


Abstract
In human cells APE1 is the major AP endonuclease and it has been reported to have no functional mitochondrial targeting sequence (MTS). We found that APE2 protein possesses a putative MTS. When its N-terminal 15 amino acid residues were fused to the N-terminus of green fluorescent protein and transiently expressed in HeLa cells the fusion protein was localized in the mitochondria. By electron microscopic immunocytochemistry we detected authentic APE2 protein in mitochondria from HeLa cells. Western blotting of the subcellular fraction of HeLa cells revealed most of the APE2 protein to be localized in the nuclei. We found a putative proliferating cell nuclear antigen (PCNA)-binding motif in the C-terminal region of APE2 and showed this motif to be functional by immunoprecipitation and in vitro pull-down binding assays. Laser scanning immunofluorescence microscopy of HeLa cells demonstrated both APE2 and PCNA to form foci in the nucleus and also to be co-localized in some of the foci. The incubation of HeLa cells in HAT medium containing deoxyuridine significantly increased the number of foci in which both molecules were co-localized. Our results suggest that APE2 participates in both nuclear and mitochondrial BER and also that nuclear APE2 functions in the PCNA-dependent BER pathway.
INTRODUCTION
DNA carrying the genetic information of living organisms is known to be attacked by various DNA-damaging agents such as reactive oxygen species (ROS) generated during normal cellular metabolism, ionizing radiation and chemicals from the environment. As a result, various damage occurs in DNA due to alterations in the chemistry of bases, sugars and phosphates (1). Such DNA damage may result in mutagenesis or cell death, because of alterations in the base pairing properties of damaged bases, with the formation of mismatched base pairs, or the consequent inhibition of DNA replication and transcription.
To counteract such deleterious effects of DNA damage, cells are equipped with several types of DNA repair systems. Damaged bases in DNA with relatively small chemical alterations are mainly repaired by the base excision repair (BER) system, which is initiated by excision of damaged bases by specific DNA glycosylases resulting in formation of base loss sites called apurinic or apyrimidinic (AP) sites (2–4). AP sites are also formed by spontaneous base loss.
BER in mammalian cells is classified into two pathways according to the repair patch size, namely short patch and long patch BER; the former replaces 1 nt while the latter replaces oligonucleotides during repair synthesis (2–4). In both pathways incision of DNA 5′ to AP sites is an essential step to generate accessible 3′-OH termini prior to repair synthesis by DNA polymerase and is catalyzed by class II AP endonuclease. In short patch BER, DNA polymerase β removes the 5′-deoxyribose 3′ to the nick due to its deoxyribose phosphatase (dRPase) activity, which generates a 1 nt gap, and then inserts the correct nucleotide; then the nick is sealed by DNA ligase I or III. In long patch BER DNA polymerase δ or ɛ extends the DNA strand from the 3′-OH terminus for several nucleotides and displaces the strand containing the 5′-deoxyribose phosphate (dRP). The resulting flap structure is removed by specific flap endonuclease (FEN1) and DNA ligase I or III joins the last newly incorporated nucleotide to the extant polynucleotide chain.
In human cells APE1 (HAP1/APEX/REF-1) is the major class II AP endonuclease in the nucleus (5–8), which is considered to be involved in both the short patch and long patch BER pathways for nuclear DNA.
On the other hand, eukaryotic cells have mitochondrial DNA (mtDNA) in addition to nuclear DNA. mtDNA is more susceptible to attack by ROS than nuclear DNA since it is located in the vicinity of the mitochondrial respiratory chain, where ROS are continuously generated (9). It has recently been noted that damaged bases in mtDNA appear to be efficiently repaired by BER (10). We have previously demonstrated that human cells possess nuclear and mitochondrial DNA glycosylases for oxidized bases, 8-oxoguanine (8-oxoG) and 2-hydroxyadenine (2-OH-A) or adenine opposite 8-oxoG, which are encoded by alternatively spliced forms of the OGG1 and MYH transcripts (11,12). The human UNG gene encodes both nuclear (UNG2) and mitochondrial (UNG1) forms of uracil DNA glycosylase (13,14). Another human DNA glycosylase encoded by the NTH1 gene was also reported to have the potential to be localized in the mitochondria (15), while thymine-glycol DNA glycosylase activity, which is similar to that of NTH1, was purified from rat mitochondria (16). In addition to these DNA glycosylases, DNA polymerase γ and DNA ligase III are considered to participate in mitochondrial BER (17–19). Although AP endonuclease activities were also detected and partially purified from mitochondria isolated from Xenopus oocytes and a mouse cell line, no gene responsible for them has yet been identified (19,20).
Based on genome databases from various organisms a novel group of AP endonucleases has recently been reported (21–24). Among them APE2 protein is the second human AP endonuclease that has been shown to have a weak class II AP endonuclease activity and was also suggested to be localized in the nuclei.
In the present study we have performed a homology search of various genome databases to retrieve candidates for human mitochondrial AP endonuclease and found APE2 to possess a putative mitochondrial targeting sequence (MTS). We examined the subcellular localization of authentic APE2 protein in cultured human cells and APE2 was thus demonstrated to be localized in both nuclei and in mitochondria. Furthermore, we also showed that APE2 protein forms foci and is associated with PCNA in the nucleus of human cells.
MATERIALS AND METHODS
Nucleotide and amino acid sequence accession numbers
The cDNA and amino acid sequences for APE2 have been deposited in DDBJ/EMBL/GenBank with accession nos ABO049211 and BAB13764, respectively.
Chemicals and others
Recombinant Taq DNA polymerase, restriction enzymes and T4 DNA ligase were all obtained from Takara Shuzo (Kyoto, Japan), New England Biolabs and Toyobo Co. (Osaka, Japan). The DNA ladder (1 kb), prestained protein molecular weight standards, DMEM and fetal bovine serum (FBS) were purchased from Life Technologies. Most of the chemicals were obtained from Wako Pure Chemical Industries (Osaka, Japan) and Sigma-Aldrich. The sources of all other materials are indicated at the appropriate places in the text.
Synthetic oligonucleotides and peptides
Synthetic oligonucleotides used as PCR primers were purchased from Greiner Japan (Tokyo, Japan) and Hokkaido System Science (Sapporo, Japan).
Two synthetic peptides labeled with biotin at their N-termini, wild-type (YF, biotin-SGSGSRGQKNLKSYFQPSPSCPQA) and mutant (AA, biotin-SGSGSRGQKNLKSAAQPSPSCPQA), were obtained from Wako Pure Chemical Industries.
DNA sequencing
The nucleotide sequence was determined using either Dye Terminator or Dye Primer Cycle Sequencing FS Ready Reaction Kits and a model 373A automated DNA sequencer (Perkin Elmer-Cetus) according to the manufacturer’s instructions.
Isolation of APE2 cDNA
We searched the genome database to retrieve candidate genes for novel AP endonucleases using the tBLASTn program (25) and then the MitoProt II program (26) was applied to the hypothetical proteins encoded by the retrieved sequences in order to examine their potential to be located in mitochondria. First, we found a cosmid clone (DDBJ/EMBL/GenBank accession no. Z956290) of Schizosaccharomyces pombe genomic DNA that encodes a hypothetical protein (S.pombe APN2/ETH1, accession no. CAB09119) homologous to human APE1 and Escherichia coli XTH and which possesses a putative MTS in its N-terminal region. Next, using the SpAPN2 amino acid sequence, we retrieved a clone of human genomic DNA sequence (accession no. Z83821). By analyzing the DNA sequence using the SPL program (Weizmann Institute of Science Bioinfomatics Unit) (27) we predicted a gene consisting of six exons encoding a hypothetical human homolog of SpAPN2. Then, once again using the MitoProt II program, we predicted that its N-terminal sequence likely functions as an MTS. cDNA fragments for the hypothetical protein were amplified by PCR from the human cDNA library (human leukemia MATCHMAKER cDNA library, HL4015AB; Clontech Laboratories) using two primers, 5′-2 (5′-GTGAGCTGGAACATCAATGG-3′) and 3′-2 (5′-GTAAACTCATCCATGTTTCC-3′), corresponding to the sequences in the first and third exons of the putative gene in combination with two other primers, GAD2 (5′-GTTTGGAATCACTACAGGGATGT-3′) and GAD3 (5′-CAGTATCTACGATTCATAGATCTG-3′), derived from the vector itself. Amplified cDNA fragments were subcloned into the pT7Blue(R)T vector (Novagen) and sequenced. The cDNA sequence was almost identical to that registered in DDBJ/EMBL/GenBank as APE2/XTH2/APEXL2 (accession nos. AF119046, AJ011311 and AB021260, respectively).
Plasmid construction
To construct pYN3103:TrpE-APE2 encoding the fusion protein TrpE–APE2, an APE2 cDNA fragment encoding amino acids 1–518 was inserted into the PstI and BamHI sites of YN3103:TrpE (28) with the aid of PCR. A NcoI–BamHI fragment prepared from pYN3103:TrpE-APE2 was transferred into the NcoI and BamHI sites of pET3d and thus construct pET3d:TrpE–APE2 was obtained for expression of TrpE–APE2 protein in E.coli. Plasmid pET32a:APE2 encoding the fusion protein TrxA–APE2 was constructed by inserting the APE2 cDNA fragment into the NcoI and BamHI sites of pET32a(+) (Novagen) with the aid of PCR. Plasmid pET3228D:hPCNA was constructed by inserting human PCNA cDNA (amino acids 1–216) from pT7-PCNA (29), a generous gift from Dr T.Tsurimoto (Nara Institute of Advanced Science and Technology, Japan), into the NdeI and HincII sites of pET3228D, in which the lacI gene was deleted from pET32a(+) and its XbaI–BlpI region was replaced with a XbaI–BlpI fragment coding an N-terminal His tag derived from pET28b(+) (Novagen).
The mammalian expression plasmid pcDEBΔ:APE2-HA, encoding APE2 with a C-terminal influenza hemagulutinin epitope (HA) tag, was constructed by inserting APE2 cDNA (amino acids 1–517) fused to a DNA sequence for the HA tag (RAQAYPYDVPDYAIN) with the aid of PCR into the HindIII and BamHI sites of pcDEBΔ carrying the SRα promoter (30). A cDNA fragment encoding enhanced green fluorescent protein (EGFP) was amplified by PCR from pEGFP-N1 (Clontech Laboratories). A cDNA encoding EGFPm, in which the first methionine was substituted by isoleucine, was prepared by PCR to prevent an internal initiation of translation when an MTS was fused to its N-terminus. These cDNA fragments were transferred into the HindIII and BamHI sites of pcDEBΔ to construct pcDEBΔ:EGFP and pcDEBΔ:EGFPm. A cDNA encoding the N-terminal 15 amino acid residues of APE2 and a linker sequence (Ala-Ala-Ala) was prepared by PCR using pcDEBΔ:APE2-HA as template and inserted into the HindIII and NotI sites of pcDEBΔ:EGFPm to construct pcDEBΔ:APE2-EGFPm. A plasmid pcDEBΔ:COX8-EGFPm was prepared by inserting a cDNA encoding the N-terminal 34 amino acid residues of cytochrome c oxidase subunit VIII (31) into the HindIII and NotI sites of pcDEBΔ:EGFPm.
Antibodies
Anti-PCNA mouse monoclonal antibody (PC10) was a product of Santa Cruz Biotechnology. Both highly cross-adsorbed Alexa Fluor 488–goat anti-rabbit IgG (H+L) conjugate and Alexa Fluor 594–goat anti-mouse IgG (H+L) conjugate were purchased from Molecular Probes.
Rabbit polyclonal antibodies against the fusion protein TrpE–APE2 were prepared as previously described (28). The antibodies were purified with the aid of antigen affinity columns (TrpE–APE2–Sepharose and TrpE–Sepharose columns) (28,30). Briefly, antiserum against TrpE–APE2 was loaded onto the TrpE–APE2–Sepharose column and the column was washed with Low E buffer (50 mM sodium phosphate buffer pH 7.6, 0.1% Triton X-100) and 10 mM sodium phosphate buffer (pH 7.6). The bound antibodies were then eluted using buffer E (0.2 M glycine–HCl pH 2.3, 0.1 M NaCl, 0.1% Triton X-100). The eluted fraction was dialyzed against TBS (10 mM Tris–HCl pH 7.5, 0.9% NaCl) and applied to the TrpE–Sepharose column to remove any non-specific antibodies. The flow-through fraction was collected and the purified antibodies were designated anti-APE2.
Pre-adsorption of anti-APE2 was performed as follows. TrxA–APE2 and TrxA proteins were purified with the aid of Ni–NTA–agarose (Qiagen) and then coupled with activated CH-Sepharose 4B (Amersham Pharmacia Biotech) in order to prepare the antigen columns (TrxA–APE2–Sepharose and TrxA–Sepharose). Anti-APE2 (20 µg) was incubated overnight at 4°C with TrxA–APE2–Sepharose or TrxA–Sepharose (1 mg antigen equiv.) in 2 ml of phosphate-buffered saline (PBS) (pH 7.4) containing 0.1% bovine serum albumin (BSA), with gentle shaking. Pre-adsorbed anti-APE2 was recovered and used for western blotting and immunocytochemistry.
RT–PCR analysis
RT–PCR for APE2 mRNA was performed as follows. Total cellular RNA was prepared as previously described (32). First strand cDNA, prepared using First-Strand cDNA Synthesis Kits (Amersham Pharmacia Biotech) according to the manufacturer’s instructions, was subjected to PCR. PCR was performed in 20 µl of a reaction mixture containing 10 mM Tris–HCl pH 8.3, 50 mM KCl and 1.5 mM MgCl2, 0.5 µl of the first strand cDNA, 0.4 U recombinant Taq DNA polymerase, 0.2 µM each primer and 200 µM each deoxynucleoside triphosphate. The initial denaturation was performed at 95°C for 2 min and amplification was performed by 30 cycles of denaturation at 95°C for 20 s, annealing at 55°C for 20 s and extension at 72°C for 40 s, followed by an additional extension at 72°C for 5 min.
Cell cultures, DNA transfection and subcellular fractionation
HeLa MR cells (33) were maintained in DMEM supplemented with 100 µg/ml streptomycin, 100 U/ml penicillin and 10% FBS. Transfection of plasmid DNA into HeLa MR cells was performed with LipofectAmine Reagent (Life Technologies) according to the manufacturer’s instructions. To establish stable cell lines transfectants were selected in medium containing 150 µg/ml hygromycin B and the colonies obtained were replated twice for purification and then were maintained in medium containing 100 µg/ml hygromycin B. Stable cell lines transfected with pcDEBΔ and pcDEBΔ:APE2-HA were designated HeLa MRV and HeLa MR:APE2-HA, respectively.
Exponentially growing cells were harvested by treatment with 0.25% trypsin/0.02% EDTA (Cosmo Bio Co., Japan), washed with PBS three times and subjected to either immunoprecipitation or peptide pull-down.
Preparations of nuclear, crude cytoplasmic and mitochondrial fractions were performed as previously described by Ishibashi et al. (33) and Kang et al. (34).
Western blotting
Protein samples were separated by SDS–PAGE and transferred to Immobilon-P membrane (Millipore Inc.) for western blot analysis. Blocking of the membranes was performed by incubation for 1 h at room temperature in TBST (10 mM Tris–HCl pH 7.5, 0.9% NaCl, 0.1% Tween-20) containing 5% non-fat dried milk. The membranes were incubated in TBST containing 2.0 µg/ml anti-APE2 or 2.5 µg/ml anti-PCNA overnight at 4°C, with gentle shaking. The membranes were washed with TBST and incubated in TBST containing 0.01% horseradish peroxidase-labeled protein A (Amersham Pharmacia Biotech) for 1 h at room temperature. After washing with TBST the antibodies bound to each blot were detected by the chemiluminescence method with an ECL-Plus Kit (Amersham Pharmacia Biotech). Digitized images were obtained with a LAS1000 Plus (Fuji Film, Tokyo, Japan) and processed for publication using the Adobe Photoshop 5.5J software package (Adobe Systems).
Immunoprecipitation
All procedures were performed at 4°C. HeLa MRV or HeLa MR:APE2-HA cells (2.7 × 108 cells) were suspended in 5 ml of lysis buffer containing 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 0.1% Triton X-100, 2.5 mM MgCl2, 1 mM phenylmethanesulfonyl fluoride (PMSF), 1 µg/ml leupeptin, 1 µg/ml pepstatin A and 1 µg/ml chymostatin and disrupted by sonication. Cell lysates were passed through 21 gauge needles several times and centrifuged at 15 000 g for 20 min to remove any cell debris. Then 0.5 ml of the supernatant was incubated for 1.5 h with 10 µl of a 50% suspension of protein A–Sepharose (Amersham Pharmacia Biotech) in lysis buffer and centrifuged. The supernatant was incubated with 3 µg anti-PCNA or anti-β-galactosidase (Promega) for 1.5 h with gentle shaking, then 10 µl of a 50% suspension of protein A–Sepharose was added and incubated further for 1.5 h. After centrifugation at 200 g for 1 min the protein A–Sepharose was washed four times with lysis buffer. The proteins bound to protein A–Sepharose were subjected to SDS–PAGE and western blot analysis with anti-APE2 or anti-PCNA.
His tag protein pull-down binding assays
BL21(DE3) E.coli cells carrying plasmid pET32a(+), pET3228D:PCNA or pET32a(+):APE2, which express His tagged proteins TrxA, His–PCNA and TrxA–APE2, respectively, were cultured in 250 ml of M9ZB medium (28) until the OD600 reached 0.6 and then incubated with 1 mM isopropyl β-d-thiogalactoside for a further 2 h. Cells were harvested by centrifugation, resuspended in 25 ml of lysis buffer [50 mM sodium phosphate buffer pH 7.0, 300 mM NaCl, 1% Triton X-100, 8 M urea, 1 mM 2-mercaptoethanol (2-ME), 1 mM PMSF, 1 µg/ml leupeptin, 1 µg/ml pepstatin A and 1 µg/ml chymostatin] and disrupted by sonication. Triton X-100 and urea were omitted from the lysis buffer for PNCA extraction. Cell lysates were clarified by centrifugation at 100 000 g for 20 min at room temperature. Two milliliters of a 50% suspension of TALON superflow metal affinity resin (Clontech Laboratories) equilibrated with lysis buffer was mixed with each cell lysate and gently rotated for 1 h. Then the resin was washed five times with binding buffer (20 mM sodium phosphate buffer pH 7.4, 0.1% BSA) containing 50 mM NaCl and used for pull-down binding assays. Purity and amount of protein fixed on each resin were determined by SDS–PAGE with a combination of GelCode Blue Stain Reagent (Pierce Chemical Co.) and BSA as the standard. Nearly homogeneous preparations of TrxA (1.2 µg/µl bed volume) and His–PCNA (1.3 µg/µl) were immobilized, while ~30% of the fixed TrxA–APE2 (1.6 µg/µl) remained intact.
HeLa MR:APE2-HA cells (4.0 × 108 cells) were sonicated in 20 ml of binding buffer containing 1 mM 2-ME, 1 mM PMSF, 1 µg/ml leupeptin, 1 µg/ml pepstatin A and 1 µg/ml chymostatin. After centrifugation at 100 000 g for 20 min at 4°C, 1 ml of the supernatant was incubated with 100 µl of a 10% suspension of TrxA resin or His–PCNA resin for 1 h at 4°C, with gentle shaking and the resin was washed three times with binding buffer. The proteins bound to 2 µl of each resin (bed volume) were subjected to western blotting with anti-APE2.
Purified recombinant human PCNA protein (300 ng), a generous gift from Dr T. Tsurimoto, was incubated with 100 µl of a 10% suspension of TrxA or TrxA–APE2 resin in 1 ml of binding buffer containing 50, 150 or 500 mM NaCl for 1 h at 4°C. The proteins bound to each resin were subjected to western blotting with anti-PCNA, as described above.
Peptide pull-down binding assay
Whole cell extract from HeLa MRV cells was prepared as described above. One nanomole of wild-type (YF) or mutant (AA) peptide were mixed with 10 µl of 50% streptavidin–agarose (Life Technologies) in 1 ml of lysis buffer and rotated for 1 h at room temperature. The agarose recovered by centrifugation at 200 g for 1 min was mixed with 1.0 ml of cell extract and incubated for 1 h at 4°C with gentle shaking. After centrifugation at 200 g for 1 min the agarose was washed four times with lysis buffer and subjected to SDS–PAGE and western blotting with anti-PCNA.
Laser scanning fluorescence microscopy
HeLa MRV or HeLa MR:APE2-HA cells were plated in an 8-well collagen I-coated chamber slide (Becton Dickinson) and cultured for 48 h. Six hours prior to fixation, HAT (100 µM hypoxanthine, 10 µM methotrexate and 5 µM thymidine) and 1 mM deoxyuridine (dUrd) were added to the culture when necessary. The cells were washed in PBS and fixed with methanol/acetone (1:1) for 10 min at room temperature. The slides were washed in PBS and incubated in PBS containing 0.1% Tween-20, 3% BSA and 10% normal goat serum (Vector Laboratories) for 1 h at 37°C in a humidified atmosphere. Next, the slides were incubated with anti-APE2 overnight at 4°C and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG for 1 h at room temperature. The slides were further incubated for 1 h with anti-PCNA and also with Alexa Fluor 594-conjugated goat anti-mouse IgG for an additional 1 h at room temperature. All antibodies were used at a concentration of 2.0 µg/ml and the slides were washed with PBS containing 0.1% Tween-20 after each incubation with antibodies. For nuclear staining the slides were incubated in 2.4 nM TOTO-3 (Molecular Probes) for 20 min. Washed slides were mounted and observed under an Axiovert 135M (Carl Zeiss Co.) equipped with a Radiance (2000) laser scanning fluorescence microscope system (Bio-Rad Laboratories). Digitized images were obtained and processed for publication using Adobe Photoshop 5.5J.
To observe living HeLa MR cells expressing fused EGFP proteins the cells were plated in a collagen I-coated chamber and cultured for 24 h. The cells were transfected with pcDEBΔ:EGFP, pcDEBΔ:APE2-EGFPm or pcDEBΔ:COX8-EGFPm for transient expression of fused EGFP and cultured for an additional 24 h. The cells were incubated for 1 h in the presence of 2 µM MitoTracker Red CM-H2XRos (Molecular Probes) and fluorescence signals in living cells were observed as described above with a long distance objective lens (LAD-Plan 40×Ph2; Carl Zeiss Co.).
Electron microscopic immunocytochemistry
Thin sections of isolated mitochondria were prepared and processed for electron microscopic immunocytochemistry as previously described (34). Briefly, mitochondrial fractions purified from HeLa MRV and HeLa MR:APE2-HA cells were fixed in PBS containing 8% paraformaldehyde and embedded in LR White resin (London Resin Co.) at 50°C. Thin sections were prepared from these samples and treated with anti-APE2 and protein A–gold (EY Laboratories). Gold signals were observed using electron microscopy.
The statistical analysis
We performed the statistical analysis using StatView v.5.0 (SAS Institute).
RESULTS
Human APE2 protein is a member of the novel AP endonuclease family with a MTS and PCNA-binding motif
Alignment of APE2 cDNA and human genomic clone Z83821 revealed that the human APE2 gene is located between the 5-aminolevulinate synthase (ALAS2) and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 (PFKFB2) genes on the X chromosome (Xp11.21) and consists of six exons. The APE2 protein consists of 518 amino acids, whose N-terminal region (amino acids 1–313) is homologous to the XTH-like AP endonuclease family, including E.coli XTH and human APE1 (Fig. 1A).
Figure 1.

APE2 and other members of the APE2/APN2 family. (A) Structures of APE2/APN2 family proteins, E.coli XTH and human APE1 are shown. The regions conserved in the XTH family (hatched box) and the unique C-terminal region of the APE2/APN2 family are indicated. Colored boxes indicate the three unique subregions found in the APE2/APN2 family. Red, mitochondrial targeting sequence (MTS); blue, PCNA-binding motif; green, TOP3 homologous subregion. (B) Predicted MTS in APE2 protein and comparison of the MTS found in APE2/APN2 family proteins. The MitoProt II program predicted that the first 18 residues of APE2 protein form an amphipathic helix with three arginine residues but no acidic residue, as shown in the left panel. No putative cleavage site was predicted. Basic residues are shown in circles and hydrophobic residues are in boxes. Each value for µH and Hmax calculated for COX8 and various repair enzymes, including the APE2/APN2 family present in mitochondria, by the MitoProt II program are plotted on the right. Red circles indicate the APE2/APN2 family proteins. Green, blue and black triangles show UNG1 (13), MYHα3 (12,43) and OGG1-2a (11), respectively. Green and blue squares show DNA polymerase γ (Polγ) (17) and DNA ligase III (LigIII) (18), respectively. An inverted triangle and a rhombus show MTH1d (accession no. BAA83791) and COX8 (31), respectively. The thick and thin lines indicate the upper and lower cut values of µH and Hmax, respectively. (C) Conserved PCNA-binding motifs in the APE2/APN2 family are shown and compared with those of other BER-related proteins and p21/Cip1/Waf1, which were pointed out by Warbrick (35), as well as putative motifs in OGG1 (11) and MYH (accession no. AAC50618). The first glutamine residue in the consensus is altered to an asparagine residue in putative motifs found in ScAPN2 and OGG1. In the motif consensus h indicates residues with moderately hydrophobic side chains, such as leucine, isoleucine or methionine, and a indicates residues with highly hydrophobic aromatic side chains, e.g. phenylalanine and tyrosine. (D) TOP3 homologous sequences of the APE2/APN2 family are aligned with the C-terminal repeats found in human TOP3α protein (accession no. Q13427).
We retrieved several candidate genes for novel AP endonucleases homologous to E.coli XTH and human APE1 from nucleotide sequence databases. Application of the MitoProt II program (26) revealed that human APE2 protein, S.pombe APN2/ETH1 protein (SpAPN2) and Arabidopsis thaliana APN2 like protein (AtAPN2, accession no. CAA18499) possess putative MTS at their N-termini (Fig. 1B). We predicted that the MTS in APE2 consists of residues 1–18 at the N-terminal region and may not be processed after being translocated into mitochondria, as shown in Figure 1B.
The C-terminal region (amino acids 314–518) of APE2 is not homologous to XTH or APE1, however, this C-terminal region is partially homologous to the corresponding regions of Saccharomyces cerevisiae APN2/ETH1 protein (ScAPN2, accession no. CAB09119), SpAPN2 and AtAPN2 (Fig. 1A). We found a putative PCNA-binding motif (Qxxhxxaa) in this C-terminal region of APE2 (Fig. 1C; 35,36). Such motifs are also found in the same region of the other three APE2/APN2 family proteins. The glutamine residue is substituted by an asparagine residue in ScAPN2, however, the same substitution has also been reported in S.pombe RFC-1 protein (37), thus indicating that the glutamine residue in the PCNA-binding motif can be substituted by an asparagine residue.
There is another subregion conserved in the C-terminal regions of all APE2/APN2 family proteins. This subregion contains a sequence homologous to a tandem repeat sequence found in the C-terminal region of DNA topoisomerase III (TOP3) family proteins (Fig. 1A and D)
Expression of the APE2 gene and APE2 protein in cultured human cells and human tissues
Expression of APE2 mRNA in human cells was confirmed by amplifying a 347 bp cDNA fragment corresponding to the APE2 coding region (nt 13–359) from total RNA prepared from HeLa and Jurkat cells and human tissues by RT–PCR (Fig. 2A). Substantial levels of expression were also detected in samples from adult kidney and brain, but the level was much less in fetal brain.
Figure 2.

Expression of APE2 mRNA and APE2 protein in cultured human cells and human tissues. (A) RT–PCR for APE2 mRNA. A 347 bp cDNA fragment corresponding to the APE2 coding region (nt 13–359) was amplified from total RNA prepared from HeLa and Jurkat cells and human tissues by RT–PCR. Human β-actin mRNA was amplified as an internal control. (B) Immunological detection of APE2 polypeptide in cultured human cells. Authentic and recombinant APE2 proteins were examined by western blot analysis using anti-APE2 pre-adsorbed with TrxA–APE2–Sepharose or TrxA–Sepharose. Whole cell lysate of HeLa MRV (5.0 × 105 cells in lane 1) or HeLa MR:APE2-HA (1.0 and 0.25 × 105 cells in lanes 2 and 3, respectively) were subjected to the analysis. (C) Whole cell lysate (lane W), crude isolated nuclei (lane N) and crude cytoplasmic fraction (lane C) prepared from 5.0 × 105 HeLa MRV cells were subjected to western blotting with anti-APE2.
For immunological detection of human APE2 protein, rabbit polyclonal antibodies (anti-APE2) were raised against a TrpE–APE2 fusion protein and purified using an antigen affinity column. The anti-APE2 specifically detected 0.1 ng of TrxA–APE2 fusion protein in western blot analyses (data not shown). Whole cell extracts prepared from HeLa MRV and HeLa MR:APE2-HA cells expressing HA tagged APE2 protein (532 amino acids, calculated molecular weight 59 103) were subjected to western blotting. A single 60 kDa polypeptide (p60) was detected in the sample from HeLa MRV cells and its size is close to the predicted molecular weight of APE2 protein (518 amino acids, 57 363) (Fig. 2B). A 62 kDa polypeptide (p62), corresponding to the recombinant fusion protein APE2–HA, was detected in a sample from HeLa MR:APE2-HA cells and its amount was estimated to be 10 times higher than that of authentic p60. To confirm that both p60 and p62 are specifically detected by anti-APE2 we used anti-APE2 pre-adsorbed to TrxA–APE2–Sepharose in western blot analysis. With this preparation neither p60 nor p62 was detected on the blot, which means that these peptides are authentic APE2 (p60) and recombinant APE2–HA (p62). Using purified recombinant TrxA–APE2 as the quantitative standard in western blot analysis we estimated that a single cell of HeLa MRV contains 3.2 × 104 molecules of authentic APE2 (data not shown).
Isolated nuclei and a cytoplasmic fraction prepared from HeLa MRV cells were subjected to western blot analysis with anti-APE2. Most APE2 protein was recovered in the nuclear fraction and a band corresponding to p60 was barely detected in the cytoplasmic fraction (Fig. 2C).
Mitochondrial localization of APE2
To examine whether the N-terminal sequence of APE2 functions as a MTS a plasmid encoding the 15 amino acid leader sequence from APE2 fused to EGFP (APE2–EGFPm) was constructed and introduced into HeLa MR cells. A plasmid encoding EGFP fused to the MTS from cytochrome c oxidase subunit VIII (38) (COX8–EGFPm) was also introduced into HeLa MR cells. Living cells incubated in the presence of a mitochondrial fluorescent marker, MitoTracker, were subjected to laser scanning fluorescence microscopy (Fig. 3A). Green fluorescence was evenly distributed in cells expressing EGFP itself and there was no apparent localization of EGFP in the mitochondria based on the findings with MitoTracker (Fig. 3Ac, Af and Ai). However, an exclusive mitochondrial localization of green fluorescence, mostly merged with MitoTracker, was evident in cells expressing COX8–EGFPm (Fig. 3Ab, Ae and Ah), thus indicating that EGFP can be targeted to the mitochondria by fusion with a functional MTS. As seen in Figure 3Aa, in cells expressing APE2–EGFPm a relatively intense degree of fibrous fluorescence was observed in the cytoplasm, which was co-localized with MitoTracker and, in addition, a weak fluorescence was seen to be evenly distributed in both the nucleus and cytoplasm (Fig. 3Ad and Ag). These results indicate that the first 15 amino acid sequence of human APE2 protein functions as an MTS but its potential is relatively lower than that of the MTS from COX8. We also analyzed the 20 amino acid leader sequence from APE2 protein, but it had a much lower MTS potential than the 15 amino acid sequence (data not shown).
Figure 3.

Mitochondrial localization of APE2. (A) The N-terminal 15 amino acid sequence of APE2 functions as a mitochondrial targeting sequence. Laser scanning fluorescence microscopy of cells expressing APE2–EGFPm (a, d and g), COX8–EGFPm (b, e and h) or EGFP (c, f and i) were performed after incubation with MitoTracker Red CM-H2Ros for 1 h. Signals for EGFP and MitoTracker are shown in green (a–c) and red (d–f), respectively. In merged images (g–i) the co-localized signals for EGFP and MitoTracker are shown in yellow. (B) Submitochondrial localization of human APE2 protein, determined by electron microscopic immunocytochemistry. Mitochondria were prepared from HeLa MRV (a and b) and HeLa MR:APE2-HA (c and d) cells. APE2 signals were examined by electron microscopic immunocytochemistry, using anti-APE2 pre-adsorbed to TrxA (a and c) or TrxA–APE2–Sepharose (b and d) in combination with protein A–gold. A signal for authentic APE2 is shown with an arrowhead in (a). (C) Distribution of APE2 signals in a mitochondrial section determined by electron microscopic immunocytochemistry. APE2 signals detected in each mitochondrial section as seen in (B) were counted for 10 sections in each sample prepared from HeLa MRV (solid column and open column) or HeLa MR:APE2-HA (gray and hatched columns), which were treated with anti-APE2 pre-adsorbed to TrxA–Sepharose (solid and gray columns) or TrxA–APE2–Sepharose (open and hatched columns) and the number of sections (y-axis) with a given number of gold signals in each section (x-axis) are shown in the histogram.
We next prepared thin sections from fixed mitochondria isolated from HeLa MRV and HeLa MR:APE2-HA cells and the mitochondrial localizations of authentic APE2 and recombinant APE2–HA were analyzed by electron microscopic immunocytochemistry, using anti-APE2 in combination with protein A–gold (Fig. 3B). In each of the mitochondrial sections from the HeLa MRV cells one or two specific signals were observed adjacent to the low electron-dense areas (Fig. 3Ba), which correspond to the inner membrane of mitochondria. An apparently increased number of gold signals were detected in sections from HeLa MR:APE2-HA cells (Fig. 3Bc). When anti-APE2 pre-adsorbed to TrxA–APE2–Sepharose was applied to the sections no signal was observed (Fig. 3Bb and Bd). We randomly picked 10 sections from each sample shown in Figure 3B and the number of gold signals in each section were counted and are shown as a histogram (Fig. 3C). From the information in the histogram we conclude that the gold signals in mitochondria from HeLa MR and HeLa MR:APE2-HA cells represent authentic APE2 and recombinant APE2–HA, respectively.
APE2 protein forms foci in the nucleus of human cells
The intracellular distribution of APE2 protein was examined by laser scanning fluorescence microscopy, using anti-APE2 and Alexa Fluor 488-labeled second antibody. In HeLa MRV cells green fluorescence signals were detected mostly in the nuclei as multiple foci and a fluorescent signal was barely seen in the cytoplasm (Fig. 4Aa). A similar but apparently increased number of fluorescent foci were observed in the nuclei of HeLa MRV:APE2-HA cells and the fluorescence in the cytoplasm also increased (Fig. 4Ac). The signals detected in both cells were completely diminished by pre-adsorption of anti-APE2 to TrxA–APE2–Sepharose (Fig. 4Ab and Ad).
Figure 4.
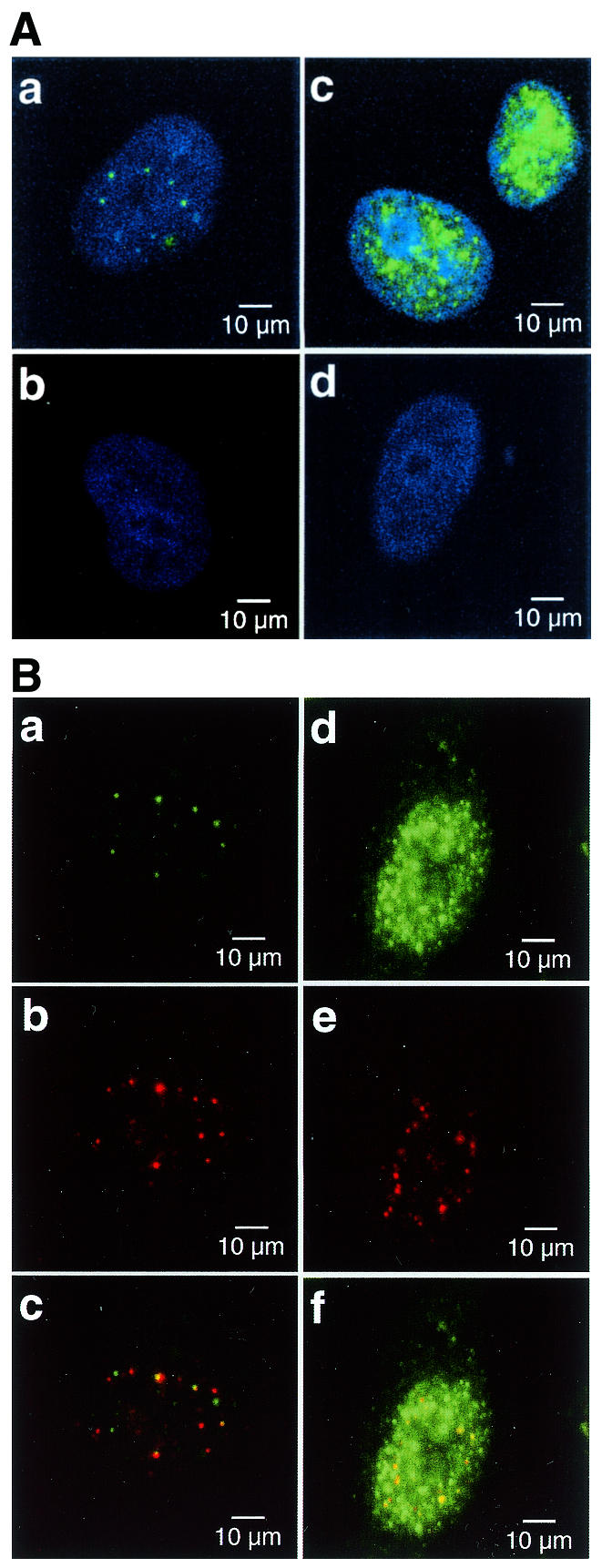
Distribution of APE2 protein in the nuclei of human cells. (A) APE2 forms nuclear foci in cultured human cells. APE2 protein in HeLa MRV (a and b) and HeLa MR:APE2-HA cells (c and d) were examined by laser scanning fluorescence microscopy, using anti-APE2 pre-adsorbed to TrxA–Sepharose (a and c) or TrxA–APE2–Sepharose (b and d), in combination with Alexa Fluor 488-labeled second antibody. Nuclei were counterstained with TOTO-3 (a–d). Alexa Fluor 488 and TOTO-3 signals are shown in green and blue, respectively. (B) Co-localization of APE2 and PCNA in nuclear foci. HeLa MRV (a–c) and HeLa MR:APE2-HA (d–f) cells were immunostained with anti-APE2/Alexa Fluor 488-labeled second antibody and anti-PCNA/Alexa Fluor 594-labeled second antibody and were subjected to laser scanning fluorescence microscopy. APE2 and PCNA signals are shown in green (a and d) and red (b and e), respectively. In merged images in (c) and (f) co-localized signals are shown in yellow.
APE2 and PCNA are partly co-localized in nuclear foci
We expected that APE2 protein might be associated with PCNA in the nucleus because APE2 has a putative PCNA-binding motif (Fig. 1C). We thus examined whether APE2 is co-localized with PCNA in the nuclei of human cells. HeLa MRV cells fixed in methanol/acetone were incubated with anti-APE2/Alexa Fluor 488-labeled second antibody and anti-PCNA/Alexa Fluor 594-labeled second antibody. The localization of APE2 and PCNA in the nuclei was analyzed by laser scanning fluorescence microscopy. Red fluorescence signals for PCNA were exclusively detected in the nuclei of HeLa MRV cells as multiple foci, as well as APE2 foci shown by green fluorescence. About one-tenth of the APE2 foci were co-localized with the PCNA foci (Fig. 4Ba–Bc). On the other hand, most of the PCNA foci were co-localized with APE2 signals in the nuclei of HeLa MR:APE2-HA cells, thus reflecting overexpression of APE2–HA (Fig. 4Bd–Bf).
Molecular interaction between APE2 and PCNA
To obtain direct evidence for the association of APE2 with PCNA whole cell extracts prepared from HeLa MRV and HeLa MR:APE2-HA cells were subjected to immunoprecipitation with anti-PCNA and co-precipitation of APE2 protein in the immune complex was examined by western blotting (Fig. 5A). A 60 kDa polypeptide corresponding to authentic APE2 protein was barely detectable in a sample precipitated from HeLa MRV cell lysate, but a substantial amount of p62 corresponding to recombinant APE2–HA protein was co-precipitated by anti-PCNA from HeLa MR:APE2-HA cell lysate. We repeated the same experiment six times and obtained similar results as shown in Figure 5A in three independent experiments. However, no apparent co-precipitation of APE2 was seen in the other three experiments. We also tried to co-precipitate PCNA with APE2 by anti-APE2, but no PCNA was detectable in the samples from HeLa MRV or HeLa MR:APE2-HA cells (data not shown).
Figure 5.

Molecular interaction of APE2 and PCNA. (A) Co-immunoprecipitation of APE2 with anti-PCNA. Whole cell extracts from HeLa MRV and HeLa MR:APE2-HA cells were incubated with anti-PCNA or anti-β-galactosidase in combination with protein A–agarose for immunoprecipitation. APE2 and PCNA antigens in the immune complex were examined by western blotting with anti-APE2 (upper) and anti-PCNA (lower). (B) Interaction of recombinant PCNA immobilized on resin with APE2–HA protein expressed in HeLa MR:APE2-HA cells. His–PCNA and TrxA proteins immobilized on TALON resin were incubated with whole cell extract prepared from HeLa MR:APE2-HA cells. APE2–HA bound to each resin (2 µl bed volume with 2.6 µg His–PCNA or 2.4 µg TrxA, respectively) were detected by western blotting with anti-APE2. (C) In vitro interaction of TrxA–APE2 protein immobilized on resin and purified recombinant PCNA. TrxA–APE2 and TrxA immobilized on TALON resin (10 µl bed volume with 4.8 µg intact TrxA–APE2 or 12 µg TrxA, respectively) were incubated with 300 ng purified recombinant PCNA (rPCNA) in binding buffer containing 50, 150 or 500 mM NaCl. PCNA bound to 2 µl of each resin was subjected to western blotting. (D) Peptides with the sequence of the PCNA-binding motif found in APE2 interact with PCNA. Whole cell extracts prepared from HeLa MRV cells were incubated with synthetic peptides carrying the PCNA-binding motif in human APE2, wild-type (YF, biotin-SGSGSRGQKNLKSYFQPSPSCPQA) or mutant (AA, biotin-SGSGSRGQKNLKSAAQPSPSCPQA), and the complex was recovered with the aid of streptavidin–agarose.
To further confirm the direct interaction between PCNA and APE2 proteins we performed in vitro pull-down binding assays using immobilized PCNA or TrxA–APE2, respectively. First, PCNA or TrxA protein immobilized on TALON resin was incubated with whole cell extract prepared from HeLa MR:APE2-HA cells and APE2–HA protein bound to each resin was quantified by western blotting. Nearly 50% of the input APE2–HA bound to the PCNA resin, while there was no detectable binding of APE2–HA protein to the TrxA resin (Fig. 5B). Secondly, TrxA–APE2 resin was incubated with a purified preparation of recombinant PCNA. As shown in Figure 5C, a substantial amount of PCNA bound to the resin and the interaction of PCNA with TrxA–APE2 was apparently increased in the presence of 500 mM NaCl. Again, there was no detectable binding of PCNA to the TrxA resin. These results indicate that human APE2 protein specifically binds to PCNA.
To demonstrate that the putative PCNA-binding motif found in APE2 protein is functional by itself we prepared two synthetic biotin-labeled peptides, with a wild-type sequence (YF) or a mutant sequence (AA), corresponding to the PCNA-binding motif of APE2. In mutant (AA) the tyrosine and phenylalanine residues in the wild-type sequence were replaced by alanine residues. Whole cell extract from HeLa MRV was incubated with wild-type or mutant peptide conjugated to streptavidin–agarose and bound fractions were subjected to western blotting using anti-PCNA. As shown in Figure 5D, a substantial amount of PCNA was detected in a sample pulled down with the wild-type peptide but not with the mutant peptide. As a result, the PCNA-binding motif of APE2 is considered to be functional.
Misincorporation of uracil in nuclear DNA increases association of APE2 and PCNA in the nuclear foci
Supplemention of the culture medium with HAT and dUrd was reported by Ingraham et al. (39) to dramatically elevate the dUTP content in hamster lung fibroblasts. They assumed that the elevated dUTP level increased uracil misincorporation during genomic DNA replication since DNA fragmentation was observed in cells treated with HAT and dUrd. It has been established that misincorporated uracil is repaired via post-replicative BER (40,41). We thus added subtoxic doses of these supplements to the culture medium of HeLa MRV cells to induce post-replicative BER. After incubation in the medium with or without the supplements for 6 h, HeLa MRV cells were fixed and subjected to laser scanning fluorescence microscopy, as described above (Fig. 6). The number of nuclear foci doubly labeled with both green and red fluorescence, which are shown in yellow in Figure 6, apparently increased in cells incubated in the presence of HAT and dUrd compared to the cells incubated in the absence of the supplements. The number of APE2 foci, PCNA foci and dual positive foci, in which APE2 and PCNA were co-localized, was counted in 40 randomly selected cells incubated with or without the supplements and the percentages of dual positive foci for each protein were also calculated (Table 1). The number of dual positive foci but not that of all the foci of each protein increased 3-fold in cells treated with HAT and dUrd in comparison to the cells without treatment and a statistical analysis demonstrated the increase to be significant (Fisher’s exact probability test, P < 0.0001) (Table 1).
Figure 6.
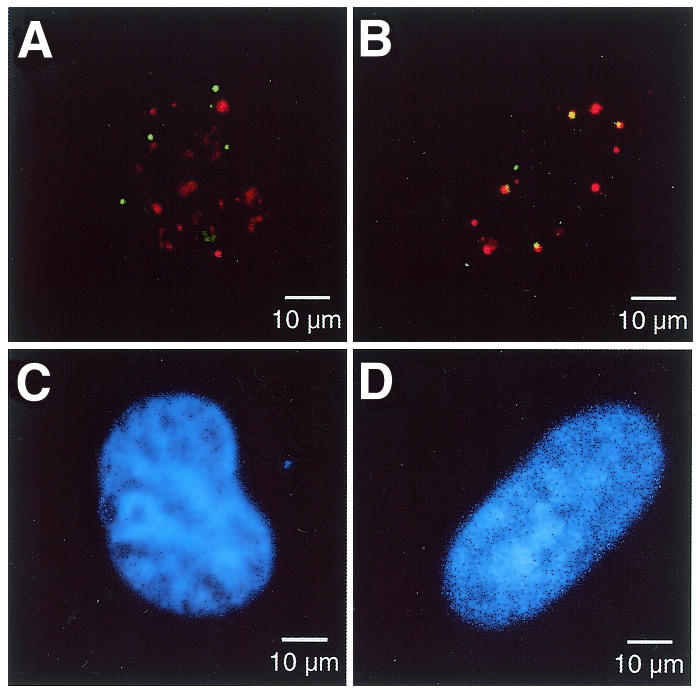
Increased misincorporation of dUMP into DNA promotes the association of APE2 and PCNA in nuclear foci. HeLa MRV cells were incubated in medium supplemented with (B and D) or without (A and C) HAT and dUrd for 6 h. The cells were immunostained with anti-APE2/Alexa Fluor 488-labeled second antibody and anti-PCNA/Alexa Fluor 594-labeled second antibody and were subjected to laser scanning fluorescence microscopy. The APE2 and PCNA signals are shown in green and red, respectively, and the co-localized signals are shown in yellow (A and B). The nuclei were counterstained with TOTO-3 (C and D).
Table 1. Treatment with HAT and dUrd significantly increased the co-localization of APE2 and PCNA in the nuclear foci.
| Foci/40 cells | Supplement | Fisher’s exact probability | |
| |
None |
HAT+dUrd |
|
| APE2 single positive foci | 147 | 110 | |
| Dual positive foci | 16 (9.8%)a | 46 (29.5%) | P < 0.0001 |
| Total | 163 | 156 | |
| PCNA single positive foci | 181 | 147 | |
| Dual positive foci | 16 (8.1%) | 46 (23.8%) | P < 0.0001 |
| Total | 197 | 193 |
aPercentages of dual positive foci for each protein.
DISCUSSION
In the present study we detected APE2 protein in cultured human cells. This is the first time that endogenous APE2 protein has been demonstrated. We examined its subcellular localization, in relation to the putative MTS and PCNA-binding motif, and came to the two major conclusions described below. Most human APE2 protein is localized in the nuclei, but some is in the mitochondria and such a distribution is partly determined by the N-terminal MTS, thus suggesting that APE2 plays an important role in both nuclear and mitochondrial BER. Furthermore, APE2 forms nuclear foci in human cells and some of them are associated with PCNA. Pull-down binding assays indicated that APE2 directly binds PCNA in vitro and their association is likely to be mediated by the PCNA-binding motif present in the C-terminal region of APE2. We thus propose the existence of a post-replicative BER complex in which APE2 plays a crucial role in a PCNA-dependent manner. In vitro pull-down binding assays demonstrated that the interaction between APE2 and PCNA is rather stable even in the presence of 500 mM NaCl. However, co-immunoprecipitation of authentic APE2 by anti-PCNA was not quantitative and co-immunoprecipitation of PCNA by anti-APE2 was hardly achieved. It is likely that both anti-PCNA and anti-APE2 may interfere with the interaction between PCNA and APE2, thus destabilizing the complex.
It has been believed for a long time that mitochondria have no functional DNA repair system for damage in its genome, since Clayton et al. (42) reported that pyrimidine dimers in mammalian mtDNA are not repaired. However, recent studies by several groups have shown the existence of several repair enzymes in mitochondria, especially those involved in BER (10–16,18–20,43). In BER an incision of the DNA strand by a class II AP endonuclease following base excision by DNA glycosylase is an essential step in the initiation of DNA repair synthesis by DNA polymerase. However, APE1, the major AP endonuclease of human cells, has no MTS and a APE1–EGFP fusion protein has been reported not to be localized in the mitochondria (15), thus indicating that APE1 is not involved in BER in the mitochondria. Tomkinson et al. (20) partially purified AP endonuclease activities from the mitochondria of a mouse cell line and reported that an ∼65 kDa peptide in the purified fraction cross-reacted with polyclonal anti-APE1 antibodies. Fung et al. (44) also reported co-localization of immunofluorescent signals with polyclonal anti-APE1 antibodies and anti-mitochondrial protein in a human cell line. These results suggest that a protein(s) sharing an epitope(s) with APE1 protein is (are) present in the mitochondria of human cells.
We demonstrated that the N-terminal 15 amino acid sequence of APE2 protein is a functional MTS but its potential is relatively weaker than that of COX8 and that endogenous and recombinant APE2 proteins are localized in the mitochondria by electron microscopic immunocytochemistry. We also isolated a cDNA encoding mouse APE2 protein and found that mouse APE2 protein has over 20% identity to APE1 and its estimated molecular weight is 57 352 (our unpublished data). The 65 kDa polypeptide reacting with polyclonal anti-APE1 antibodies present in the mitochondrial AP endonuclease fraction from mouse cells (20) may be mouse APE2.
Human APE2 protein was detected adjacent to the mitochondrial inner membrane by electron microscopic immunocytochemistry. We previously reported that human OGG1 and MYH are also localized adjacent to the mitochondrial inner membrane (11,12). It is likely that a repair complex containing the entire machinery essential for BER is associated with the inner membrane and mitochondrial DNAs as expected for the two DNA glycosylases and APE2. As a result, efficient repair by the complex thus probably helps to maintain the functions and the integrity of mtDNA, even if threatened by attack of ROS produced during oxidative phosphorylation.
Most APE2 proteins are localized as foci in the nuclei of human cells. Although APE2 protein has no typical nuclear localization signal, an NNCN analysis using the PSORT II program was able to predict its nuclear localization because of its amino acid content (reliability 70.6) (45). Hadi et al. reported that EGFP fused with the C-terminal region (amino acids 313–518) of APE2 was localized in the nuclei (22). The C-terminal region (amino acids 314–518, pI 10.56) of APE2, uniquely conserved in the APE2/APN2 family as shown in Figure 1A, is far more basic than its N-terminal region (amino acids 1–313, pI 5.14) and the basic property of this region is likely responsible for its nuclear localization. Furthermore, the C-terminal regions of the APE2/APN2 family contain a consensus sequence for a PCNA-binding motif that in human APE2 has been proven to be functional.
Unk et al. (24) reported that the C-terminal region of ScAPN2 is completely dispensable for its AP endonuclease activity measured in vitro, but not for in vivo suppression of the increased sensitivity of an apn1Δ/apn2Δ double mutant to methyl methanesulfonate. A mutant ScAPN2 lacking a region to the TOP3 homologous sequence but not to the PCNA-binding motif from its C-terminus was able to complement the double mutant. This observation strongly suggested that the PCNA-binding motifs found in the APE2/APN2 family proteins are indeed essential for their in vivo function.
PCNA forms a homotrimer with a ring structure and functions as a DNA sliding clamp, which is an accessory factor for a number of proteins related to DNA replication and DNA repair, including DNA polymerase δ/ɛ, FEN1, DNA ligase I, etc. (46,47). Matsumoto et al. (48–50) reconstituted a novel BER pathway dependent on PCNA using purified enzyme components. The PCNA-dependent BER pathway may have two biological roles. First of all, PCNA-dependent BER may repair modified AP sites which cannot be processed by the dRPase activity of DNA polymerase β, i.e. FEN1 cleaves the flapped ends generated during elongation of the DNA strand by DNA polymerase δ/ɛ. Secondly, post-replicative BER for efficient repair of dUMP misincorporated during DNA replication is also likely to be PCNA dependent, as proposed by Nilsen et al. (40) and Otterlei et al. (41). A nuclear form of uracil DNA glycosylase (UNG2) possesses a PCNA-binding motif and it may initiate post-replicative BER in association with PCNA once dUMP is misincorporated during DNA replication.
Oxidized nucleotides such as 8-oxo-dGTP and 2-OH-dATP, which are known to be substrates for oxidized purine nucleoside triphosphatase, MTH1 (51,52), may also be incorporated during DNA replication (53). dAMP may be misincorporated opposite 8-oxoG in template DNA. These misincorporated nucleotides can be repaired by human OGG1 or MYH proteins (11,12) and post-replicative BER dependent on PCNA is likely to promote efficient and well-coordinated repair of these misincorporated nucleotides during DNA replication. OGG1 and MYH proteins also have putative PCNA-binding motifs, thus indicating the importance of PCNA-dependent post-replicative BER (Fig. 1C). Recently MYH protein was reported to bind to PCNA (54). In the PCNA-dependent BER pathway most steps are likely to be processed dependent on a DNA clamp, PCNA. Already, several catalytic components in the pathway have been demonstrated to have a PCNA-binding motif, i.e. their functions are somehow PCNA dependent (48–50). Among them only APE1 lacks a PCNA-binding motif. Since APE2 has a functional PCNA-binding motif and is actually associated with PCNA in nuclei, APE2 is likely to be the AP endonuclease responsible for the PCNA-dependent BER pathway. The increased co-localization of APE2 and PCNA in nuclear foci after treatment with HAT and dUrd supports the involvement of APE2 in the post-replicative BER complex.
In the C-terminal regions of APE2/APN2 family proteins another subregion is also conserved. This subregion contains a sequence homologous to the C-terminal repeats found in TOP3 family proteins. This subregion may also play a role in the interaction with other proteins related to BER.
In mitochondria, DNA polymerase γ accessory subunit p55 was reported to function as a DNA clamp on mtDNA, like PCNA (55,56). It is not likely that PCNA is localized in the mitochondria and thus accessory subunit p55 may function as a DNA clamp for APE2 in mitochondria, if necessary. Reconstituting mitochondrial BER with recombinant proteins is expected to help us answer this question.
APE2 and ScAPN2 have been reported to have class II AP endonuclease activities in vitro, however, their activities were much weaker than those of APE1 and XTH (22,24). The recombinant APE2 proteins expressed in E.coli and insect cells were mostly insoluble (our unpublished data). These observations suggest that the interaction of APE2 with other cellular components such as PCNA and DNA may facilitate its proper folding or enhance its catalytic function. It is possible that APE2/APN2 family proteins may exert their AP endonuclease activities only toward DNA with AP sites on the newly synthesized strand.
Acknowledgments
ACKNOWLEDGEMENTS
We extend our special thanks to Dr T.Tsurimoto for providing pT7-PCNA and PCNA protein, to Drs Y.Matsumoto, M.Furuichi and Y.Tominaga for their excellent technical assistance and to Dr B.Quinn for useful comments on this manuscript. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
DDBJ/EMBL/GenBank accession nos AB049211 and BAB13764
References
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC.
- 2.Krokan H.E., Nilsen,H., Skorpen,F., Otterlei,M. and Slupphaug,G. (2000) Base excision repair of DNA in mammalian cells. FEBS Lett., 476, 73–77. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. (2000) Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat. Res., 462, 129–135. [DOI] [PubMed] [Google Scholar]
- 4.Memisoglu A. and Samson,L. (2000) Base excision repair in yeast and mammals. Mutat. Res., 451, 39–51. [DOI] [PubMed] [Google Scholar]
- 5.Demple B., Herman,T. and Chen,D.S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robson C.N. and Hickson,I.D. (1991) Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E.coli xth (exonuclease III) mutants. Nucleic Acids Res., 19, 5519–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seki S., Hatsushika,M., Watanabe,S., Akiyama,K., Nagao,K. and Tsutsui,K. (1992) cDNA cloning, sequencing, expression and possible domain structure of human APEX nuclease homologous to Escherichia coli exonuclease III. Biochim. Biophys. Acta, 1131, 287–299. [DOI] [PubMed] [Google Scholar]
- 8.Xanthoudakis S. and Curran,T. (1992) Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J., 11, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richter C., Park,J.W. and Ames,B.N. (1988) Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl Acad. Sci. USA, 85, 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bogenhagen D.F. (1999) Repair of mtDNA in vertebrates. Am. J. Hum. Genet., 64, 1276–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishioka K., Ohtsubo,T., Oda,H., Fujiwara,T., Kang,D., Sugimachi,K. and Nakabeppu,Y. (1999) Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell, 10, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohtsubo T., Nishioka,K., Imaiso,Y., Iwai,S., Shimokawa,H., Oda,H., Fujiwara,T. and Nakabeppu,Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res., 28, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nilsen H., Otterlei,M., Haug,T., Solum,K., Nagelhus,T.A., Skorpen,F. and Krokan,H.E. (1997) Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res., 25, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slupphaug G., Markussen,F.H., Olsen,L.C., Aasland,R., Aarsaether,N., Bakke,O., Krokan,H.E. and Helland,D.E. (1993) Nuclear and mitochondrial forms of human uracil-DNA glycosylase are encoded by the same gene. Nucleic Acids Res., 21, 2579–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stierum R.H., Croteau,D.L. and Bohr,V.A. (1999) Purification and characterization of a mitochondrial thymine glycol endonuclease from rat liver. J. Biol. Chem., 274, 7128–7136. [DOI] [PubMed] [Google Scholar]
- 17.Ropp P.A. and Copeland,W.C. (1996) Cloning and characterization of the human mitochondrial DNA polymerase, DNA polymerase γ. Genomics, 36, 449–458. [DOI] [PubMed] [Google Scholar]
- 18.Lakshmipathy U. and Campbell,C. (1999) The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol., 19, 3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinz K.G. and Bogenhagen,D.F. (1998) Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol. Cell. Biol., 18, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomkinson A.E., Bonk,R.T. and Linn,S. (1988) Mitochondrial endonuclease activities specific for apurinic/apyrimidinic sites in DNA from mouse cells. J. Biol. Chem., 263, 12532–12537. [PubMed] [Google Scholar]
- 21.Bennett R.A. (1999) The Saccharomyces cerevisiaeETH1 gene, an inducible homolog of exonuclease III that provides resistance to DNA-damaging agents and limits spontaneous mutagenesis. Mol. Cell. Biol., 19, 1800–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hadi M.Z. and Wilson,D.M. (2000) Second human protein with homology to the Esherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen., 36, 312–324. [PubMed] [Google Scholar]
- 23.Johnson R.E., Torres-Ramos,C.A., Izumi,T., Mitra,S., Prakash,S. and Prakash,L. (1998) Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1 and its role in the repair of abasic sites. Genes Dev., 12, 3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unk I., Haracska,L., Johnson,R.E., Prakash,S. and Prakash,L. (2000) Apurinic endonuclease activity of yeast Apn2 protein. J. Biol. Chem., 275, 22427–22434. [DOI] [PubMed] [Google Scholar]
- 25.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 26.Claros M.G. and Vincens,P. (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem., 241, 779–786. [DOI] [PubMed] [Google Scholar]
- 27.Solovyev V.V., Salamov,A.A. and Lawrence,C.B. (1994) Predicting internal exons by oligonucleotide composition and discriminant analysis of spliceable open reading frames. Nucleic Acids Res., 22, 5156–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakabeppu Y. and Nathans,D. (1991) A naturally occurring truncated form of FosB that inhibits Fos/Jun transcriptional activity. Cell, 64, 751–759. [DOI] [PubMed] [Google Scholar]
- 29.Fukuda K., Morioka,H., Imajou,S., Ikeda,S., Ohtsuka,E. and Tsurimoto,T. (1995) Structure–function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J. Biol. Chem., 270, 22527–22534. [DOI] [PubMed] [Google Scholar]
- 30.Nakabeppu Y., Oda,S. and Sekiguchi,M. (1993) Proliferative activation of quiescent Rat-1A cells by delta FosB. Mol. Cell. Biol., 13, 4157–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Kuilenburg A.B., Muijsers,A.O., Demol,H., Dekker,H.L. and Van Beeumen,J.J. (1988) Human heart cytochrome c oxidase subunit VIII. Purification and determination of the complete amino acid sequence. FEBS Lett., 240, 127–132. [DOI] [PubMed] [Google Scholar]
- 32.Chirgwin J.M., Przybyla,A.E., MacDonald,R.J. and Rutter,W.J. (1979) Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry, 18, 5294–5299. [DOI] [PubMed] [Google Scholar]
- 33.Ishibashi T., Nakabeppu,Y., Kawate,H., Sakumi,K., Hayakawa,H. and Sekiguchi,M. (1994) Intracellular localization and function of DNA repair methyltransferase in human cells. Mutat. Res., 315, 199–212. [DOI] [PubMed] [Google Scholar]
- 34.Kang D., Nishida,J., Iyama,A., Nakabeppu,Y., Furuichi,M., Fujiwara,T., Sekiguchi,M. and Takeshige,K. (1995) Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J. Biol. Chem., 270, 14659–14665. [DOI] [PubMed] [Google Scholar]
- 35.Warbrick E. (1998) PCNA binding through a conserved motif. Bioessays, 20, 195–199. [DOI] [PubMed] [Google Scholar]
- 36.Warbrick E., Heatherington,W., Lane,D.P. and Glover,D.M. (1998) PCNA binding proteins in Drosophila melanogaster: the analysis of a conserved PCNA binding domain. Nucleic Acids Res., 26, 3925–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds N., Warbrick,E., Fantes,P.A. and MacNeill,S.A. (2000) Essential interaction between the fission yeast DNA polymerase delta subunit Cdc27 and Pcn1 (PCNA) mediated through a C-terminal p21(Cip1)-like PCNA binding motif. EMBO J., 19, 1108–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rizzuto R., Brini,M., Pizzo,P., Murgia,M. and Pozzan,T. (1995) Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr. Biol., 5, 635–642. [DOI] [PubMed] [Google Scholar]
- 39.Ingraham H.A., Dickey,L. and Goulian,M. (1986) DNA fragmentation and cytotoxicity from increased cellular deoxyuridylate. Biochemistry, 25, 3225–3230. [DOI] [PubMed] [Google Scholar]
- 40.Nilsen H., Rosewell,I., Robins,P., Skjelbred,C.F., Andersen,S., Slupphaug,G., Daly,G., Krokan,H.E., Lindahl,T. and Barnes,D.E. (2000) Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell, 5, 1059–1065. [DOI] [PubMed] [Google Scholar]
- 41.Otterlei M., Warbrick,E., Nagelhus,T.A., Haug,T., Slupphaug,G., Akbari,M., Aas,P.A., Steinsbekk,K., Bakke,O. and Krokan,H.E. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clayton D.A., Doda,J.N. and Friedberg,E.C. (1974) The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc. Natl Acad. Sci. USA, 71, 2777–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takao M., Zhang,Q.M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fung H., Kow,Y.W., Van Houten,B., Taatjes,D.J., Hatahet,Z., Janssen,Y.M., Vacek,P., Faux,S.P. and Mossman,B.T. (1998) Asbestos increases mammalian AP-endonuclease gene expression, protein levels and enzyme activity in mesothelial cells. Cancer Res., 58, 189–194. [PubMed] [Google Scholar]
- 45.Reinhardt A. and Hubbard,T. (1998) Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res., 26, 2230–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hingorani M.M. and O’Donnell,M. (2000) Sliding clamps: a (tail)ored fit. Curr. Biol., 10, R25–R29. [DOI] [PubMed] [Google Scholar]
- 47.Tsurimoto T. (1999) PCNA binding proteins. Front. Biosci., 4, D849–D858. [DOI] [PubMed] [Google Scholar]
- 48.Gary R., Kim,K., Cornelius,H.L., Park,M.S. and Matsumoto,Y. (1999) Proliferating cell nuclear antigen facilitates excision in long-patch base excision repair. J. Biol. Chem., 274, 4354–4363. [DOI] [PubMed] [Google Scholar]
- 49.Matsumoto Y., Kim,K. and Bogenhagen,D.F. (1994) Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol. Cell. Biol., 14, 6187–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsumoto Y., Kim,K., Hurwitz,J., Gary,R., Levin,D.S., Tomkinson,A.E. and Park,M.S. (1999) Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins. J. Biol. Chem., 274, 33703–33708. [DOI] [PubMed] [Google Scholar]
- 51.Fujikawa K., Kamiya,H., Yakushiji,H., Fujii,Y., Nakabeppu,Y. and Kasai,H. (1999) The oxidized forms of dATP are substrates for the human MutT homologue, the hMTH1 protein. J. Biol. Chem., 274, 18201–18205. [DOI] [PubMed] [Google Scholar]
- 52.Sakumi K., Furuichi,M., Tsuzuki,T., Kakuma,T., Kawabata,S., Maki,H. and Sekiguchi,M. (1993) Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem., 268, 23524–23530. [PubMed] [Google Scholar]
- 53.Kamiya H. and Kasai,H. (1995) 2-Hydroxyadenine (isoguanine) as oxidative DNA damage: its formation and mutation inducibility. Nucleic Acids Symp. Ser., 34, 233–234. [PubMed] [Google Scholar]
- 54.Parker A., Gu,Y., Mahoney,W., Lee,S.H., Singh,K.K. and Lu,A.L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long-patch DNA base excision repair. J. Biol. Chem., 276, 5547–5555. [DOI] [PubMed] [Google Scholar]
- 55.Fan L., Sanschagrin,P.C., Kaguni,L.S. and Kuhn,L.A. (1999) The accessory subunit of mtDNA polymerase shares structural homology with aminoacyl-tRNA synthetases: implications for a dual role as a primer recognition factor and processivity clamp. Proc. Natl Acad. Sci. USA, 96, 9527–9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim S.E., Longley,M.J. and Copeland,W.C. (1999) The mitochondrial p55 accessory subunit of human DNA polymerase γ enhances DNA binding, promotes processive DNA synthesis and confers N-ethylmaleimide resistance. J. Biol. Chem., 274, 38197–38203. [DOI] [PubMed] [Google Scholar]