


Abstract
Extracts of the human glioma cell line A1235 (lacking O6-methylguanine-DNA methyltransferase) are known to restore a G:T mismatch to a normal G:C pair in a G:T-containing model (45 bp) DNA substrate. Herein we demonstrate that substitution of G:T with O6-methylguanine:T (m6G:T) results in extract-induced intra-strand incision in the DNA at an efficiency comparable to that of complete repair of the G:T-containing substrate, although the m6G:T mispair serves as a poor substrate for later repair steps (e.g. gap filling, as judged by defective DNA repair synthesis). The A1235 extract, when supplemented with ATP and the four normal dNTPs, incises 5′ to the mismatched T, as inferred by the generation of a single-stranded 20mer fragment. Unlike its parental (A1235) counterpart, an extract of the alkylation-tolerant derivative cell line A1235-MR4 produces no 20mer fragment, even when thymine-DNA glycosylase (TDG) is added to the reaction mixture. In contrast, the A1235 extract, when augmented with TDG, catalyzes enhanced incision at m6G:T in the 45 bp DNA, yielding 5–10-fold greater 20mer than that of either extract or TDG alone. Interestingly, the absence of m6G:T incision activity in the A1235-MR4 extract is similar to that seen for extracts of several known mismatch repair-deficient cell lines of colon tumor origin. Together these results suggest that derivative A1235-MR4 cells are defective in m6G:T incision activity and that the efficiency of this activity in the parental (A1235) cells may depend on the presence of several ill-defined mismatch repair recognition proteins along with TDG and ATP.
INTRODUCTION
In living cells base mismatches can arise in DNA, for example from errors occurring during semi-conservative replication and genetic recombination and from spontaneous deamination of 5-methylcytosine (m5C) (1,2). In Escherichia coli base mismatches produced during replication and recombination are corrected by the MutHSL repair pathway (2). Accordingly, all eight possible types of the normal base mismatches are restored to Watson–Crick base pairs in a ‘long repair patch’ mode, which entails removal of the mismatched base followed by replacement of up to 3000 nt (3). In Homo sapiens an analogous pathway has been characterized, both genetically and mechanistically. In vitro studies of the latter pathway have revealed varying levels of repair specificity for the 8 base mismatches (4–7), as found earlier in the bacterial system (2). Anomalies in the human mismatch repair process have been linked to cancer predisposition (8–12), as well as to elevated rates of spontaneous mutation and pronounced microsatellite instability (13–15). Moreover, human mismatch repair-deficient (MMR–) cells are known to display varying levels of tolerance to DNA-damaging agents, such as N-methyl-N′-nitro-nitrosoguanidine (MNNG), and aberrant repair events are thought to be causally associated with enhanced cell killing by these agents (13,14).
In contrast, spontaneous hydrolytic deamination of m5C and cytosine in cellular DNA results in G:T and G:U mispairs, respectively, independent of semi-conservative replication or genetic recombination. In vertebrates CpG dinucleotides are exclusive sites of deoxycytidine methylation (16) and thus G:T mispairs arising from spontaneous hydrolytic deamination of m5C occur solely at CpG sites. In E.coli G:T mispairs formed in this fashion are acted upon by a mismatch-specific endonuclease, Vsr (17), whereas in human cells these errors are operated on by a G:T/G:U-recognizing G:T-specific thymine-DNA glycosylase (TDG) (18,19). A 55 kDa glycosylase displaying the latter activity has been purified from HeLa cells (20) and the cognate cDNA has been isolated (21). This TDG reacts with G:T mispairs in CpG sequences so as to remove the mismatched T, thereby creating an AP site in DNA (22). The enzyme also acts on a variety of other inappropriate base pair structures in DNA, such as O6-methylguanine (m6G):T and 2-amino-6-(methylamino)purine (AMAP):T (22). The in vitro G:T mismatch repair system likewise corrects 6-thioguanine (6TG):T (23) and 2,6-diaminopurine (DiAP):T lesions (24) and a purified human 55 kDa protein removes 3,N4-ethenocytosine and mismatched thymine from G:T mispairs as well (25). Another protein that contains a methyl CpG-binding domain, denoted MBD4, can also remove mismatched thymine from methyl CpG/GpT sequences (26). The precise mechanism by which TDG recognizes a mismatched T and its subsequent removal through N-glycosidic bond hydrolysis are not well understood. The enzyme is known to bind DNA containing a G:U mispair, as revealed by a DNase I footprint, and interaction between the N7-guanine moiety 3′ to the mismatched U and the protein is inferred from a methylation interference assay (27). Cell-free systems have been reported to possess G:T incision activity towards DNA substrates containing analogs of guanine and thymine involved in mispairing, suggesting that 2-aminoguanine and 4-ketothymine are both recognized by the enzyme (24). The purified human TDG is known to catalyze excision of uracil from a G:U mispair at a rate significantly higher than the excision of mismatched T from a G:T mispair (28).
Herein we have explored the possibility that human G:T repair activity requires one or more mismatch recognition proteins in order for TDG to incise efficiently at G:T mismatches. The human glioma cell line A1235 and its MNNG-resistant derivative line A1235-MR4 (29) have been utilized to test this hypothesis. The A1235 and A1235-MR4 cells are known to be mismatch repair-proficient (MMR+) and MMR–, respectively (R.S.Day and T.A.Kunkel, unpublished data) and the unidentified MMR protein defective in the latter cells may be necessary to facilitate G:T mismatch repair in addition to the G:T recognition protein TDG. In short, we have examined the G:T mismatch repair pathway by comparing the extent of incision of a 45 bp DNA containing a single m6G:T mispair (hereafter referred to as m6G:T* substrate) by an A1235 cell-free extract (henceforth denoted A1235 extract) with A1235-MR4 cell-free extract (referred to as A1235-MR4 extract). The m6G:T* substrate was incised in the strand containing and 5′ to the mismatched T base when incubated with the A1235 extract supplemented with ATP and the four normal dNTPs, whereas the m6G:T* substrate was not incised by the A1235-MR4 extract under similar conditions. This lack of incision activity by the latter extract cannot be attributed to a lack of TDG as exogenously added TDG was ineffective in restoring incision activity. Furthermore, both A1235 and A1235-MR4 extracts in the absence of ATP showed weak incision activity at m6G:T sites, reflecting low endogenous TDG activity. In addition, the A1235-MR4 extract behaved like extracts from colon tumor cell lines HCT15 and HCT116, with known defects in MMR recognition proteins. Collectively these results suggest that the A1235-MR4 extract is deficient in an ATP-dependent m6G:T-specific incision activity possibly due to a malfunctional MMR recognition protein.
MATERIALS AND METHODS
Preparation of cell-free extracts and enzymes
Cell line A1235 (Mer–) and its alkylation-resistant derivative line A1235-MR4 (Mer–) were cultured in DMEM or Ham’s F12 medium supplemented with 15% fetal bovine serum, 50 U/ml penicillin and 50 mg/ml streptomycin. Human colon tumor cell lines HT29, HCT116, HCT15, LoVo and DLD-1 (15) were also grown as described above. For each cell line confluent cultures, in ten 150 mm dishes, were harvested by standard cell trypsinization. Extracts (each equivalent to 2–5 × 108 cells) were prepared by the method of Manley et al. (30), snap frozen in liquid nitrogen and stored at –70°C. Each extract contained 15–20 mg protein/ml. Human recombinant TDG was a kind gift of Dr Josef Jiricny (Institute of Medical Radiobiology, Zurich, Switzerland). T4 polynucleotide kinase and the restriction enzymes HpaII and NarI were purchased from Pharmacia Biotech (Piscataway, NJ).
Preparation of DNA substrates
A phosphoroamidite derivative of m6G, supplied by American Bionetics (Emeryville, CA), was used in the synthesis of the m6G-containing 45mer oligonucleotide, which was performed by the Regional DNA Synthesis Laboratory (Calgary, Alberta, Canada) (31). All other oligonucleotides, including those containing a mismatched T, were prepared by the DNA Synthesis Laboratory, University of Alberta (Edmonton, Alberta, Canada). All oligonucleotides were purified by 12% sequencing PAGE followed by electroelution. Where indicated, DNA strands were labeled at the 5′-end using T4 polynucleotide kinase and [γ-32P]ATP and model duplex DNA substrates were prepared as described previously (31). DNA termini were always labeled prior to forming DNA duplexes, which were subsequently purified by 12% non-denaturing PAGE before use in DNA repair assays (see below). [α-32P]dCTP and [α-32P]TTP were both purchased from Amersham International (Little Chalfont, UK).
TDG incision assay
The TDG incision assay was carried out as described earlier (22,31). Briefly, labeled m6G:T* substrate or G:T-containing substrate (hereafter called G:T* substrate) (2 ng) was incubated (30°C for 12–24 h) with a cell-free extract or recombinant TDG in a buffer (total volume 50 µl) containing 20 mM HEPES, pH 7.9, 0.5 mM EDTA, 1 mM DTT, 0.01 mM ZnCl2 and 100 µg/ml BSA. Unlabeled 45 bp DNA (∼1 ng) containing no mismatched pair was included in the reaction mixture to serve as a competitive normal substrate for any non-specific strand incision activity. When the reaction contained purified TDG in place of a cell-free extract the substrate was subsequently treated with 0.1 N NaOH at 90°C for 30 min so as to convert any TDG-generated AP sites into single-strand breaks. The appearance of the resulting breaks was detected by 12% sequencing PAGE followed by autoradiography on X-ray film (22).
G:T/m6G:T mismatch-specific DNA synthesis assay
The repair synthesis assay was performed using a cell-free extract and the G:T* substrate. We followed both: (i) incorporation of radioactive [α-32P]dCMP or [α-32P]TMP into the unlabeled substrate; and (ii) repair of the mismatch-specific incision site by extract-mediated DNA synthesis (gap filling) and strand ligation, thus reflecting restoration of the 45 bp DNA substrate to a normal structure. Under similar conditions repair of the m6G:T* substrate was monitored by measuring appearance of the mismatch-specific incision (20mer) fragment, as conversion of the incised site into a fully repaired DNA duplex did not occur to any significant extent. Therefore, the repair assay involving the latter substrate is also referred to as the m6G:T incision assay. In short, the DNA repair synthesis reaction (total volume 50 µl) contained 20 mM HEPES, pH 7.9, 0.5 mM EDTA, 1 mM DTT, 50 mM KCl, 1 mM MgCl2, 0.01 mM ZnCl2, 2 µM ATP, 40 µg phosphocreatine/U creatine phosphokinase, 100 µg/ml BSA, 100 µM each of the four normal dNTPs (henceforth denoted dNTPs), either G:T* or m6G:T* substrate (2 ng) and cell extract (20 µg). When the substrates were unlabeled either dCTP or TTP was replaced by its radioactive deoxynucleoside triphosphate counterpart. Likewise, ddNTP was replaced by the corresponding dNTP when monitoring repair of the labeled G:T* or m6G:T* substrate. The reaction mixture was incubated at 30°C for 12–24 h and the DNA reaction products were analyzed by electrophoresis on 12% sequencing gels, as noted above (22).
RESULTS
Repair of G:T and m6G:T mispairs in a model DNA substrate
In order to measure G:T mismatch repair activity in the A1235 extract we first constructed a 45 bp heteroduplex, which contained either a single G:T or m6G:T mismatch at the same predetermined site and was labeled at the 5′-terminus in the strand harboring the mismatched T (Fig. 1). Results of the TDG incision and DNA repair synthesis assays are presented in Figure 2. As shown in lane 1, incubation of the G:T* substrate with the extract alone produced a labeled single-stranded fragment which migrated as a 20mer under the electrophoresis conditions employed. The size of the incision product is consistent with introduction of an intra-strand nick immediately 5′ to the mismatched T base that presumably had been released by a TDG-type activity (22,31). In lane 2 absence of the 20mer fragment suggests that the G:T* substrate was restored to normalcy by post-incision DNA repair synthesis and ligation in the presence of ATP and dNTPs, i.e. the multistep repair process has gone to completion. The amount of 20mer in lane 3, as revealed by HpaII digestion, is comparable to that seen in lane 1. This result implies that repair of the model substrate did indeed occur when the extract was supplemented with ATP and dNTPs, since failure to convert a G:T mismatch to a G:C pair in the 45 bp DNA would have blocked the endonucleolytic action of HpaII (32). To determine the specificity of the base incorporated during DNA repair synthesis (gap filling) at the mispair site, dCTP was replaced by the chain terminator ddCTP (100 µM) in the repair assay buffer. The repair activity in the A1235 extract produced predominantly a 21mer fragment (lane 4), which suggests that one nucleotide (i.e. dCMP) was added to the 20mer incision fragment by 5′→3′ DNA synthesis activity residing in the extract. In the presence of ddTTP (100 µM) instead of TTP, ddTMP was not incorporated at the incision site since no 21mer was observed (lane 5). Addition of ddATP and ddGTP to the reaction mixture likewise failed to yield a 21mer (data not shown). Hence, the addition of one specific nucleotide, namely dCMP, and the lack of any ladder formation greater in length than the 21mer indicate that only 1 nt (containing the mismatched thymine) was removed in the initial stages of repair, after which dCMP was inserted opposite the G template by a G:T mismatch repair synthesis activity and, finally, the strand was sealed to restore the intact duplex.
Figure 1.

The structure of the 45 bp model DNA substrate. The locations of the G:T or m6G:T mismatch and the HpaII and NarI restriction sites are indicated.
Figure 2.
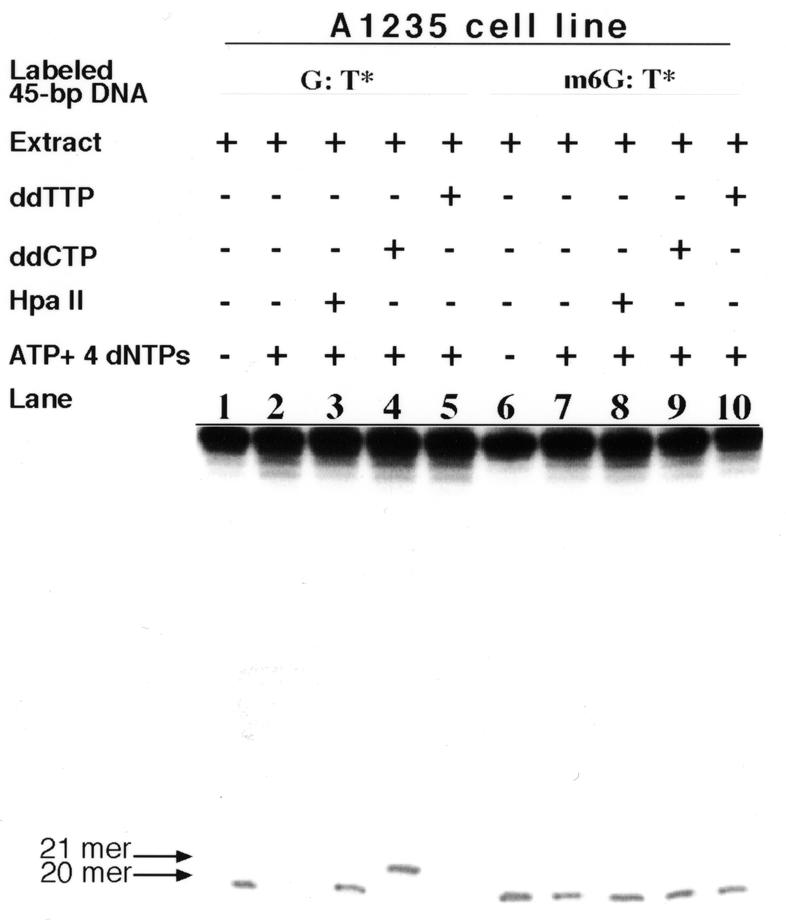
Repair of G:T and m6G:T mismatches in the model substrate by the A1235 extract. The extract (20 µg protein) and model substrate (2 ng), labeled at the 5′-end of the bottom strand (see Fig. 1), were incubated at 30°C for 18 h to conduct TDG incision and repair synthesis assays, as described in Materials and Methods. Lanes 1 and 6, TDG-induced incision of the G:T* and m6G:T* substrates (indicated as G:T* and m6G:T*, respectively); lanes 2–5, repair synthesis on the G:T* substrate; lanes 7–10, repair synthesis on the m6G:T* substrate under the assay conditions indicated. The TDG-induced incision product of the G:T* substrate yielded a labeled 20mer oligonucleotide (lane 1), which has been described elsewhere (31). The 21mer fragment in lane 4 arose from the addition of 1 nt (ddCMP) to the 20mer (see Results).
We next assessed the A1235 extract for its ability to repair the m6G:T* substrate. As shown in Figure 2, lane 6, the extract alone was proficient at incising m6G:T sites, as judged by the abundant yield of the 20mer fragment, inferring that the early reactions leading to intra-strand cleavage at the mismatched T site had taken place normally. It is also evident in lanes 7–10 that the addition of ATP and dNTPs to the extract did not eliminate appearance of the 20mer product. The latter observation demonstrates that the A1235 extract was defective in executing later repair steps (e.g. gap filling).
Figure 3 illustrates repair of the unlabeled G:T* substrate by the A1235 extract supplemented with radioactive dCTP, the three remaining (non-radioactive) dNTPs and ATP. Lane 2 reveals that a full-length 45mer duplex and a 21mer fragment containing the radioactive nucleotide were produced in the aforementioned reaction mixture. The 21mer product was presumably formed by the gap filling addition of one labeled dCMP to the 20mer incision fragment (shown in lane 1), whereas the full-length DNA presumably resulted from completion of the entire repair process. To confirm this latter possibility the reaction mixture-treated DNA was digested with HpaII and NarI. As the G:T mispair in the model substrate was located at overlapping HpaII and NarI sites, 5′-GGCGCCGG-3′/3′-CCGCGGTC-5′ (see Fig. 1), conversion of the G:T mispair to a normal G:C pair would be required in order to observe restriction enzyme digestion of the substrate. The fact that digestion of the extract-treated substrate with HpaII and NarI specifically produced labeled 25mer and 23mer fragments, respectively (lanes 3 and 4), indicates that the extract-treated DNA contained a G:C pair (dCMP as a radioactive derivative) in place of a G:T mispair, given that the G:T* substrate had been shown earlier to be resistant to HpaII/NarI digestion (32). In vitro repair of the unlabeled m6G:T* substrate by the extract in the presence of radioactive dCTP or TTP did not give rise to a labeled oligonucleotide corresponding to either a 21mer or 23mer and 25mer restriction products (Fig. 3, lanes 5–7). These latter findings are in complete agreement with the results described above. Hereafter, characterization of general G:T mismatch repair in the extracts was confined to following the initial incision of only one substrate, namely m6G:T*.
Figure 3.
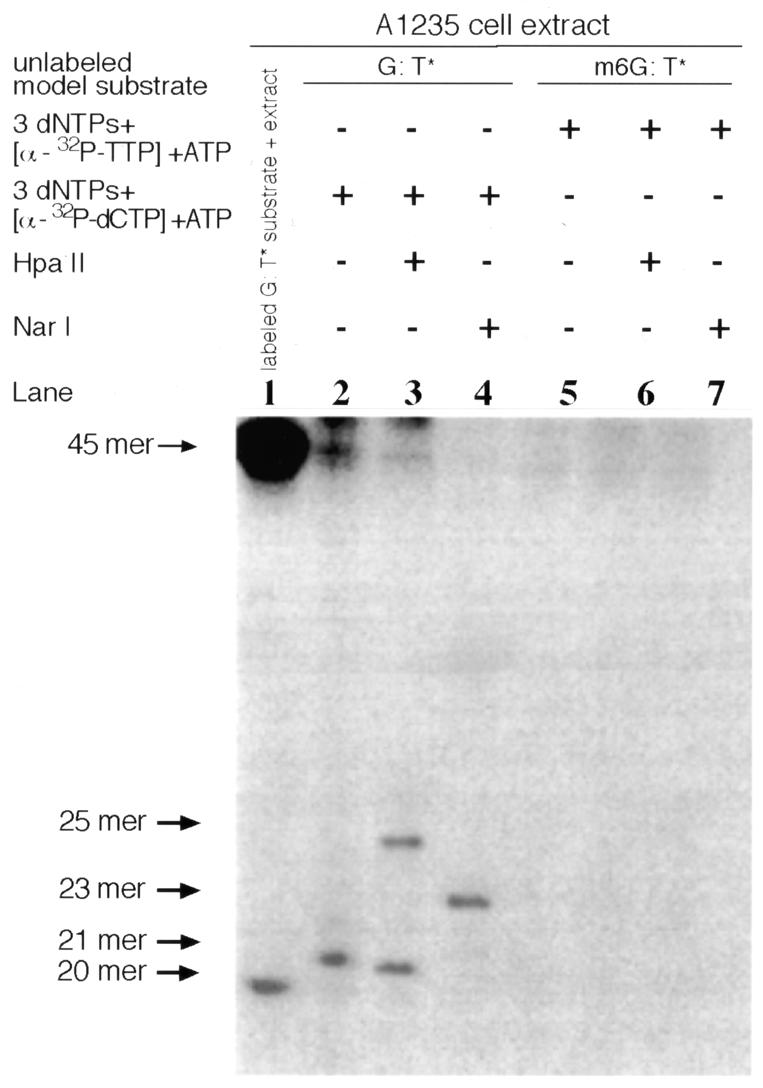
DNA repair synthesis performed by the A1235 extract on the G:T* or m6G:T* substrate. The A1235 extract (20 µg protein) and each unlabeled substrate (2 ng) were incubated (30°C for 12 h) with labeled deoxynucleoside triphosphates, as indicated at the top of the figure, whereupon the resulting radioactively labeled DNA was analyzed by restriction digestion. Unlabeled G:T* substrate (designated G:T*) was converted to labeled full-length DNA and an unligated 21mer (lane 2); the corresponding labeled substrate gave rise to a 25mer restriction fragment following HapII digestion (lane 3) and a 23mer following NarI digestion (lane 4). Lane 1, TDG-mediated incision of the labeled G:T* substrate; lanes 5–7, DNA repair synthesis performed by the extract on the m6G:T * substrate under the conditions indicated.
Effect of ATP on the rate of extract-mediated incision at a m6G:T mispair site
As shown in Figure 4, the A1235 extract when supplemented with ATP and dNTPs acted efficiently on the m6G:T* substrate, yielding a labeled 20mer fragment arising from the introduction of an intra-strand nick immediately 5′ to the mismatched T (compare lanes 1 and 2). In some, but not all, experiments the 20mer is accompanied by a limited amount of 21mer, implying addition of a single nucleotide. The incision rate was reduced drastically when the substrate was incubated with the A1235-MR4 extract augmented with ATP and dNTPs (lane 6). To confirm that exogenous ATP was indeed responsible for the differential m6G:T incision activity exhibited by the two extracts the reaction conditions were monitored as follows. The substrate was first mixed with ATP and intra-strand incision was initiated 10 min thereafter by extract addition. In this case the 20mer fragment was generated when the reaction mixture contained the A1235 extract (lane 3) but not the A1235-MR4 extract (lane 7). Similarly, incubation of the A1235 extract with ATP for 10 min followed by addition of the model substrate produced a 20mer fragment; however, very little product was observed with the A1235-MR4 extract (compare lanes 4 and 8). In contrast, incision of the m6G:T* substrate occurred to a similar extent with both extracts when the incision reaction was performed in the absence of ATP and dNTPs (lane 5 versus 9). One plausible interpretation of these surprising results is that ATP and dNTPs serve to inhibit an m6G:T mismatch incision activity in the A1235-MR4 extract.
Figure 4.
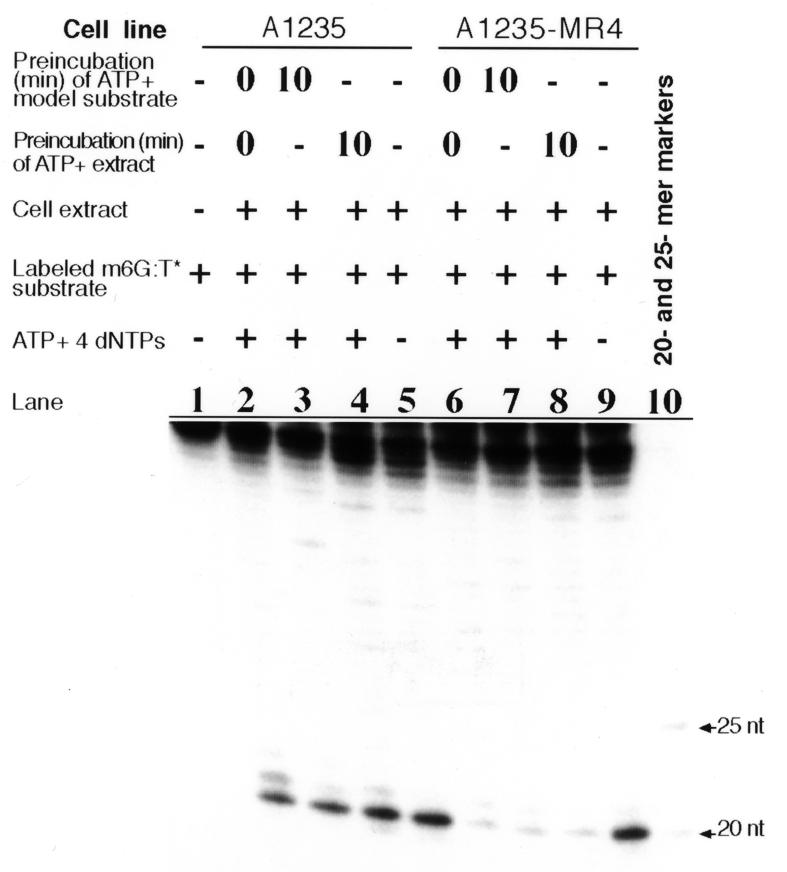
Effect of the addition of ATP on the ability of cell-free extracts to incise the m6G:T* substrate. The A1235 or A1235-MR4 extract (20 µg protein) was incubated at 30°C for 24 h with the labeled model substrate in repair synthesis assay buffer. Analysis of the DNA reaction products was performed as described in Materials and Methods. Lane 1, only m6G:T* substrate in the assay buffer; lanes 2–5, incision of the m6G:T* substrate by the A1235 extract; lanes 6–8, incision of the same substrate by the A1235-MR4 extract under the conditions indicated; lanes 5 and 9, incision by the A1235 and A1235-MR4 extracts, respectively, in the absence of ATP and dNTPs. Markers in lane 10 were produced by HpaII digestion of the model substrate containing the normal G:C pair instead of the G:T mispair.
Effect of dNTPs on extract-induced incision at a m6G:T mispair site
Next we wished to preclude the distinct possibility that the A1235-MR4 extract acts aberrantly on the m6G:T* substrate, introducing an incision proximal to the mispair, followed by nick translation to yield a full-length 45 bp duplex. To this end we assayed m6G:T incision activity in both A1235 and A1235-MR4 extracts when supplemented with two or more of the four dNTPs. As shown in Figure 5, lane 2, the A1235 extract-containing mixture operated on the m6G:T* substrate so as to produce a 20mer fragment, which is not seen in lane 5 as a result of no such activity in the A1235-MR4 extract. The A1235 extract with ATP and only three dNTPs (i.e. missing TTP) gave rise to a 20mer fragment (lane 3). In contrast, under the same conditions the A1235-MR4 extract did not produce a 20mer or any higher order fragment as a truncated nick translation product (lane 6). Similarly, extracts containing ATP and only two dNTPs (i.e. without dCTP and TTP) were tested for incision activity at a m6G:T mispair. Under these conditions the A1235 extract (lane 4) incised at a m6G:T site to give a 20mer fragment, the efficiency of which was comparable to that of the assay results in lane 2, while the A1235-MR4 extract failed to generate any 20mer product (lane 7). These combined results demonstrate that a 20mer product was not formed with the m6G:T* substrate when the reaction mixture contained the A1235-MR4 extract.
Figure 5.
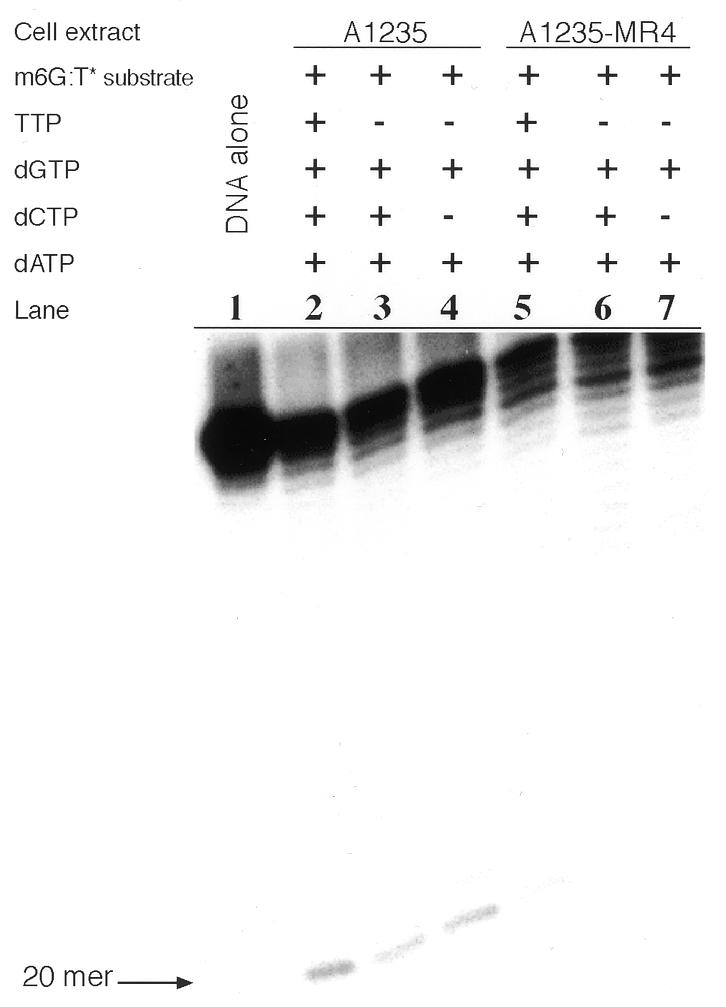
Effect of the presence of two or more dNTPs on the ability of the A1235 and A1235-MR4 extracts to incise the m6G:T* substrate. Each extract (20 µg protein) was incubated (30°C for 16 h) with labeled m6G:T* substrate (2 ng) in DNA repair synthesis buffer. Strand incision was assayed using either the A1235 extract (lanes 2–4) or the A1235-MR4 extract (lanes 5–7) supplemented with different combinations of dNTPs, as indicated, and the products were analyzed as described in Figure 4. Lane 1, sham-treated DNA; the 20mer reaction product is identified by an arrow.
Effect of TDG on extract-mediated incision in the m6G:T* substrate
We questioned whether the lack of incision at a m6G:T mismatch by the A1235-MR4 extract in comparison to the parental extract was attributable to a defect in the G:T mismatch recognition protein TDG, which has been implicated in repair of a G:T mispair in human cells (21,22). First, the activity of human TDG protein was measured by the TDG incision assay employing varying amounts of enzyme (0.02–2 µg/ml TDG) and the m6G:T* substrate. As shown in lane 1 of Figure 6A, the substrate when not treated with NaOH following incubation with TDG contained an insignificant amount of incision sites, but a substantial quantity of 20mer was formed after NaOH treatment (lane 2). These observations are consistent with the TDG activity first removing the mismatched T base, whereupon the resulting AP site is converted to a single-strand break by the alkali treatment (22). TDG (40 and 20 ng/ml) acted very weakly, if at all, on the m6G:T* substrate, as the 20mer fragment was not seen in lanes 3–6. Therefore, TDG at a concentration of 20 ng/ml was added to extracts in the investigation of m6G:T mismatch repair described below.
Figure 6.
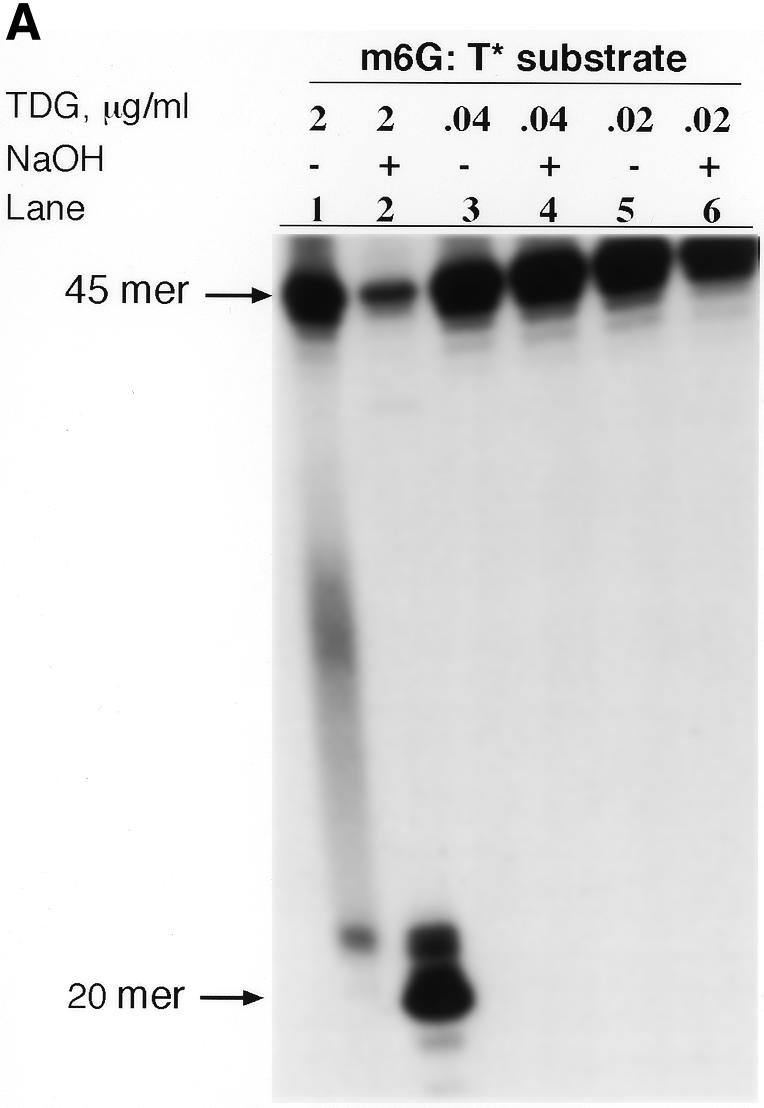
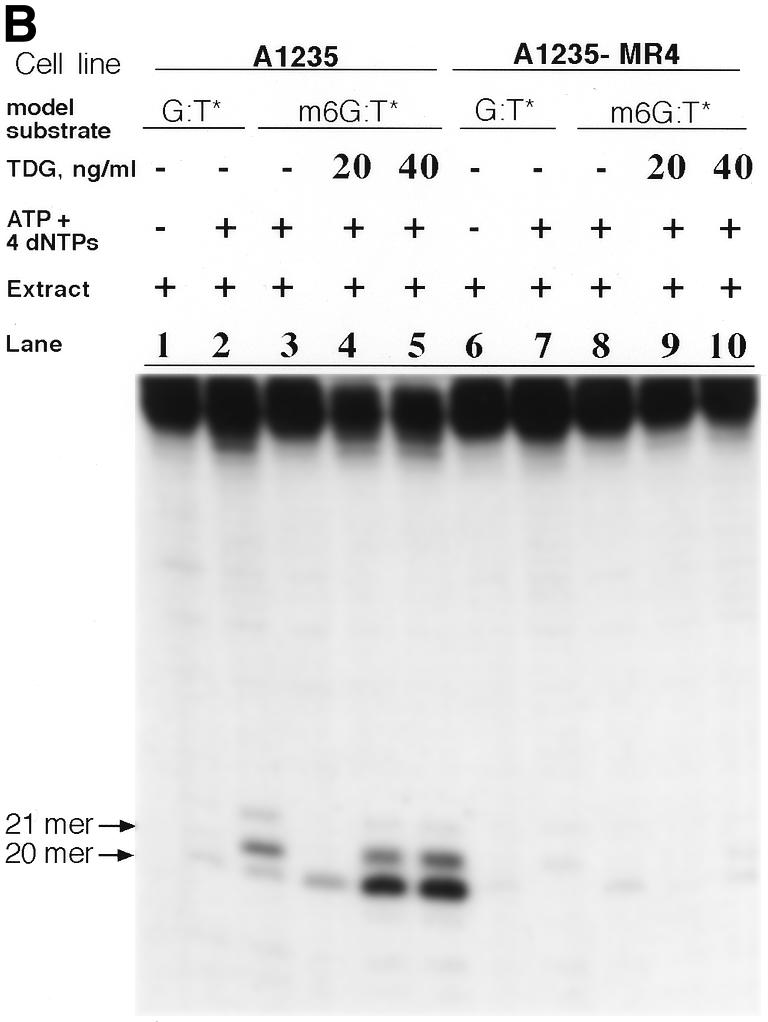
(A) TDG assay of the m6G:T* substrate. Purified TDG (0.02–2 µg protein/ml) was incubated with labeled substrate (2 ng) for 12 h at 30°C. DNA reaction products, which were either treated (+) or not (–) with NaOH, are shown in the respective lanes. The predominant incision product in lane 2 is a 20mer, as reported elsewhere (22). (B) Effect of the presence of purified human TDG on the ability of the A1235 and A1235-MR4 extracts to repair the m6G:T* substrate. Each extract (20 µg protein), supplemented with 0, 20 or 40 ng/ml TDG, and labeled substrate (2 ng) were used in the DNA incision and repair synthesis assays as noted in the figure. Each assay was carried out for 12 h at 30°C and product analysis was performed as described in Materials and Methods. Lanes 1–2 and 6–7, incision and gap filling of the G:T* substrate by the A1235 and A1235-MR4 extracts, respectively. Results of the m6G:T incision assay are also shown: lane 3, substrate acted on by the A1235 extract in the absence of TDG; lanes 4 and 5, substrate acted on by the extract and 1 or 2 ng TDG, respectively; lanes 8–10, repair of the m6G:T* substrate by the A1235-MR4 extract containing 0, 1 and 2 ng TDG, respectively. Note also that ATP and dNTPs were present in the assay mixtures, except for lanes 1 and 6. Arrows indicate the size of incision products.
An equal amount of purified human TDG was added to either A1235 or A1235-MR4 extract and the TDG incision and repair synthesis assays were performed. As shown in Figure 6B (lanes 1 and 2), the A1235 extract alone produced both a 20mer and a 21mer, in keeping with the presence of both G:T incision (TDG) and G:T repair synthesis activities in the A1235 extract. As expected, incubation of the m6G:T* substrate with the A1235 extract augmented with ATP and dNTPs generated a 20mer fragment (lane 3). Addition of TDG at 20 and 40 ng/ml to this reaction mixture resulted in relatively more extensive incision in the m6G:T* substrate. In fact, not only did the amount of 20mer appear to increase with increasing amounts of TDG in the reaction mixture (lanes 4 and 5), but a substantial quantity of 21mer was also formed. It is noteworthy that the yield of the 20mer fragment in lanes 4 and 5 was ∼5–10-fold greater than that produced by either the extract alone (lane 3) or TDG alone (see Fig. 6A, lanes 3–6). Similarly, the A1235-MR4 extract when assayed for TDG incision and repair synthesis activities towards the G:T* substrate displayed relatively poor responses, as indicated by the drastically reduced yield of 20mer and 21mer fragments, respectively (Fig. 6B, lanes 6 and 7). Surprisingly, the weak incision activity of the A1235-MR4 extract towards the m6G:T* substrate (see the 20mer band in lane 8) was not stimulated by the addition of TDG (20 and 40 ng/ml) to the reaction mixture, as the yield of 20mer in lanes 9 and 10 is unchanged from that found in lane 8. These results suggest that the lack of incision at the m6G:T site by the A1235-MR4 extract can be ascribed to a deficiency of a protein(s) which serves as a critical component of the m6G:T incision apparatus. Intriguingly, the efficiency of the parental (A1235) extract, which presumably contains a fully functional m6G:T incision complex, is enhanced by addition of TDG.
Incision at a m6G:T mismatch by known MMR– cell extracts
Because A1235-MR4 cells are resistant to killing by MNNG compared to parental (A1235) cells (29), the former cells are phenotypically reminiscent of colon carcinoma cell lines harboring a deficiency in one or more known MMR factors (4–7). We therefore monitored the efficiency of m6G:T-specific incision by several colon carcinoma cell extracts, which were prepared in a manner identical to the A1235-MR4 extract (30). Each extract was supplemented with 20 ng/ml TDG, ATP and dNTPs and the reaction mixture was incubated with the m6G:T* substrate. The results of the incision assay are shown in Figure 7. The appearance of a 20mer, which is indicative of m6G:T-specific incision activity in a given cell extract, is listed in Table 1, along with other pertinent properties of the corresponding cell line. As can be seen in Figure 7, the A1235 extract (lane 2), but not the A1235-MR4 extract (lane 3), yielded a 20mer fragment following incubation with the m6G:T* substrate, as found previously in Figure 6B. The finding that extracts of colon carcinoma lines HCT15 and HCT116 did not produce a characteristic 20mer fragment after incubation with the DNA substrate (lanes 5 and 6) is identical to that of the A1235-MR4 extract (lane 3) with respect to the absence of m6G:T-specific incision activity. Unlike the A1235-MR4 extract, however, the MMR– extracts of cell lines DLD-1and LoVo were able to generate a 20mer by incising the substrate (lanes 7 and 8). Here again, as illustrated earlier in Figure 6B, the addition of TDG to an extract containing proficient m6G:T incision activity generated a 21mer fragment, in addition to the 20mer fragment. Consequently, MMR– extracts, as a group, do not consistently lack ATP-dependent m6G:T incision activity (Table 1). Finally, the HT29 extract, like the A1235 extract, proved to be proficient in nicking the m6G:T* substrate (lane 4), suggesting that MMR+ cell extracts possess ATP-dependent m6G:T incision activity (Table 1).
Figure 7.
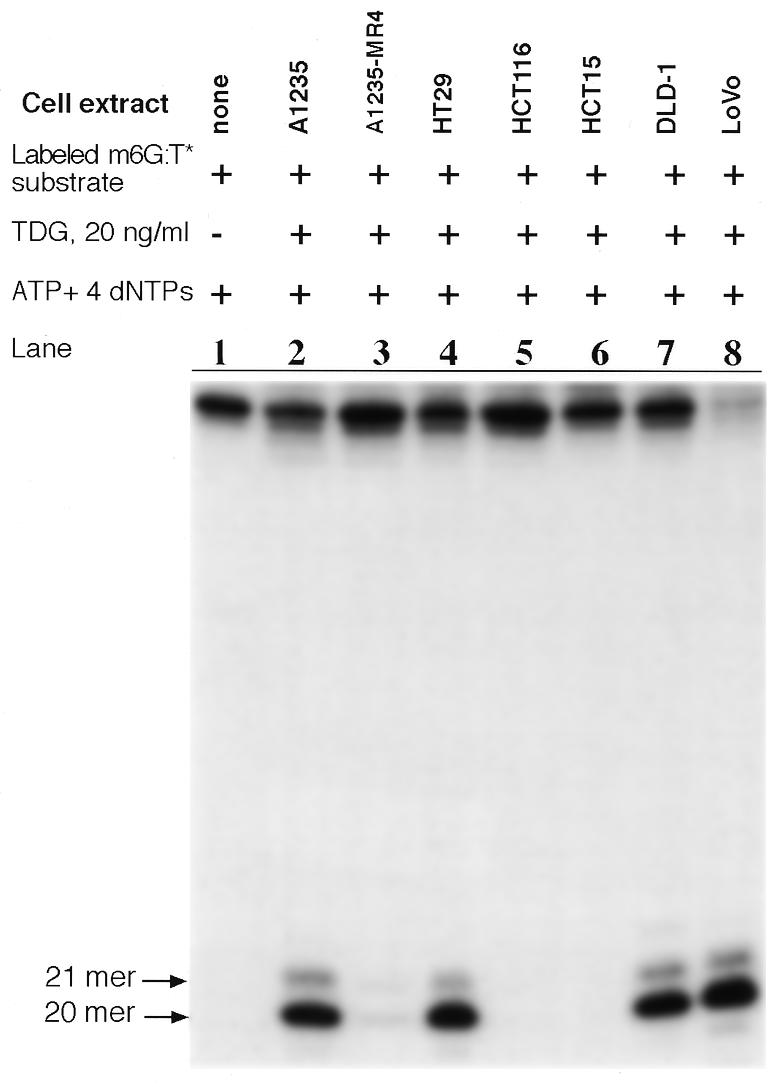
Capacity of extracts of different human tumor cell lines to incise the m6G:T* substrate. To an extract (20 µg protein) of each indicated cell line was added an equal quantity of purified TDG and each mixture was then incubated with labeled substrate (2 ng). After performing the DNA repair synthesis assay (12 h at 30°C) the extent of incision was analyzed as described in Materials and Methods. The appearance of a 20mer as a product of the m6G:T incision activity is indicated by an arrow. Note that in lanes 2, 4, 7 and 8 extracts produced 21mer as a minor product, possibly due to incorporation of a single dCMP or TMP opposite m6G in the m6G:T* substrate following the action of the ATP-dependent m6G:T incision activity.
Table 1. Comparison of ATP-dependent m6G:T mismatch incision activity in a cell-free extract with various conventional mismatch repair hallmarks for various human cell lines.
| Cell line |
ATP-dependent m6G:T incision activitya (20mer fragment) |
Mismatch repair activityb |
Mutator phenotypec |
Mismatch repair gene mutationd |
| HT29 | + | ND | – | ND |
| LoVo | + | – | + | MSH2, Δ exons 3–8 |
| HCT15 | – | – | + | MSH6, 1,5 bp del at 222, 1103 |
| HCT116 | – | – | + | MLH1, TCA→TAA at 252 |
| DLD-1 | + | – | + | MSH6, 1,5 bp del at 222, 1103 |
| A1235 | + | + | – | ND |
| A1235-MR4 | – | – | + | ND |
ND, not determined.
aATP-dependent m6G:T incision activity as measured in extracts supplemented with purified TDG (see Figs 6B and 7).
bMismatch repair activity in extracts of LoVo, DLD-1 and HCT15 (37,38), HCT116 (39) and A1235-MR4 (R.S.Day and T.A.Kunkel, unpublished data) cells.
cHT29, LoVo, HCT15, HCT116 and DLD-1(15) and A1235 and A1235-MR4 (R.S.Day et al., unpublished work).
dReported mutations in the mismatch repair genes: MSH2 in LoVo (37), MSH6 in DLD-1 and HCT15 (36) and MLH1 in HCT116 (39).
Detection of ATP-independent m6G:T incision activity
Finally, we assayed m6G:T incision activity in the absence of ATP and dNTPs in colon tumor and glioma (A1235 and A1235-MR4) cell extracts. To this end the m6G:T* substrate was incubated with each extract and in all cases a 20mer fragment was generated, as shown in Figure 8. The size of the incision fragment corresponded to the introduction of a nick 5′ to the mismatched T, suggesting that TDG activity was present at significant levels in all seven cell extracts (31). Thus, A1235-MR4, HCT15 and HCT116 extracts contained similar levels of ATP-independent incision activity and yet m6G:T incision activity was abolished in these same three extracts when ATP was present in the reaction mixture.
Figure 8.
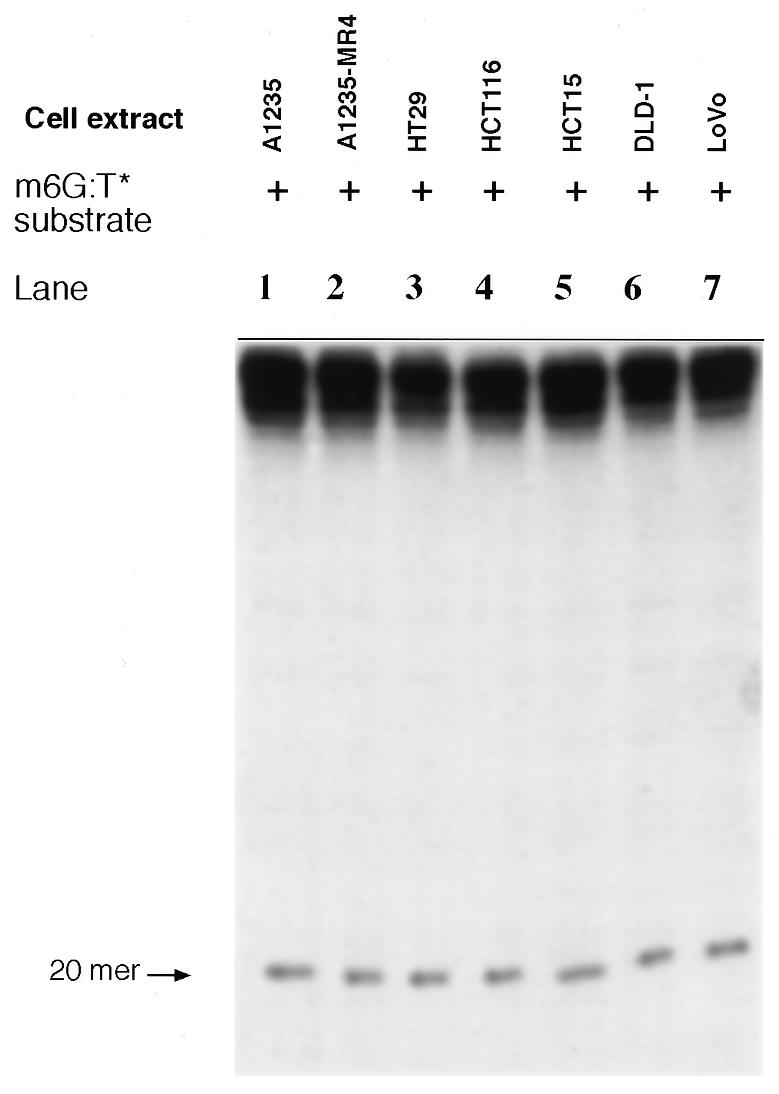
TDG-induced incision of the m6G:T* substrate. An extract of each indicated cell line (20 µg protein) was incubated with the labeled substrate (2 ng) in TDG incision assay buffer. After incubation for 16 h at 30°C DNA reaction products were analyzed as described in Materials and Methods.
DISCUSSION
Data presented above indicate that the A1235 extract of human glioma origin catalyzes both intra-strand incision immediately 5′ to the mismatched T at a G:T mispair and subsequent replacement of the mismatched T nucleotide with dCMP in the model substrate (Figs 2 and 3). This finding is in keeping with the working hypothesis that TDG removes the mismatched T base, whereupon endonuclease APE1 in the extract ruptures the phosphodiester linkage immediately 5′ to the resulting AP site to create a 1 nt gap in the DNA (31–34). This enzymatically modified substrate with the 1 nt gap is then filled by DNA polymerase β activity (33) and a DNA ligase seals the nick to complete restoration of a G:T mispair to a normal G:C pair in the DNA.
Incision of the m6G:T* or G:T* substrate occurred at a rate corresponding to that of appearance of TDG incision products (compare Fig. 2, lanes 1 and 6 with Fig. 6A, lane 2). Incision efficiency at a m6G:T mispair by the A1235 extract (Fig. 2, lanes 7–10) was comparable to the overall repair efficiency of the G:T* substrate under the same assay conditions (Fig. 2, lanes 2 and 3). These findings support the contention that a m6G:T or G:T mispair in the model substrate is acted upon by one and the same G:T mismatch incision activity (see also 31). Moreover, the negligible DNA repair synthesis at a m6G:T site relative to efficient repair of G:T→G:C by the A1235 extract (Figs 2 and 3) is consistent with the fact that the post-incision DNA synthesis activity, mediated by DNA polymerase β (33), is known to act with a substantially reduced efficiency on DNA containing a small single-strand gap with m6G, as opposed to G, opposite the gap (35).
The demonstration that purified TDG elicited two different responses with respect to incision of the m6G:T* substrate after addition to the parental and derivative cell extracts is worth emphasizing. On the one hand, the data in Figure 6B (20mer band, lane 3 versus 4 and 5) clearly depict an interaction between the parental (A1235) extract and TDG, as revealed by the several-fold increase in m6G:T incision activity in the presence of ATP. Paradoxically, augmentation of the derivative (A1235-MR4) extract with ATP blocked incision completely (Fig. 6B, lane 8 versus 9 and 10). On the other hand, when ATP was absent from the m6G:T incision assay mixture the A1235-MR4 extract incised at m6G:T sites at a rate comparable to that of the A1235 extract, as shown in Figure 4 (see 20mer in lanes 5 and 9). The most plausible interpretation of these unexpected findings is that human cells may possess two m6G:T mismatch incision activities, namely an ATP-dependent m6G:T incision activity and an ATP-independent TDG-like incision activity, with the former activity being malfunctional in the derivative extract but not the parental extract.
As shown in Table 1, extracts of three of the seven cell lines tested, namely HCT15, HCT116 and A1235-MR4, were found to lack detectable ATP-dependent m6G:T incision activity. Tumor cell line HCT15, which is known to harbor deletion mutations in one of the human mutS gene homologs, MSH6 (also denoted GTBP), is deficient in in vitro repair of heteroduplexes containing all eight types of single normal base mismatches (Table 1; 36–38). HCT116 contains no functional MLH1 (one of the human mutL homologs) protein (Table 1; 11,39). The A1235 and A1235-MR4 cell lines both lack detectable O6-methylguanine-DNA methyltransferase (MGMT) (our unpublished data); however, the former line is MMR proficient whereas the latter is MMR deficient (R.S.Day and T.A.Kunkel, unpublished data). Defects in mismatch repair genes and loss of mismatch repair activity correlate closely with loss of ATP-dependent m6G:T incision activity in some of the cell lines, i.e. HCT15, HCT116 and A1235-MR4 (see Table 1). We therefore hypothesize that a mismatch repair gene(s) may be mutated in A1235-MR4 cells, given that ATP-dependent m6G:T incision activity in the corresponding extract is undetectable. This deficiency in the A1235-MR4 extract was corrected by addition of the fully functional A1235 extract. However, our initial attempt to restore activity to the A1235-MR4 extract by mixing with both of the well-defined MMR– extracts (HCT15 and HCT116) proved unsuccessful, possibly suggesting that the human m6G:T incision machinery is simply too complex to permit functional complementation between crude cell-free extracts.
It should be mentioned that the MutSα (MSH2:MSH6) mismatch-binding complex may not be responsible for targeting the ATP-dependent m6G:T incision complex to one of our model substrates (i.e. m6G:T*), since activity of the latter complex is detectable in the LoVo extract (Table 1). This extract is known to be defective in MutSα as a result of dysfunctional mutations in the MSH2 gene (36,38). As shown in Table 1, detection of constitutional ATP-dependent m6G:T incision activity in the DLD-1, but not the HCT15, extract is intriguing, given that the MSH6 gene is defective in both cell lines. It is uncertain whether MSH6 protein is stable in the absence of functional MSH2 in LoVo cells or is in fact partially functional in DLD-1 cells due to the cognate gene being present in the heterozygous state. The answer must await further investigation.
In summary, this study provides evidence for the existence of a novel ATP-dependent repair activity in human cell extracts in which incision at m6G:T (G:T) sites requires one or more MMR recognition factors in addition to TDG. Our findings that (i) an ATP-dependent m6G:T incision activity is defective in many MMR– cell extracts and (ii) ATP constitutes a requisite cofactor, serve to distinguish this activity from the known TDG-like incision activity. In addition, the present study describes, for the first time, an assay system well-suited to characterize the early steps and the participating MMR proteins in the enzymatic repair of m6G:T mismatches. Exploitation of this novel assay system promises to elucidate the functional inter-relationship between m6G:T mismatch repair and the more general MMR machinery in human cells.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Dr Sultan Al-Sedairy for his encouragement and support. We acknowledge Dr Abdelilah Aboussekhra for his constructive commentary on the manuscript. This work was supported by Research Proposal No. 2000 006 from the Research Advisory Council, KFSH&RC, Saudi Arabia.
References
- 1.Duncan B.K. and Miller,J.H. (1980) Mutagenic deamination of cytosine residues in DNA. Nature, 287, 560–561. [DOI] [PubMed] [Google Scholar]
- 2.Modrich P. (1991) Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet., 25, 229–253. [DOI] [PubMed] [Google Scholar]
- 3.Wagner R.Jr and Messelson,M. (1976) Repair tracts in mismatched DNA heteroduplex. Proc. Natl Acad. Sci. USA, 73, 4135–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiricny J. (1998) Eukaryotic mismatch repair: an update. Mutat. Res., 409, 107–121. [DOI] [PubMed] [Google Scholar]
- 5.Kolodner R. (1996) Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev., 10, 1433–1442. [DOI] [PubMed] [Google Scholar]
- 6.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 7.Modrich P. (1997) Strand-specific mismatch repair in mammalian cells. J. Biol. Chem., 272, 24727–24730. [DOI] [PubMed] [Google Scholar]
- 8.Leach F.S., Nicolaides,N.C., Papadopoulos,N., Liu,B., Jen,J., Parsons,R., Peltomäki,P., Sistonen,P., Aaltonen,L.A., Nyström-Lahti,M., Guan,X.-Y., Zhang,J., Meltzer,P.S., Yu,J.-W., Kao,F.-T., Chen,D.J., Cerosaletti,K.M., Fournier,R.E.K., Todd,S., Lewis,T., Leach,R.J., Naylor,S.L., Weissenbach,J., Mecklin,J.-P., Järvinen,H. et al. (1993) Mutations of a MutS homolog in hereditary nonpolyposis colorectal cancer. Cell, 75, 1215–1225. [DOI] [PubMed] [Google Scholar]
- 9.Parsons R., Li,G.-M., Longley,M.J., Fang,W.-H., Papadopoulos,N., Jen,J., de la Chapelle,A., Kinzler,K.W., Vogelstein,B. and Modrich,P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell, 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 10.Fishel R., Lescoe,M.K., Rao,M.R.S., Copeland,N.G., Jenkins,N.A., Garber,J., Kane,M. and Kolodner,R. (1993) The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell, 75, 1027–1038. [DOI] [PubMed] [Google Scholar]
- 11.Papadopoulos N., Nicolaides,N.C., Wei,Y.-F., Ruben,S.M., Carter,K.C., Rosen,C.A., Hasteline,W.A., Fleischmann,R.D., Fraser,C.M., Adams,M.D., Venter,J.C., Hamilton,S.R., Petersen,G.M., Watson,P., Lynch,H.T., Peltomäki,P., Mecklin,J.-P., de la Chapelle,A., Kinzler,K.W. and Vogelstein,B. (1994) Mutation of mutL homolog in hereditary colon cancer. Science, 263, 1625–1629. [DOI] [PubMed] [Google Scholar]
- 12.Bronner C.E., Baker,S.M., Morrison,P.T., Warren,G., Smith,L.G., Lescoe,M.K., Kane,M., Earabino,C., Lipford,J., Lindblom,A., Tannergard,P., Bollag,R.J., Godwin,A.R., Ward,D.C., Nordenskjold,M., Fishel,R., Kolodner,R. and Liskay,R.M. (1994) Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature, 368, 258–261. [DOI] [PubMed] [Google Scholar]
- 13.Branch P., Aquilina,G., Bignami,M. and Karran,P. (1993) Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature, 362, 652–654. [DOI] [PubMed] [Google Scholar]
- 14.Kat A., Thilly,W.G., Fang,W.-H., Longley,M.J., Li,G.-M. and Modrich,P. (1993) An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc. Natl Acad. Sci. USA, 90, 6424–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhattacharyya N.P., Skandalis,A., Ganesh,A., Groden,J. and Meuth,M. (1994) Mutator phenotypes in human colorectal carcinoma cell lines. Proc. Natl Acad. Sci. USA, 91, 6319–6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razin A. and Riggs,A.D. (1980) DNA methylation and gene function. Science, 210, 604–610. [DOI] [PubMed] [Google Scholar]
- 17.Hennecke F., Kolmer,H., Bründl,K. and Fritz,H.-J. (1991) The vsr gene product of E. coli K12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature, 353, 776–778. [DOI] [PubMed] [Google Scholar]
- 18.Brown T.C. and Jiricny,J. (1987) A specific mismatch repair event protects mammalian cells from loss of 5-methylcytosine. Cell, 50, 945–950. [DOI] [PubMed] [Google Scholar]
- 19.Wiebauer K. and Jiricny,J. (1989) In vitro correction of G.T mispairs to G.C pairs in nuclear extracts from human cells. Nature, 339, 234–236. [DOI] [PubMed] [Google Scholar]
- 20.Neddermann P. and Jiricny,J. (1993) The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J. Biol. Chem., 268, 21218–21224. [PubMed] [Google Scholar]
- 21.Neddermann P., Gallinari,P., Lettieri,J., Schmid,D., Truong,O., Hsuan,J.J., Wiebauer,K. and Jiricny,J. (1996) Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J. Biol. Chem., 271, 12767–12774. [DOI] [PubMed] [Google Scholar]
- 22.Sibghat-Ullah, Gallinari,P., Xu,Y.-Z., Goodman,M.F., Bloom,L.B., Jiricny,J. and Day,R.S. (1996) Base analog and neighboring base effects on substrate specificity of recombinant human G:T mismatch-specific thymine DNA-glycosylase. Biochemistry, 35, 12926–12932. [DOI] [PubMed] [Google Scholar]
- 23.Griffin S., Branch,P., Xu,Y.-Z. and Karran,P. (1994) DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA methylation damage. Biochemistry, 33, 4787–4793. [DOI] [PubMed] [Google Scholar]
- 24.Sibghat-Ullah, Xu,Y.-Z. and Day,R.S. (1995) Incision at diaminopurine:thymine base pairs but not at guanine:O4-methylthymine base pairs in DNA by extracts of human cells. Biochemistry, 34, 7438–7442. [DOI] [PubMed] [Google Scholar]
- 25.Hang B., Medina,M., Fraenkel-Conrat,H. and Singer,B. (1998) A 55-kDa protein isolated from human cells shows DNA glycosylase activity towards 3,N4-ethenocytosine and the G/T mismatch. Proc. Natl Acad. Sci. USA, 95, 13561–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hendrich B., Hardeland,U., Ng,H.-H., Jiricny,J. and Bird,A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- 27.Schärer O.D., Kawate,T., Gallinari,P., Jiricny,J. and Verdine,G.L. (1997) Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc. Natl Acad. Sci. USA, 94, 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waters T.R. and Swann,P.F. (1998) Kinetics of the action of thymine DNA glycosylase. J. Biol. Chem., 273, 20007–20014. [DOI] [PubMed] [Google Scholar]
- 29.Yarosh D.B., Ziolkowski,C.H.J. and Day,R.S. (1984) Conversion of human cells to carcinogen resistance by DNA transfection. In Bishop,J.M., Rowley,J.D. and Greaves,M. (eds) Genes and Cancer. Allan R. Liss, New York, NY, pp. 69–78.
- 30.Manley J.L., Fire,A., Cano,A., Sharp,P.A. and Gefter,M.L. (1980) DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc. Natl Acad. Sci. USA, 77, 3855–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sibghat-Ullah, L. and Day,R.S. (1992) Incision at O6-methylguanine:thymine mispairs in DNA by extracts of human cells. Biochemistry, 31, 7998–8008. [DOI] [PubMed] [Google Scholar]
- 32.Sibghat-Ullah, L. and Day,R.S. (1993) DNA-substrate sequence specificity of human G:T mismatch repair activity. Nucleic Acids Res., 21, 1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiebauer K. and Jiricny,J. (1990) Mismatch-specific thymine DNA glycosylase and DNA polymerase β mediate the correction of G.T mispairs in nuclear extracts from human cells. Proc. Natl Acad. Sci. USA, 87, 5842–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindahl T. and Wood,R.D. (1999) Quality control by DNA repair. Science, 286, 1897–1905. [DOI] [PubMed] [Google Scholar]
- 35.Reha-Krantz L.J., Nonay,R.L., Day,R.S. and Wilson,S.H. (1996) Replication of O6-methylguanine-containing DNA by repair and replicative DNA polymerases. J. Biol. Chem., 271, 20088–20095. [DOI] [PubMed] [Google Scholar]
- 36.Papadopoulos N., Nicolaides,N.C., Liu,B., Parsons,R., Lengauer,C., Palombo,F., D’Arrigo,A., Markowitz,S., Willson,J.K., Kinzler,K.W., Jiricny,J. and Vogelstein,B. (1995) Mutations of GTBP in genetically unstable cells. Science, 268, 1915–1917. [DOI] [PubMed] [Google Scholar]
- 37.Umar A., Boyer,J.C., Thomas,D.C., Nguyen,D.C., Risinger,J.I., Boyd,J., Ionov,Y., Perucho,M. and Kunkel,T.A. (1994) Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J. Biol. Chem., 269, 14367–14370. [PubMed] [Google Scholar]
- 38.Drummond J.T., Li,G.-M., Longley,M.J. and Modrich,P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 39.Li G.-M. and Modrich,P. (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human mutL homologs. Proc. Natl Acad. Sci. USA, 92, 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]