

Abstract
The Chinese crocodile lizard, Shinisaurus crocodilurus, is the only living representative of the monotypic family Shinisauridae under the order Squamata. It is an obligate semi-aquatic, viviparous, diurnal species restricted to specific portions of mountainous locations in southwestern China and northeastern Vietnam. However, in the past several decades, this species has undergone a rapid decrease in population size due to illegal poaching and habitat disruption, making this unique reptile species endangered and listed in the Convention on International Trade in Endangered Species of Wild Fauna and Flora Appendix II since 1990. A proposal to uplist it to Appendix I was passed at the Convention on International Trade in Endangered Species of Wild Fauna and Flora Seventeenth meeting of the Conference of the Parties in 2016. To promote the conservation of this species, we sequenced the genome of a male Chinese crocodile lizard using a whole-genome shotgun strategy on the Illumina HiSeq 2000 platform. In total, we generated ∼291 Gb of raw sequencing data (×149 depth) from 13 libraries with insert sizes ranging from 250 bp to 40 kb. After filtering for polymerase chain reaction–duplicated and low-quality reads, ∼137 Gb of clean data (×70 depth) were obtained for genome assembly. We yielded a draft genome assembly with a total length of 2.24 Gb and an N50 scaffold size of 1.47 Mb. The assembled genome was predicted to contain 20 150 protein-coding genes and up to 1114 Mb (49.6%) of repetitive elements. The genomic resource of the Chinese crocodile lizard will contribute to deciphering the biology of this organism and provides an essential tool for conservation efforts. It also provides a valuable resource for future study of squamate evolution.
Keywords: Chinese crocodile lizard, Shinisaurus crocodilurus, sequencing, genome assembly, annotation
Data Description
Background
The Chinese crocodile lizard, Shinisaurus crocodilurus (NCBI taxonomy ID 52224) (Fig. 1), was first collected in 1928. In 1930, to accommodate the monotypic genus and species, Ahl established Shinisauridae as a new family under the order Squamata [1]. The species usually is found along slow-flowing rocky streams in montane evergreen forests [2] and is distributed in the east part of the Guangxi Zhuang Autonomous Region, the west and north parts of Guangdong Province in southern China, and in mountainous areas of northern Vietnam [3]. It is a semi-aquatic diurnal predator and a strong swimmer, preying on fish, tadpoles, and aquatic insects. It is ovoviviparous and breeds in July and August in the wild [1, 4, 5]. A variety of anthropogenic hazards have caused severe population declines within the last decades. Illegal poaching for the international pet trade, traditional medicine, and food represents the main driver fueling the ongoing population decline [2]. A quantitative survey on the species was carried out in 1978, which estimated the total known population at 6000 individuals, and in 2008 it estimated a total population of 950 animals [5, 6]. The species was listed in the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES) Appendix II in 1990 [7] and in the IUCN Red List of Threatened Species in 2014 [6], and a proposal to uplist it to Appendix I was passed at the CITES Seventeenth meeting of the Conference of the Parties in 2016 [8].
Figure 1:

Example of a Chinese crocodile lizard, Shinisaurus crocodilurus (image from Wong Sai Lok).
Sample collection and sequencing
The genomic DNA of the Chinese crocodile lizard was extracted from the blood collected from the tail vein of a single adult male lizard in Ocean Park Hong Kong, which is a zoological theme park in Hong Kong. The venipuncture procedure was identical to that used for routine clinical blood draws in lizards in compliance with the Animal Welfare and Use Guidelines of Ocean Park. This lizard was alive in the collection of Ocean Park Hong Kong at the time of manuscript submission (Animal ID: MIG12–30 061 867; the CITES license to possess number is APO/PL 266/12). Three standard DNA libraries with short-insert sizes (250, 500, and 800 bp) and 10 mate-paired libraries with long-insert sizes (2 kb × 2, 5 kb × 2, 10 kb × 2, 20 kb × 2, and 40 kb × 2) were constructed with the standard protocol provided by Illumina (San Diego, CA, USA). Paired-end sequencing was performed for all the 13 libraries on the HiSeq 2000 platform according to the manufacturer's instructions (Illumina, San Diego, CA, USA). The sequenced read length was 150 bp for the short-insert libraries and 49 bp for the long-insert libraries. In total, about 290.85 Gb (×149) of raw reads were eventually produced (Table 1).
Table 1:
Statistics of the Chinese crocodile lizard genome sequencing
| Raw data | Clean data | |||||||
|---|---|---|---|---|---|---|---|---|
| Insert size (bp) | Library | Reads length (bp) | Total data (Gb) | Sequence coverage (×) | Physical coverage (×) | Total Data (Gb) | Sequence coverage (×) | Physical coverage (×) |
| 250 | 1 | 150 | 54.16 | 27.78 | 23.15 | 41.99 | 23.15 | 20.32 |
| 500 | 1 | 150 | 54.67 | 28.04 | 46.73 | 39.27 | 46.73 | 39.5 |
| S800 | 1 | 150 | 15.68 | 8.04 | 21.45 | 11.82 | 21.45 | 18.31 |
| 2000 | 2 | 49 | 34.93 | 17.92 | 365.62 | 13.99 | 7.18 | 146.48 |
| 5000 | 2 | 49 | 34.35 | 17.62 | 899.1 | 13.60 | 6.97 | 355.83 |
| 10 000 | 2 | 49 | 33.74 | 17.3 | 1765.7 | 9.48 | 4.86 | 496.14 |
| 20 000 | 2 | 49 | 30.64 | 15.71 | 3207.13 | 3.25 | 1.66 | 340.01 |
| 40 000 | 2 | 49 | 32.68 | 16.76 | 6842.73 | 3.33 | 1.71 | 697.58 |
| Total | 13 | - | 290.85 | 149.17 | 13 171.61 | 136.73 | 70.12 | 2114.17 |
Coverage calculation was based on the estimated genome size of 1.95 Gb. Sequence coverage is the average number of times a base is read, while physical coverage is the average number of times a base is spanned by mate-paired reads.
Read filtering and genome size estimation
We obtained 136.73 Gb of clean data from the raw data by removing duplicated reads arising from polymerase chain reaction amplification during library construction, adapter-contaminated reads with ≥10 bp aligned to adapter sequence, low-quality reads that contain >5% “Ns” for the short-insert (250, 500, and 800 bp) data or >20% for the long-insert (2, 5, 10, 2, and 40 kb) data, and low-quality reads that contain ≥40 low-quality (Illumina phred quality score ≤ 7) bases for the short-insert data or ≥30 bases for the long-insert data using SOAPfilter, an application included in the SOAPdenovo package (SOAPdenovo2, RRID:SCR_014986) (Table 1) [9]. We obtained about 136.73 Gb of clean reads from about 290.85 Gb of raw reads; the total number of clean reads is 1 494 896 603, and the total number of raw reads is 3631.968,900. Then we used the clean data from the 3 short-insert (250, 500, and 800 bp) libraries to estimate the genome size of Chinese crocodile lizard with a 17-mer analysis [10]. A k-mer is related to an artificial sequence division of K nucleotides iteratively from sequencing reads [11]. We selected a fragment length of 17; the fragment is called 17-mer. When a certain coverage was reached, k-mer frequencies were plotted against the sequence depth gradient following a Poisson distribution [12], then the genome length could be estimated from the number and depth of kmer by the following formula: genome size = (Kmer number)/(Peak depth). According to that prediction, the Chinese crocodile lizard genome is estimated to be 1.95 Gb in size (Table 2; Fig. 2).
Table 2:
Statistics of 17-mer analysis
| Genome | Kmer | Kmer number | Peak depth | Estimated genome size (bp) | Used base (bp) |
|---|---|---|---|---|---|
| Shinisaurus crocodilurus | 17 | 68 234 898 814 | 35 | 1 949 568 537 | 78 251 030 750 |
The genome size was estimated according to the following formula: genome size = (Kmer number)/(Peak depth).
Figure 2:
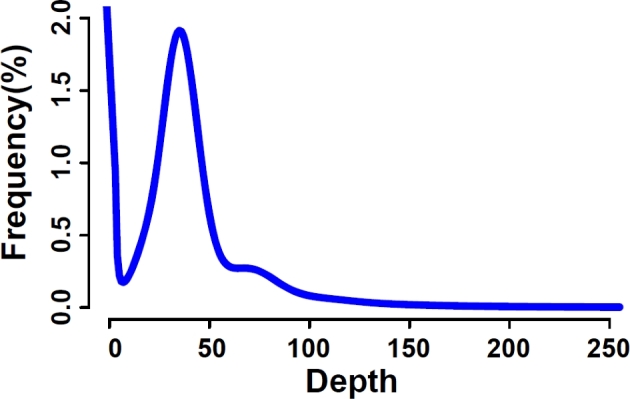
17-mer depth distribution. The 17-mer analysis was employed by using 250, 500, and 800 bp short-insert size libraries. The peak depth was ×35. The total number of 17-mer present in this subset was 68 234 898 814. The genome size was estimated to be 1.95 Gb according to the following formula: genome size = (Kmer number)/(Peak depth).
Genome assembly and completeness estimation
We employed the SOAPdenovo package (version 2.04.4) for genome assembly (SOAPdenovo2, RRID:SCR_014986) [9]. Briefly, the sequences derived from the short-insert libraries were first decomposed into k-mers to construct the de Bruijn graph, which was simplified to allow connection of the k-mers into a contiguous sequence (contigs). We tested a series of kmer lengths ranging from 31 to 81 bp, and the 69-mer was finally selected to generate a contig assembly with the longest N50 value. We then aligned the paired-end reads from both the small- and large-insert libraries to the contigs, calculated the support for relationships between contigs, and constructed scaffolds using distance information from read pairs. We required at least 3 and 5 read pairs to form a reliable connection between 2 contigs for short-insert and large-insert data, respectively. Finally, Kgf (version 1.16) [9] and GapCloser (GapCloser, RRID:SCR_015026; version 1.10.1) [9] were employed to close intra-scaffold gaps using paired-end reads from the small-insert libraries. The end result was a genome assembly with a total length of 2.24 Gb, scaffold and contig N50s of 1470 kb and 11.7 kb, respectively, and unclosed gap regions representing 7.98% of the assembly, which is comparable to the previously published reptile genome assemblies (Table 3).
Table 3:
Comparison of genome assembly and gene number for 15 reptiles with published genomes
| Species | Common name | Sequencing platform | Sequence coverage (×) | Assembled genome size (Gb) | Contig N50 (kb) | Scaffold N50 (kb) | Gap ratio (%) | Gene number | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Alligator mississippiensis | American alligator | NGS | 156.0 | 2.17 | 7.0 | 509 | 2.09 | 23 323 | [40] |
| Alligator sinensis | Chinese alligator | NGS | 109.0 | 2.30 | 23.4 | 2188 | 3.17 | 22 200 | [41] |
| Anolis carolinensis | Green anole lizard | Sanger | 6.0 | 1.78 | 79.9 | 4033 | 4.49 | 17 472 | [42] |
| Chrysemys picta bellii | Western painted turtle | Sanger + NGS | 18.0 | 2.59 | 11.9 | 5212 | 7.64 | 21 796 | [43] |
| Chelonia mydas | Green sea turtle | NGS | 82.3 | 2.24 | 20.4 | 3778 | 4.33 | 19 633 | [44] |
| Crocodylus porosus | Saltwater crocodile | NGS | 74.0 | 2.12 | 32.8 | 205 | 5.30 | 13 321 | [40] |
| Deinagkistrodon acutus | Five-pacer viper | NGS | 114.2 | 1.47 | 22.4 | 2122 | 5.29 | 21 194 | [45] |
| Eublepharis macularius | Leopard gecko | NGS | 135.8 | 2.02 | 20.0 | 664 | 1.76 | 24 755 | [26] |
| Gavialis gangeticus | Indian gharial | NGS | 81.0 | 2.88 | 14.2 | 127 | 2.22 | 14 043 | [40] |
| Gekko japonicus | Japanese gecko | NGS | 131.3 | 2.55 | 21.1 | 685 | 3.54 | 22 487 | [46] |
| Ophiophagus hannah | King cobra | NGS | 28.0 | 1.66 | 4.0 | 226 | 13.5 | 18 579 | [19] |
| Pelodiscus sinensis | Soft-shell turtle | NGS | 105.6 | 2.21 | 21.9 | 3331 | 4.35 | 23 649 | [44] |
| Pogona vitticeps | Australian dragon lizard | NGS | 179.1 | 1.82 | 31.3 | 2290 | 3.78 | 19 406 | [20] |
| Python molurus bivittatus | Burmese python | NGS | 20.0 | 1.44 | 10.7 | 208 | 3.52 | 25 385 | [18] |
| Shinisaurus crocodilurus | Chinese crocodile lizard | NGS | 149 | 2.24 | 11.7 | 1470 | 7.98 | 20 150 |
We then employed Benchmarking Universal Single-Copy Orthologs (BUSCO; version 3.0.0) to evaluate the completeness of the assembly using 2586 vertebrata expected genes (BUSCO, RRID:SCR_015008) [13]. This analysis showed that 2391 (92.5%) and 125 (4.8%) of the 2586 expected vertebrata genes were identified as complete and fragmented, respectively, while 70 (2.7%) genes were considered missing in the assembly. We ran the same version of BUSCO to the other 14 retile genomes, respectively; the completeness of the Chinese crocodile lizard assembly was also comparable to other published reptile genome assemblies (Table 4).
Table 4:
The percentages of complete, fragmented, and missing genes out of the 2586 expected vertebrata genes in 15 reptile genomes based on the BUSCO assessment
| Species | Common name | Complete single-copy (%) | Complete duplicated (%) | Fragmented (%) | Missing (%) |
|---|---|---|---|---|---|
| Alligator mississippiensis | American alligator | 95.0 | 0.6 | 3.1 | 1.3 |
| Alligator sinensis | Chinese alligator | 94.4 | 0.7 | 3.2 | 1.7 |
| Anolis carolinensis | Green anole lizard | 88.1 | 0.8 | 5.6 | 5.5 |
| Chelonia mydas | Green sea turtle | 93.9 | 0.8 | 3.7 | 1.6 |
| Chrysemys picta bellii | Western painted turtle | 75.5 | 0.8 | 3.3 | 20.4 |
| Crocodylus porosus | Saltwater crocodile | 94.1 | 0.6 | 2.1 | 3.2 |
| Deinagkistrodon acutus | Five-pacer viper | 94.5 | 0.6 | 2.4 | 2.5 |
| Eublepharis macularius | Leopard gecko | 94.0 | 1.2 | 3.3 | 1.5 |
| Gavialis gangeticus | Indian gharial | 85.2 | 0.5 | 11.6 | 2.7 |
| Gekko japonicus | Japanese gecko | 89.8 | 1.1 | 6.3 | 2.8 |
| Ophiophagus hannah | King cobra | 86.6 | 0.6 | 8.6 | 4.2 |
| Pelodiscus sinensis | Soft-shell turtle | 93.5 | 0.5 | 3.8 | 2.2 |
| Pogona vitticeps | Australian dragon lizard | 94.3 | 0.6 | 3.1 | 2.0 |
| Python molurus bivittatus | Burmese python | 91.0 | 0.7 | 5.4 | 2.9 |
| Shinisaurus crocodilurus | Chinese crocodile lizard | 91.6 | 0.9 | 4.8 | 2.7 |
Repeat annotation
Repetitive elements in the Chinese crocodile lizard genome were identified by homology searches against known repeat databases and de novo predictions. Briefly, we identified known transposable elements (TEs) by using RepeatMasker (version 3.3.0; RepeatMasker, RRID:SCR_012954) [14] to search against the Repbase TE library (RepBase16.10) [14] and RepeatProteinMask within the RepeatMasker package to search against the TE protein database. We then employed RepeatModeler (version 1.0.5; RepeatModeler, RRID:SCR_015027) [15] and LTR_FINDER (version 1.0.5) [16] for de novo prediction of TEs. RepeatModeler was first used to construct a de novo crocodile lizard repeat library, which was subsequently used by RepeatMasker to annotate repeats in the crocodile lizard genome. LTR_FINDER was used to search the whole genome for a characteristic structure of the full-length long terminal repeat retrotransposons (LTRs), mainly based on their ∼18 bp terminal sequences being complementary to the 3’ tail of some tRNAs [16]. We provided LTR_FINDER with all eukaryotic tRNAs to search for LTRs. Finally, we searched the genome for tandem repeats using Tandem Repeats Finder (TRF; version 4.04) [17]. The results from different methods were presented in Table 5. Overall, a total of 1114 Mb of non-redundant repetitive sequences were identified, accounting for 49.6% of the Chinese crocodile lizard genome (Table 5), and by using RepeatMasker, we found that long interspersed elements are the most predominant elements in de novo predictions, which accounted for 10% of the genome. This lizard genome, in terms of repeat content, is similar to the known genomes of the Burmese python [18], king cobra [18, 19], Australian dragon lizard [20], and green anole lizard (Table 6) [21].
Table 5:
The statistics of repeats annotated by different methods in the Chinese crocodile lizard genome
| Method | Total repeat length (bp) | Percentage of genome |
|---|---|---|
| TRF | 35 995 906 | 1.74 |
| Repeatmasker | 199 442 776 | 9.65 |
| Proteinmask | 164 914 070 | 7.98 |
| RepeatModeler | 938 017 292 | 41.79 |
| LTR_FINDER | 235 204 092 | 10.48 |
| Total | 1 113 900 339 | 49.62 |
Table 6:
Breakdown of repeat content for 5 reptile genomes estimated by RepeatMasker
| Repeat type | The Burmese python (%) | The king cobra (%) | The green anole lizard (%) | The Australian dragon lizard (%) | The Chinese crocodile lizard (%) |
|---|---|---|---|---|---|
| DNA | 3.45 | 3.49 | 8.71 | 3.26 | 3.80 |
| LINE | 8.57 | 10.55 | 12.19 | 10.93 | 10.20 |
| SINE | 1.60 | 2.09 | 5.11 | 3.14 | 2.72 |
| LTR | 0.85 | 1.75 | 2.94 | 0.92 | 1.52 |
| Unknown | 12.61 | 12.87 | 7.49 | 16.23 | 23.95 |
| Total | 31.82 | 35.22 | 33.82 | 35.93 | 41.79 |
Gene prediction
We combined homology-based and de novo methods to build consensus gene models of the reference genome. In the homology-based method, protein sequences of Anolis carolinensis, Gallus gallus, and Homo sapiens derived from the Ensembl database (release-67; Ensembl, RRID:SCR_002344) were first mapped to the Chinese crocodile lizard genome using TBLASTN (version 2.2.23; TBLASTN, RRID:SCR_011822) [22] with an E-value cutoff of 1e-5, and the BLAST hits were linked into candidate gene loci with GenBlastA (version 1.0.4) [23]. Then the genomic sequences of the candidate loci together with 2 kb flanking sequences were extracted, and gene structures were determined by aligning the homologous proteins to these extracted genomic sequences using GeneWise (version 2.2; GeneWise, RRID:SCR_015054) [24]. In the de novo method, we randomly chose 1000 homology-based gene models with intact open reading frames and an aligning rate of 100% (i.e., query protein length/predicted protein length) to train Augustus (version 2.5.5) (Augustus: Gene Prediction, RRID:SCR_008417) [25] in order to obtain gene parameters appropriate to the Chinese crocodile lizard genome. Then we performed de novo prediction on the repeat-masked genome using Augustus with the obtained gene parameters. Finally, gene models from these 2 methods were combined into a non-redundant gene set of 20 150 genes in the Chinese crocodile lizard using a similar strategy as Xiong et al. (Table 3) [26].
Gene function annotation
Gene names were assigned according to the best hit of the alignments to the SwissProt and TrEMBL databases (Uniprot release 2011–06; UniProt, RRID:SCR_002380) [27] using BLASTP (version 2.2.3). The motifs and domains of genes were determined by InterProScan (version 4.7; InterProScan, RRID:SCR_005829) [28] against the InterPro protein signature databases including ProDom (ProDom, RRID:SCR_006969) [29], PRINTS (PRINTS, RRID:SCR_003412) [30], Pfam (Pfam, RRID:SCR_004726) [31], SMART (SMART, RRID:SCR_005026) [32], PANTHER (PANTHER, RRID:SCR_004869) [33], and (PROSITE PROSITE, RRID:SCR_003457) [34]. Gene ontology (GO; GO, RRID:SCR_002811) terms for each gene were obtained from the corresponding InterPro entries [35], and 14 518 genes were assigned to 1 or more GO terms. All genes were aligned against KEGG proteins (release 58; KEGG, RRID:SCR_012773) [36], and the pathway in which the gene might be involved was derived from the matched genes in KEGG. Overall, we inferred 20 010 (99.31%) genes annotated from the results of the 4 databases (Table 7).
Table 7:
Number and percentage of genes with functional annotation
| Number | Percentage (%) | |
|---|---|---|
| SwissProt | 18 817 | 93.38 |
| TrEMBL | 9675 | 48.01 |
| InterPro | 17 589 | 87.29 |
| KEGG | 15 791 | 78.37 |
| GO | 14 518 | 72.05 |
| Combined | 20 010 | 99.31 |
Conclusion
Here we report the first annotated Chinese crocodile lizard genome sequence assembly. This research yielded a draft genome assembly with a total length of 2.24 Gb and an N50 scaffold size of 1.47 Mb. The assembled genome was predicted to contain 20 150 protein-coding genes and up to 1114 Mb (49.6%) of repetitive elements. The draft genome and annotation will provide valuable data for the captive breeding and aid research into the phylogeny and biological features such as ovoviviparity, venom glands, etc. [37, 38].
Abbreviations
BUSCO: Benchmarking Universal Single-Copy Orthologs; CITES: the Convention on International Trade in Endangered Species of Wild Fauna and Flora; Gb: Gigabase; GO: gene ontology; LTRs: terminal repeat retrotransposons; TE: transposable element.
Funding
This work was funded by the China National Genebank, and the Project U1301252 was supported by National Natural Science Foundation of China. The sample was supplied by the Genome 10K (G10K) consortium. We would like to thank the faculty and staff in BGI-Shenzhen, who contributed to the sequencing of the Chinese crocodile lizard genome.
Availability of supporting data
Supporting data for this Data Note are available in the GigaScience database (GigaDB) [39]. Raw data are available in the SRA database with number SRA492579 and Bioproject accession PRJNA353147.
Competing interests
The authors declare that they have no competing interests.
Author contributions
Supplementary Material
Acknowledgments
G.Z. and Q.L. conceived and supervised the project. P.M. prepared the Chinese crocodile lizard sample. Z.W. and J.G. performed genome assembly, repeat annotation, gene annotation, and gene function annotation. J.G. conducted assessment of assembly quality. J.W. and H.Y. contributed reagents/materials/analysis tools. J.G. drafted the manuscript with contributions from Z.W., Y.Z., F.L., and Z.X. Q.L. revised the manuscript. All of the authors read and approved the final manuscript.
References
- 1. Ahl E. Beitrage zur Lurch und Kriechtierfauna Kwangsi: Section 5, Eidechsen. Sitzungsberichte der Gesellschaft der naturforschenden Freunde zu Berlin 1930;1930:326–31. [Google Scholar]
- 2. van Schingen M, Ihlow F, Nguyen TQ et al. . Potential distribution and effectiveness of the protected area network for the crocodile lizard, Shinisaurus crocodilurus (Reptilia: Squamata: Sauria). Salamandra 2014;50(2):71–76. [Google Scholar]
- 3. Huang H, Wang H, Li L et al. . Genetic diversity and population demography of the Chinese crocodile lizard (Shinisaurus crocodilurus) in China. PLoS One 2014;9(3):e91570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bever GS, Bell CJ, Maisano JA. The ossified braincase and cephalic osteoderms of Shinisaurus crocodilurus (Squamata, Shinisauridae). Palaeonto Electronica 2005;8(1):1–36. [Google Scholar]
- 5. Huang C, Yu H, Wu Z et al. . Population and conservation strategies for the Chinese crocodile lizard (Shinisaurus crocodilurus) in China. Anim Biodivers Conserv 2008;31(2):63–70. [Google Scholar]
- 6. Nguyen TQ, Hamilton P, Ziegler T. Shinisaurus crocodilurus. The IUCN Red List of Threatened Species. 2014. http://dx.doi.org/10.2305/IUCN.UK.2014-1.RLTS.T57287221A57287235.en. (12 July 2017 date last accessed). [Google Scholar]
- 7. Ziegler T, Le KQ, Hendrix R et al. . A comparative study of crocodile lizards (Shinisaurus crocodilurus AHL, 1930) from Vietnam and China. Raffles Bull Zool 2008;56:181–7. [Google Scholar]
- 8. Appendices I, II and III. Convention on International trade in endangered species of wild fauna and flora. 2011. https://cites.org/eng/app/appendices.php (4 April 2017 date last accessed). [DOI] [PubMed]
- 9. Luo R, Liu B, Xie Y et al. . SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 2012;1(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li R, Fan W, Tian G et al. . The sequence and de novo assembly of the giant panda genome. Nature 2010;463(7279):311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song L, Bian C, Luo Y et al. . Draft genome of the Chinese mitten crab, Eriocheir sinensis. Gigascience 2016;5(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Havlak P, Chen R, Durbin KJ et al. . The Atlas Genome Assembly System. Genome Res 2004;14(4):721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simao FA, Waterhouse RM, Ioannidis P et al. . BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015;31(19): 3210–2. [DOI] [PubMed] [Google Scholar]
- 14. Smit A, Hubley R, Green P. RepeatMasker. 1996. http://repeatmasker.org/. (12 July 2017 date last accessed).
- 15. Smit A, Hubley R. RepeatModeler-1.0.5. Institute for Systems Biology.2012. http://www.repeatmasker.org/RepeatModeler/. (12 July 2017 date last accessed). [Google Scholar]
- 16. Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 2007;35(Web Server issue):W265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999;27(2):573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Castoe TA, De Koning AJ, Hall KT et al. . The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A 2013;110(51):20645–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vonk FJ, Casewell NR, Henkel CV et al. . The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci U S A 2013;110(51):20651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Georges A, Li Q, Lian J et al. . High-coverage sequencing and annotated assembly of the genome of the Australian dragon lizard Pogona vitticeps. Gigascience 2015;4(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alföldi J, Di Palma F, Grabherr M et al. . The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011;477(7366):587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res 2002;12(4):656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. She R, Chu JS-C, Wang K et al. . GenBlastA: enabling BLAST to identify homologous gene sequences. Genome Res 2009;19(1):143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res 2004;14(5):988–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stanke M, Keller O, Gunduz I et al. . AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 2006;34(Web Server issue):W435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong Z, Li F, Li Q et al. . Draft genome of the leopard gecko, Eublepharis macularius. Gigascience 2016;5(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 2000;28(1):45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001;17(9):847–8. [DOI] [PubMed] [Google Scholar]
- 29. Corpet F, Servant F, Gouzy J et al. . ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res 2000;28(1):267–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Attwood TK, Bradley P, Flower DR et al. . PRINTS and its automatic supplement, prePRINTS. Nucleic Acids Res 2003;31(1):400–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bateman A, Coin L, Durbin R et al. . The Pfam Protein Families Database. Nucleic Acids Res 2004;32(suppl 1):D138–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 2012;40(D1):D302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mi H, Dong Q, Muruganujan A et al. . PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 2009:38(Database issue):D204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sigrist CJ, Cerutti L, De Castro E et al. . PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 2010;38(suppl 1):D161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ashburner M, Ball CA, Blake JA et al. . Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25(1):25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fry BG, Vidal N, Norman JA et al. . Early evolution of the venom system in lizards and snakes. Nature 2006;439(7076):584–8. [DOI] [PubMed] [Google Scholar]
- 38. Fry BG, Winter K, Norman JA et al. . Functional and structural diversification of the Anguimorpha lizard venom system. Mol Cell Proteomics 2010;9(11):2369–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Q, Wang Z, Zhou Y et al. . The genome of the Chinese crocodile lizard; Shinisaurus crocodilurus. GigaScience Database 2017. http://dx.doi.org/10.5524/100315. [DOI] [PMC free article] [PubMed]
- 40. Green RE, Braun EL, Armstrong J et al. . Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 2014;346(6215):1254449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wan QH, Pan SK, Hu L et al. . Genome analysis and signature discovery for diving and sensory properties of the endangered Chinese alligator. Cell Res 2013;23(9):1091–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alfoldi J, Di Palma F, Grabherr M et al. . The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011;477(7366):587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Minx P, Delehaunty KD, Fronick CC et al. . The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol 2013;14:R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Z, Pascual-Anaya J, Zadissa A et al. . The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet 2013;45(6):701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yin W, Wang ZJ, Li QY et al. . Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat Commun 2016;7:13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y, Zhou Q, Wang Y et al. . Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nat Commun 2015;6:10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.