followed by live/dead staining. B-cell subsets were sorted on a FACSAria II (Becton Dickinson) into FBS on ice. Sorted B cells of defined phenotype were assessed for viability, then plated at 5 or 50 cells in complete RPMI in individual wells in a 96-well plate that were seeded with a CD40-L expressing cell line, MS40L at 3,000 cells/well without or with the TLR9 agonist, CpG2006 (1 μg/mL) + IL-21 for 1–12 days. PBMC from seropositive RA patients and healthy control B cells were sorted into CD27+/IgD− (switched memory), CD27+IgD+ (unswitched memory), CD27− IgD− (double negative), and CD27− IgD+ (antigen-inexperienced transitional/naïve) populations. Supernatants were prescreened by ELISA for IgG CCP3 reactivity, followed by multiplex analysis of all positive and select negative wells from each patient. Supernatants were diluted at 1:10, 1:100, and 1:1000 and assayed for total IgG, and 1:2 for multiplex analyses.
Supplementary Figure 3. In seropositive RA serum, IgG ACPA reactivity by ELISA correlated with results from the multiplex bead-based assay for CCP3 and other cyclic citrullinated peptide epitopes. Using a biotinylated version of the CCP3 peptide (INOVA), we compared IgG ACPA levels assessed by CCP3 peptide ELISA and compared to values for CCP3 reactivity from the multiplex bead-based assay (MFI) in sera at a 1:1000 dilution. Similarly, we tested for binding to other Cit/non-Cit ligands by both methods (see Supplementary Table 2 for list of citrullinated and native peptides/proteins). Significant correlations were determined by Spearman correlation.
Supplementary Figure 4. Patterns of epitope-reactive ACPA from stimulated PBMC correlated with serum IgG ACPA reactivity. 1 × 106 PBMC were stimulated with CpG2006/IL-21/sCD40L for 6 days, and culture supernatants were tested for epitope-specific ACPA by multiplex bead-based array. For testing, sera were diluted 1:1000. For each peptide tested, Spearman correlations were performed. Assays included testing for tetanus toxoid (TT) and phosphorylcholine conjugated to albumin (PC-BSA).