

Abstract
Progressive myoclonus epilepsies (PMEs) are disorders characterized by myoclonic and generalized seizures with progressive neurological deterioration. While several genetic causes for PMEs have been identified, the underlying causes remain unknown for a substantial portion of cases. Here we describe several affected individuals from a large, consanguineous family presenting with a novel PME in which symptoms begin in adolescence and result in death by early adulthood. Whole exome analyses revealed that affected individuals have a homozygous variant in GPR37L1 (c.1047G>T [Lys349Asp]), an orphan G protein-coupled receptor (GPCR) expressed predominantly in the brain. In vitro studies demonstrated that the K349N substitution in GPR37L1 did not grossly alter receptor expression, surface trafficking or constitutive signaling in transfected cells. However, in vivo studies revealed that a complete loss of Gpr37L1 function in mice results in increased seizure susceptibility. Mice lacking the related receptor Gpr37 also exhibited an increase in seizure susceptibility, while genetic deletion of both receptors resulted in an even more dramatic increase in vulnerability to seizures. These findings provide evidence linking GPR37L1 and GPR37 to seizure etiology and demonstrate an association between a GPR37L1 variant and a novel progressive myoclonus epilepsy.
Keywords: Brain, Epilepsy, Disease, Receptors, Mutation, Mutant, Orphan GPCR
Introduction
Several forms of epilepsy are highly heritable, but underlying mutations have been identified in only a small fraction of epilepsy patients1–7. The identification of new epilepsy-associated gene variants is important as it can potentially lead to novel targets and approaches for treating epilepsy. Progressive myoclonus epilepsies (PMEs) are rare epilepsy syndromes that result in myoclonic and generalized seizures with progressive neurological deterioration. PMEs typically present in late childhood and/or adolescence. They are among the most devastating forms of epilepsy as they are often associated with debilitating disease progression and poor outcomes. Most PMEs are inherited in an autosomal recessive (AR) manner8. For example, Unverricht-Lundborg disease (MIM 254800), the most common PME, is caused by AR mutations in CSTB (MIM 601145)8–10, the gene encoding cystatin B. In contrast, Lafora disease (MIM 254780), another form of PME, is caused by AR mutations in EPM2A (MIM 607566) and NHLRC1 (MIM 608072), the genes encoding laforin and malin, respectively5; 11; 12. Thus, although the genetic bases for several PMEs are known, additional genes remain to be identified for a substantial number of other cases5.
Exome and genome sequencing are powerful tools that have dramatically increased disease gene discovery13. Here we present a large family of Iraqi descent with five children affected with PME. Previous clinical testing was negative and the family was therefore enrolled in a research exome study. A novel homozygous variant, c.1047G>T [Lys349Asp], of unknown significance was identified in GPR37L1, an orphan G protein-coupled receptor (GPCR) expressed predominantly in the brain14–16. GPR37L1 and its closest relative, GPR37, are class A, rhodopsin-like family members. These receptors share their strongest sequence similarity to the endothelin and bombesin receptors and other peptide-activated GPCRs and are thought to be activated by a yet-to-be-identified peptide ligand. Little is known about the function of GPR37L1, indeed, to date there is only one published report assessing the role of GPR37L1 in vivo17. In that study, mice lacking Gpr37L1 were found to have a modest alteration in postnatal cerebellar development, characterized by reduced granule cell precursor proliferation and early maturation of Bergmann glia and Purkinje cells17. In contrast, GPR37 has been extensively studied for its association with Parkinson’s disease and role in dopaminergic signaling18–24. Expression of both GPR37L1 and GPR37 has been shown to be protective in models of cell stress in vitro25; 26, and thus it is possible that these receptors also play protective roles in vivo. To investigate the potential role of GPR37L1 in epilepsy, functional characterization of the seizure-associated GPR37L1 K349N variant was performed and complemented by in vivo studies on mice lacking Gpr37L1 and/or Gpr37 function to assess the potential roles of these receptors in modulating seizure susceptibility.
Materials and methods
Study Subjects
The proband, VI:9 was the sixth child born to a consanguineous couple of Iraqi descent (Figure 1). She presented to clinic at approximately 11 years of age with complaints of recurrent headaches. Subsequently, she developed visual hallucinations, in the form of colors and lines and myoclonic seizures. Prior to the development of recurrent headaches, she had normal growth and development. All of her symptoms were progressive in nature, becoming more frequent and severe. In addition to epileptic seizures, the proband developed psychogenic non-epileptic seizures. By 13 years of age, she began to demonstrate signs of dementia with cognitive decline and mood disturbances.
Figure 1. Consanguineous family with multiple affected children presenting with a PME.
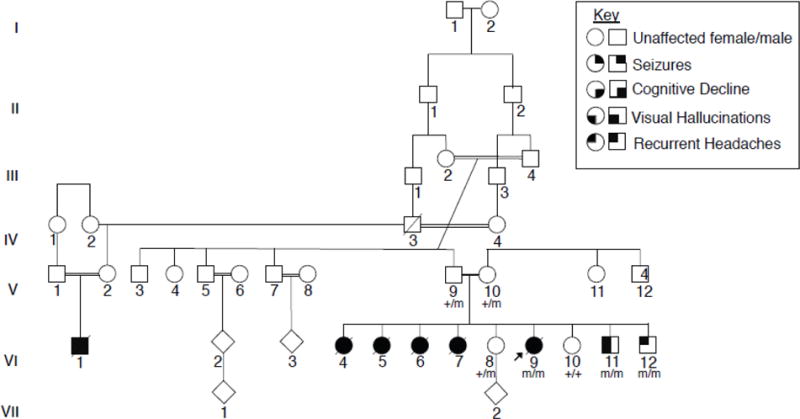
The proband (VI:9) was the sixth child born to a consanguineous couple of Iraqi descent, four older sisters, VI:4, VI:5, VI:6, and VI:7, were described as having a very similar clinical presentation. Affected children presented around the age of puberty with recurrent headaches, followed by visual hallucinations and seizures. Individuals eventually presented with cognitive decline and developed a progressive myoclonic epilepsy. Exome sequencing was completed for V:14, VI:8, VI:9., and VI:11. The proband was found to have a homozygous missense variant of unknown significance, GPR37L1: c.1047G>T (p.K349N). Sanger sequencing was completed for all available family members and confirmed that both parents are heterozygous for the variant, and the unaffected sisters are either heterozygous or wild type. The two younger brothers were found to be homozygous for the K349N variant. Each copy of the K349N mutant is labeled with an “m” in the pedigree shown here, as opposed to a “+” for the wild-type variant, in all individuals available for genetic testing.
EEG analyses were performed at three different time points. The first was done at age 10 and the third was done at age 18, late in the course of the illness. All three EEG studies showed a consistent absence of an occipital dominant rhythm with a background dominated by a mixture high voltage delta and theta activity. Over the course of the three EEG studies, this background activity progressively worsened and became more disorganized. Overlying this background were frequent generalized 4–6hz spike and polyspike and slow wave discharges. MRI and MRS of the brain at 18 years of age detected only a mild prominence of the subarachnoid spaces and were otherwise unremarkable. The patient’s symptoms continued to worsen with generalized and diffuse myoclonic jerks and fasciculations over much of her body, including her face and limbs. Her epilepsy and dementia also gradually worsened and she required multiple hospitalizations in the last year of her life. The patient’s seizures became intractable and she died at 20 years of age from aspiration pneumonia.
Four older sisters, VI:4, VI:5, VI:6, and VI:7, were described as having a similar clinical presentation. They first presented around the age of puberty with headaches, followed by visual hallucinations and eventually developed progressive myoclonic epilepsy. All four sisters died from complications of their disease. Two sisters, VI:8 and VI:10, have reported headaches but no documented seizure activity. They are currently 24 and 18 years old, respectively, and in good health. A younger brother, VI:11, developed headaches and visual hallucinations in the form of black spots at 11 years of age. An EEG showed generalized spike and wave complexes in addition to occipital sharp waves, but no seizure activity was recorded. He was initially treated with anticonvulsants, but medications were subsequently stopped, and he has remained seizure free for 10 months. The youngest child, VI:12, currently 11 years of age, also recently presented with headaches but has had no documented seizure activity. The extended family history was positive for a maternal first cousin, VI:1, who also died prematurely after being diagnosed with headaches and seizures. Additional clinical details are not available.
Human Exome Sequencing
The study was approved by the Institutional Review Board (IRB) at Children’s Mercy Hospital (CMH). Informed written assent/consent to participate in this study was obtained from all available family members. DNA was extracted from peripheral blood from the five living siblings and their parents. Exome sequencing was completed for V:14, VI:8, VI:9., and VI:11. DNA was not available from the four affected siblings who had died prior to moving to the United States. Library preparation was performed utilizing the KAPA Biosystems kit (KAPA Biosystems, Woburn, MA.) Enrichment using Illumina TruSeq Exome enrichment (Illumina, San Diego, CA) was performed according to manufacturers’ instructions. Samples were sequenced on an Illlumina HiSeq 2500 instrument with TruSeq v3 reagents, paired ~100 nucleotide reads. Alignment and variant calling was performed as previously reported27; 28 Exome-enriched DNA was sequenced to a depth of 10.6Gb resulting in median target coverage of 67x. Variants were filtered to 1% minor allele frequency (MAF) in an internal database of 3974 samples, then prioritized by the American College of Medical Genetics (ACMG) categorization29.
Sanger Confirmation
Sanger sequencing was completed for all available family members. Sequencing confirmed that both parents are heterozygous for the GPR37L1 variant, and the unaffected sisters (VI:8 and VI:10) are heterozygous and wild-type, respectively. Like the proband (VI:9), the two younger brothers (VI:11 and VI:12) were found to be homozygous for the K349N GPR37L1 variant.
Cell culture
HEK-293T/17 and NIH-3T3 cells were acquired from ATCC (Manassas, VA) and maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humid, 5% CO2, 37°C incubator. Cells were transfected using Mirus TransITLT1 (Madison, WI) per the manufacturer’s protocol.
Constructs
The Flag-GPR37L1 construct was purchased commercially (Multispan, Inc.) and contains an N-terminal Flag tag. The Flag-K349N construct was created with a single point mutation of the Flag-GPR37L1 construct using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent). The following primers were used to induce a mutation from a lysine to an asparagine at the 349th amino acid residue, forward: CACACTGCTCGTGATTGCTGGCCCTGCAC and reverse: GTGCAGGGCCAGCAATCACGAGCAGTGTG. The mutation was confirmed via DNA sequencing. GPR37L1-GFP and GPR37L1-K349N-GFP constructs (containing C-terminal GFP tags) and were created via gene synthesis performed by Genscript.
Western Blotting
Protein samples were reduced and denatured in Laemmli buffer, loaded into 4–20% Tris-Glycine gels (Bio-Rad) for SDS-PAGE, and then transferred to nitrocellulose membranes (Bio-Rad). Blots were blocked with 5% milk (in 50mM NaCl, 10mM HEPES, pH 7.3 with 0.1% Tween-20 (Sigma)) and incubated with primary antibodies overnight at 4°C. Flag-tagged GPR37L1 and K349N constructs are were detected with mouse HRP-conjugated anti-Flag (Sigma). Protein quantification was done using densitometry, performed with ImageJ software.
Cell Surface Biotinylation
HEK-293T/17 cells were transfected with 2 μg of DNA (empty vector or receptor). At 24 h post-transfection, cells were placed on ice and washed with ice-cold PBS+Ca2+. Cells were then incubated with 10 mM Sulfo-NHS-Biotin (Thermo Scientific) in PBS+Ca2+ on ice for 1 h and then washed three more times with PBS+Ca2+ + 100 mM glycine to quench. Cells were harvested in 500 μl of lysis buffer (1% Triton X-100, 10 mM Hepes, 50 mM NaCl, 5 mM EDTA, and protease inhibitor cocktail (Roche Diagnostics) and lysed by end-over-end rotation for 30 min at 4°C. Cell debris was cleared by centrifugation, and soluble cell lysates were incubated with 50 μl of streptavidin agarose beads (Thermo Scientific) for 1 h at 4°C. Beads were washed 3 times with lysis buffer and resuspended in 100 μl of Laemmli buffer. Biotinylated proteins were detected via Western blot, as described above.
Confocal Microscopy
NIH-3T3 cells transiently transfected with GFP-tagged constructs were grown on collagen I-coated culture slides (BD Biosciences), and 48 h after transfection cells were fixed with 4% PFA at room temperature (RT) for 10 minutes. Following one wash with PBS+Ca2+, cells were blocked in blocking buffer (PBS+Ca2+ + 1% goat serum (Invitrogen) and 0.1% Triton-X-100) for 30 minutes at RT. Cilia were labeled with rabbit anti-Arl13b antibody (1:500; ProteinTech) overnight in blocking buffer. Following primary antibody incubation, cells were washed three times in blocking buffer and incubated with Alexa 546 fluorophore (red) conjugated anti-rabbit secondary antibody (Invitrogen) in blocking buffer for 1 h at RT. After a single rinse, cell nuclei were stained with DAPI (USB Affymetrix) for 10 min. Following two additional rinses, cells were mounted onto slides using Vectashield mounting medium (Vector Laboratories) and sealed. Images were captured using an Olympus FV1000 confocal microscope (Olympus).
ERK phosphorylation assays
HEK-293T/17 cells were plated in 6-well plates (Corning) 20–24 h prior to transfection. Each well was transfected with 0.33 μg of receptor plasmid or empty vector. After 36–48 h, cells were serum-starved for 2 h before drug treatment by replacing complete growth medium with DMEM. To initiate stimulation, half (1 ml) of the DMEM was removed and slowly replaced with 1 mL fresh DMEM, containing a 2X concentration of vehicle, prosaptide, or head activator (HA) peptide. Plates were carefully returned to a 37°C incubator for 10 min. Following stimulation, cells were rapidly harvested in Laemmli buffer. Samples were sonicated and loaded into 4–20% Tris-Glycine gels (Bio-Rad) for SDS-PAGE, and then transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked via shaking at RT for 30 min in Odyssey blocking buffer and incubated overnight with shaking at 4°C with mouse anti-phospho ERK (Santa Cruz) and rabbit anti-total ERK (Cell Signaling Technologies) in antibody buffer (equal parts blocking buffer and PBS + 0.1% Tween-20). Membranes were then washed three times in a wash buffer (PBS with 0.1% Tween-20) and incubated with Alexa-fluor anti-mouse 700-nm conjugated secondary antibody (1:20,000; Invitrogen) and anti-rabbit 800-nm conjugated secondary antibody (1:20,000; Li-Cor) for 30 min in antibody buffer. Blots were washed three times and rinsed in PBS until they were visualized on an Odyssey Imaging System (Li-Cor). Protein quantification was done using densitometry, performed with ImageJ software.
Luciferase reporter assays
HEK-293T/17 cells were seeded in 96-well plates 20–24 h prior to transfection. Each well was transfected with 50 ng of firefly reporter, 1 ng of Renilla luciferase, and 10 ng of either receptor plasmid or empty vector (EV). Reporter constructs (CRE: pGL4.29, Renilla pRLSV40) were acquired from Promega (Madison, WI). After 24–48 hrs, DualGlo luciferase assays (Promega) were performed according to the manufacturer’s protocol and plates were read on a BMG Omega plate reader. Results were calculated for each assay by determining the luminescence ratio of firefly:Renilla luciferase counts, normalized to EV transfected wells. Error bars for all EV-transfected conditions were represented as the standard errors of the normalized raw value means.
Ubiquitination Assays
HEK-293T/17 cells were plated and transfected as described above with 2 μg of receptor and 1 μg of HA-ubiquitin. At 24 h post-transfection, cells were treated overnight with 100nM MG-132 (Tocris) to inhibit the proteasome. The following day, cells were washed, harvested, and solubilized as described above. Cleared lysates were incubated with anti-HA agarose beads (Sigma) for 1h, washed, and eluted in Laemmli buffer. Tagged constructs were detected via Western blotting with mouse HRP-conjugated anti-Flag (Sigma) rabbit HRP-conjugated anti-HA (Abcam).
Generation of Knockout Mice and Maintenance of Mouse Colony
Gpr37 knockout (Gpr37−/−) mice were obtained from Jackson Laboratory (strain Gpr37tm1Dgen, stock number 005806) and Gpr37L1 knockout mice (Gpr37L1−/−) were obtained from the NIH Mutant Mouse Regional Resource Centers (strain Gpr37l1tm1Lex, stock number 011709-UCD). These mouse lines were backcrossed with wild-type C57BL/6J mice (Jackson Laboratory) for 10 generations each to ensure uniformity of genetic background. Following this backcrossing, homozygous Gpr3T−/− and homozygous Gpr37L1−/− mice were crossed to generate the Double knockout (DKO) line of mice. Genetic deletion of Gpr37 and/or Gpr37L1 was confirmed by DNA sequencing, and loss of GPR37 and/or GPR37L1 protein expression was confirmed by Western blotting of brain tissue samples with specific anti-GPR37 and anti-GPR37L1 antibodies (MAb Technologies).
All mice were maintained on a C57BL/6J background and housed on a 12-h light/dark cycle, with food and water available ad libitum. All experiments were conducted during the light cycle prior to 4:00 p.m. and were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of Emory University. Wild-type (WT) littermates were used as controls for all single mutant experiments, and age-matched WT littermates from each single mutant line were combined and used as controls for experiments involving DKO mice. Male mice were used in all seizure induction paradigms. In all experiments, the experimenter was blinded to the genotypes of the animals.
6 Hz Seizure Induction
Seizures were induced using the 6 Hz paradigm, as previously described30–32. Briefly, 30 minutes prior to seizure induction, a topical anesthetic (0.5% tetracaine hydrochloride) was applied to the cornea. Each mouse was manually restrained during corneal stimulation (6 Hz, 0.2-ms pulse, 3 s) using a constant current device (ECT Unit 57800; Ugo Basile, Comerio, Italy). Behavioral seizures were scored based on a modified Racine Scale (RS): RS0 = no abnormal behavior; RS1 = immobile for ≥3 s, then resumption of normal behavior; RS2 = forelimb clonus, paw waving; RS3 = generalized tonic-clonic seizure with rearing and falling31. Male mice were tested at three current intensities: 18 mA (N = 11–16/group), 22 mA (N = 16–22/group), and 27 mA (N = 16–22/group) with a one-week recovery between each test session.
Flurothyl Seizure Induction
Seizures were induced in male mice using the chemiconvulsant flurothyl (2,2,2-trifluroethylether; Sigma-Aldrich). Each mouse was placed in a clear, Plexiglas chamber contained within a chemical fume hood, and flurothyl was introduced into the chamber (20 μl/min) such that it vaporized and filled the chamber. Latencies to the first myoclonic jerk and generalized tonic-clonic seizure (GTCS) were recorded33; 34 Mice were removed from the chamber immediately after the GTCS.
EEG Analyses
Male Gpr37L1−/−, Gpr3T−/−, and DKO mice (N = 5/group) were implanted with four cortical EEG recording electrodes at the following coordinates, relative to bregma: anterior-posterior (AP) 0.5 mm, medial-lateral (ML) −2.2 mm; AP −3.5 mm, ML −2.2 mm; AP 2.0 mm, ML 1.2 mm; AP −1.5 mm, ML 1.2 mm. Two wires were also implanted into the neck muscle to obtain EMG recordings. Mice were allowed to recover for one-week prior to EEG analyses.
Continuous video and EEG analyses (24 hours/day) were performed over a two-week period. Using Harmonie software for rodent studies (Stellate), EEG/EMG signals were analyzed utilizing a notch filter (60 Hz) and a low- and high-pass filter of 5 and 35 Hz, respectively. Seizures were manually identified and characterized as high-frequency electrographic signals with an amplitude at least twice the background. Synchronous video recordings were used to observe behavior during electrographic activity.
Results
A novel GPR37L1 variant is associated with a progressive myoclonus epilepsy
In a highly consanguineous family (Figure 1) with multiple siblings affected by intractable, progressive myoclonus epilepsy (PME), exome sequencing revealed a homozygous variant (c.1047G>T [Lys349Asn] [Genebank NM_004767.3]) in GPR37L1. Unaffected parents were carriers, and healthy siblings, of sufficient age to be symptomatic, were found to lack the variant or have a single copy. All affected individuals presented with recurrent headaches and visual hallucinations in the form of colors and lines in early adolescence (Table 1), followed by onset of myoclonic seizures. Symptoms progressively worsened and were accompanied by gradual and continuous cognitive decline culminating in death in late adolescence. The variant was not found in 3970 in-house (Center for Pediatric Genomic Medicine, Kansas City, MO) control exomes but was identified in a heterozygous state in 3 European (Non-Finnish) individuals from a total of 121,328 alleles (minor allele frequency = 0.002%), per the ExAC Browser. The variant was predicted to be pathogenic by three web-based prediction tools, SIFT35, Polyphen36, and MutationTaster37.
Table 1.
Comparison of Family Members with GPR37L1-K349N Variant
| VI: 1* | VI:4* | VI:5* | VI:6 | VI:7 | VI:8 | VI:9 | VI:10 | VI:11 | VI:12 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age of Clinical Presentation (Years) | 14 | 14 | 13 | 12 | 10 | n/a | 13 | n/a | 11 | 12 |
| Age of Death (Years) | 19 | 19 | 18 | 18 | 16 | n/a | 20 | n/a | n/a | n/a |
| Current Age (Years) | n/a | n/a | n/a | n/a | n/a | 24 | n/a | 18 | 15 | 12 |
| Sex | male | female | female | female | female | female | female | female | male | male |
| K349N mutation | Not tested | Not tested | Not tested | Not tested | Not tested | Heterozygous | c.1047G>T (Lys349Asp) | WT | c.1047G>T (Lys349Asp) | c.1047G>T (Lys349Asp) |
| Recurrent Headaches | + | + | + | + | + | − | + | − | + | + |
| Progressive Myoclonic Epilepsy | + | + | + | + | + | − | + | − | − | − |
| Seizures | + | + | + | + | + | − | + | − | − | − |
| Visual Hallucinations | + | + | + | + | + | − | + | − | + | − |
| Cognitive Decline | + | + | + | + | + | − | + | − | − | − |
| Brain Imaging | mild prominence of the subarachnoid spaces; otherwise unremarkable | |||||||||
| EEG | intermittent superimposed posterior 5–6 Hz background activity, recurrent generalized high voltage 4–6 Hz spike, polyspike and slow wave discharges with occasional abortive generalized high voltage spike and slow wave discharges | generalized spike and wave complexes; occipital sharp waves |
Denotes individuals for whom clinical information was relayed by family members
Functional analysis of the GPR37L1 K349N mutant
GPR37L1 is an orphan G protein-coupled receptor (GPCR) expressed predominantly in the brain, with highest expression in astrocytes and Bergmann glia, as well as expression in some populations of neurons and oligodendrocytes14–16, 38–40. While Gpr37L1 has been shown to play a role in postnatal cerebellar development in a mouse model17, the overall function of this receptor is unknown. To assess the effect of the K349N mutation on the ability of GPR37L1 to express and traffic to the cell surface, we transiently expressed Flag-tagged versions of GPR37L1 wild-type (WT) or the K349N mutant receptor in HEK293T cells. Total expression of K349N was equal to GPR37L1 WT (Figure 2A & B, upper panels). The predicted molecular weight for GPR37L1 WT and the K349N mutant is ~53 kDa. Both versions of the receptor have several predicted glycosylation sites, and thus the full-length receptor likely runs at ~70–75 kDa (arrowhead) with higher order bands representing oligomeric forms of the receptor and the ~37 kDa band representing a cleaved form of the receptor. To determine whether the K349N mutation might alter trafficking to the plasma membrane, surface proteins were labeled with a membrane-impermeant biotinylation reagent and pulled down with streptavidin beads. The K349N mutant receptor trafficked to the cell surface to a similar extent as GPR37L1 WT (Figure 2A & B, lower panels). In these studies, the cleaved form of both GPR37L1 and the K349N mutant appeared to be enriched at the cell surface, but the physiological relevance of this observation is unknown. As Marazziti and colleagues17 reported GPR37L1 to be localized to cilia in cerebellar Bergmann glial cells, we assessed the localization of GFP-tagged versions of each receptor in NIH-3T3 cells (Figure 2C), a cell type commonly used for studying cilia41. Both versions of the receptor were found predominantly on the cell surface and exhibited generally similar patterns of subcellular localization in confocal microscopy analyses. However, in the NIH-3T3 cells, no co-localization of GPR37L1 WT or the K349N mutant with the cilia marker Arl13b was observed (Figure 2C). The lack of cilial targeting may be due to the differences in the cell type and/or culture method used in our studies versus the studies of Marazziti and colleagues17.
Figure 2. Expression and localization of GPR37L1 wild-type and K349N mutant are equivalent.
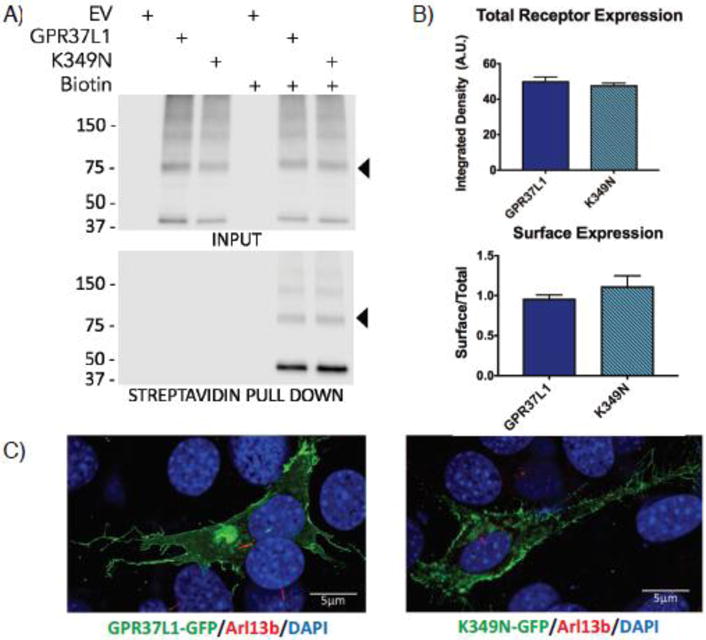
(A & B) Transient transfection of Flag-tagged versions of GPR37L1 wild-type or K349N mutant in HEK293T cells yielded comparable levels of receptor expression (upper panels) and trafficking to the cell surface (lower panels). Full-length GPR37L1 runs on SDS-PAGE gels at ~70–75 kDa (arrow head) with higher order bands representing oligomeric forms of the receptor and the ~37 kDa band representing a cleaved form of the receptor. Results are from three independent experiments (± SEM shown). (C) Confocal microscopy analysis of GFP-tagged versions of GPR37L1 wild-type (left panel) and K349N mutant (right panel) revealed a similar pattern of subcellular localization in NIH-3T3 cells, with both receptor variants being predominantly localized to the plasma membrane and neither version of the receptor exhibiting significant ciliary localization. Cilia were labeled using an anti-Arl13b antibody, indicated in red.
Previous reports have described prosaptide, a peptide fragment of prosaposin25; 42; 43, and the Hydra undecapeptide head activator (HA)44; 45 as potential ligands of GPR37 and GPR37L1. To assess the signaling activity of GPR37L1 WT vs. the K349N mutant, induction of ERK phosphorylation was measured, as signaling from both GPR37 and GPR37L1 has been shown to stimulate ERK phosphorylation25. Indeed, transfection of HEK-293T cells with either Flag-GPR37L1 WT or the Flag-K349N mutant raised phospho-ERK levels to a similar extent (Fig. 3B). However, neither prosaptide nor HA stimulation significantly increased ERK phosphorylation further (Figure 3A & B). Similarly, transfection with either Flag-GPR37L1 WT or the Flag-K349N mutant resulted in comparable increases in constitutive signaling to CRE luciferase (Figure 3C), another readout that has been reported to be downstream of GPR37L146. Again, neither prosaptide nor HA increased GPR37L1 signaling to CRE luciferase. Finally, as GPR37 is known to be ubiquitinated18 and ubiquitination occurs on lysine residues, we investigated whether Flag-GPR37L1 might also be ubiquitinated and whether mutation of lysine 349 might alter this ubiquitination. As shown in Figure 3D, Flag-GPR37L1 was indeed found to be robustly ubiquitinated, but Flag-GPR37L1 WT and the Flag-K349N mutant were ubiquitinated to a similar extent (Figure 3D).
Figure 3. No difference in constitutive signaling activity or ubiquitination between GPR37L1 wild-type and K349N mutant.
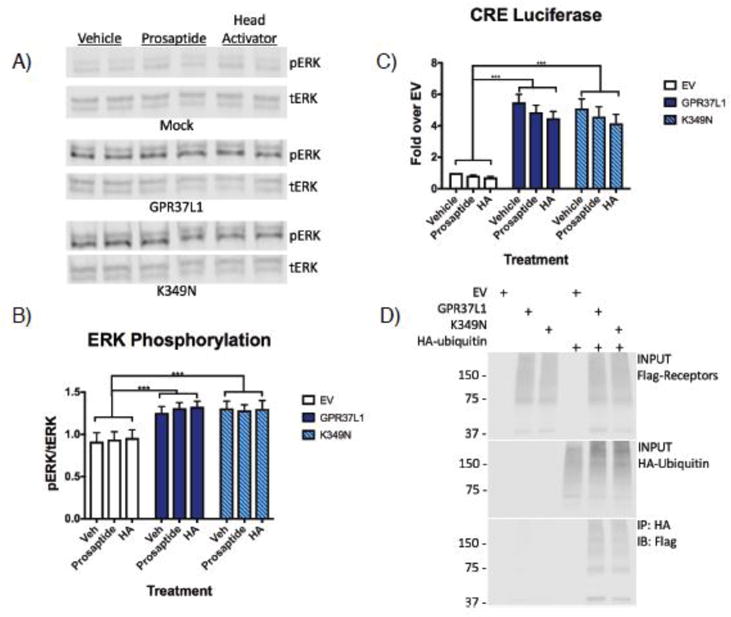
(A & B) HEK-293T cells transiently transfected with GPR37L1 or K349N mutant exhibited increased levels of ERK phosphorylation compared to cells transfected with empty vector (EV). At 48 h post transfection, cells were treated with 100 nM prosaptide, 100 nM head activator peptide (HA) or vehicle for 10 minutes at 37°C. Neither treatment significantly increased ERK phosphorylation. (C) GPR37L1 wild-type and the K349N mutant exhibited significant constitutive signaling activity to CRE luciferase but neither prosaptide or HA modulated this signaling. (D) Loss of the lysine residue in the K349N mutant did not affect ubiquitination of the receptor. All experiments were performed in HEK293T cells and the results shown are from 3–5 independent experiments (± SEM shown, ***p< .001).
Loss of Gpr37L1 in vivo increases seizure susceptibility
Although no striking differences were observed in the fundamental trafficking and constitutive signaling properties of GPR37L1 WT vs. the K349N variant in vitro, it is possible that the K349N variant results in a significant change in ligand-induced signaling, resulting in a reduction or loss of receptor function. Without a functional ligand, it is not feasible to address this possibility in vitro. Thus, to shed light on whether loss of GPR37L1 function can affect seizure susceptibility in vivo, male mice lacking a single copy (Gpr37L1+/−) or both copies of Gpr37L1 (Gpr37L1−/−) were evaluated in two seizure-induction paradigms. As GPR37L1 shares 68% similarity with GPR37, it is likely that these two receptors share the same ligand and perhaps also share a number of redundant functions in vivo. Thus, in parallel experiments, mice lacking a single copy (Gpr37+/−) or both copies of Gpr37 (Gpr3T−/−), and double-knockout mice lacking both receptors (DKO), were also assessed in the same paradigms.
The effects of Gpr37 and/or Gpr37L1 deletions were first examined using the 6 Hz psychomotor seizure induction model. Interestingly, a greater number of Gpr37L1−/− mice exhibited more severe seizures compared to WT and Gpr37L1−/− littermates at 27 mA (Figure 4A). Of the Gpr37L1−/− mice tested at 27mA, 5 did not seize (RS0), and among the other mice, 2 RS1, 11 RS2 and 3 RS3 seizures were observed. In contrast, for the WT littermates, the Racine scores were 9 RS0, 7 RS1,5 RS2 and 1 RS3. Similarly, for the Gpr37L1−/− mice, the Racine scores were 10 RS0, 5 RS1,6 RS2, and 1 RS3. All 3 genotypes had similar seizure responses at 18 and 22 mA, although 3 Gpr37L1−/− exhibited RS3 seizures at 22 mA.
Figure 4. Loss of Gpr37L1 and/or Gpr37 in vivo increases susceptibility to 6 Hz-induced seizures.
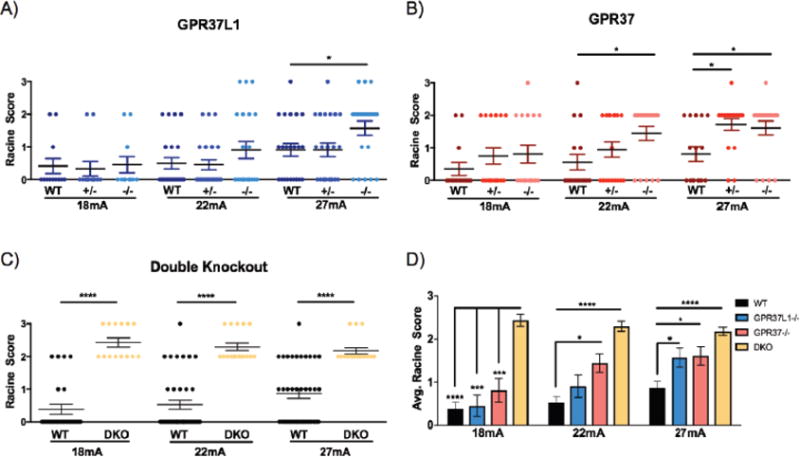
(A) Gpr37L1−/− mice exhibited increased susceptibility to 6-Hz-induced seizures compared to wild-type (WT) and Gpr37+/− littermates at 27 mA (N = 11–22/genotype/current). (B) Gpr3T−/− mice exhibited significantly increased seizure susceptibility compared to their WT and Gpr37+/− littermate controls at 22 mA, whereas both Gpr37+/− and Gpr3T−/− mice had significantly higher susceptibility to 6 Hz-induced seizures at 27 mA (N = 16–18/genotype/current). (C) Mice lacking both receptors (DKO) exhibited increased seizure susceptibility compared to age-matched WT mice at all currents tested (N = 14–38/genotype/current) (D) DKO mice exhibited increased susceptibility to 6 Hz-induced seizures compared to WT controls and homozygous mutants at 18 mA. Gpr3T−/− and DKO mice displayed higher susceptibility compared to WT controls at 22 mA. All homozygous mutants and DKO mice exhibited increased seizure susceptibility compared to WT at 27 mA. One-way ANOVA followed by Dunn’s multiple comparisons post-hoc analyses. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < .0001.
Gpr3T−/− mice were significantly more susceptible to 6 Hz-induced seizures compared to WT and Gpr37+/− littermates at 22 and 27 mA (Figure 4B). At 22 mA, 13/18 Gpr3T−/− mice exhibited RS2 seizures while 5 did not seize (RS0). In contrast, 13/18 WT littermates did not have a seizure. While not statistically significant, a greater number of Gpr37−/− exhibited more severe RS2 seizures compared to WT littermates at 18 and 22 mA. Both Gpr37−/− and Gpr3T−/− mice exhibited increased seizure susceptibility (p< 0.05) compared to WT littermates at the highest test current (27 mA) although susceptibility was comparable between the two groups of mutants. At 27 mA, 16/18 Gpr37−/− and 14/18 Gpr3T−/− exhibited a seizure whereas only 8/16 WT littermates exhibited a seizure, 3 of which were mild (RS1). Strikingly, at all three test currents, mice lacking both Gpr37L1 and Gpr37 (DKO) exhibited either RS2 or RS3 seizures and were therefore significantly more susceptible compared to age-matched WT mice (Figure 4C). Furthermore, compared to Gpr37L1−/− and Gpr3T−/− animals, DKO mice also demonstrated higher average Racine scores at each test current (Figure 4D).
Given that the most dramatic differences in the 6 Hz paradigm were observed with the homozygous knockout mice, we next compared susceptibility to flurothyl-induced seizures in homozygous knockout mice and WT littermates. Gpr37L1−/− and DKO mice exhibited significantly decreased latencies to both the first myoclonic jerk (MJ) (Figure 5A) and generalized tonic-clonic seizure (GTCS) (Figure 5B). In contrast, Gpr3T−/− mice exhibited latencies comparable to WT littermates at each seizure event.
Figure 5. Loss of Gpr37L1 but not Gpr37 in vivo increases susceptibility to flurothyl-induced seizures.
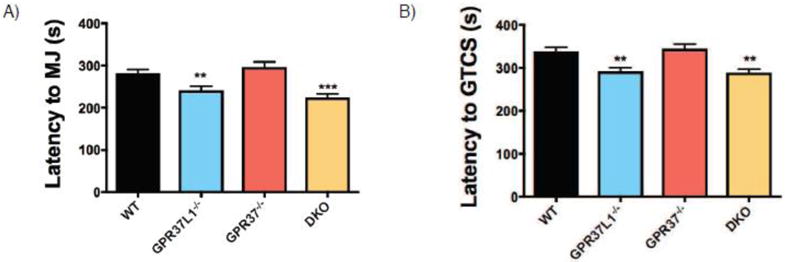
(A & B) Gpr37L1−/− mice exhibited decreased latency to first myoclonic jerk (MJ) and generalized tonic-clonic seizure (GTCS) compared to WT littermates. Latency to the MJ and GTCS were comparable between Gpr37L1−/− and DKO mice. Gpr3T−/− mice were not significantly different from WT littermates. One-way ANOVA, Dunnett’s post hoc, *p < 0.001, 10–42 animals/genotype.
Finally, to assess whether the knockout mice exhibit spontaneous seizures, we obtained two weeks of continuous video/EEG recordings for Gpr37L1−/−, Gpr3T−/−, and DKO mice. Spontaneous seizures were not observed in the five Gpr37L1−/− mice examined. However, seizures were detected in 2/5 Gpr3T−/− and 4/5 DKO mice (Figure 6A). Furthermore, the average duration of the seizures was significantly longer in mice lacking both receptors (Figure 6B). Examination of video recordings revealed that spontaneous seizures were accompanied by abnormal behaviors, which began with rearing, paw waving, and head bobbing at seizure onset with subsequent progression to generalized tonic-clonic seizure and loss of posture. Motor activity during the seizure is indicated by increased EMG activity (Figure 6C).
Figure 6. Gpr3T−/− and DKO mice exhibit spontaneous seizures.
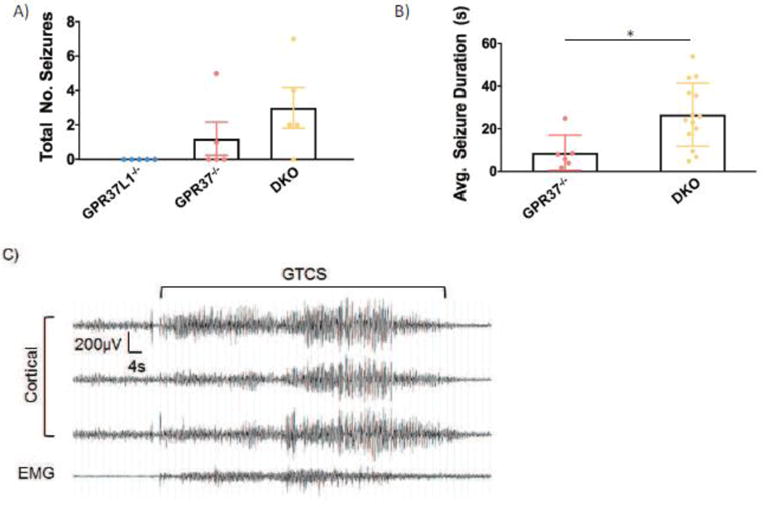
(A) Spontaneous seizures were observed in Gpr3T−/− and DKO mice (N = 5/genotype). (B) Mice lacking both receptors exhibited significantly longer spontaneous seizure duration compared to Gpr3T−/− mice. (C) Representative EEG trace of a spontaneous seizure. One-way ANOVA, Dunnett’s post hoc, *p < .05.
Discussion
In this study, we describe the identification of a GPR37L1 variant in a family with a novel progressive myoclonus epilepsy (PME). GPR37L1 is an orphan G protein-coupled receptor that has not previously been associated with epilepsy. The homozygous autosomal recessive variant (c.1047G>T, [Genebank NM_004767.3]) identified in the proband (VI:9), results in a K349N substitution in the third intracellular loop of the receptor. Individuals VI:8 and VI:10, found to be heterozygous and wild type, respectively, are well beyond the age of presentation of all their siblings and are considered clinically unaffected. The youngest members, and the only males in the sibship (VI:11 and VI:12), were also found to be homozygous for the GPR37L1 variant. To date, these youngest individuals have not had a clinical presentation of PME but have exhibited abnormal neurological symptoms: VI:11 has had daily recurrent headaches, two abnormal EEGs, and visual disturbances, while VI:12 has reported daily recurrent headaches (no EEG performed). Interestingly, VI:11, who at the time of the present study is 16 years of age, is beyond the average age of seizure onset of his sisters and cousin. While there are well-known neurodevelopmental differences between males and females47, and previous studies have identified GPR37L1 as differentially expressed between the sexes48, further observation of the youngest siblings will be required to determine if any sex differences exist in this case.
GPR37L1 is expressed predominantly in the brain14–16 and is most closely related to GPR37, which is also known as the “parkin-associated endothelin receptor-like receptor” (Pael-R). Much of the research on these receptors over the past two decades has focused on the role of GPR37 in Parkinson’s disease18; 20; 21; 26; 49–51 and regulation of the dopamine system19; 21–23 In the absence of parkin, GPR37 has a propensity to misfold and aggregate, resulting in dopaminergic cell death18; 49 GPR37 aggregates have also been found in inclusion bodies in patients with Parkinson’s disease20. However, as GPR37L1 is not ubiquitinated by parkin18, it has not been associated with Parkinson’s pathology. GPR37L1 is highly expressed in the Bergmann glia of the cerebellum14; 17 and mice lacking Gpr37L1 show alterations in postnatal cerebellar development17. GPR37L1 is also highly expressed in astrocytes and certain populations of neurons, and both GPR37 and GPR37L1 are widely expressed throughout the brain14–16; 38–40 Thus, it is likely that GPR37L1 and GPR37 have additional functions outside the cerebellum and dopaminergic system, respectively. We and others have shown that expression of GPR37L1 and/or GPR37 can mediate cytoprotective effects in vitro25; 26 Therefore, it is possible that these receptors play important roles in mediating protective effects following insult or injury in vivo.
No differences were observed in the present study between GPR37L1 WT and the K349N mutant in terms of receptor expression, localization or constitutive signaling in HEK293T or NIH-3T3 cell lines in vitro. The natural ligand for GPR37L1 remains uncertain, and in the studies described here, we assessed two peptides that have been proposed as ligands for GPR37L1 and/or GPR37: prosaptide25; 42; 43 and head activator (HA)44; 45 However, no significant increases in receptor signaling activity were observed with either peptide, and therefore the previously-reported agonistic actions of these two peptides could not be confirmed here. It is possible that the regulation of GPR37/GPR37L1 signaling activity by these peptides is dependent on cellular context and/or other variables, although another recent study also reported a lack of activation of GPR37L1 signaling by prosaptide46. Thus, while the in vitro data described here cannot confirm the pathogenicity of the K349N variant, it is plausible that functional differences between GPR37L1 WT and the K349N mutant may be observed only with agonist-induced signaling. More generally, it is possible that the K349N variant may perturb GPR37L1 function in ways that are not easily testable at present using in vitro models of receptor trafficking and signaling activity. Therefore, even though no striking effects of the K349N mutation on receptor function were observed in the in vitro studies, we assessed the importance of GPR37L1 for seizure susceptibility in vivo.
Until the present study, seizure susceptibility had not been evaluated in mice lacking Gpr37L1 and/or Gpr37. Indeed, mice lacking both receptors had not previously been described at all. In the 6 Hz seizure induction paradigm, Gpr37L1−/− mice displayed increased seizure susceptibility at the highest test current (27 mA). However, Gpr37L1+/− and WT littermates had comparable seizure responses at each test current, which indicates that having a single copy of GPR37L1 is sufficient to maintain normal susceptibility to 6 Hz-induced seizures. In contrast, Gpr37+/− and Gpr3T−/− exhibited significantly increased seizure susceptibility at 27 mA; therefore, reduced GPR37 expression results in altered seizure susceptibility. Given this finding, it is interesting to note that GPR37 is one of six genes located within a heterozygous deletion that has been reported in an epilepsy patient52. Another point of interest in the present study is that mice lacking both receptors exhibited markedly increased seizure susceptibility compared to WT controls at each test current. These results suggest that loss of both Gpr37L1 and Gpr37 has an additive effect in the DKO mice. This is particularly clear at 18 mA, where DKO mice exhibit increased susceptibility to 6 Hz-induced seizures compared to WT controls, but both single homozygous mutants have normal seizure susceptibility.
In the flurothyl seizure induction paradigm, Gpr37L1−/− and DKO mice exhibited decreased latencies to both the MJ and GTCS, whereas Gpr3T−/− mice were not significantly different from WT littermates. This result suggests that loss of Gpr37L1 drives the decreased susceptibility to flurothyl-induced seizures in the mice lacking both receptors. Considered together with the 6 Hz data, these findings from the flurothyl studies suggest that Gpr37L1 and Gpr37 alter seizure susceptibility via different mechanisms. The 6 Hz paradigm is a model of limbic seizures, whereas flurothyl-induced seizures are generalized. Finally, spontaneous seizures were observed in both Gpr3T−/− and DKO mice, which is consistent with the 6 Hz results in which GPR37 appeared to be a major contributor to the dramatic increase in seizure susceptibility in the mice deficient for both receptors.
Conclusions
Taken together, the data presented here represent the first evidence linking GPR37L1 and GPR37 to seizure etiology. Future work will focus on elucidating the cellular mechanism(s) underlying the observed increases in seizure susceptibility and assessing cell-specific effects of the GPR37L1 K349N mutation in native cell types. The findings presented here set the stage for future studies that will shed light on the functional roles of these receptors in the brain and provide new insights into the pathogenesis and potential treatment of progressive myoclonus epilepsies.
Highlights.
-
▪
A GPR37L1 variant was linked with progressive myoclonic epilepsy in a large family
-
▪
No striking differences observed between GPR37L1 WT & variant in transfected cells
-
▪
Gpr37L1−/− mice were more susceptible to seizures than WT mice in 2 seizure models
-
▪
Gpr37−/− mice were also more susceptible that WT mice to seizures
-
▪
Mice lacking both Gpr37 & Gpr37L1 were extremely susceptible to seizures
Acknowledgments
We would like to acknowledge the family for their participation in this study. This work was supported by grants R01-NS088413 and R21-NS091986 (RAH), R01-NS090319 (AE), and T32-NS00748016 (JCW) from the National Institutes of Health. This study was also supported in part by the Emory University Integrated Cellular Imaging Microscopy Core and the Emory Rodent Behavioral Core, which are subsidized by the Emory University School of Medicine and are part of the Emory Integrated Core Facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 2.Heinzen EL, Depondt C, Cavalleri GL, Ruzzo EK, Walley NM, Need AC, Ge D, He M, Cirulli ET, Zhao Q, et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Consortium E, Consortium EM, Steffens M, Leu C, Ruppert AK, Zara F, Striano P, Robbiano A, Capovilla G, Tinuper P, et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet. 2012;21:5359–5372. doi: 10.1093/hmg/dds373. [DOI] [PubMed] [Google Scholar]
- 4.Epi, K.C.E.a.e.k.c.e., and Epi, K.C. De Novo Mutations in SLC1A2 and CACNA1A Are Important Causes of Epileptic Encephalopathies. Am J Hum Genet. 2016;99:287–298. doi: 10.1016/j.ajhg.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franceschetti S, Michelucci R, Canafoglia L, Striano P, Gambardella A, Magaudda A, Tinuper P, La Neve A, Ferlazzo E, Gobbi G, et al. Progressive myoclonic epilepsies: definitive and still undetermined causes. Neurology. 2014;82:405–411. doi: 10.1212/WNL.0000000000000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulley JC, Mefford HC. Epilepsy and the new cytogenetics. Epilepsia. 2011;52:423–432. doi: 10.1111/j.1528-1167.2010.02932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasperaviciute D, Catarino CB, Heinzen EL, Depondt C, Cavalleri GL, Caboclo LO, Tate SK, Jamnadas-Khoda J, Chinthapalli K, Clayton LM, et al. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain. 2010;133:2136–2147. doi: 10.1093/brain/awq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol. 2005;4:239–248. doi: 10.1016/S1474-4422(05)70043-0. [DOI] [PubMed] [Google Scholar]
- 9.Lehesjoki AE, Gardiner M. Progressive myoclonus epilepsy: Unverricht-Lundborg disease and Neuronal ceroid lipofuscinoses. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- 10.Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, Miao J, D’Amato E, Ramirez L, Faham M, Koskiniemi M, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science. 1996;271:1731–1734. doi: 10.1126/science.271.5256.1731. [DOI] [PubMed] [Google Scholar]
- 11.Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–174. doi: 10.1038/2470. [DOI] [PubMed] [Google Scholar]
- 12.Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC, Avanzini G, Elia M, Ackerley CA, Jovic NJ, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125–127. doi: 10.1038/ng1238. [DOI] [PubMed] [Google Scholar]
- 13.Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 14.Valdenaire O, Giller T, Breu V, Ardati A, Schweizer A, Richards JG. A new family of orphan G protein-coupled receptors predominantly expressed in the brain. FEBS Lett. 1998;424:193–196. doi: 10.1016/s0014-5793(98)00170-7. [DOI] [PubMed] [Google Scholar]
- 15.Leng N, Gu G, Simerly RB, Spindel ER. Molecular cloning and characterization of two putative G protein-coupled receptors which are highly expressed in the central nervous system. Brain Res Mol Brain Res. 1999;69:73–83. doi: 10.1016/s0169-328x(99)00092-3. [DOI] [PubMed] [Google Scholar]
- 16.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marazziti D, Di Pietro C, Golini E, Mandillo S, La Sala G, Matteoni R, Tocchini-Valentini GP. Precocious cerebellum development and improved motor functions in mice lacking the astrocyte cilium-, patched 1-associated Gpr37l1 receptor. Proc Natl Acad Sci U S A. 2013;110:16486–16491. doi: 10.1073/pnas.1314819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 19.Imai Y, Inoue H, Kataoka A, Hua-Qin W, Masuda M, Ikeda T, Tsukita K, Soda M, Kodama T, Fuwa T, et al. Pael receptor is involved in dopamine metabolism in the nigrostriatal system. Neurosci Res. 2007;59:413–425. doi: 10.1016/j.neures.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 20.Murakami T, Shoji M, Imai Y, Inoue H, Kawarabayashi T, Matsubara E, Harigaya Y, Sasaki A, Takahashi R, Abe K. Pael-R is accumulated in Lewy bodies of Parkinson’s disease. Ann Neurol. 2004;55:439–442. doi: 10.1002/ana.20064. [DOI] [PubMed] [Google Scholar]
- 21.Marazziti D, Golini E, Mandillo S, Magrelli A, Witke W, Matteoni R, Tocchini-Valentini GP. Altered dopamine signaling and MPTP resistance in mice lacking the Parkinson’s disease-associated GPR37/parkin-associated endothelin-like receptor. Proc Natl Acad Sci U S A. 2004;101:10189–10194. doi: 10.1073/pnas.0403661101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marazziti D, Mandillo S, Di Pietro C, Golini E, Matteoni R, Tocchini-Valentini GP. GPR37 associates with the dopamine transporter to modulate dopamine uptake and behavioral responses to dopaminergic drugs. Proc Natl Acad Sci U S A. 2007;104:9846–9851. doi: 10.1073/pnas.0703368104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marazziti D, Di Pietro C, Mandillo S, Golini E, Matteoni R, Tocchini-Valentini GP. Absence of the GPR37/PAEL receptor impairs striatal Akt and ERK2 phosphorylation, DeltaFosB expression, and conditioned place preference to amphetamine and cocaine. FASEB J. 2011;25:2071–2081. doi: 10.1096/fj.10-175737. [DOI] [PubMed] [Google Scholar]
- 24.Mandillo S, Golini E, Marazziti D, Di Pietro C, Matteoni R, Tocchini-Valentini GP. Mice lacking the Parkinson’s related GPR37/PAEL receptor show non-motor behavioral phenotypes: age and gender effect. Genes Brain Behav. 2013;12:465–477. doi: 10.1111/gbb.12041. [DOI] [PubMed] [Google Scholar]
- 25.Meyer RC, Giddens MM, Schaefer SA, Hall RA. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc Natl Acad Sci U S A. 2013;110:9529–9534. doi: 10.1073/pnas.1219004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundius EG, Stroth N, Vukojevic V, Terenius L, Svenningsson P. Functional GPR37 trafficking protects against toxicity induced by 6-OHDA, MPP+ or rotenone in a catecholaminergic cell line. J Neurochem. 2013;124:410–417. doi: 10.1111/jnc.12081. [DOI] [PubMed] [Google Scholar]
- 27.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon JB, Miller NA, Thiffault I, Dinwiddie DL, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, Soden SE, Cakici JA, Herd SM, Twist G, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3:377–387. doi: 10.1016/S2213-2600(15)00139-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–227. doi: 10.1016/s0920-1211(01)00302-3. [DOI] [PubMed] [Google Scholar]
- 31.Gilchrist J, Dutton S, Diaz-Bustamante M, McPherson A, Olivares N, Kalia J, Escayg A, Bosmans F. Nav1.1 modulation by a novel triazole compound attenuates epileptic seizures in rodents. ACS Chem Biol. 2014;9:1204–1212. doi: 10.1021/cb500108p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong JC, Dutton SB, Collins SD, Schachter S, Escayg A. Huperzine A Provides Robust and Sustained Protection against Induced Seizures in Scn1a Mutant Mice. Front Pharmacol. 2016;7:357. doi: 10.3389/fphar.2016.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szot P, Weinshenker D, White SS, Robbins CA, Rust NC, Schwartzkroin PA, Palmiter RD. Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. J Neurosci. 1999;19:10985–10992. doi: 10.1523/JNEUROSCI.19-24-10985.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cubells JF, Schroeder JP, Barrie ES, Manvich DF, Sadee W, Berg T, Mercer K, Stowe TA, Liles LC, Squires KE, et al. Human Bacterial Artificial Chromosome (BAC) Transgenesis Fully Rescues Noradrenergic Function in Dopamine beta-Hydroxylase Knockout Mice. PLoS One. 2016;11:e0154864. doi: 10.1371/journal.pone.0154864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, Bork P. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. doi: 10.1093/hmg/10.6.591. [DOI] [PubMed] [Google Scholar]
- 37.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaboub LS, Manalo JM, Lee HK, Glasgow SM, Chen F, Kawasaki Y, Akiyama T, Kuo CT, Creighton CJ, Mohila CA, et al. Temporal Profiling of Astrocyte Precursors Reveals Parallel Roles for Asef during Development and after Injury. J Neurosci. 2016;36:11904–11917. doi: 10.1523/JNEUROSCI.1658-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy AS, O’Brien D, Pisat N, Weichselbaum CT, Sakers K, Lisci M, Dalal JS, Dougherty JD. A Comprehensive Analysis of Cell Type-Specific Nuclear RNA From Neurons and Glia of the Brain. Biol Psychiatry. 2017;81:252–264. doi: 10.1016/j.biopsych.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alieva IB, Gorgidze LA, Komarova YA, Chernobelskaya OA, Vorobjev IA. Experimental model for studying the primary cilia in tissue culture cells. Membr Cell Biol. 1999;12:895–905. [PubMed] [Google Scholar]
- 42.Meyer RC, Giddens MM, Coleman BM, Hall RA. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 2014;1585:1–12. doi: 10.1016/j.brainres.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundius EG, Vukojevic V, Hertz E, Stroth N, Cederlund A, Hiraiwa M, Terenius L, Svenningsson P. GPR37 protein trafficking to the plasma membrane regulated by prosaposin and GM1 gangliosides promotes cell viability. J Biol Chem. 2014;289:4660–4673. doi: 10.1074/jbc.M113.510883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rezgaoui M, Susens U, Ignatov A, Gelderblom M, Glassmeier G, Franke I, Urny J, Imai Y, Takahashi R, Schaller HC. The neuropeptide head activator is a high-affinity ligand for the orphan G-protein-coupled receptor GPR37. J Cell Sci. 2006;119:542–549. doi: 10.1242/jcs.02766. [DOI] [PubMed] [Google Scholar]
- 45.Gandia J, Fernandez-Duenas V, Morato X, Caltabiano G, Gonzalez-Muniz R, Pardo L, Stagljar I, Ciruela F. The Parkinson’s disease-associated GPR37 receptor-mediated cytotoxicity is controlled by its intracellular cysteine-rich domain. J Neurochem. 2013;125:362–372. doi: 10.1111/jnc.12196. [DOI] [PubMed] [Google Scholar]
- 46.Coleman JL, Ngo T, Schmidt J, Mrad N, Liew CK, Jones NM, Graham RM, Smith NJ. Metalloprotease cleavage of the N terminus of the orphan G protein-coupled receptor GPR37L1 reduces its constitutive activity. Sci Signal. 2016;9:ra36. doi: 10.1126/scisignal.aad1089. [DOI] [PubMed] [Google Scholar]
- 47.Giatti S, Boraso M, Melcangi RC, Viviani B. Neuroactive steroids, their metabolites, and neuroinflammation. J Mol Endocrinol. 2012;49:R125–134. doi: 10.1530/JME-12-0127. [DOI] [PubMed] [Google Scholar]
- 48.Shi L, Zhang Z, Su B. Sex Biased Gene Expression Profiling of Human Brains at Major Developmental Stages. Sci Rep. 2016;6:21181. doi: 10.1038/srep21181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- 50.Omura T, Kaneko M, Okuma Y, Orba Y, Nagashima K, Takahashi R, Fujitani N, Matsumura S, Hata A, Kubota K, et al. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J Neurochem. 2006;99:1456–1469. doi: 10.1111/j.1471-4159.2006.04155.x. [DOI] [PubMed] [Google Scholar]
- 51.Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- 52.Heinzen EL, Radtke RA, Urban TJ, Cavalleri GL, Depondt C, Need AC, Walley NM, Nicoletti P, Ge D, Catarino CB, et al. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86:707–718. doi: 10.1016/j.ajhg.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]