

Abstract
Influenza virus inflicts a heavy death toll annually and resistance to existing antiviral drugs has generated interest in the development of agents with novel mechanisms of action. Favipiravir is an antiviral drug that acts by increasing the genome-wide mutation rate of influenza A virus (IAV). Potential synergistic benefits of combining oseltamivir and favipiravir have been demonstrated in animal models of influenza, but the population-level effects of combining the drugs are unknown. In order to elucidate the underlying evolutionary processes at play, we performed genome-wide sequencing of IAV experimental populations subjected to serial passaging in vitro under a combined protocol of oseltamivir and favipiravir. We describe the interplay between mutation, selection, and genetic drift that ultimately culminates in population extinction. In particular, selective sweeps around oseltamivir resistance mutations reduce genome-wide variation while deleterious mutations hitchhike to fixation given the increased mutational load generated by favipiravir. This latter effect reduces viral fitness and accelerates extinction compared with IAV populations treated with favipiravir alone, but risks spreading both established and newly emerging mutations, including possible drug resistance mutations, if transmission occurs before the viral populations are eradicated.
Keywords: influenza, population genetics, genetic hitchhiking, mutational meltdown
Introduction
Influenza A virus (IAV) inflicts a heavy disease burden worldwide, thus developing effective drugs remains a public health priority. The most frequently used drug, oseltamivir, was designed as a competitive inhibitor of the viral surface neuraminidase (NA) glycoprotein responsible for binding host cell sialic acid to enable the release of virus progeny (Moscona 2005). Oseltamivir binding requires altering a hydrophobic pocket in the NA region and can be destabilized by mutations near the active site (Varghese et al. 1998; Collins et al. 2008). Early studies in vitro and in vivo identified high fitness costs associated with such mutations, which lent support to the view that the development of resistance was unlikely in clinical settings (Ives et al. 2002). The most common resistance mutation in H1N1 strains, NA H275Y, was initially observed infrequently during clinical testing (Gubareva et al. 2001) but spread rapidly worldwide during the 2007/2008 influenza season (Moscona 2009) and continues to be a clinical concern (Ghedin et al. 2012; Meijer et al. 2014; Takashita et al. 2015). H275Y confers resistance to oseltamivir but lowers viral fitness by reducing the amount of NA that reaches the cell surface (Bloom et al. 2010). The higher than expected fitness of mutants carrying H275Y is likely due to the presence of compensatory mutations that increase cell surface expression and enzymatic activity of NA (Bloom et al. 2010; Bouvier et al. 2012; Ginting et al. 2012; Butler et al. 2014).
Oseltamivir resistance has increased interest in developing drugs with an alternative mechanism of action and a lower likelihood of resistance. Favipiravir is a mutagenic drug that inhibits the viral RNA-dependent RNA polymerase (RdRp) and dramatically increases the IAV mutation rate, ideally driving the virus towards extinction (Baranovich et al. 2013; Furuta et al. 2013). Over the past few decades, several studies have shown the impact of mutagenic drugs on RNA virus extinction (e.g., foot-and-mouth disease virus [Sierra et al. 2000; Pariente et al. 2001], HIV-1 [Loeb et al. 1999; Loeb and Mullins 2000], and lymphocytic choriomeningitis virus [Grande-Pérez et al. 2002]). Favipiravir is effective against a range of RNA viruses including influenza and is currently in phase III clinical trials. It is safe for use in humans in part because human proteins do not contain RdRp domains, and it has a distinct mode of action from both oseltamivir and M2 inhibitor drugs. Most importantly, no resistance mutations have been functionally validated to date, in part because the development of resistance may involve multiple mutational steps, or perhaps because viral extinction occurs too rapidly for resistance to evolve.
Under serial passage in cell culture with an escalating concentration of favipiravir, IAV populations steadily accumulate an increasing mutation load (Bank et al. 2016), leading to an eventual sharp population collapse and extinction. Results fit predictions of the mutational meltdown model (Lynch et al. 1993). Specifically, Muller’s ratchet (the stochastic loss of the fittest class of haplotypes and resulting reduction in fitness) (Muller 1964; Felsenstein 1974) and Hill–Robertson interference (the reduction in the efficacy of selection owing to linkage between selected sites) (Hill and Robertson 1966) have been argued to be the key drivers of this process. Notably, in cell culture experiments with IAV, extinction depends on a high concentration of favipiravir; when the drug concentration was held constant at low levels, or withdrawn, a reduction in the negative growth rate (relative to the no-drug control) was observed in some populations (Bank et al. 2016).
Using drugs in combination is an established clinical strategy aimed at preventing or delaying resistance by rapidly depleting pathogen populations before resistance can emerge (Mitchison 2012). Synergistic benefits have been observed using favipiravir and oseltamivir in combination against pandemic influenza A/California/04/2009 (H1N1 pdm) as well as against oseltamivir resistant (H275Y NA mutation) influenza in mice in vivo (Smee et al. 2013). However, given the two very different mechanisms of action, the effects of combining these drugs on the emergence of drug resistance are unknown. To assess the potential advantage of a combined protocol, we treated IAV populations with escalating concentrations of oseltamivir and favipiravir (“combined drug” populations) over ten passages in cell culture, and compared these with three paired control replicate IAV populations treated with oseltamivir only, as well as with our previously generated favipiravir only and no-drug control populations. This experimental set-up offered an excellent platform to dissect the complex dynamics contributing to mutational meltdown, and in particular the roles of genetic hitchhiking, Muller’s ratchet, and Hill–Robertson interference. In addition, the generation of replicates allows us to assess the reproducibility of the observed evolutionary phenomena.
Fundamentally, our results demonstrated that viral growth in oseltamivir-treated replicates remains high and stable throughout all passages due to the emergence of resistance mutations, whereas the combined drug populations become extinct or near extinct over ten passages (i.e., ∼130 generations). Important differences were observed in the underlying dynamics compared with the favipiravir-alone population. Intriguingly, extinction proceeded more quickly in the combined drug compared with the favipiravir-alone population despite a lower number of segregating mutations. Results suggest that this is due to the hitchhiking of deleterious mutations with strongly selected oseltamivir resistance mutations, a process that reduces viral fitness and accelerates extinction. These selective sweeps depressed genetic variation, providing an explanation for the lower number of segregating mutations. Additionally, evidence of a small and rapidly declining effective population size supports a role for Muller’s ratchet. Finally, evidence of Hill–Robertson interference specific to the combined drug and favipiravir populations emerged.
We did not find evidence here that the oseltamivir resistance mutation NA H275Y arose earlier in combined oseltamivir + favipiravir drug replicates than in oseltamivir replicates. Rather, our results suggest that the mutational effect of favipiravir combined with oseltamivir more broadly explores sequence space, clustering beneficial mutations on the same haplotype and generating alternative NA mutations, although at the cost of a high linked mutation load. Thus, the combined drug protocol potentially drives an earlier extinction point for viral populations compared with the favipiravir protocol alone, but at the risk of first introducing novel mutations that may be beneficial for resistance into the population.
Materials and Methods
Experimental Details
We compared the evolution of influenza A/Brisbane/59/2007 (H1N1) in Madin–Darby canine kidney cells under treatment with oseltamivir alone or under treatment with a combination of oseltamivir carboxylate (F. Hoffmann-La Roche Ltd, Basel, Switzerland) and favipiravir (FUJIFILM Pharmaceutical USA, Inc.) over a total of 10 passages. In the first three passages, IAV was adapted from chicken egg and serially amplified in the MDCK cells with no treatment, as part of an earlier experiment (Foll et al. 2014). Stock viral populations from an earlier experiment were used to seed passage 4 to ensure that replicates are identical before administration of the drug treatment. In passages 4–10, three replicates of IAV were exposed to increasing concentrations of a combination of oseltamivir and favipiravir (fig. 1). While such increasing concentrations do not directly relate to patient-treatment strategies, this approach was chosen simply to study the adaptive potential of the experimental populations. The replicates were paired with three populations exposed to increasing doses of oseltamivir only as a control. This ensured that the oseltamivir only replicates are subject to the same experimental conditions as the populations exposed to the combination treatment. A multiplicity of infection (MOI) of 0.01 was used for each passage in all replicates, except at passage 10 (MOI = 0.005) in replicate 1. As in Foll et al. (2014), 13 viral generations are assumed to occur during each passage. Details of drug treatment, MOI, and output plaque forming units (PFU) for each replicate are shown in supplementary table 1, Supplementary Material online. These results were compared with results obtained from a previous experiment, where two populations of IAV were exposed to favipiravir alone and to a no drug control over passages 4–15 (Bank et al. 2016). The favipiravir-only population was treated with 2 µM of the drug from passage 4, and the concentration was doubled at every passage, which represents twice the level of drug administered in the combined drug replicates (1 µM favipiravir in passage 4 and doubling of the dose thereafter). At the end of each passage, samples from each replicate were sequenced using high coverage, whole genome high throughput population sequencing as previously described (Bank et al. 2016).
Fig. 1.
—Experimental set-up. In each of three replicates, influenza A virus was serially passaged in MDCK cells and exposed to increasing concentrations of either oseltamivir only, or of oseltamivir and favipiravir combined, from passage 4 onwards. The multiplicity of infection (MOI) used to seed each passage is shown on the right hand side. ED50 represents a 50% effective dose for drug-naïve virus. The MOIs are valid for all replicates except combined drug and oseltamivir replicate 1 passage 10, where an MOI of 0.005 was applied.
WFABC Analysis
In oseltamivir-treated replicates, the software developed by Foll et al. (2014) was used to estimate global effective population size Ne and site-specific selection coefficients s from time sampled data using an approximate Bayesian computation approach. Because the frequency of the third most frequent mutation is very low in this data set, all sites are treated as bi-allelic. Sites with coverage >100 were randomly (hypergeometrically) down-sampled to a sample size of 100. Only trajectories with a down-sampled frequency >2.5% were kept for the analysis, to ensure that these are above the estimated sequencing error of 1%. Following Foll et al. (2014), mutations with a Bayesian posterior distribution for s excluding zero of <0.5% (P(s < 0|x)<0.5%) were identified as being putatively positively selected.
Population Size Estimates
WFABC implements a block bootstrap approach to obtain a distribution P(Ne|T(X)) for the effective population size Ne, where T(X) is a single statistic based on the temporal method of (Jorde and Ryman 2007). This method uses Fs’, which is a measure of the variance in allele frequencies between two time points adjusted for sampling bias to calculate Ne
where Fs is the estimator for allele frequency variance before adjusting for sampling bias, x and y are the allele frequencies at the two time points, txy is the number of generations between the two time points, z is the average frequency where z = (x + y)/2, and ñ is the harmonic mean of the sample sizes nx and ny at each time point. Ne is calculated as 1/Fs’.
A second method for calculating effective population size uses the harmonic mean of census sizes (Ewens 1967).
Population Dynamics
Absolute growth rates for all replicates were obtained from the starting and final population sizes at each passage. The underlying assumption is that each viral plaque is the result of a single infective particle, based on the low MOI used for each passage. Following Foll et al. (2014) and Bank et al. (2016), we assume 13 generations of viral populations per passage and calculate the Malthusian growth rate r per passage as
where t is the number of generations, N(t) is the population size at time t, and N1 is the initial population size at the start of each passage.
Hierarchical Clustering Analysis
The hierarchical clustering analysis is based on the squared Euclidian distance between allele frequency trajectories of the candidate mutations using Ward’s (1963) minimum variance criterion, starting from the time point where the frequency was higher than the estimated sequencing error of 1%. Additionally, pairwise correlations between allele trajectories were calculated and are reported in supplementary table 4, Supplementary Material online.
Results and Discussion
Rapid Extinction of Combined Drug Replicates
We first confirmed that the combination of favipiravir and oseltamivir leads to the extinction of virus populations under serial passaging (see experimental design in fig. 1), consistent with the effect of favipiravir shown in our previous study (Bank et al. 2016). All combined drug replicates reached extinction or near extinction by passage 10 (i.e., after ∼130 generations). The output number of plaque-forming units per ml (PFU/ml) tracks census viral population size at the end of each passage. The output PFU/ml declined to 3.0×102 for replicate 1, 8.0×103 for replicate 2, and 2.4×104 for replicate 3 (supplementary table 1, Supplementary Material online). In contrast, the output PFU/ml for oseltamivir-only replicates remained high and stable throughout all passages (6.0×105 PFU/ml for replicate 1, 5.0×106 PFU/ml for replicate 2 and 7.0×106 PFU/ml for replicate 3), indicating that the virus developed resistance to this drug (see section on resistance to oseltamivir for further details).
We calculated relative viral growth per passage (a measure of the fitness of the viral population) as output/input PFU/ml and show its progression in figure 2A. The relative growth for combined drug replicates declined more rapidly than for the population treated with favipiravir alone, despite the lower concentration of favipiravir. The relative growth for combined replicate 1 showed a sharp recovery at passage 7 followed by a rapid decline, whereas the relative growth for the other two replicates declined steadily. Relative growth for the oseltamivir replicates remained stable, although below the level exhibited by the control population.
Fig. 2.
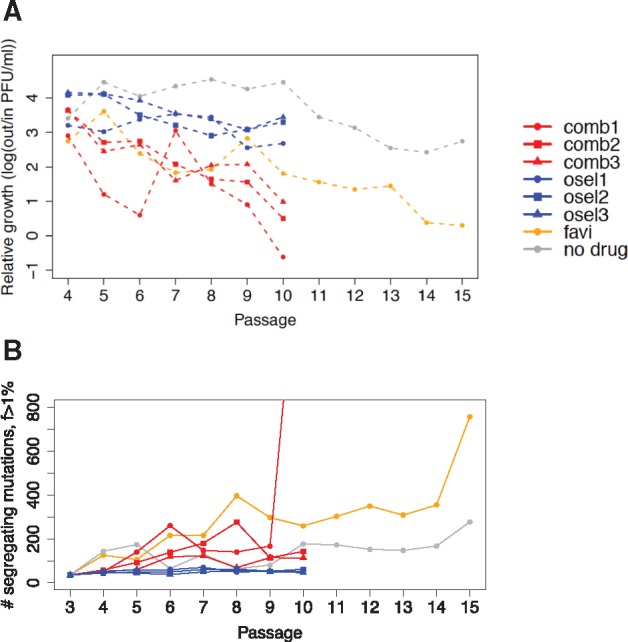
—Relative growth and total number of segregating sites in IAV treated with zero, one (“osel” for oseltamivir), or two antiviral agents (“comb” for combined favipiravir and oseltamivir). (A) Relative growth, a measure of viral fitness, is calculated as log10(output/input PFU) for each passage. A more rapid decline in relative growth was observed in the combined drug replicates (red), than in the favipiravir-only population (yellow). Relative growth for the oseltamivir-only replicates (blue) remains relatively stable, although below the level exhibited by the control population (gray). (B) The number of segregating mutations represents all sites segregating at > 1% DAF at each passage. Despite the more rapid decline in relative growth, the number of segregating sites for the combined drug replicates (red) is lower than for the favipiravir population (orange), indicating a lower segregating mutation load. The number of segregating sites observed in the oseltamivir replicates is low, likely owing to selective sweeps around the oseltamivir resistance mutations, as well as to the absence of favipiravir’s mutagenic effect.
We estimated the total number of sites segregating above a 1% derived allele frequency (DAF) (the estimated sequencing error) in the populations at each passage (fig. 2B), to explore whether a higher segregating mutation load was responsible for the rapid decline of combined drug replicates (see Materials and Methods). Intriguingly, the number of segregating mutations was lower for the combined drug replicates than for the favipiravir-only population. Further, peaks in the number of segregating mutations at passage 6 for the combined drug replicate 1 and passage 8 for the combined drug replicate 2, followed by a rapid reduction, were likely hallmarks of genetic hitchhiking (see below). In contrast, oseltamivir-only replicates exhibited constantly low mutation loads and stable viral population sizes, confirming that these populations remained in mutation–selection–drift balance after achieving resistance.
Combined drug replicate 1 provided the strongest evidence of transition into a phase of rapid population collapse (negative relative growth) and escalating mutation accumulation, similar to the dynamics observed in passage 14 and 15 of the favipiravir population. The total number of segregating mutations increased nearly 9-fold, specifically from 202 in passage 9 to 1,800 in passage 10. For replicates 2 and 3, a slowing of relative growth and an increase in the number of segregating sites above the levels exhibited by oseltamivir-only control replicates was observed, but the end dynamics indicated that these replicates had not yet transitioned into population collapse.
Evidence for the Role of Muller’s Ratchet
An explanation for the rapid decline in relative viral growth in combined drug replicates as opposed to individual drug application is the accelerated action of Muller’s ratchet, and generally less efficient selection, in smaller populations. Under Muller’s ratchet, the mean number of deleterious mutations per individual accumulates at a constant rate (Muller 1964; Felsenstein 1974), assuming a static mutation rate and constant population size. The rate of this process—the speed of the ratchet—increases exponentially with mutation rate and decreases with population size and with the selection strength of deleterious mutations (Haigh 1978; Gordo and Charlesworth 2000a, 2000b). In combined drug and favipiravir-only treated populations subject to an influx of mainly deleterious and neutral mutations, we would expect an acceleration in the rate of Muller’s ratchet, particularly in the case of declining population size. These evolutionary processes are determined by effective population size Ne rather than census size (Wright 1931; Charlesworth 2009). The serial passaging of virus populations created a series of bottlenecks followed by exponential growth (or contraction) over 13 viral generations at each passage, which depressed effective population size. Strong purifying selection in IAV populations (evidenced by low average expected heterozygosity and a left skew in the site frequency spectrum (SFS), supplementary figs. 1 and 2, Supplementary Material online) also acted to reduce effective population sizes (Charlesworth et al. 1993; Gordo and Charlesworth 2001). Thus, we explored whether differences in effective population size exist between the favipiravir-only and combined drug replicates, and used this as a means to indirectly assess the contribution of Muller’s ratchet, and to quantify the rate of mutation accumulation.
Theoretical work and simulations have shown that the fixation of neutral and weakly deleterious mutations is a robust indicator of the loss of the least loaded classes and therefore of the speed of Muller’s Ratchet, assuming a haploid asexual population under an influx of deleterious mutations with the same selection coefficient (Charlesworth and Charlesworth 1997; Bergstrom and Pritchard 1998; Gordo and Charlesworth 2001). However, because of strong purifying selection (supplementary figs. 1 and 2, Supplementary Material online), here we observed a relatively small number of mutations segregating above a DAF of 40% and fixing in the population (supplementary table 2, Supplementary Material online), most of which can be attributed to genetic hitchhiking (discussed below). Therefore, the rate of fixation of deleterious mutations in this case is rather conflated between the rate of genetic hitchhiking and the speed of the ratchet.
Global estimates of effective population size were calculated to enable a comparison between the favipiravir-only treated population and the combined drug replicates (fig. 3). These were obtained 1) by calculating the effective population size at each passage based on the harmonic mean of the estimated population size for each generation (assuming exponential growth) and 2) by calculating the harmonic mean of the estimates per passage (Ewens 1967). However, this method ignores differences in virion budding, which create skewed offspring distributions (Irwin et al. 2016) consistent with evidence that only a few virions seed subsequent generations (Grenfell et al. 2004). Calculations of effective population size are therefore likely to be overestimated. We found that the estimated effective population size for the combined drug replicate 1 (Ne=2,750) was similar to that for the favipiravir-only population over 13 passages (Ne=3,200), consistent with the observation of a collapse in both of these populations. Estimated effective population sizes in combined replicates 2 and 3 were also low and below estimates for oseltamivir-only and no-drug populations. A second estimate of effective population size is obtained from Wright–Fisher approximate Bayesian computation, or WFABC (supplementary fig. 3, Supplementary Material online), which leverages the variance in allele frequencies between time points (Jorde and Ryman 2007). This method is anticipated to be downwardly biased owing to the increasing mutation rate through time in this experiment. Using this method, we also found that estimates of effective population size for combined drug replicates (Ne= 239.16 for replicate 1, Ne= 216.19 for replicate 2 and Ne= 161.64 for replicate 3) were similar to the estimate for the favipiravir-only population (Ne= 209.15) and were lower than estimates for oseltamivir-only replicates. Thus, the small effective population size processes described were likely equally at play in precipitating extinction in combined drug replicates as well as in the favipiravir-alone populations.
Fig. 3.
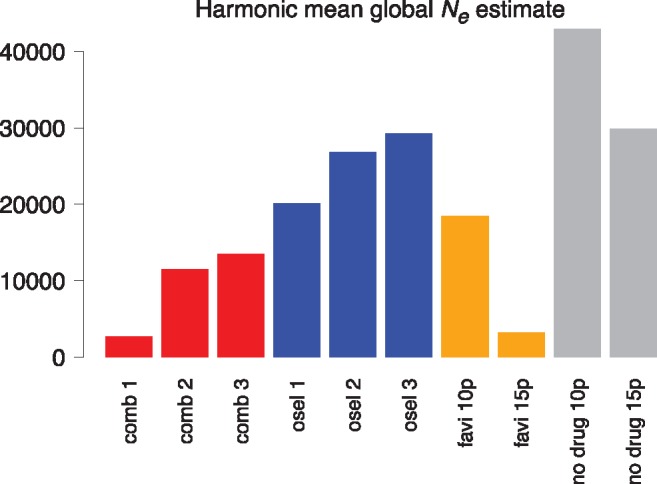
—Global effective population sizes. Effective population sizes at each passage are calculated as the harmonic mean of estimated population sizes at each generation (assuming exponential growth over 13 generations for each passage). Estimates are calculated over eight passages (passages 3–10) for the combined drug and oseltamivir treated replicates and over 13 passages (passages 3–15) for the favipiravir and no drug populations (“favi 15p” and “no drug 15p”). For comparison purposes, effective population sizes are also calculated over eight passages (passages 3–10) for the favipiravir and no drug populations (“favi 10p” and “no drug 10p”).
Mutations Putatively Evolving under Positive Selection
To identify positively selected mutations, the posterior distributions for estimates of s were utilized (i.e., with a posterior density interval for the selection coefficient s excluding zero of <0.5%, P(s < 0|x) < 0.005). WFABC differentiates trajectories of mutations under selection from those due to genetic drift under the assumption that all sites are unlinked and independently selected, and that the mutation rate is static. Because many of the trajectories in the oseltamivir-only, combined drug, and favipiravir-only replicates exhibited nonstandard trajectories (i.e., trajectories that have an exceedingly small probability under any single selection coefficient), and because the assumptions of a constant mutation rate and of unlinked sites did not always hold, we also tracked trajectories that exceed a DAF of 40% at any time point.
We identified 11 mutations that potentially evolved under positive selection in oseltamivir-only replicates (see fig. 4A–C), including the known resistance mutation NA H275Y (based on N1 numbering). Four of these mutations were synonymous and seven were nonsynonymous. The seven nonsynonymous contending mutations were also the only ones that arose in more than one replicate (outside of the NA region) in either the combined drug replicates or in the favipiravir-only treated population. Neutrality was rejected for all of these mutations in at least one oseltamivir-only replicate (except for NP D101N, which only exhibited a “standard” trajectory in the favipiravir-only population) and WFABC was used to estimate selection coefficients and Bayesian P values (table 1). The varied trajectories and limited clustering of these mutations in oseltamivir-only replicates supports the assumption that these are unlinked, independently selected sites, although epistatic interactions cannot be excluded.
Fig. 4.

—Putatively beneficial mutations. The contending beneficial mutations (table 1) are tracked in the oseltamivir-only replicates (A–C), in the combined drug replicates (D–F) and in the favipiravir-only and no drug populations (G and H). The key to these mutations is given below, with the NA mutations in red. Clustering is observed amongst these beneficial mutations in combined drug replicates. All other segregating mutations (i.e., arising in only one replicate) are assumed to be neutral or deleterious and are plotted in gray. In combined drug replicates, this class of mutations also shows evidence of genetic hitchhiking with the resistance mutations, or with HA E78G (a ubiquitous contending beneficial mutation in all oseltamivir and combined drug replicates, except for combined drug replicate 2) (D–F). In (G) and (H), the contending beneficial mutations are tracked in the favipiravir-only and no-drug control populations, with longer trajectories (weaker selection coefficients) and less evidence of clustering.
Table 1.
Mutations Inferred to Be Evolving under Positive Selection
| Seg | Pos | Ref Base | Mut Base | Type S/NS | SNP | WFABC sb | Bayesian P Value (P < 0) | Prev id | Replicate | Functional Interpretation |
|---|---|---|---|---|---|---|---|---|---|---|
| PA | 199 | G | T | S | G58G | 0.026 | 0.017 | Y | All | Cell adaptation |
| PA | 225 | C | T | S | D67D | N | osel3 | Synonymous | ||
| HA | 1,280 | G | A | S | L73L | 0.099 | 0** | N | osel1 | Synonymous |
| HA | 1,294 | A | G | NS | E78G | 0.114(1) | 0** | N | osel123 and comb13 | Possible epistasis with H275Y |
| 0.117(3) | 0** | |||||||||
| HA | 1,395 | G | A | NS | D112N | 0.126 | 0** | Y | osel2 and no drug | Cell adaptation |
| NP | 346 | G | A | NS | D101N | 0.027(1) | 0.0349 | Y | all except no drug | Cell adaptation |
| NA | 403 | A | G | NS | E128G | a | N | comb3 | Possible resistance mutation | |
| NA | 843 | C | T | NS | H275Y | 0.125(1) | 0** | Y | osel123 and comb13 | Known resistance mutation |
| 0.209(2) | 0** | |||||||||
| 0.218(3) | 0** | |||||||||
| NA | 998 | G | T | S | P326P | 0.075 | 0.002** | N | osel2 | Synonymous |
| NA | 1,381 | C | T | NS | A454V | a | N | comb23 | Possible resistance mutation | |
| MP1 | 92 | G | C | NS | E23Q | 0.057(1) | 0** | Y | All | Compensatory mutation |
| MP2 | 848 | C | G | NS | A37G | 0.038(3) | 0.039 | N | osel23 and comb12 | Compensatory mutation |
| NS1 | 399 | G | A | NS | D125N | 0.036 | 0.03 | N | osel2 and no drug | Cell adaptation |
In combined drug replicates only, strength not estimated (the assumptions of a constant mutation rate and of unlinked sites do not hold).
The numbers in brackets indicate the oseltamivir replicate used for the estimation (where the mutation arises in several replicates).
significant (P < 0.005).
In contrast, three of the four synonymous mutations to reach a DAF >40% arose in one oseltamivir-only replicate; two cluster with the strongly selected H275Y (HA L73L in oseltamivir 1 and NA P326P in oseltamivir 2) suggesting probable genetic hitchhiking of neutral variants. The third (PA D67D) had a nonstandard trajectory and its functional significance is unknown. The fourth synonymous mutation, PA G58G, arose in all oseltamivir and combined drug replicates, as well as in the favipiravir population. It fixed in oseltamivir replicate 1 (along with MP1 E23Q) but exhibits nonstandard trajectories in other oseltamivir and combined drug replicates, as well as in favipiravir-only and no-drug replicates, where it clustered with MP1 E23Q, suggesting both a cell adaptation function and a possible epistatic interaction with MP1 E23Q.
Figure 4 tracks these contending mutations potentially evolving under positive selection in combined drug replicates (fig. 4D–F) and in favipiravir-only and no-drug treated populations (fig. 4G and H). In addition to H275Y, two other nonsynonymous mutations (A454V and E128G) in the NA region that is critical for oseltamivir resistance arose in the combined drug replicates only; one of these mutations, NA A454V arose in both combined drug replicates 2 and 3. In addition to the 11 mutations described earlier, this gives a new total of 13 contending beneficial candidates that are tracked in the combined drug (fig. 4D–F) and in the favipiravir-only (fig. 4G) and no-drug (fig. 4H) populations (table 1). Outside of the NA region, the HA region contains the mutations with the highest selection coefficient estimates: D112N and E78G. This is consistent with studies showing that changes in the HA region may counter the deleterious growth effects of H275Y (Bloom et al. 2010; Ginting et al. 2012). The HA D112N mutation was previously identified by Foll et al. (2014) and has been described in other influenza strains and HA serotypes (Daniels et al. 1985; Reed et al. 2009): it acts by inducing a pH change at the point of endosome and viral fusion, thereby improving IAV infectivity (Thoennes et al. 2008). Here it was significant in oseltamivir replicate 2 (s = 0.126, table 1) and in the no-drug comparison population, suggesting an additional role in cell adaption. A newly identified HA mutation, E78G, was present in all oseltamivir and combined drug replicates where H275Y was present (five out of six populations), but not in combined drug replicate 2 where H275Y was absent, indicating a possible epistatic interaction. E78G was significant in oseltamivir replicate 1 and 3 (s = 0.114 and s = 0.117 respectively, table 1) and fixed in combined replicate 3, whereas in oseltamivir 2 and combined replicate 1 its trajectory suggests clonal interference (see section on Hill–Robertson Interference).
Mutations in the M1 region have been suggested to have compensatory benefits upon interacting with H275Y by improving the process of virion budding and helping to overcome the fitness cost of the H275Y mutation, reflected in the lower amount of NA to reach the cell surface (Jin et al. 1997; Noton et al. 2007; Rossman and Lamb 2011). E23Q was identified in both previous sets of experiments (Foll et al. 2014; Bank et al. 2016) and was significant in oseltamivir 1 (s = 0.057), favipiravir-only, and no-drug populations, with trajectories typical of clonal interference in the other replicates. A37G was very near to the previously identified A41V and may serve a similar function in improving virion budding; it clustered with D101N in some replicates and E23Q in others and arose in oseltamivir 2 and 3 (s = 0.038) and combined 1 and 2. NP mutation D101N has been previously screened as a resistance mutation to the mutagenic drug ribavirin (Cheung et al. 2014) with inconclusive results; here, we found it to be present in all populations except the no-drug control, including the oseltamivir-only populations, and therefore may have a role in improving the formation of infective virions (Noton et al. 2009).
Although H275Y did not appear more rapidly in combined drug populations than in oseltamivir-only replicates, the two other NA nonsynonymous mutations described earlier (A454V and E128G) only appeared in combined drug replicates (fig. 4) and exhibit trajectories characteristic of strong selection coefficients. We put forward the hypothesis that the mutational effect of favipiravir allowed the virus to rapidly explore sequence space for alternative resistance solutions within the limits of the time to extinction imposed by the increasing mutational load burden.
Effects of Genetic Hitchhiking
Under a model of genetic hitchhiking, neutral, or weakly selected sites in physical linkage to strongly beneficial mutations rise in frequency (Maynard Smith and Haigh 1974). Here, we found that the positively selected nonsynonymous mutations identified in the oseltamivir-only replicates showed strong pairwise correlations in their allele trajectories in the combined drug populations (supplementary table 4, Supplementary Material online), which provides greater evidence of clustering in the combined drug populations (fig. 4D–F) than in the oseltamivir-only populations (fig. 4A–C). This clustering of beneficial mutations may be seen around the strongly selected NA mutations H275Y, A454V, and E128G (fig. 5 and supplementary table 4, Supplementary Material online). Potential beneficial mutations for cell adaptation including D101N and A37G (with trajectories characterized by a low selection coefficient in some replicates) are rapidly driven to fixation by association with H275Y in combined replicate 1 and A454V in combined replicate 2. In contrast, genetic hitchhiking in the oseltamivir-only replicates is of synonymous (presumed neutral) variants (HA L73L with H275 in replicate 1 and NA P326P with H275Y in replicate 2). This suggests that, in addition to generating possible alternative oseltamivir resistance mutations, the enhanced mutational input of favipiravir may also serve to optimize combinations of beneficial mutations on single haplotypes.
Fig. 5.

—Hierarchical cluster analysis on combined drug trajectories. Ward’s minimum variance criterion (Ward 1963) was used to cluster allele-frequency trajectories. The dissimilarity distances are shown in panels (A) and the details of the clusters in subsequent panels (B–D) for replicates 1 and 2, and (B–G) for replicate 3. We observe hitchhiking patterns suggesting that either NA mutations (H275Y, A454V, or E128G) or HA E78G sweep other beneficial and neutral/deleterious mutations to fixation. Other cluster groups containing contending beneficial mutations do not fix. On panel (A), synonymous mutations are shown in gray font and nonsynonymous mutations in black. The pairwise correlations matrix in supplementary table 4, Supplementary Material online, supports the hierarchical cluster analysis.
Tracking the remaining mutations specific to the combined drug replicates and segregating in excess of 40% DAF, shows that these also cluster with the strongly selected NA mutations and with the ubiquitous HA mutation E78G (H275Y and E78G in replicate 1, A454V in replicate 2, and A454V, E128G, and HA E78G in replicate 3, see fig. 4D–F), with hierarchical clustering analysis (fig. 5) and pairwise correlations (supplementary table 4, Supplementary Material online) confirming this association. These mutations are unique to each combined drug replicate and are therefore assumed to be largely the result of hitchhiking of neutral and deleterious variants. The pairwise allele trajectory correlations are particularly high 1) in combined replicate 1 within cluster D (fig. 5) that contains H275Y (0.965–0.997 pairwise correlations between NA H275Y and the mutations in cluster D, shown in blue in supplementary table 4, Supplementary Material online) and are reasonably high within cluster B that contains HA E78G (0.708–0.849 pairwise correlations between HA E78G and the mutations in cluster B, shown in green in supplementary table 4, Supplementary Material online); 2) in combined replicate 2, within cluster B that contains NA A454V (0.814–0.991 pairwise correlations between NA A454V and the other mutations in cluster B, shown in green in supplementary table 4, Supplementary Material online); in combined replicate 3 within cluster F that contains HA E78G (0.980–0.994 pairwise correlations between HA E78G and the mutations in cluster F, in orange in supplementary table 4, Supplementary Material online), within cluster G that contains NA A454V and NA E128G (0.970–1.0 pairwise correlations between NA A454V and the mutations in cluster G, shown in pink in supplementary table 4, Supplementary Material online), and within cluster C (0.88 pairwise correlation between NA H275Y and the other mutation in cluster C, shown in blue in supplementary table 4, Supplementary Material online). These clusters of mutations mostly fix by passage 10. Pairwise correlations outside of the clusters are lower than within the clusters and support the hierarchical cluster analysis.
Thus, our analyses suggest that in the combined drug replicates the strongly selected beneficial mutations hitchhike not only other beneficial mutations but also a high mutation load to fixation. The timing of the selective sweeps coincides with a sharp decrease in the total number of segregating mutations after passage 6 in combined replicate 1 and after passage 8 in combined replicate 2 (fig. 2B), suggesting that the selective sweeps reduce genome-wide variation but at the cost of fixing deleterious mutations. A likely explanation is that the rapid trajectory to fixation of the beneficial mutations does not allow sufficient time to purge the linked deleterious mutations, particularly as effective population size has diminished and selection is therefore less efficient. In addition, there is a rapid and constant input of new deleterious mutations segregating at a low frequency from the impact of favipiravir. Ultimately this combined high load burden accelerates the decline in viral fitness and precipitates the population towards extinction (supplementary fig. 4, Supplementary Material online).
Thus, the strength of beneficial mutations (as indicated by the shape of their trajectory and the corresponding WFABC estimates) governed the size of the linked mutation load and potentially accelerated the process of extinction. As shown, the trajectories of mutations in the favipiravir-treated population were more random and diffuse (fig. 4G), with fewer obvious clusters. The tracked beneficial mutations in the favipiravir-only population-mediated cell adaptation and were less strongly selected than NA mutations in the oseltamivir and combined drug replicates with longer trajectories, giving purifying selection more time to act. The absence of strong selective sweeps reducing genome-wide variation was reflected in the high and escalating number of segregating mutations for favipiravir-only populations observed in figure 2B. Notably, the favipiravir population reached extinction by passage 15 (fig. 2A), compared with passage 10 for combined replicate 1, suggesting a tentative hypothesis that the weaker hitchhiking dynamics were at least partly responsible for the later point of collapse.
Effects of Hill–Robertson Interference
In a nonrecombining, asexual population such as IAV, strongly selected beneficial alleles arising on different haplotypes are in competition for fixation (Fisher 1930; Muller 1932; Barton 2010). There is a build-up in negative linkage disequilibrium between these “repulsion haplotypes” (Hill and Robertson 1966; McVean and Charlesworth 2000) (i.e., the beneficial mutations and their linked variants are found associated less frequently than by chance) and a reduction in the efficacy of selection. It is important to note here that while IAV does not recombine, it does reassort, thus these strong linkage effects will persist within segments, but are not expected genome-wide. In the combined drug replicates, we find examples of nonstandard trajectories of beneficial mutations (known to be mutations under weak positive selection in the oseltamivir-only replicates), which were characterized by a rapid rise and decline. These trajectories suggested patterns of clonal interference between beneficial mutations: haplotypes carrying the strongly selected NA mutations H275Y or A454V outcompete haplotypes carrying weaker mutations mediating cell adaptation, in cases where these weaker mutations did not hitchhike with the resistance mutations. Indeed, in the combined drug replicates, these weak beneficial mutations only fixed if they were associated with the NA mutations.
For example, in combined replicate 1, the H275Y haplotype swept the associated NP D101N and A37G to fixation; its rise coincided with the decline of haplotypes carrying E23Q. In combined replicate 2, the rise of the haplotype carrying NA A454V coincided with the extinction of MP1 E23Q, MP2 A37G, PA G58G, and NP D101N, until NP D101N was reshuffled onto the A454V background and fixed. In combined replicate 3, the rapid spread and fixation of haplotypes carrying NA A454V, NA E128G, and HA E78G coincided with the decline of MP1 E23Q. Some evidence of these effects is present in the oseltamivir-only replicates but not in the favipiravir-alone or no-drug replicates, where the spread of beneficial mutations mediating cell adaptation followed more standard trajectories characteristic of weakly selected mutations (fig. 4D–F).
In the favipiravir population, we observed the influence of a different form of interference between linked weakly selected beneficial and deleterious mutations (weak-selection Hill–Robertson Interference) (Hill and Robertson 1966; McVean and Charlesworth 2000). There was limited clustering and significant variance in allele trajectories over the longer lifespan of this population (fig. 4G). Linkage was likely between weakly selected mutations mediating cell adaptation and the increasing influx of slightly deleterious mutations. The high variance in allele trajectories was not observed to the same extent in the control (fig. 4H), the oseltamivir-only populations (fig. 4A–C), or in the combined drug populations (fig. 4D–F). Hitchhiking of beneficial, deleterious, and neutral variants with the resistance mutations accounts for almost all of the trajectories in the mutations in the combined drug populations, with the exception of combined replicate 1 passage 10 where genetic drift (i.e., Muller’s ratchet) led to the fixation of many neutral and deleterious mutations owing to population collapse.
Conclusion
To our knowledge, this represents the first genome-wide examination of the combined effects of oseltamivir and favipiravir treatment on virus populations. We found evidence of mutational meltdown in combined drug populations leading to extinction. Further, global estimates of effective population size were consistent between the favipiravir-only and combined drug replicates, and support an important role for Muller’s ratchet in facilitating this extinction—where the loss of the least loaded class is being driven both by genetic drift as well as the elevated mutation rate. Intriguingly, despite a lower mutation load, we observed a more rapid decline in relative growth rate in the combined drug population relative to the favipiravir-treated population. Strongly selected beneficial mutations influence the evolutionary dynamics in combined drug replicates by sweeping deleterious mutations to fixation. The timing of these sweeps coincides with sharp reductions in the number of segregating mutations after passage 6 in replicate 1 and passage 8 in replicate 2 (fig. 2B). This striking evolutionary dynamic is not apparent in the favipiravir-only population, where the identified beneficial mutations only mediate cell adaptation (and no resistance mutations are identified). Thus, fundamentally, these results suggest an interesting evolutionary trade-off. On the one hand, the combined drug effect may speed extinction owing to the stronger association between oseltamivir resistance mutations and the deleterious mutations induced by favipiravir treatment. On the other hand, the sequence space underlying oseltamivir resistance is explored rapidly under favipiravir treatment, thus allowing for higher segregating frequencies of a range of NA mutations in the population.
If indeed the hitchhiking of deleterious mutations is the key factor accelerating extinction, it is noteworthy that the strongly selected mutations required for hitchhiking need not be oseltamivir resistance mutations per se—and that in this experimental set-up those are simply the only beneficial mutations of necessary effect size. This suggests an interesting avenue of exploring combination treatments that may similarly invoke these hitchhiking effects to speed extinction under favipiravir treatment, but perhaps combined with a partner for which beneficial mutations would not be as clinically problematic as oseltamivir resistance.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Medical Research Program (Award No. W81XWH-15-1-0317) by the Defense Advanced Research Projects Agency (DARPA) Prophecy Program, Defense Sciences Office (DSO), Contract No. HR0011-11-C-0095, and D13AP00041, and support from MediVector, as well as funding from the European Research Council (ERC) to JDJ. We also thank Fuji Films for providing favipiravir. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Literature Cited
- Bank C, et al. 2016. An experimental evaluation of drug-induced mutational meltdown as an antiviral treatment strategy. Evolution 70:2470–2484. [DOI] [PubMed] [Google Scholar]
- Baranovich T, et al. 2013. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J Virol. 87:3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH. 2010. Genetic linkage and natural selection. Philos Trans R Soc Lond B Biol Sci. 365:2559–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom CT, Pritchard J.. 1998. Germline bottlenecks and the evolutionary maintenance of mitochondrial genomes. Genetics 149:2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Gong LI, Baltimore D.. 2010. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328:1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier NM, Rahmat S, Pica N.. 2012. Enhanced mammalian transmissibility of seasonal influenza A/H1N1 viruses encoding an oseltamivir-resistant neuraminidase. J Virol. 86:7268–7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J, et al. 2014. Estimating the fitness advantage conferred by permissive neuraminidase mutations in recent oseltamivir-resistant A(H1N1)pdm09 influenza viruses. PLoS Pathog. 10:e1004065.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 10:195–205. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D.. 1997. Rapid fixation of deleterious alleles can be caused by Muller’s ratchet. Genet Res. 70:63–73. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D.. 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134:1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung PP, et al. 2014. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat Commun. 5:4794.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins PJ, et al. 2008. Crystal structures of oseltamivir-resistant influenza virus neuraminidase mutants. Nature 453:1258–1261. [DOI] [PubMed] [Google Scholar]
- Daniels RS, et al. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431–439. [DOI] [PubMed] [Google Scholar]
- Ewens WJ. 1967. The probability of survival of a mutant. Heredity 22:307–310. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1974. The evolutionary advantage of recombination. Genetics 78:737–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. 1930. The genetical theory of natural selection. Oxford: Oxford University Press. [Google Scholar]
- Foll M, et al. 2014. Influenza virus drug resistance: a time-sampled population genetics perspective. PLoS Genet. 10:e1004185.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, et al. 2013. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Res. 100:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E, et al. 2012. Presence of oseltamivir-resistant pandemic A/H1N1 minor variants before drug therapy with subsequent selection and transmission. J Infect Dis. 206:1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginting TE, et al. 2012. Amino acid changes in hemagglutinin contribute to the replication of oseltamivir-resistant H1N1 influenza viruses. J Virol. 86:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B.. 2000a. On the speed of Muller’s ratchet. Genetics 156:2137–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B.. 2000b. The degeneration of asexual haploid populations and the speed of Muller’s ratchet. Genetics 154:1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordo I, Charlesworth B.. 2001. The speed of Muller’s ratchet with background selection, and the degeneration of Y chromosomes. Genet Res. 78:149–161. [DOI] [PubMed] [Google Scholar]
- Grande-Pérez A, Sierra S, Castro MG, Domingo E, Lowenstein PR.. 2002. Molecular indetermination in the transition to error catastrophe: systematic elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc Natl Acad Sci U S A. 99:12938–12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell BT, et al. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327–332. [DOI] [PubMed] [Google Scholar]
- Gubareva LV, Kaiser L, Matrosovich MN, Soo-Hoo Y, Hayden FG.. 2001. Selection of influenza virus mutants in experimentally infected volunteers treated with oseltamivir. J Infect Dis. 183:523–531. [DOI] [PubMed] [Google Scholar]
- Haigh J. 1978. The accumulation of deleterious genes in a population–Muller’s Ratchet. Theor Popul Biol. 14:251–267. [DOI] [PubMed] [Google Scholar]
- Hill WG, Robertson A.. 1966. The effect of linkage on limits to artificial selection. Genet Res. 8:269–294. [PubMed] [Google Scholar]
- Irwin KK, et al. 2016. On the importance of skewed offspring distributions and background selection in virus population genetics. Heredity 117:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives JA, et al. 2002. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Res. 55:307–317. [DOI] [PubMed] [Google Scholar]
- Jin H, Leser GP, Zhang J, Lamb RA.. 1997. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 16:1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorde PE, Ryman N.. 2007. Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, et al. 1999. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proc Natl Acad Sci U S A. 96:1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Mullins JI.. 2000. Lethal mutagenesis of HIV by mutagenic ribonucleoside analogs. AIDS Res Hum Retroviruses 16:1–3. [DOI] [PubMed] [Google Scholar]
- Lynch M, Bürger R, Butcher D, Gabriel W.. 1993. The mutational meltdown in asexual populations. J Hered. 84:339–344. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J, Haigh J.. 1974. The hitch-hiking effect of a favourable gene. Genet Res. 23:23–35. [PubMed] [Google Scholar]
- McVean GA, Charlesworth B.. 2000. The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics 155:929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer A, et al. 2014. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2012-2013. Antiviral Res. 110:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison DA. 2012. Prevention of drug resistance by combined drug treatment of tuberculosis. Handb Exp Pharmacol. 211:87–98. [DOI] [PubMed] [Google Scholar]
- Moscona A. 2005. Oseltamivir resistance–disabling our influenza defenses. N Engl J Med. 353:2633–2636. [DOI] [PubMed] [Google Scholar]
- Moscona A. 2009. Global transmission of oseltamivir-resistant influenza. N Engl J Med. 360:953–956. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1932. Some genetic aspects of sex. Am Nat. 66:118–138. [Google Scholar]
- Muller HJ. 1964. The relation of recombination to mutational advance. Mutat Res. 106:2–9. [DOI] [PubMed] [Google Scholar]
- Noton SL, et al. 2007. Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J Gen Virol. 88:2280–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noton SL, et al. 2009. Studies of an influenza A virus temperature-sensitive mutant identify a late role for NP in the formation of infectious virions. J Virol. 83:562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariente N, Sierra S, Lowenstein PR, Domingo E.. 2001. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J Virol. 75:9723–9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed ML, et al. 2009. Amino acid residues in the fusion peptide pocket regulate the pH of activation of the H5N1 influenza virus hemagglutinin protein. J Virol. 83:3568–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman JS, Lamb RA.. 2011. Influenza virus assembly and budding. Virology 411:229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra S, Dávila M, Lowenstein PR, Domingo E.. 2000. Response of foot-and-mouth disease virus to increased mutagenesis: influence of viral load and fitness in loss of infectivity. J Virol. 74:8316–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Tarbet EB, Furuta Y, Morrey JD, Barnard DL.. 2013. Synergistic combinations of favipiravir and oseltamivir against wild-type pandemic and oseltamivir-resistant influenza A virus infections in mice. Future Virol. 8:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashita E, et al. 2015. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2013-2014. Antiviral Res. 117:27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoennes S, et al. 2008. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 370:403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese JN, et al. 1998. Drug design against a shifting target: a structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase. Structure 6:735–746. [DOI] [PubMed] [Google Scholar]
- Ward J., Jr 1963. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 58:236–244. [Google Scholar]
- Wright S. 1931. Evolution in Mendelian populations. Genetics 16:97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.