

Abstract
Dysregulation of the mechanistic target of rapamycin complex 1 (mTORC1)-p70 ribosomal protein kinase 1 (S6K1) signaling pathway occurs frequently in acute myeloid leukemia (AML) patients. This pathway also plays a critical role in maintaining normal cellular processes. Given the importance of leukemia stem cells (LSC) in the development of minimal residual disease (MRD), it is critical to use therapeutic interventions that target LSC population to prevent disease relapse. mTORC1-S6K1 pathway has been identified as an important regulator of hematopoietic stem cell (HSC) and LSC functions. Both HSC and LSC functions require regulation of key cellular processes including proliferation, metabolism and autophagy, which are regulated by mTORC1 pathway. Despite mTORC1-S6K1 pathway being a critical regulator of AML initiation and progression, inhibitors of this pathway alone have yielded mixed results in clinical studies. Recent studies have identified strategies to develop new mTORC1-S6K1 inhibitors like RapaLink-1, which could circumvent the drug resistance observed in AML cells as well as in LSC. In this article, we review recent advances made in identifying the role of different components of this pathway in the regulation of HSC and LSC along with possible therapeutic approaches.
Keywords: Hematopoietic stem cells, Leukemia stem cells, mTORC1, S6K1, Acute myeloid leukemia
Introduction
The mTORC1-S6K1 pathway regulates multiple cellular functions including cell cycle, apoptosis, glucose metabolism, protein synthesis and autophagy. This pathway has also been identified as one of the critical regulators of HSC and AML cells. Earlier studies have primarily examined different activators or repressors of mTORC1 activity in the context of hematopoiesis and leukemogenesis (1-4). More recently, multiple studies have identified the role(s) of distinct components of mTORC1 and S6K1 in hematopoiesis and leukemogenesis including the role of mTORC-S6K1 in regulating self-renewal of LSC (5-8). Here, we review the role of mTORC1 and S6K1 in hematopoiesis and AML, and discuss current progress towards pharmacological targeting of mTORC1 and S6K1.
Components and signaling of mTORC1-S6K1 pathway
mTOR is a serine/threonine complex that can be divided in two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (9). mTORC1 has six and mTORC2 has seven protein components (9). Both complexes have the catalytic mTOR subunit, mammalian lethal with sec-13 protein 8 (mLST8) and the DEP domain containing mTOR-interacting protein (DEPTOR) in common (9) (Figure 1). Furthermore, the telomere maintenance 2 (Tel2) and Tel2 interacting protein 1 (Tti1) complex is also necessary for assembly of both mTORC1 and mTORC2 (10) (Figure 1). Regulatory-associated protein of mammalian target of rapamycin (Raptor) and proline-rich Akt substrate 40 kDa (PRAS40) are specific components of mTORC1, whereas rapamycin-insensitive companion of mTOR (rictor), mammalian stress-activated map kinase-interacting protein 1 (mSin1), and protein observed with rictor 1 and 2 (protor1/2) are part of mTORC2 (9) (Figure 1).
Figure 1. Components of mTORC1 and mTORC2.
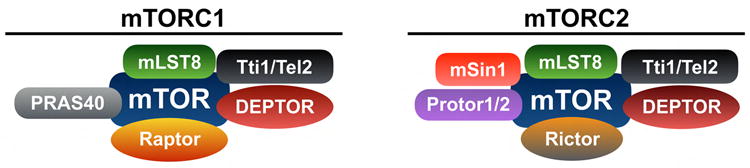
mTORC1 has six and mTORC2 has seven protein components. Both complexes have the catalytic mTOR subunit, mLST8, DEPTOR, and the Tti1-Tel2 complex in common. Raptor and PRAS40 are part of mTORC1 whereas Rictor, mSin1 and Protor1/2 are part of mTORC2. mTOR acts as a catalytic serine/threonine kinase while DEPTOR is an inhibitor of mTOR activity. The Tti1-Tel2 complex acts as a scaffolding protein, which regulates the assembly and stability of mTORC1 and mTORC2. mLST8 binds to mTOR near its kinase domain and regulates its function. PRAS40 binds to mTORC1 and acts as an inhibitor of mTORC1 activity. Raptor is a scaffolding protein, and it regulates assembly, localization and substrate binding of mTORC1. Rictor acts as the scaffolding protein in mTORC2 and regulates the assembly and substrate binding of mTORC2. Protor and mSin1 regulate mTORC2's interaction with SGK1. Moreover, mSin1 acts as a scaffolding protein regulating the assembly of mTORC2. Mechanistic target of rapamycin complex 1, mTORC1; mammalian lethal with sec-13 protein 8, mLST8; DEP domain containing mTOR-interacting protein, DEPTOR; Tel2 interacting protein 1, Tti1; telomere maintenance 2, Tel2; regulatory-associated protein of mammalian target of rapamycin, Raptor; proline-rich Akt substrate 40 kDa, PRAS40; rapamycin-insensitive companion of mTOR, Rictor; mammalian stress-activated map kinase-interacting protein 1, mSin1; protein observed with rictor 1 and 2, Protor1/2; serum- and glucocorticoid-regulated kinase 1, SGK1.
mTORC1 is activated by multiple external inputs including growth factors, nutrients and the cellular energy status. Growth factors like insulin activate mTORC1 through the PI3K-Akt pathway. Following stimulation by insulin, Akt undergoes PDK1-mediated activation. Activated Akt phosphorylates TSC2 and inhibits the formation of TSC complex (11). TSC complex acts as a GTPase-activating protein (12) (12) for the small GTPase Ras homologue enriched in brain (Rheb), which resides on lysosomal surface (13-15) (Figure 2). Following inhibition of TSC complex, GTP-bound Rheb binds to the catalytic domain of mTOR to activate mTORC1 (16). Insulin can also activate mTORC1 through Akt-mediated phosphorylation of PRAS40 at Thr-246 (17). In contrast, amino acids induce activation of mTORC1 through a different pathway. Amino acids facilitate translocation of mTORC1 to the lysosomal surface through Rag heterodimer-dependent and independent pathways (Figure 2) (18-23). Following translocation of mTORC1 from the cytoplasm to lysosome (24), Rheb activates mTORC1 (25). The cellular levels of ATP and AMP are also a critical regulator of mTORC1 activation. When cellular energy level is low, the AMP: ATP ratio remains high. During high AMP: ATP levels, AMPK phosphorylates TSC2 (14) and Raptor (26) to inhibit mTORC1 activity. Conversely, a high ATP:AMP level inhibits activation of AMPK (27) (Figure 2). Activated mTORC1 regulates the activity of 4E-BP1 and S6K1 by phosphorylating them (Figure 2). mTORC1 phosphorylates 4E-BP1 thus causing its dissociation from eukaryotic translation initiation factor 4E (eIF4E). Following dissociation of 4E-BP1, eIF4G binds to eIF4E and initiates 5′ cap-dependent translation (27). Activated mTORC1 phosphorylates S6K1, which is a member of cAMP-dependent protein kinases A, cGMP-dependent protein kinases G, and phospholipid-dependent protein kinases C (AGC) subfamily of serine-threonine kinases (28), at Thr-389 and activates it (29) (Figure 2). Following its activation, S6K1 phosphorylates multiple substrates including ribosomal protein S6 (RPS6) (30), eukaryotic translation elongation factor 2 kinase (eEF2K) (31), S6K1 Aly ⁄ REF-like substrate (SKAR) (32), eukaryotic translation initiation factor 4B (eIF4B) (33), and programmed cell death 4 (PDCD4) (34) (Figure 2). Moreover, S6K1 can also act as a repressor of mTORC1 pathway by creating a negative feedback loop through its regulation of mTORC2 activity (Figure 3). Activated mTORC2 phosphorylates and activates Akt at Ser-473. Absence of mTORC2 activity results in reduced phosphorylation of Akt at Ser-473 (35). mSin1 and Rictor, two components of mTORC2, are required for mTORC2-dependent phosphorylation of Akt at Ser-473 (36). S6K1-mediated phosphorylation of mSIN1 at Thr-86 and Thr-398 results in dissociation of mSIN1 from mTORC2 thus impairing the overall mTORC2 activity (Figure 3) (35). S6K1 can also regulate mTORC2 activity by phosphorylating Rictor, another component of mTORC2. Following mTORC1-dependent activation, S6K1 phosphorylates Rictor at Thr-1135 thus inhibiting mTORC2-mediated Akt activation (37). In addition, S6K1 also inhibits Akt-mTORC1 activity by phosphorylating IRS-1 at Ser-302. In response to insulin, insulin receptor substrate-1 (IRS-1) recruits and subsequently activates phosphoinositide 3-OH kinase (PI3K), an upstream regulator of mTORC1-S6K1. Phosphorylation at Ser-302 of IRS-1 by S6K1 results in disruption of its interaction with insulin receptor and inhibits PI3K-mediated Akt activation (Figure 3) (38).
Figure 2. Activators and inhibitors of mTORC1-S6K1 signaling pathway.
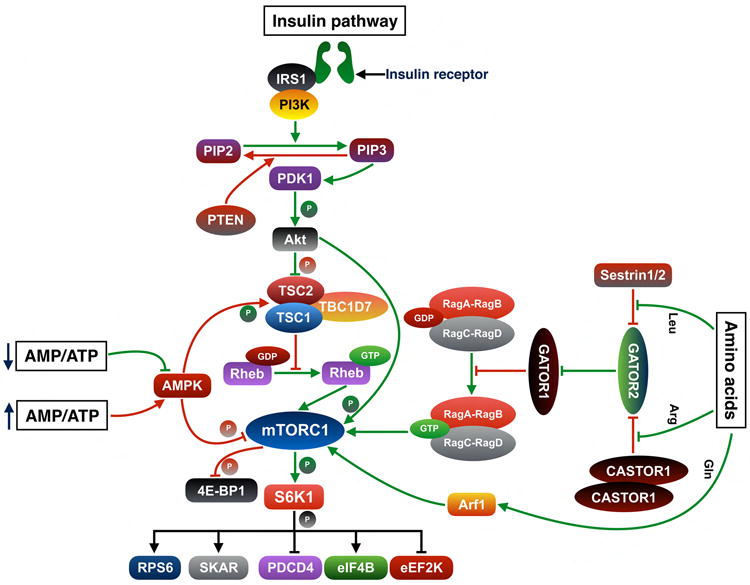
Growth factors such as insulin activate mTORC1 through the PI3K-Akt pathway. Following activation by insulin, insulin receptors stimulate and activate IRS-bound PI3K. Activated PI3K converts PIP2 to PIP3 and subsequently binding of PIP3 recruits Akt to the plasma membrane. The binding of Akt to PIP3 allows PDK1 to bind with Akt and phosphorylate it at Thr-308. Activated Akt phosphorylates TSC2 at multiple sites and inhibits its GAP activity on Rheb. Following inhibition of TSC2, Rheb is converted into an active GTP-bound state and activates mTORC1. mTORC1 could also be activated by Akt-mediated phosphorylation of PRAS40, a component of mTORC1. In presence of amino acids, mTORC1 translocates to the lysosomal surface and activated by Rheb. Rag GTPases, which form heterodimeric complexes comprised of RagA or RagB bound to RagC or RagD, are critical for recruitment of mTORC1 to lysosomal surface. GATOR1 acts as a GAP for RagA/B and inhibits the translocation of mTORC1 to lysosome. GATOR2 acts as an upstream inhibitor of GATOR1 and subsequently facilitates mTORC1 activation through Rag heterodimers. CASTOR1 homodimer and Sestrin2 are negative regulators of GATOR2. Sestrin2 binds to GATOR2 and inhibits mTORC1 activation. Following binding of leucine to Sestrin2, GATOR2 dissociates from Sestrin2. Similarly, CASTOR1 homodimers bind to GATOR2 thus exerting an inhibitory effect. Arginine binds to CASTOR1 homodimers and dissociates it from GATOR. Following its dissociation from inhibitory regulators, GATOR2 inhibits GATOR1 activity and facilitates mTORC1 activation. Glutamine regulates mTORC1 translocation through Arf1 GTPases, a process which is independent of the Rag GTPases. In response to low cellular energy level, AMPK inhibits mTORC1 pathway to limit energy consumption as well as to facilitate recovery of cellular energy materials. AMPK phosphorylates TSC2 at Ser-1387 and activates its GAP activity, which subsequently converts Rheb into an inactive GDP-bound state. AMPK also phosphorylates Raptor at Ser-722 and Ser-792, which results in binding of 14-3-3 protein to Raptor and subsequent inactivation of mTORC1. Conversely, a high cellular ATP level inhibits activation of AMPK by AMP, which results in withdrawal of AMPK-mediated inhibitory signals on mTORC1 activation. Activated mTORC1 regulates the activity of 4E-BP1 and S6K1 by phosphorylating them. Following its activation, S6K1 phosphorylates multiple substrates including RPS6, eEF2K, SKAR, eIF4B and PDCD4. Insulin receptor substrate 1, IRS1; phosphoinositide 3-kinase, PI3K; phosphatidylinositol (4,5)-bisphosphate, PIP2; phosphatidylinositol (3,4,5)-trisphosphate, PIP3; phosphatase and tensin homologue, PTEN; 3-phosphoinositide-dependent protein kinase 1, PDK1; tuberous sclerosis complex, TSC; TBC1 domain family member 7, TBC1D7; Ras homologue enriched in brain, Rheb; p70 ribosomal protein kinase 1, S6K1; eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1, 4E-BP1; ribosomal protein S6, RPS6; eukaryotic translation elongation factor 2 kinase, eEF2K; S6K1 Aly ⁄ REF-like substrate SKAR; eukaryotic translation initiation factor 4B, eIF4B; programmed cell death 4, PDCD4; adenosine monophosphate, AMP; adenosine triphosphate, ATP; AMP-activated protein kinase, AMPK; GAP activity towards Rags, GATOR; CASTOR; Leucine, Leu; Arginine, Arg; Glutamine, Gln; ADP-ribosylation factor 1, Arf1. Adapted and modified from Shimobayashi et al (Ref. 27).
Figure 3. Inhibition of mTORC1 activity by S6K1.
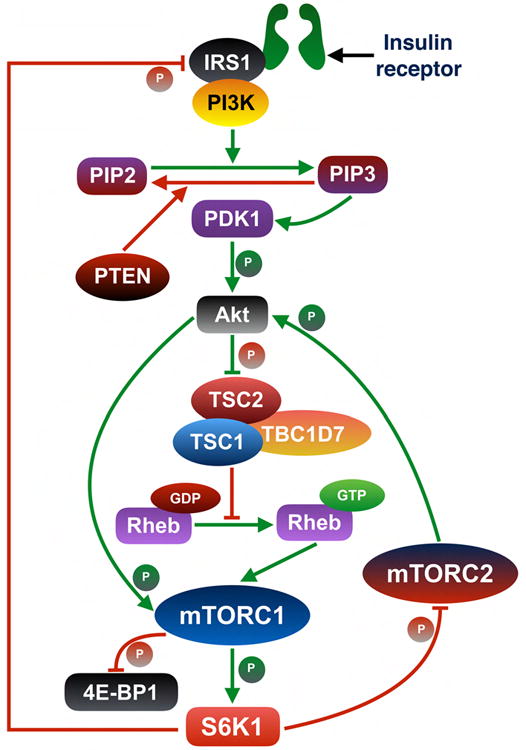
S6K1 acts as a negative regulator of mTORC1 activity by creating negative feedback loops. mSin1 and Rictor, two components of mTORC2, are required for mTORC2-dependent phosphorylation of Akt at Ser-473. S6K1-mediated phosphorylation of mSin1 at Thr-86 and Thr-398 results in dissociation of mSin1 from mTORC2 thus impairing the overall mTORC2 activity and subsequently diminishing Akt activation level. S6K1 could also regulate mTORC2 activity by phosphorylating Rictor, another component of mTORC2. Following mTORC1-dependent activation, S6K1 phosphorylates Rictor at Thr-1135. Phosphorylation of Rictor at Thr-1135 results in inhibition of Akt activity. S6K1 also inhibits Akt-mTORC1 activity by phosphorylating IRS-1 on Ser-302, which results in disruption of the interaction between IRS-1 and insulin receptor and subsequent inhibition of PI3K-mediated Akt activation. Inhibition of Akt activity, an upstream positive regulator of mTORC1, results in a reduction in mTORC1 activation.
Pharmacological inhibitors of mTORC1-S6K1
As mTORC-S6K1 pathway is hyperactivated in multiple cancers including hematologic malignancies, it has been a critical target for development of pharmacological inhibitors. In 1999, rapamycin, an allosteric inhibitor of mTORC1, was approved for use as an immunosuppressant to prevent rejection following organ transplant (39). In mammalian cells, rapamycin interacts with immunophilin FKBP12. The carboxy-terminal region of mTOR contains the FKBP-rapamycin binding (FRB) domain and the kinase domain. The FKBP12-rapamycin complex docks to the FRB domain and allosterically inhibits the kinase activity of mTORC1 (40). Identification of rapamycin as a clinically relevant compound has led to the development of semi-synthetic analogues, which are collectively called rapalogues. Temsirolimus, everolimus and ridaforolimus are some of the rapalogues, and considered as the first generation mTORC1 inhibitors (41-43).
The second generation mTORC1 inhibitors are ATP analogues, which compete with ATP for binding to the kinase domain of mTOR and subsequently inhibit the kinase function of mTOR. These ATP analogues can inhibit the function of both mTORC1 and mTORC2. Moreover, due to the structural similarity between kinase domains of mTOR and PI3K, some of the ATP analogues inhibit functions of both mTOR and PI3K. Based on their activity against mTOR and PI3K, ATP analogues can be divided into two distinct groups. The first group is comprised of inhibitors which show similar efficacy in inhibiting the function of both mTOR and PI3K. NVPBEZ235, XL765, GSK2126458, SF1126, BGT226, GDC0980, PKI587, PF04691502, GSK2126458 are the inhibitors that belong to the first group (40). The second group of inhibitors are more specific towards inhibiting mTOR activity at a significantly lower IC50 than that for PI3K. PP242, INK128 (a derivative of PP242), CC223, OSI027, AZD8055, AZD2014, Palomid 529 are the ATP analogues with higher efficacy towards inhibiting mTOR activity (40). However, prolonged treatment with either rapalogues or ATP analogues led to development of drug resistance in malignant cells (44, 45). To overcome drug resistance, a new study has proposed to exploit the juxtaposition of both drug-binding pockets in mTORC1 to create a bivalent interaction (46). RapaLink-1 is a bivalent mTORC1 inhibitor, which was created by linking MLN0128, a structural analogue of PP242, using a 39-heavy-link atom linker, with rapamycin (46). Treatment of mice bearing either rapalogue-resistant or ATP analogue- resistant tumors with RapaLink-1 results in reduced tumor growth. RapaLink-1 can inhibit phosphorylation of both S6K1 and Akt (Ser-473), thus inhibiting both mTORC1 and mTORC2 activity.
One of the major drawback of rapalogues is that they also inhibit the negative feedback loop that S6K1 exerts on IRS-1 (Figure 3), leading to an increase in Akt activation (47). This provides strong rationale for development of inhibitors, which can inhibit both mTORC1 activity and Akt activity simultaneously. To this end, dual S6K1/Akt inhibitors could be more useful in treating malignancies. Treatment of breast cancer cells with M2698, a dual S6K1/Akt inhibitor, results in the inhibition of both S6K1 and Akt activity (48). Treatment with M2698 induces increased phosphorylation of Akt in breast cancer cells due to withdrawal of the negative feedback loop exerted by S6K1. However, phosphorylation of PRAS40, a substrate of Akt, was not increased under these conditions, suggesting possible inhibition of Akt activity. In addition to the dual S6K1/Akt inhibitors, PF-4708671, a specific inhibitor of S6K1 activity, also inhibited hyperphosphorylation of Akt following S6K1 inhibition (49).
Effect of mTORC1 on steady state hematopoiesis
Multiple studies have established that activity of mTORC1 is critical for the maintenance of steady-state hematopoiesis. Deletion of mTOR, a component of both mTORC1 and mTORC2, results in reduced white blood cell (WBC) counts in mice, which is associated with a reduction in neutrophils and monocytes. Furthermore, deficiency of mTOR also causes reduction in platelets and erythrocytes in peripheral blood. Conditional deletion of mTOR in HSC led to a reduction in BM cellularity (8). The decrease in BM cellularity was due to a reduction in both myeloid and lymphoid cells. Both the myeloid and lymphoid cells in BM of mTOR-deficient mice show an increase in apoptosis compared to controls. At the molecular level, lineage committed cells in the BM of mTOR-deficient mice display reduced expression of Mcl-1, an anti-apoptotic protein.
The role of Raptor, a specific component of mTORC1, in regulating hematopoiesis has been studied using two different models (6, 7). Interestingly, effect of Raptor deletion on HSC differed depending on the model. In a tamoxifen-inducible model, deletion of Raptor caused decreased BM cellularity (6). Differentiated hematopoietic cells (Lin+ and Mac1+Gr1+) showed an increase in apoptosis following loss of Raptor. Following administration of tamoxifen, Raptor-deficient mice also succumbed to death within 17 days. Moreover, Raptor deletion did not affect cell cycle state of any hematopoietic population (6). In contrast, conditional deletion of Raptor using polyinosinic-polycytidylic acid (pI:pC) did not affect the BM cellularity (7). In this model, Raptor-deficient mice displayed pancytopenia and extramedullary hematopoiesis in the spleen. Raptor-deletion also led to an accumulation of monocytes as well as increased level of pro-B cells in mice. Deletion of Raptor specifically increased the frequency and proliferation of short-term HSC (ST-HSC) compared to long-term HSC (LT-HSC) (7). Raptor-deficient LSK cells displayed elevated level of AMP, NADP+ and other intermediates involved in lipid metabolism. In addition, Raptor-null HSC show decreased level of metabolites involved in nitrogen metabolism (7). Thus, Raptor deficiency affects the cellular nutrient status as well as metabolic pathways in HSC. Downstream of mTORC1, deficiency of S6K1 in the HSC also results in reduced BM cellularity (5). Moreover, the number of HSC are also reduced in S6K1-/- mouse. It is possible that S6K1 is a key substrate of mTORC1 in maintaining BM cellularity and deficiency of either Raptor or mTORC1 negatively affects BM cellularity.
One of the key properties of HSC is that they reside mostly in a quiescent state (50). This is a protective mechanism, which helps to preserve the functional stem cell pool by preventing their exhaustion. In HSC, either mTOR or S6K1 activity is required to maintain their quiescence. Deficiency of either mTOR or S6K1 in HSC results in reduced frequency of HSC in G0 phase of cell cycle (5, 8). Mechanistically, deficiency of mTOR activity in HSC-enriched population results in reduced activation of S6K1 and increased activation of Akt at Ser-473 (8). Moreover, increased activation of Akt is associated with an increase in proliferation of HSC (4). As S6K1 exerts an inhibitory feedback loop on Akt, it is possible that deficiency of S6K1 activity in HSC causes increased activation of Akt, which subsequently leads to a decrease in cellular quiescence. Interestingly, in contrast to lineage-committed cells, Mcl-1 expression leveli significantly increased in HSC following mTOR deletion (8). Moreover, mTORC1 has differential activity in different hematopoietic populations. Phosphorylation levels of both S6, a downstream substrate of S6K1, and 4E-BP1 are low in HSC and high in multipotent progenitors (MPP), common myeloid progenitors (CMP) and granulocyte macrophage progenitors (GMP). In contrast, S6 and 4E-BP1 phosphorylation levels were low in B lymphocytes (6). These results suggest that mTORC1 signaling might have a differential effect on the same substrate in different hematopoietic populations. Furthermore, evidence suggests that mTORC1 might act on different substrates depending on hematopoietic subsets. Deficiency of Raptor results in increased phosphorylation of Akt in relatively mature hematopoietic population only (7). This indicates that the S6K1-mediated negative feedback loop might exist in specific cell type of the BM, which is withdrawn following deletion of Raptor. Multiple studies have demonstrated the association of quiescence with functional defects and exhaustion of HSC. S6K1-/- HSC have reduced Cdkn1a (the gene encoding p21) expression compared to controls (5). Cdkn1a is a critical mediator of HSC quiescence and Cdkn1a-/- HSC undergo increased proliferation and exhaust their functional potential upon serial transplantation (51). Taken together, it is probable that S6K1-/- HSC are less quiescent due do decrease in Cdkn1a expression level. Functionally, HSC are defined by their ability to reconstitute the hematopoietic system of irradiated hosts following serial transplantation. Conditional deletion of mTOR or Raptor results in reduced engraftment of HSC in primary transplant recipients (7, 8). However, loss of expression of S6K1 does not affect the long-term engraftment in primary recipients, but results in reduced self-renewal of HSC in secondary and tertiary recipients (5). As activation of mTORC1 results in inactivation of 4E-BP1 and activation of S6K1, it is conceivable that both events are required for engraftment of HSC in primary recipients. However, S6K1 might be the critical downstream substrate of mTORC1 and regulates self-renewal of HSC. Expression level of Cdkn1a was significantly down-regulated in S6K1-/- LSKs isolated from BM of secondary recipients (5). Cdkn1a-deficient HSC exhaust upon serial transplant, which suggests a decline in self-renewal potential (51). Given that S6K1-/- HSC shows a reduction in Cdkn1a expression and also mirrors the functional ability of Cdkn1a-/- HSC, Cdkn1a could be a possible target of mTORC1-S6K1 signaling in HSC.
By using genetic models, it has been established that loss of mTORC1 activity negatively impacts engraftment and self-renewal of HSC in mice (5, 7). However, inhibition of mTORC1 activity in HSC by using pharmacological inhibitors in human and murine HSC have yielded opposite results compared to genetic approaches. Phenotypically defined murine HSC-enriched population is expanded following treatment with rapamycin (52). Further, rapamycin treatment increases the long-term engraftment of murine HSC (52). Similarly, rapamycin treatment also affects the function of human HSC. Human UCB CD34+ cells display an increase in engraftment and self-renewal following treatment with rapamycin (53). Simultaneous pharmacological inhibition of GSK3 and mTORC1 also increases the self-renewal and engraftment of human HSC (54). Moreover, human HSC could maintain their function in cytokine-free culture following pharmacological inhibition of GSK3 and mTORC1 (54). It is possible that in HSC, in vitro treatment with rapamycin might activate or repress other pathways resulting in increased engraftment following transplantation.
Effect of mTORC1 activity in AML
AML is characterized by clonal expansion of early myeloid progenitors. AML patients have poor long-term overall survival (OS) and for older patients, the median OS is one year (55). One of the major reason for poor outcome in AML is the relapse of the disease. LSC are a small population of cells, which can give rise to identical daughter cells as well as differentiated cells. LSC have the potential to initiate and maintain AML through serial transplantation (56). Recent studies have identified mTORC1-S6K1 pathway as a key regulator of LSC maintenance. Rheb1, an activator of mTORC1, is overexpressed in AML patients (57). AML patients with increased Rheb1 expression have decreased mean survival time compared to patients with low level of Rheb1. Deletion of Rheb1 in a murine model of leukemia results in impaired LSC activity. In LSC-enriched population, deletion of Rheb1 results in reduced mTORC1 activity (57). Moreover, S6K1 and 4E-BP1, the downstream substrates of mTORC1, are constitutively phosphorylated in 60% of AML cells isolated from patients (58). The phosphorylation level of S6K1 in AML blasts is decreased following inhibition of mTORC1 activity, suggesting that S6K1 is a target of mTORC1 in AML cells (59). Deficiency of Raptor in AML cells prolonged the survival of mice, which suggests that Raptor regulates leukemia initiation. Raptor deletion selectively causes apoptosis in differentiated cells. In addition, following serial transplantation of AML cells, Raptor deficiency resulted in prolonged survival of recipient mice suggesting that Raptor is a regulator of LSC self-renewal. PTEN deletion in HSC results in hematologic malignancies. Deletion of Raptor in PTEN-deficient HSC prolongs the survival of mice (7) indicating mTORC1 activity is required for PTEN-deletion induced leukemogenesis as well. Deficiency of S6K1 does not affect leukemia initiation or progression. However, S6K1 deficiency negatively affects the self-renewal potential of LSC and increases the median survival time after serial transplantation (5). In contrast to HSC, deficiency of S6K1 results in increased quiescence of LSC-enriched population (5). Mechanistically, the phosphorylation of 4E-BP1 is significantly decreased in S6K1 deficient AML cells indicating S6K1 might regulate mTORC1 activity (5). These findings suggest that S6K1 might affect LSC function by inhibiting the activity of mTORC1.
Pharmacological inhibition of mTORC1-S6K1 activity in AML
Studies have shown that rapalogues display potent anti-leukemic activity. However, in a clinical setting, treatment with rapalogue alone failed to display significant effect in AML patients. In a phase I trial of deferolimus, none of the AML patients responded to the drug (60). In another phase I/II trial, everolimus also failed to elicit any response in AML patients. The rapalogues are considered to be a specific inhibitor of mTORC1 (61). However, when rapalogues were used in combination with chemotherapy, a significant improvement in median disease free survival and median overall survival was reported (62, 63). In contrast, a recent study has demonstrated that activation of mTORC1 in AML cells could be used as a therapeutic strategy. Sustained mTORC1 activation led to increased AMPK activation-mediated cytotoxicity in AML cells (64). The authors proposed that AMPK and mTORC1 contribute toward a synthetic lethal interaction in AML cells.
One of the major roadblocks in using rapalogues alone as a therapeutic strategy to treat AML is that it leads to withdrawal of S6K1-mediated negative feedback loop involving mTORC2. Although mTORC2 is active in AML cells, efficacy of rapalogues in inhibiting mTORC2 activity in AML cells has been inconclusive. Treatment of AML patients with either everolimus or temsirolimus results in reduced Akt phosphorylation at Ser-473, suggesting inhibition of mTORC2 activity (65). Conversely, in another study, treatment of primary leukemic blasts with rapamycin failed to attenuate mTORC2 activity (66). The differences in the outcomes between these studies could be due to the time of exposure of leukemic cells to rapalogues; as studies have shown that only long-term treatment with rapalogues results in reduced mTORC2 activity (67). To circumvent mTORC2 activation following mTORC1 inhibition, dual inhibitors of mTORC1 and mTORC2 have been developed. SNS-032, a dual inhibitor of mTORC1/2, induced cytotoxicity in AML blasts isolated from patients (68). However, a group of patients were also not responsive to SNS-032. AZD8055, another dual mTORC inhibitor, blocked protein translation and proliferation in AML cells (69). AZD8055 also induced autophagy in human AML cells and inhibited the initiation of leukemia in vivo (69). In a recent study, treatment of AML cells with an anti-CD44 monoclonal antibody showed a decrease in cellular proliferation and blocked mTORC1 and mTORC2 activation (70). Inhibition of mTORC1 activity by rapalogues also led to upregulation of insulin-like growth factor-1 (IGF-1)-mediated activation of PI3-K-Akt pathway (71). Another obstacle of treatment with rapalogues is the insensitivity of 4E-BP1 towards rapalogue treatment compared to S6K1. Long-term rapamycin treatment causes rephosphorylation of 4E-BP1, which could possibly counteract the effect of mTORC1 inhibition in malignant cells. However, recent data suggest that in AML cells, specific inhibition of S6K1 activity could result in decreased phosphorylation of 4E-BP1. Treatment of human AML cells expressing MLL-AF9 with PF-4708671 results in reduced proliferation in vitro and in vivo (5). Phosphorylations of both mTOR and 4E-BP1 were also decreased in human AML cells treated with PF-4708671 (5). These data argue that as 4E-BP1 is a substrate of mTORC1, it is possible that S6K1 also acts as an activator of mTORC1 in AML cells. Given the complex nature of feedback signaling loops in mTORC1 pathway, mTORC1 inhibitors could be a better therapeutic option when used in combination with another inhibitor targeting the negative feedback loops.
Future Directions
The mTORC1-S6K1 pathway plays an important role in leukemia initiation, progression and as a critical regulator of LSC. This could potentially lead to targeting of this pathway in eliminating LSC in AML. However, given the critical role of this pathway in hematopoiesis, it is necessary to develop therapeutic strategies, which will not perturb normal hematopoiesis but will only target LSC. Moreover, given the existence of inhibitory feedback loops in this pathway, it is necessary to take into consideration the impact of prolonged inhibition of this pathway in AML cells. In clinical setting, combined inhibition of mTORC1-S6K1 pathway along with another target has been proven to be more successful. Moreover, mTORC-S6K1 pathway has been shown to be a critical regulator of integral cellular processes like metabolism and autophagy. Recent findings show that metabolism (72, 73) and autophagy (74-76) are critical regulators of LSC maintenance. Additionally, mTORC1-S6K1 pathway could also reprogram metabolic pathways in malignant cells to help the cells escape glycolysis dependency and become resistance to inhibition of cellular glycolysis (77). Moreover, from recent literature, one could argue that mTORC1 and S6K1 signaling could be differentially targeted on the basis of the cell type in question, which makes it imperative to design therapeutic strategies that would allow targeting of specific cell populations. Additionally, S6K1 could act differentially from mTORC1 towards regulating similar cellular processes, suggesting that S6K1 might have mTORC1-independent functions. For example, mTORC1 acts as an inhibitor of autophagy (78, 79). However, the role of S6K1 in regulating autophagy depends on the cellular context (80-84). Additionally, the role of S6K1 in regulation of autophagy and metabolism in HSC and LSC is not known. In view of the above facts, it is important to determine how the components of mTORC1-S6K1 pathway regulate these processes in both HSC and LSC.
Highlights.
mTORC1-S6K1 pathway is dysregulated in acute myeloid leukemia.
mTORC1 and S6K1 regulates function of leukemia stem cells (LSC).
mTORC1 and S6K1 are also critical regulators of hematopoietic stem cell (HSC) function.
New generation of mTORC1 inhibitors like RapaLink-1 could be a useful therapeutic tool against rapamycin-resistant leukemic cells.
Acknowledgments
This work was supported in part by grants from National Institutes of Health (R01HL077177 to RK; R01HL075816 to RK; R01HL081111 to RK, R01CA173852 to R.K; and R01CA134777 to RK).
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haneline LS, White H, Yang FC, Chen S, Orschell C, Kapur R, et al. Genetic reduction of class IA PI-3 kinase activity alters fetal hematopoiesis and competitive repopulating ability of hematopoietic stem cells in vivo. Blood. 2006;107(4):1375–82. doi: 10.1182/blood-2005-05-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115(20):4030–8. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–82. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 4.Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115(7):1406–15. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh J, Kobayashi M, Ramdas B, Chatterjee A, Ma P, Mali RS, et al. S6K1 regulates hematopoietic stem cell self-renewal and leukemia maintenance. J Clin Invest. 2016;126(7):2621–5. doi: 10.1172/JCI84565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshii T, Tadokoro Y, Naka K, Ooshio T, Muraguchi T, Sugiyama N, et al. mTORC1 is essential for leukemia propagation but not stem cell self-renewal. J Clin Invest. 2012;122(6):2114–29. doi: 10.1172/JCI62279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C, et al. mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell. 2012;11(3):429–39. doi: 10.1016/j.stem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo F, Zhang S, Grogg M, Cancelas JA, Varney ME, Starczynowski DT, et al. Mouse gene targeting reveals an essential role of mTOR in hematopoietic stem cell engraftment and hematopoiesis. Haematologica. 2013;98(9):1353–8. doi: 10.3324/haematol.2012.080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010;285(26):20109–16. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002;277(38):35364–70. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 12.Lai KP, Leong WF, Chau JF, Jia D, Zeng L, Liu H, et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010;29(17):2994–3006. doi: 10.1038/emboj.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 15.Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47(4):535–46. doi: 10.1016/j.molcel.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15(8):702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 17.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 18.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell. 2016;165(1):153–64. doi: 10.1016/j.cell.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saxton RA, Chantranupong L, Knockenhauer KE, Schwartz TU, Sabatini DM. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature. 2016;536(7615):229–33. doi: 10.1038/nature19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science. 2016;351(6268):53–8. doi: 10.1126/science.aad2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–8. doi: 10.1126/science.aab2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100–6. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jewell JL, Kim YC, Russell RC, Yu FX, Park HW, Plouffe SW, et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347(6218):194–8. doi: 10.1126/science.1259472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roccio M, Bos JL, Zwartkruis FJ. Regulation of the small GTPase Rheb by amino acids. Oncogene. 2006;25(5):657–64. doi: 10.1038/sj.onc.1209106. [DOI] [PubMed] [Google Scholar]
- 26.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15(3):155–62. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]
- 28.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 29.Isotani S, Hara K, Tokunaga C, Inoue H, Avruch J, Yonezawa K. Immunopurified mammalian target of rapamycin phosphorylates and activates p70 S6 kinase alpha in vitro. J Biol Chem. 1999;274(48):34493–8. doi: 10.1074/jbc.274.48.34493. [DOI] [PubMed] [Google Scholar]
- 30.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24(8):3112–24. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001;20(16):4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson CJ, Broenstrup M, Fingar DC, Julich K, Ballif BA, Gygi S, et al. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol. 2004;14(17):1540–9. doi: 10.1016/j.cub.2004.08.061. [DOI] [PubMed] [Google Scholar]
- 33.Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24(19):2274–80. doi: 10.1016/j.cub.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314(5798):467–71. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 35.Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat Cell Biol. 2013;15(11):1340–50. doi: 10.1038/ncb2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20(20):2820–32. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30(4):908–21. doi: 10.1128/MCB.00601-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–23. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson CJ, Friend PJ, Jamieson NV, Frick TW, Alexander G, Gimson AE, et al. Sirolimus: a potent new immunosuppressant for liver transplantation. Transplantation. 1999;67(4):505–9. doi: 10.1097/00007890-199902270-00002. [DOI] [PubMed] [Google Scholar]
- 40.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–80. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 41.Mita M, Sankhala K, Abdel-Karim I, Mita A, Giles F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Investig Drugs. 2008;17(12):1947–54. doi: 10.1517/13543780802556485. [DOI] [PubMed] [Google Scholar]
- 42.Gabardi S, Baroletti SA. Everolimus: a proliferation signal inhibitor with clinical applications in organ transplantation, oncology, and cardiology. Pharmacotherapy. 2010;30(10):1044–56. doi: 10.1592/phco.30.10.1044. [DOI] [PubMed] [Google Scholar]
- 43.Rini BI. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin Cancer Res. 2008;14(5):1286–90. doi: 10.1158/1078-0432.CCR-07-4719. [DOI] [PubMed] [Google Scholar]
- 44.Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371(15):1426–33. doi: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vicier C, Dieci MV, Andre F. New strategies to overcome resistance to mammalian target of rapamycin inhibitors in breast cancer. Curr Opin Oncol. 2013;25(6):587–93. doi: 10.1097/CCO.0000000000000014. [DOI] [PubMed] [Google Scholar]
- 46.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–6. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mei H, Wang Y, Fan J, Lin Z. Alternative splicing of S6K1 promotes non-small cell lung cancer survival. Tumour Biol. 2016;37(10):13369–76. doi: 10.1007/s13277-016-5253-1. [DOI] [PubMed] [Google Scholar]
- 48.Machl A, Wilker EW, Tian H, Liu X, Schroeder P, Clark A, et al. M2698 is a potent dual-inhibitor of p70S6K and Akt that affects tumor growth in mouse models of cancer and crosses the blood-brain barrier. Am J Cancer Res. 2016;6(4):806–18. [PMC free article] [PubMed] [Google Scholar]
- 49.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431(2):245–55. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 50.Nakamura-Ishizu A, Takizawa H, Suda T. The analysis, roles and regulation of quiescence in hematopoietic stem cells. Development. 2014;141(24):4656–66. doi: 10.1242/dev.106575. [DOI] [PubMed] [Google Scholar]
- 51.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–8. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 52.Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, et al. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014;97(1):20–9. doi: 10.1097/TP.0b013e3182a7fcf8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rohrabaugh SL, Campbell TB, Hangoc G, Broxmeyer HE. Ex vivo rapamycin treatment of human cord blood CD34+ cells enhances their engraftment of NSG mice. Blood Cells Mol Dis. 2011;46(4):318–20. doi: 10.1016/j.bcmd.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang J, Nguyen-McCarty M, Hexner EO, Danet-Desnoyers G, Klein PS. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med. 2012;18(12):1778–85. doi: 10.1038/nm.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roboz GJ. Current treatment of acute myeloid leukemia. Curr Opin Oncol. 2012;24(6):711–9. doi: 10.1097/CCO.0b013e328358f62d. [DOI] [PubMed] [Google Scholar]
- 56.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 57.Gao Y, Gao J, Li M, Zheng Y, Wang Y, Zhang H, et al. Rheb1 promotes tumor progression through mTORC1 in MLL-AF9-initiated murine acute myeloid leukemia. J Hematol Oncol. 2016;9:36. doi: 10.1186/s13045-016-0264-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Recher C, Dos Santos C, Demur C, Payrastre B. mTOR, a new therapeutic target in acute myeloid leukemia. Cell Cycle. 2005;4(11):1540–9. doi: 10.4161/cc.4.11.2159. [DOI] [PubMed] [Google Scholar]
- 59.Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102(3):972–80. doi: 10.1182/blood-2002-11-3429. [DOI] [PubMed] [Google Scholar]
- 60.Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, Strair R, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2008;14(9):2756–62. doi: 10.1158/1078-0432.CCR-07-1372. [DOI] [PubMed] [Google Scholar]
- 61.Yee KW, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, Ferrajoli A, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2006;12(17):5165–73. doi: 10.1158/1078-0432.CCR-06-0764. [DOI] [PubMed] [Google Scholar]
- 62.Amadori S, Stasi R, Martelli AM, Venditti A, Meloni G, Pane F, et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: results of a phase II GIMEMA study (AML-1107) Br J Haematol. 2012;156(2):205–12. doi: 10.1111/j.1365-2141.2011.08940.x. [DOI] [PubMed] [Google Scholar]
- 63.Park S, Chapuis N, Saint Marcoux F, Recher C, Prebet T, Chevallier P, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia. 2013;27(7):1479–86. doi: 10.1038/leu.2013.17. [DOI] [PubMed] [Google Scholar]
- 64.Sujobert P, Poulain L, Paubelle E, Zylbersztejn F, Grenier A, Lambert M, et al. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep. 2015;11(9):1446–57. doi: 10.1016/j.celrep.2015.04.063. [DOI] [PubMed] [Google Scholar]
- 65.Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109(8):3509–12. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ, et al. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res. 2011;17(13):4378–88. doi: 10.1158/1078-0432.CCR-10-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 68.Meng H, Jin Y, Liu H, You L, Yang C, Yang X, et al. SNS-032 inhibits mTORC1/mTORC2 activity in acute myeloid leukemia cells and has synergistic activity with perifosine against Akt. J Hematol Oncol. 2013;6:18. doi: 10.1186/1756-8722-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2012;26(6):1195–202. doi: 10.1038/leu.2011.339. [DOI] [PubMed] [Google Scholar]
- 70.Gadhoum SZ, Madhoun NY, Abuelela AF, Merzaban JS. Anti-CD44 antibodies inhibit both mTORC1 and mTORC2: a new rationale supporting CD44-induced AML differentiation therapy. Leukemia. 2016;30(12):2397–401. doi: 10.1038/leu.2016.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tamburini J, Chapuis N, Bardet V, Park S, Sujobert P, Willems L, et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood. 2008;111(1):379–82. doi: 10.1182/blood-2007-03-080796. [DOI] [PubMed] [Google Scholar]
- 72.Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell. 2016;19(1):23–37. doi: 10.1016/j.stem.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17(5):585–96. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sumitomo Y, Koya J, Nakazaki K, Kataoka K, Tsuruta-Kishino T, Morita K, et al. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood. 2016;128(12):1614–24. doi: 10.1182/blood-2015-12-684696. [DOI] [PubMed] [Google Scholar]
- 75.Altman JK, Szilard A, Goussetis DJ, Sassano A, Colamonici M, Gounaris E, et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014;20(9):2400–9. doi: 10.1158/1078-0432.CCR-13-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Q, Chen L, Atkinson JM, Claxton DF, Wang HG. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016;7(9):e2361. doi: 10.1038/cddis.2016.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, et al. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell. 2016;29(4):548–62. doi: 10.1016/j.ccell.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 78.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yorimitsu T, Zaman S, Broach JR, Klionsky DJ. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2007;18(10):4180–9. doi: 10.1091/mbc.E07-05-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hac A, Domachowska A, Narajczyk M, Cyske K, Pawlik A, Herman-Antosiewicz A. S6K1 controls autophagosome maturation in autophagy induced by sulforaphane or serum deprivation. Eur J Cell Biol. 2015;94(10):470–81. doi: 10.1016/j.ejcb.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 82.Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7(2):167–78. doi: 10.1016/j.devcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 83.Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM, Sinclair DA. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging (Albany NY) 2009;1(6):515–28. doi: 10.18632/aging.100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shin JH, Min SH, Kim SJ, Kim YI, Park J, Lee HK, et al. TAK1 regulates autophagic cell death by suppressing the phosphorylation of p70 S6 kinase 1. Sci Rep. 2013;3:1561. doi: 10.1038/srep01561. [DOI] [PMC free article] [PubMed] [Google Scholar]