


Abstract
Despite its relatively streamlined genome, there are important examples of regulated RNA splicing in Saccharomyces cerevisiae, such as splicing of meiotic transcripts. Like other eukaryotes, S. cerevisiae undergoes a dramatic reprogramming of gene expression during meiosis, including regulated splicing of a number of crucial meiosis-specific RNAs. Splicing of a subset of these is dependent upon the splicing activator Mer1. Here we show a crucial role for the chromatin remodeler Swi/Snf in regulation of splicing of meiotic genes and find that the complex affects meiotic splicing in two ways. First, we show that Swi/Snf regulates nutrient-dependent downregulation of ribosomal protein encoding RNAs, leading to the redistribution of spliceosomes from this abundant class of intron-containing RNAs (the ribosomal protein genes) to Mer1-regulated transcripts. We also demonstrate that Mer1 expression is dependent on Snf2, its acetylation state and histone H3 lysine 9 acetylation at the MER1 locus. Hence, Snf2 exerts systems level control of meiotic gene expression through two temporally distinct mechanisms, demonstrating that it is a key regulator of meiotic splicing in S. cerevisiae. We also reveal an evolutionarily conserved mechanism whereby the cell redirects its energy from maintaining its translational capacity to the process of meiosis.
INTRODUCTION
Saccharomyces cerevisiae, like all eukaryotes, contains genes interrupted by non-coding intron sequences. Introns are removed from the pre-messenger RNA by the spliceosome, a large macromolecular assembly of five small nuclear ribonucleoprotein complexes (snRNPs). Pre-messenger RNA splicing is critical for proper expression of eukaryotic genes. Although important insights into the mechanisms of RNA splicing have been gleaned from in vitro analysis of the reaction, over the last decade it has become increasingly clear that, in fact, assembly of the spliceosome occurs co-transcriptionally, while the RNA polymerase is actively engaged with a chromatin template. Moreover, changes in the state of the chromatin can affect splicing (1). However, understanding the nature of the causal relationships between chromatin modification and splicing has been difficult to decipher, even in an organism with a relatively streamlined genome such as S. cerevisiae.
Unlike metazoans, S. cerevisiae contains a relatively small number of introns, suggesting that those RNAs that have retained their introns may be subject to important and/or intron-tuned regulation. The largest single class of intron-containing genes (ICGs) in yeast is comprised of ribosomal protein genes (RPGs), which ensures that a critical step in gene regulation is dependent upon accurate and efficient splicing. Furthermore, splicing of RPGs can be regulated by the cell's environmental conditions (2). For example, a decrease in amino acid availability leads to a dramatic downregulation of RPG splicing (3) and hence downregulation of the energy-intensive process of translation.
In the last several years, there have been a number of other documented examples of splicing regulation in yeast (reviewed in (4)). Perhaps one of the best examples is found in the meiotic regulatory program. One of the largest classes of genes containing introns is made up of those involved in meiosis. Transcription of intron-containing meiotic transcripts and a critical meiotic splicing regulator, Mer1, is dependent upon degradation of the meiotic transcription inhibitor Ume6 (5). Mer1, in turn, regulates the splicing of four of the meiotic pre-mRNAs (MER2, MER3, AMA1 and SPO22) via its interaction with a conserved intronic enhancer sequence (5΄-AYACCCYU-3΄) (6,7). Ume6-dependent repression of meiotic gene transcription (including MER1), in part by recruiting the deacetylase Rpd3 (8), is critical for timing the onset of meiosis. However, while it is clear that cells entering meiosis rapidly adjust their gene expression profiles, neither the molecular basis of this massive reprogramming nor the specific consequences of this shift in the cell's gene expression are fully understood.
Although yeast genes typically have well-conserved splicing signals, particularly when compared to other eukaryotes, meiotic genes, in general, show lower conservation of their branch point (BP) and 5΄ splice site (SS) sequences, making these introns weak substrates for the spliceosome. However, the splicing of these introns becomes more likely when there is decreased competition for a limited pool of spliceosomes. In fact, artificially decreasing the pool of intron-containing RPGs (IC-RPGs) by treating cells with rapamycin effectively shifts the pool of spliceosomes to the other intron-containing RNAs and increases splicing of genes with non-canonical SSs, including meiotic genes (9). This is reminiscent of other examples whereby redistribution of gene expression machineries that are typically envisioned to be plentiful, allows regulated expression of the appropriate substrates under specific environmental conditions. For example, in S. cerevisiae competition for available ribosomes by different transcripts is overcome by spatial redistribution of ribosomes (10). In human cells, it has recently been shown that the seemingly abundant U1 snRNP becomes transiently limiting during neuronal activation due to transcriptional upregulation, presumably because of the high abundance of U1 substrates. As a consequence, U1-dependent repression of the use of cryptic polyadenylation signals is decreased (11,12). Such examples suggest that RNA competition for limited pools of regulatory machinery such as the ribosome or the spliceosome may be a more general mechanism whereby eukaryotic organisms regulate expression of specific transcripts in response to cellular needs (8).
Here we describe a central role for the chromatin remodeling complex Swi/Snf in meiotic splicing regulation, by controlling the competition of different transcripts for the spliceosome and regulating the expression of the meiotic splicing activator. First, we show that deletion of Snf2 activates increased use of non-canonical BPs and 5΄ SS and also causes a dramatic downregulation of RPG expression. Moreover, in the absence of Snf2 the efficiency of splicing of meiotic RNAs in particular, which are enriched in non-consensus (nc) SSs, is increased, while RPG expression is downregulated. In fact, the extent of enhanced splicing mirrors the effect of Snf2-independent RPG downregulation via treatment of cells with rapamycin. To determine if our genetic manipulation of Snf2 recapitulates what occurs under physiological conditions, we examined Snf2 levels under sporulation conditions, in both the strain background that allows for the genetic manipulation of Snf2 as well as in the SK1 background, which undergoes meiosis synchronously and efficiently. Snf2 protein levels decrease in both strain backgrounds. We also observe a corresponding decrease in RPG expression and an increase in splicing of meiotic transcripts. However, the timing of Snf2 downregulation is important. In fact, before Snf2 is downregulated, it persists in a deacetylated form and the MER1 locus is hyperacetylated creating optimal conditions for Snf2 binding to the MER1 locus and expression of the splicing activator. These data reveal that fine-tuned regulation of Snf2 activity is important for regulated splicing during meiosis.
MATERIALS AND METHODS
Yeast strains and growth
The yeast strains used in this study are listed in Table 1. All strains except the SK1 strain are derived from BY4743 and individual deletion strains were obtained from Open Biosystems. The Snf2-FLAG and Snf2K1493,1497R-FLAG strains were a kind gift from Dr Jerry Workman (described in (13)). Yeast strains were grown in Yeast extract-Peptone-Dextrose (YPD) (1% yeast extract, 2% peptone and 2% dextrose) media at 30°C. For rapamycin experiments, overnight yeast cultures were diluted in 25 ml media to OD600 of 0.1 and then grown to OD600 of 0.3. Rapamycin (Sigma) was then added to a final concentration of 200 ng/μl (Stock concentration of 1 mg/ml in Dimethyl Sulfoxide (DMSO)). Cells were precipitated after 90 min of treatment with rapamycin.
Table 1. List of yeast strains used in this study.
| Strain Number | Name | Genotype |
|---|---|---|
| TJY6724 | WT | his3Δ leu2Δ LYS2 met15Δ ura3Δ |
| TJY6727 | snf2Δ | his3Δ leu2Δ ura3Δ snf2Δ::NatMX |
| TJY6729 | ume6Δ | his3Δ leu2Δ ura3Δ ume6Δ::KanMX |
| TJY6797 | ume6Δ snf2Δ | his3Δ leu2Δ ura3Δ ume6Δ::KanMX snf2Δ::NatMX |
| TJY6726 | WT | Mata/Matα his3Δ/his3Δ leu2Δ/leu2Δ LYS2/lys2Δ met15Δ/MET15 ura3Δ/ura3 |
| TJY6856 | snf2Δ | Mata/Matα his3Δ/his3Δ leu2Δ/leu2Δ LYS2/lys2Δ met15Δ/MET15 ura3Δ/ura3 snf2Δ::NatMX/snf2Δ::NatMX |
| TJY6917 | SK1 K8409 | SK1 MATa/MAT_ HO URA3::tetO224 LEU2::tetR-GFP REC8-HA3::URA3 lys2 his3 trp1 |
| TJY7086 | Snf2-FLAG | his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 SNF2-Flag::LEU2 |
| TJY7087 | Snf2 K1493,1497R-FLAG | his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 Snf2 K1493R, K1497R-Flag::LEU2 |
RNA-seq analysis
RNA-seq libraries were prepared using Illumina Truseq® V3 kit and ribosomal RNA depletion (Ribo-Zero, Illumina). Single-end, 50 nucleotide sequence reads (HiSeq 2000) were aligned to SacCer3 and spliced transcripts from the Ares Lab Yeast Intron Database Version 3 (14) in a single step using STAR (15) with a maximum of two mismatches and a minimum overhang of four nucleotides for spliced alignments. In the case of reads aligning to multiple loci, we kept only the highest scoring alignments. Reads that aligned with the same score to more than two loci were discarded. Library depth was an average of ∼18 million reads per sample with an average of 13 million successfully aligned to annotated gene models per sample. RPKMs were computed for each gene by dividing the total number of reads that aligned entirely within the gene's exon boundaries by the gene's total exon length in kilobase pairs per million mapped reads. Reads within ICGs were categorized as exonic, spliced or unspliced. Exonic reads are those that map entirely within an exon, as defined by the Ares Lab Yeast Intron Database. Introns with annotated snoRNAs within the defined intron boundaries were disregarded in this analysis. Spliced reads are those that align with a gap that corresponds to an annotated intron and unspliced reads map partially within an exon and partially within an intron with no gap. Spliced and unspliced read counts were normalized by dividing total spliced counts by the number of potential unique alignment positions that contribute to the total. For spliced reads, this is read length minus one for every intron. For unspliced read counts, this is the length of the intron plus the read length minus one. Splicing efficiency (SE) for each intron was calculated as normalized spliced counts divided by the sum of the normalized spliced and normalized unspliced counts. Percent (%) increase in splicing efficiency was calculated as: 100 * (SE in snf2Δ − SE in WT)/(SE in WT).
To assess global splicing changes, a chi-squared test was used to measure the deviation of our RNA-seq data (‘observed’ distribution) from a random distribution of splicing efficiency changes in snf2Δ (‘expected’ distribution). Under our null model that snf2Δ does not effect splicing, there would be an equal number of introns showing increased splicing as decreased splicing. To assess enrichment of ncSSs in the genes with the greatest improvement in splicing, the two outcomes were defined as ‘all-consensus SSs’ or ‘ncSSs’ (including nc 5΄ SS, BP or both). The ‘expected’ group is populated by the products of the fraction of introns in each category (indicated in Figure 1B, left panel) and number of genes considered (n = 40). The ‘observed’ group was populated based on the RNA-seq analysis. To calculate the distribution functions of ncSSs in an unbiased manner, ICGs were ranked according to magnitude of improvement in splicing upon deletion of SNF2. The cumulative occurrence of ncSS at each position was plotted and compared against a uniform distribution function generated by averaging the total number of ncSS in the yeast genome. Significance was assessed using a one-sample non-parametric Kolmogorov–Smirnov test.
Figure 1.

Deletion of Snf2 improves splicing globally. (A) Changes in splicing efficiencies of all intron-containing genes (ICGs) upon deletion of Snf2, represented as an X–Y plot. Overall splicing improves in snf2Δ yeast over WT (Chi-squared test, approximate P-value indicated). Largest changes in splicing are observed in genes with nc 5΄SS (green dots) or nc branch point (BP) sequences (blue dots). (B) Left: distribution of non-consensus (nc) splice sites (SSs) (5΄ SS and/or BP) in Saccharomyces cerevisiae ICGs, among all ICGs expressed in our samples. Right: distribution of ncSSs among genes showing highest improvements in splicing upon deletion in Snf2. ncSS-containing introns are enriched (Chi-squared test, approximate P-value indicated). (C) Top: log2-fold change in expression of ribosomal protein genes (RPG) upon deletion of Snf2, relative to scR1. Each bar represents an individual RPG (n = 128). Bottom: log2-fold change in expression of intron-containing RPGs (IC-RPGs) upon deletion of SNF2 (n = 84). (D) Fold enrichment of occupancy of Snf2 over input by chromatin immunoprecipitation (ChIP)-Seq across the 500 nucleotides upstream and downstream of translational starts for all genes, RPGs and IC-RPGs. Dotted line represents the annotated translational start (ATG). Analysis of data from (13).
RT-PCR and real time PCR analysis
RNA was isolated from cells at indicated time points. After DNase treatment (Roche), 4 μg of total RNA from each time point was used to make cDNA using a cDNA synthesis kit (Fermentas). To detect splicing isoforms, primers flanking the intronic sequences were used for polymerase chain reaction (PCR) using 1 μl of cDNA diluted to 1:20. PCR products were then run on 6% Tris/Borate/EDTA (TBE) polyacrylamide gel. The gel was stained with SYBR green (Sigma) and imaged. qRT-PCR was done in 10 μl reaction with gene specific primers using 1 μl of cDNA diluted 1:20 using Perfecta Sybr Green Fastmix (Quanta Biosciences) and a CFX96 Touch System (BioRad). All samples were run in triplicate for each independent experiment. qRT-PCR was also performed for scR1 gene from each cDNA sample. Gene expression analysis was done by 2−ΔCt methods using scR1 as reference gene. Fold expression of mRNA was measured compared to WT by 2−ΔΔCt methods (16).
ChIP-Seq analysis
Chromatin immunoprecipitation (ChIP)-seq reads (13) were obtained (GEO accession number GSE61210) and converted to FastQ format using the NCBI SRA Toolkit. Bowtie2 was used to align FastQ reads to SacCer3 with only one reported alignment (−k 1). Genomic track files from immunoprecipitation and input alignments were created using SAMTools, BEDTools and the UCSC bedgraphToBigWig utility. After normalizing input and immunoprecipitation pileup tracks for differential read count, a ratio track was created by dividing the normalized immunoprecipitation track by the normalized input track. Pileup ratios of the region 500+/− of the Translation Start Site (TSS) of genes of interest were retrieved and were added coordinate-wise to generate a 500+/− association array for each list of genes. Association array values were normalized for the number of genes included and vertically scaled between 0 and 100 to generate the meta-association array.
Induction of sporulation
All diploid strains derived from BY4743 were grown in YPD overnight and diluted in pre-sporulation media (5% glucose, 3% Difco Nutrient Broth and 1% Yeast Extract) to a final OD of 0.1. After 4 h, the cells are pelleted, washed with SPO media (1% potassium acetate, 0.005% zinc acetate and amino acid supplements as needed) and resuspended in SPO media and grown at 25°C (17). At the indicated time points, cells were pelleted each for RNA and/or protein analysis. For SK1 strains, SPorulation Media (SPM) media (0.3% potassium acetate, 0.02% raffinose and amino acid supplements as needed) was used in place of SPO (18).
Western blot
Protein was isolated from cells pellets with FA-1 lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, 0.1% Sodium Deoxycholate, 1 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitors) using bead-beating. The buffer was supplemented with protease inhibitor cocktail tablet (Roche). Total protein was resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis. The gel was transferred to Polyvinylidene fluoride (PVDF) membrane, probed with anti-SNF2 antibody (yN-20, Santa Cruz) at a 1:200 dilution in 2% milk or anti-acetylated lysine (ST1027, Millipore) at a 1:2000 dilution in 5% milk. Signal was detected using ECL (Thermo Scientific) as per the manufacturer's instructions. For Pgk1 western blotting, the membrane was probed with anti Pgk1 antibody (Molecular probes) at a 1:3000 dilution in 5% milk.
Random spore analysis
Sporulation was set up as described above. After 7 days of sporulation (BY background), 106 cells were pelleted and resuspended in 500 μl of water containing 0.5 mg/ml Zymolyase-100T (Zymo research) and 1% β-mercaptoethanol and incubated overnight at 30°C with gentle rocking to lyse unsporulated diploid cells. A total of 200 μl of 1.5% NP-40 was added to the solution and vortexed to disrupt tetrads (as observed by microscopy). Spores were centrifuged, resuspended in water and plated on YPD plates in serial dilutions to count the number of viable spores (19,20).
Immunoprecipitation
Sporulation was induced as described earlier and 50 ml of cells at various time points were pelleted. Protein was isolated from cells pellets with FA-1 lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 1 mM PMSF and protease inhibitors) using bead-beating methods. Immunoprecipitation was performed with anti-SNF2 antibody (yN-20, Santa Cruz, 10 μl per IP) and γ-G sepharose beads and washed with the lysis buffer. Western blots were performed as described above.
Chromatin immunoprecipitation (ChIP) and qPCR
Sporulation was induced as described earlier and 50 ml of cells at various time points were crosslinked at room temperature for 15 min with formaldehyde to a final concentration of 1%. Crosslinking was quenched at room temperature for 5 min with glycine to a final concentration of 125 mM. Cells were disrupted with glass beads (0.5 mm) for 40 min at 4°C. To shear chromatin, lysates were sonicated on ice for a total of 3 min 20 sec at 15% intensity (10 s on, 15 s off and on ice). After sonication, lysates were cleared by centrifugation. Samples were then used for immunoprecipitation with anti-H3K9ac (07-352, Millipore) or anti-Snf2 antibody (yN-20, Santa Cruz). After immunoprecipitation, samples were washed with the lysis buffer and incubated overnight at 65°C to reverse crosslinking. Samples were incubated with Proteinase K (Sigma) and RNAse Cocktail (Ambion), followed by purification using a PCR product purification kit (Qiagen). DNA samples were then analyzed by real-time PCR. Input DNA was serially diluted and 1 μl of these dilutions was used in a 20 μl reaction volume to calculate ‘percentage input’. One microliter of ChIP DNA samples were used in a 20 μl reaction volume. Real-time PCR was performed using a CFX96 Touch System (BioRad) as described earlier. All samples were run in triplicate for each independent experiment. ChIP samples were quantified as a function of percentage input as indicated by standard curves generated by serial dilutions of the input. Values are reported as enrichment over a beads-only control. Reported values are averages of three independent experiments and error bars represent the standard deviation.
RESULTS
Deletion of Snf2 leads to global enhancement of splicing through RPG depletion and spliceosome redistribution
We previously demonstrated an important relationship between chromatin and splicing. For example, we have shown that chromatin modifiers are important for spliceosome recruitment and rearrangements in S. cerevisiae (21,22). These data led us to explore the possibility that other chromatin modifiers can influence splicing. So the splicing of all ICGs in S. cerevisiae was assayed in cells deleted of Snf2, the adenosine triphosphatase (ATPase) component of the Swi/Snf chromatin-remodeling complex. To our surprise, upon Snf2 deletion, we found that a majority of introns show enhanced splicing, particularly those introns with ncSSs (Figure 1A). In fact, although yeast introns generally have highly conserved intron sequences, the genes showing the largest improvement in splicing are significantly enriched in nc 5΄ SS or BP sequences (Figure 1B; Supplementary Figure S1A and B), which we confirm by RT-PCR of several candidate genes with ncSSs (Supplementary Figure S1C).
Recent studies have revealed a surprising mechanism for increased splicing of poorly recognized introns—namely by decreased expression of competing, highly expressed RPGs (9). So we considered the possibility that the change in splicing efficiency in cells deleted of Snf2 might be due to redistribution of spliceosomes away from RPGs. When we analyzed the RNA sequencing data from cells deleted of Snf2, we observed dramatic Snf2-dependent downregulation of RPGs (Figure 1C). Consistent with this, recent analysis of the promoter architecture of RPGs reveals increased MNase sensitivity at their promoters (23). Our own analysis of ChIP-Seq datasets for Snf2 occupancy (13) revealed Snf2 enrichment at the promoters and gene bodies of RPGs, and this enrichment remained consistent when the analysis was restricted to IC-RPGs (Figure 1D). Finally, the Snf2 enrichment profile persisted and was unique to RPGs, even when compared to other highly expressed genes (Supplementary Figure S1D). These data suggest that the nucleosome remodeling activity of the Swi/Snf complex at the promoters and gene bodies of RPGs leads to decreased nucleosome occupancy at these regions, and is crucial for the efficient transcription of these genes. Deletion of Snf2, on the other hand, leads to downregulation of RPGs, including the genes containing the majority of yeast introns.
Snf2-dependent RPG downregulation causes an improvement in the splicing of Mer1-regulated introns
The process of yeast meiosis or sporulation in response to nitrogen starvation, relies on numerous ICGs significantly enriched in ncSS sequences. Since introns with ncSSs appear to receive a significant benefit from cellular conditions lacking Snf2, we next evaluated whether the meiotic splicing program might be under the control of Snf2. However, the transcriptional repressor Ume6 negatively regulates expression of these transcripts until it is degraded during meiosis (24). In order to assay whether the absence of Snf2 results in an improvement in the splicing of the meiotic ICGs, the RNA-seq analysis was repeated in a ume6Δ background. While a substantial fraction of ICGs show no increase in splicing, we observe a significant improvement in the splicing of most of the meiotic ICGs upon deletion of Snf2 (Figure 2A and Table 2). Once again, genes showing the greatest improvements in splicing are significantly enriched in intron-containing nc 5΄SS or BP sequences. Interestingly, SPO22, MER2 and MER3, three of the four genes whose splicing have previously been demonstrated to be regulated by the meiosis-specific splice enhancer Mer1 (6) are the most significantly affected by the deletion of Snf2 (Figure 2B and C, Supplementary Figure S2A and Table 2). It is worth noting that although AMA1 did not make our read cutoff filters for the RNA-seq analysis, its splicing is significantly improved in the absence of Snf2 (Figure 2C). One explanation for these results could be that in the ume6Δ snf2Δ cells grown under vegetative conditions, Mer1 or other factors required for Mer1-dependent splicing such as Nam8 (6), are transcribed at higher levels. However, we observe virtually no effect on the levels of these RNAs, indicating that deletion of Snf2 under vegetative conditions affects the splicing of the Mer1 regulon via a mechanism not involving the levels of Mer1 or Nam8 (Supplementary Figure S2B).
Figure 2.

Snf2-dependent RPG downregulation activates meiotic splicing. (A) Changes in splicing efficiencies of meiotic ICGs upon deletion of SNF2 in an Ume6 null background, represented as a box-and-whisker plot (Paired two-tailed students t-test, P < 0.05). (B) Distribution of changes in splicing efficiencies upon deletion of SNF2 in a Ume6-null background. Mer1-regulated genes are represented by red bars. Meiotic ICGs whose splicing is not regulated by Mer1 are represented by teal and red-checkered bars. ncSS-containing genes that are not involved in meiosis are represented by teal bars. Black bars represent genes containing introns with all consensus sites. (C) Expression and splicing of Mer1-regulated meiotic transcripts SPO22, AMA1, MER2 and MER3 in ume6Δ and ume6Δsnf2Δ vegetative cells. Genomic DNA (gDNA) PCR products indicate the mobility of the unspliced form. PCR products representing the spliced and unspliced forms are indicated. (D) Expression and splicing of Mer1 regulated meiotic transcripts SPO22, AMA1, MER2 and MER3 in ume6Δ vegetative cells treated with rapamycin and control vehicle (DMSO). gDNA PCR products indicate the mobility of the unspliced form. PCR products representing the spliced and unspliced forms are indicated.
Table 2. Deletion of Snf2 improves splicing of meiotic ICGs.
| Gene name | Function | % increase in splicing efficiency between ume6Δ snf2Δ and ume6Δ |
|---|---|---|
| MER3 | Meiotic recombination | 508.2 |
| SPO22 | Meiotic nuclear division | 143.7 |
| MER2 | Meiotic recombination | 118.2 |
| REC102 | Chromosome synapsis | 60.5 |
| SAE3 | Meiotic recombination | 16.5 |
| SPO1 | Transcriptional regulator | 8.0 |
| DMC1 | Meiotic recombination | 7.5 |
| MEI4 | Meiotic recombination | −1.6 |
| HOP2 | Homologous chromosome pairing | −6.6 |
Percentage improvements in splicing of intron-containing meiotic genes in ume6Δsnf2Δ versus ume6Δ yeast, determined by RNA-seq. This is not an exhaustive list of meiotic ICGs, as AMA1, MND1, PCH2 and REC114 did not pass RNA-seq read count filters.
Not surprisingly, IC-RPGs are downregulated upon deletion of Snf2 in the ume6Δ strain background as well (Supplementary Figure S2C). To determine if downregulation of RPGs by another Snf2-independent method could lead to a comparable degree of splicing enhancement, ume6Δ yeast cells were treated with rapamycin to inhibit TOR (Target of Rapamycin)-dependent RPG transcription (25) (Supplementary Figure S2D and E) and observed that the splicing of the four genes of the Mer1 regulon increases significantly (Figure 2D and Supplementary Figure S2F). Intriguingly, maximum splicing efficiency achieved by Snf2 deletion is comparable to rapamycin-dependent RPG downregulation (Compare quantitation in Figures S2A and Figure S2F). This supports the model that Snf2-dependent RPG downregulation and subsequent spliceosome redistribution is a mechanism for the improved splicing of the genes of the Mer1 regulon.
Levels of Snf2 decrease under sporulation conditions
In light of the effect Snf2 has on the splicing of meiotic transcripts we predicted that Snf2 might itself be subject to regulation under sporulation conditions. To test this prediction, wild-type S288C-derived diploid yeast (in the BY strain background) were shifted to sporulation media and allowed to sporulate over 7 days, a typical time course for this strain. Snf2 protein levels are undetectable at the end of the time course and, in fact, fall below the limits of detection within 24 h of transferring cells to sporulation media (Figure 3A). This downregulation of Snf2 occurs at the level of the protein, as SNF2 mRNA levels do not drop significantly under these conditions (data not shown). These data indicate that Snf2 protein is downregulated under the conditions that support meiosis.
Figure 3.

Levels of Snf2 decrease when cells are shifted to sporulation media, corresponding with RPG downregulation and increase in meiotic splicing. (A) Western blot for levels of Snf2 protein through a time course of meiosis. Pgk1 is an internal control. (B) Quantification of total (exon 2) transcript levels for RPS16A, RPL34B and RPL19A by RT-qPCR relative to scR1 and normalized to t = 0 in WT yeast at indicated times after shifting to sporulation media. SEC65 encodes a component of the signal recognition complex and is a control. Error bars represent ± 1 Standard Deviation (SD). Levels of Snf2 protein as detected by western blot and normalized to t = 0 are indicated by the green line. (C) Expression and splicing of Mer1 regulated meiotic transcripts SPO22, AMA1, MER2 and MER3 at indicated times under sporulation conditions. PCR products representing the spliced and unspliced forms are indicated. (D) Quantification of splicing efficiencies from (C). Levels of Snf2 protein as detected by western blot and normalized to t = 0 are indicated by the green line. (E) Left panel: identical to Figure 3C, provided for side-by-side comparison. Expression and splicing of Mer1 regulated meiotic transcripts SPO22, AMA1, MER2 and MER3 at indicated times under sporulation conditions. PCR products representing the spliced and unspliced forms are indicated. Right Panel: expression and splicing of Mer1 regulated meiotic transcripts SPO22, AMA1, MER2 and MER3 in snf2Δ yeast at indicated times after the transfer into sporulation media. PCR products representing the spliced and unspliced forms are indicated. (F) Fold-change RT-qPCR measurement of MER1 transcripts between WT and snf2Δ diploid strains at indicated times after the transfer to sporulation media. Error bars represent ± 1 SD.
To determine the correlation between Snf2 downregulation and RPG expression, we measured the levels of representative IC-RPGs and the splicing of the genes of the Mer1 regulon through the 7-day sporulation time course and observed a dramatic decrease in RPG levels (Figure 3B) and a concomitant rise in splicing of the transcripts in the Mer1 regulon (Figure 3C and D).
If the only role of Snf2 in meiosis is to downregulate RPG expression, we predicted that deletion of Snf2 would allow cells to sporulate, perhaps even more efficiently. So Snf2 deficient diploid BY cells were transferred to sporulation media and sporulation was measured using random spore analysis (19,20). Diploid cells lacking Snf2 produce no viable spores (Supplementary Figure S3). Moreover, when expression and splicing of Mer1-dependent transcripts was analyzed in these diploid cells under meiotic conditions, the Mer1-dependent transcripts are expressed but are not spliced (Figure 3E). Analysis of MER1 expression in these cells revealed that in diploid cells entering meiosis, unlike haploid cells lacking Ume6, MER1 expression requires Snf2 (Figure 3F). These data suggest that Snf2 has another role in the meiotic program in S. cerevisiae in addition to regulating the levels of free spliceosomes, which appears to occur prior to its downregulation. While the S288C-derived strain (BY4743, which we refer to as ‘BY’) provides a useful tool for genetic analysis (e.g. SNF2 deletion is tolerated in these diploid cells), it was clear from these studies that a strain that was more amenable to finer temporal dissection of molecular changes under sporulation conditions was necessary. So we analyzed the meiotically synchronous, sporulation efficient SK1 strain.
Snf2 downregulation occurs in the meiotically efficient SK1 strain
In order to maintain consistency in the way SK1 and BY strains were sporulated, SK1 K8409 diploid cells were moved directly from rich media to sporulation media (0.3% KOAc, 0.02% Raffinose and amino acids as needed). The SK1 diploid yeast achieve near complete sporulation (∼100%) within 36 h of shifting to sporulation media, compared to about 30% for the BY strain under our experimental conditions.
To determine how Snf2 levels change in the SK1 strain under sporulation conditions, SK1 diploid yeast were shifted to sporulation media and allowed to sporulate over 48 h. As we previously observed in the BY strain background, Snf2 protein levels are undetectable at the end of the sporulation time course. In fact, Snf2 falls below the limits of detection within 5 h of the shift into sporulation media (Figure 4A). Similar to the BY strain, this downregulation of Snf2 occurs at the level of the protein (data not shown). Over this time course, levels of RPG RNAs were also measured and they also decrease between 4- and 6-fold (Figure 4B, three representative RPGs are shown). Interestingly, there does appear to be a slight increase in RPG expression at ∼12 h which may be indicative of a requirement for mid-meiotic translation (26). Nonetheless, RPGs remain at around 10% of their pre-sporulation levels even with this slight increase.
Figure 4.

Snf2 is downregulated in the meiotically efficient SK1 yeast strain. (A) Western blot for levels of Snf2 protein through a time course of sporulation in the SK1 strain. Pgk1 is an internal control. (B) Quantification of total (exon 2) transcript levels for RPS16A, RPL34B and RPL13A by RT-qPCR relative to scR1 and normalized to t = 0 in WT yeast (SK1 strain) at indicated times after shift to sporulation media. Error bars represent ± 1 SD. (C) Quantification of total transcript levels for Initiator of Meiotic Entry 1 (IME1) by RT-qPCR relative to scR1 and normalized to t = 0 in WT yeast (SK1 strain) at indicated times after shift to sporulation media. Error bars represent ± 1 SD. (D) Quantification of total transcript levels for MER1 by RT-qPCR relative to scR1 and normalized to t = 0 in WT yeast (SK1 strain) at indicated times after shift to sporulation media. Error bars represent ± 1 SD. (E). Expression and splicing of Mer1-regulated meiotic transcript MER2 at indicated times after shift to sporulation media. PCR products representing the spliced and unspliced forms are indicated.
With these finer time points, we noted a discrepancy between the levels of Snf2 at early time points and the downregulation of RPGs. The decrease in Snf2 is most dramatic after 3 h (Figure 4A), while RPG downregulation is initiated almost immediately after the shift to sporulation media. Furthermore, in light of the results from the BY diploid strain deleted of SNF2, which demonstrate that Snf2 is required for MER1 expression and Mer1-dependent splicing, presumably before its downregulation (Figure 3E and F), it became clear that it was important to examine more closely the activity of Snf2 in MER1 expression and RPG downregulation during this 0–3 h time period. As is often the case, different yeast strains show differential tolerance to deletion of specific genes, and deletion of SNF2 is lethal in the SK1 strain. So, Mer1-dependent splicing in snf2Δ SK1 cells could not be examined (data not shown). Nonetheless, deletion of Snf2 in the BY strain reveals a previously unexplored role for Snf2 in the expression of MER1. This led us to examine more closely how Snf2 affects RPG expression at early time points before its absence leads to decreased RPG expression (Figure 2) and to assess the mechanism by which Snf2 affects MER1 expression.
First, we assessed the meiotic state of the SK1 cells by measuring the expression of the key meiotic transcription factor Initiator of Meiotic Entry 1 (IME1) (Figure 4C) (27). Moreover, to monitor Mer1-dependent splicing, we measured the levels of MER1 by qRT-PCR (Figure 4D) as well as the splicing of MER2, one of the genes of the Mer1 regulon (Figure 4E) over 48 h. Consistent with literature that has established that Ime1 is required for MER1 transcription (28), MER1 expression and Mer1-dependent splicing follow IME1 expression. It is also evident from that timing that even after Snf2 decreases to undetectable levels, other molecular events (such as Ime1-dependent MER1 expression), support meiotic splicing. The results with both strains also show that Snf2 is not dispensable for meiosis, but may affect both the levels of the Mer1 splicing activator and the spliceosomes that the activator helps to recruit to the meiotic transcripts. Next we explored how Snf2 can perform both functions.
Snf2 levels and acetylation are temporally coordinated to direct meiosis specific splicing regulation
In light of our observation that Snf2 is required for MER1 expression in diploid cells, we next examined how Snf2 activity at the MER1 locus might be regulated. Snf2 contains a bromodomain that directs its interactions with acetylated histones (29). It has recently been shown that the Snf2 protein itself can also be acetylated. Moreover, Snf2 acetylation competes with acetylated histones for recognition by the Snf2 bromodomain. In other words, the bromodomain on acetylated Snf2 can recognize itself over acetylated histones and this effect is counteracted by Snf2 deacetylation (13). With this in mind, we hypothesized that Snf2 acetylation might change early in meiosis. Snf2 was immunoprecipitated from sporulating SK1 diploid cells and probed for acetylated lysines. Importantly, the assay was performed within the first few hours of sporulation during the period in which Snf2 levels remain high. We observed that acetylated Snf2 decreased to undetectable levels within the first 3 h of transfer to sporulation media although Snf2 levels remain high, even when Snf2 is overloaded (Figure 5A, 3-h time point). These data show that the Snf2 present in these cells is largely unacetylated.
Figure 5.

The timing of Snf2 activity is coordinated by its acetylation and acetylation of histones. (A) Immunoprecipitation of Snf2 during a time course of meiosis in WT SK1 yeast. Snf2 acetylation decreases steadily between 0 and 3 h after transfer into sporulation media, but Snf2 levels remain constant. The immunoprecipitate at the 3-h time point is slightly overloaded (compare with Figure 4A) and acetylation is still barely detectable. (B) Bar graph showing Snf2 occupancy at the MER1 promoter in the SK1 strain over time. Error bars represent ± 1 SD. Quantification from three biological replicates. (C) Bar graph showing H3K9ac occupancy at the MER1 promoter in the SK1 strain over time in sporulation media. Error bars represent ± 1 SD. Quantification from three biological replicates. (D) Bar graph showing Snf2 occupancy at the RPL13A promoter in the SK1 strain over time. Error bars represent ± 1 SD. Quantification from three biological replicates. (E) Quantification of total (exon 2) transcript levels for RPS16A, RPL34B and RPL13A by RT-qPCR relative to scR1 and normalized to t = 0 in SK1 strain at indicated times after shift to sporulation media (part of Figure 4B). (F) Relative Snf2 occupancy at the MER1 promoter to an RPG locus, upon ChIP using an anti-Snf2 antibody, in a SK1 strain. Error bars represent ± 1 SD. Quantification from three biological replicates. Relative and absolute levels of Snf2 increases at the MER1 locus after transfer to sporulation media.
We hypothesized that the early role for Snf2 in MER1 expression suggested by Figure 3 and 4 may involve this deacetylated form of Snf2. Hence we analyzed Snf2 occupancy and histone acetylation at the MER1 locus under sporulation conditions between 0 and 3 h. We find that both Snf2 (Figure 5B) and importantly, H3K9ac (Figure 5C) increase at the MER1 locus over this time course. Over this same time course, we previously found that RPG transcript levels decrease (Figure 4B). Consistent with this, Snf2 is redistributed away from RPGs (Figure 5D) in a manner concomitant with the downregulation of RPG transcript levels (Figures 5E and 4B) and the ratio of Snf2 at RPGs versus MER1 shifts over the 3-h time course, toward MER1 (Figure 5F). This redistribution of Snf2 also follows a shift in the relative H3K9ac levels from the RPGs to the MER1 locus (Supplementary Figure S4).
We also analyzed these molecular events in the BY diploid strain. Again we observed that the levels of acetylated Snf2 decrease before the levels of total Snf2, indicating that the Snf2 present in the cells is deacetylated rapidly when shifted to sporulation conditions (Supplementary Figure S5A). As is the case with the SK1 cells, we observe an increase in Snf2 occupancy at the MER1 locus (Supplementary Figure S5B and D) in a manner coincident with a change in H3K9 acetylation (Supplementary Figure S5C and E). This redistribution is due to the change in Snf2 acetylation, as demonstrated by the increased occupancy of a mutant form of Snf2 that cannot be acetylated (K1493,1497R, referred to as ‘2R’ (13) at the MER1 promoter (Supplementary Figure S6).
As described above, an intriguing difference between snf2Δ in haploid cells in which UME6 is deleted and WT diploid cells (Compare Supplementary Figure S2 to Figure 4) is that Snf2 is required for MER1 expression in the diploid cells. Previous studies have shown that Ume6 recruits the histone deacetylase Rpd3 to target promoters and represses expression of early meiotic genes; Ume6 degradation is required to relieve this inhibition (8,30). Our results suggest that the repressive conditions inhibiting MER1 expression in Ume6-expressing cells require Snf2 for them to be overcome. It is also interesting that both BY, a poorly sporulating strain and SK1, a strain that sporulates efficiently, show similar downregulation by Snf2. Although it remains unclear why BY undergoes meiosis poorly, it does not appear to be due to a defect in Snf2-mediated regulation of meiotic splicing.
These data reveal that there is a temporally regulated role for Snf2 in meiotic splicing, which is illustrated in the model in Figure 6. Under vegetative conditions, with relatively high levels of Snf2, Snf2-dependent transcription of RPGs sequesters spliceosomes away from transcripts with weak SSs, such as the meiotic transcripts. At the same time, MER1 transcription is repressed by Ume6, in part, via the activity of histone deacetylase Rpd3. Upon shift to sporulation media, Snf2 deacetylation/histone acetylation is associated with de-repression at the MER1 locus. This, combined with subsequent molecular events (e.g. Ime1 activity), allows Mer1 levels to rise. Our data suggests that the well-correlated events: meiosis-specific expression of Mer1, downregulation of Snf2, and downregulation of RPG expression create conditions whereby active spliceosomes are free to fine tune expression of the meiotic RNAs; in some cases increasing splicing efficiency by as much as 500%.
Figure 6.

Temporal control of meiotic splicing by Snf2. (A) Upon shift into sporulation media, Ume6 is degraded and Snf2 is deacetylated. This contributes to its relocalization to the MER1 promoter from the RPGs in a manner correlated with H3K9ac. Snf2 reverses Ume6-dependent repression at the MER1 promoter and MER1 mRNA levels rise. (B) Snf2 regulates RPG transcription, such that under vegetative growth conditions, high levels of RPGs titrates away the free pool of active spliceosomes. Ume6 represses expression of meiotic transcripts. Upon shift to sporulation media and the establishment of favorable conditions for MER1 transcription (see panel A), Snf2 is downregulated, which is correlated with repressed RPG expression and the freeing up of active spliceosomes to increase splicing and expression of the meiotic RNAs.
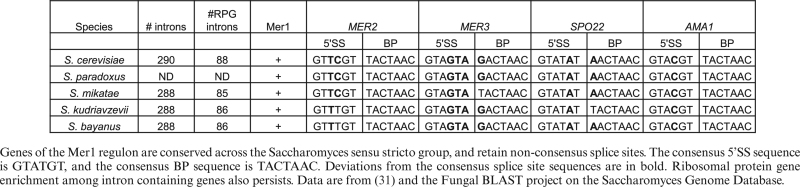
The shift between splicing that supports ribosome biogenesis/translation to splicing that allows spore formation and meiosis seems likely to be an evolutionarily conserved ‘choice’ that the cell makes and in fact, the mechanisms described here appear to be evolutionarily conserved among related Saccharomyces species separated by between 5 and 20 million years of evolution. The genome of the Saccharomyces sensu stricto group possess relatively few introns and there is a significant enrichment of the highly expressed RPGs among ICGs (Table 3). This enrichment persists in even more distant hemiascomycetous yeast as evolutionarily distant as Candida albicans (31). Additionally, all the members of the sensu stricto group possess a homolog of the Mer1 protein, as well as of the four genes of the Mer1 regulon. Importantly, all of these genes in the Mer1 regulon contain a putative Mer1 binding site and nonconsensus SSs, which can tune these introns to the activity of Mer1 and to the level of free spliceosomes. Finally, as we describe below, the Swi/Snf complex is conserved all the way to metazoans. These studies suggest an evolutionarily conserved mechanism whereby the cell redirects its energy from maintaining its translational capacity to the process of meiosis.
Table 3. The Mer1 regulon is conserved across closely related Saccharomyces species.
|
DISCUSSION
One of the outstanding challenges has been to identify specific mechanisms by which changes in chromatin can influence splicing, distinguish between direct (i.e. co-transcriptional) versus transcription-dependent effects of chromatin on splicing and identify biologically important conditions under which these mechanisms are utilized by the cell. Here we show that the chromatin remodeling complex Swi/Snf plays a previously unexplored but fundamental role in regulating meiotic splicing. We propose a model whereby the Swi/Snf complex controls meiotic splicing on multiple levels. We demonstrate that Snf2 is necessary (albeit not sufficient) for expression of MER1 in diploid Ume6-containing cells. As it was previously demonstrated that other factors contribute to MER1 expression (28), it will be important to decipher how the Swi/Snf complex may prime the chromatin environment of the MER1 promoter to facilitate MER1 mRNA expression (Figures 4 and 5, Supplementary Figure S6). Interestingly, this is not the only example of chromatin remodeling contributing to finely timed transcription factor binding and transcriptional activation in the meiotic program. For example, Inai et al. demonstrated that expression of IME2 involves chromatin remodeling to overcome HDAC-dependent nucleosome placement over the IME2 TATA element (32). Moreover, temporally regulated binding of Ime1p to the IME2 gene ensures a biologically important lag in IME2 expression (32).
We also show a crucial role for competitive acetyl marks on histone H3 and Snf2 to achieve temporal and spatial specificity of Snf2 targeting. While it is possible that there is active deacetylation of Snf2 (33), another possibility is that continued translation of SNF2 leads to a rapid replacement of the acetylated population of Snf2 with an unacetylated one, allowing the protein to be targeted to the MER1 locus. The retargeting of deacetylated Snf2 away from the RPG promoters and subsequent rapid downregulation of Snf2 corresponds to downregulation of ribosomal protein gene expression and a complete reprogramming of the cell's splicing landscape. Sustained translation and rapid turnover could also explain Snf2 downregulation a few hours after transfer to sporulation media. Once translation of SNF2 is downregulated, potentially due to decrease in ribosomal activity, the existing degradation pathway turns over the remaining protein, leading to a loss Snf2. The relationship between Snf2 translation, deacetylation and degradation is likely to be important for fine-tuned control of the protein, which we are actively exploring. Remarkably, Snf2 expression is so tightly controlled during meiosis that its overexpression (an experiment we attempted to establish causation between Snf2 levels and meiotic events) is buffered in meiotic cells (data not shown). Nonetheless, our results illustrate the principle that competition (competition amongst acetylated residues for the Snf2 bromodomain and competition amongst different introns for the spliceosome) is an essential part of cellular adaptation to various environmental and/or developmental stresses. While core gene expression machineries, such as the transcription apparatus, the spliceosome and the ribosome appear to be abundant, new models must take into account that not only are these machineries limiting, but they can be more so under specific cellular conditions and substrate induced competition for gene expression machineries plays an important role in the determining regulatory outcomes (9,10).
Although Snf2 is the core catalytic component of the Swi/Snf complex and it has not been shown to function as an independent subunit, an outstanding question is whether Snf2 is the only component of Swi/Snf that contributes to meiotic regulation. Recent studies that measured changes in gene expression upon deletion of components of the Swi/Snf complex showed that deletion of Snf2 had broader and more significant changes in cellular expression profiles than other non-essential components of the complex (34,35). Importantly, expression of genes involved in translation and ribosomal assembly is downregulated in cells deleted of SNF2, but not when genes encoding other components of the Swi/Snf complex such as SNF5 and SNF12 are deleted. It has been suggested that the peripheral components of Swi/Snf perform regulatory and/or targeting roles within the complex and are dispensable for certain functions of Snf2. It has also been suggested that Snf2 can be targeted to genes with high H3K9-ac occupancy (such as the RPGs) even when the structural integrity of the Swi/Snf complex is compromised (34). Further studies are required to assess the degree to which the other subunits of the complex contribute to Snf2 regulation and activity during meiosis.
Interestingly, there is significant evidence that the process of translation is under heavy regulatory control during meiosis, across eukaryotes even as far as humans (36), perhaps for energetic reasons. We provide evidence here that the principle effector of this coordination between translational regulation and meiosis in hemiascomycetous yeast is the chromatin remodeling complex Swi/Snf.
One striking result is that the benefits of RPG downregulation and subsequent spliceosome redistribution are preferentially reaped by the genes of the Mer1 regulon (Supplementary Figure 2B). Snf2΄s role in Mer1 expression is an important contributor to this effect since, as others have shown, Mer1 helps recruit spliceosomes to these pre-mRNAs (6). It may be unexpected, then, that the four Mer1-dependent introns all still have a range of splicing efficiency well below 100% (from about 50–75%). Other reports in the literature suggest that introns may possess intrinsic splicing properties. For example, different introns respond differently to perturbations in the splicing environment and demonstrate variable dependencies on components of the spliceosome (2). Hence, each intron may also have an intrinsic ‘maximum possible splicing efficiency,’ a parameter determined by factors specific to the intron itself, such as the nature of its SSs, its length, its genetic context and the specific environmental conditions. Consistent with this, even though rapamycin treatment downregulates RPGs to an even greater extent than the deletion of Snf2 (Supplementary Figure S2D and E), the end point in terms of splicing efficiency remains the same. Hence, even with Mer1 present to ‘attract’ spliceosomes to RNAs, splicing efficiency appears to be an intrinsic property of these ICGs.
Although it does not appear to significantly impact Snf2's RPG/Mer1-dependent mechanism of meiotic splicing regulation, we cannot exclude the possibility that Snf2 may affect spliceosome assembly co-transcriptionally. Recent observations from studies in Schizosaccharomyces pombe also describe a role for Swi/Snf in splicing. However, these studies suggest that Swi/Snf stimulates spliceosome activation via its role in nucleosome deposition and maintenance (37). There exist significant functional differences between the complexes of the two yeast species. For example, reports suggest that many of the roles that the Swi/Snf complex performs in S. cerevisiae are, in S. pombe, divided between the RSC and Snf2 complexes (38), which might serve to explain differences in the specific roles of the complexes in splicing regulation. The studies in S. pombe nevertheless implicate the Swi/Snf chromatin remodeling activity in splicing regulation, a feature that appears to be conserved among metazoans as well (39,40).
Intriguingly, Swi/Snf has been shown to have roles in metazoan meiosis, although the molecular mechanisms underlying these roles are not known. Brg1, the ATPase subunit of the mammalian Swi/Snf complex, peaks in expression during the early stages of meiosis and is turned off in maturing round spermatids. Moreover, knocking down Brg1 results in prophase arrest during meiosis I, due to a failure to complete synapsis (41,42). Another protein involved in meiosis, mammalian HFM1/Mer3, is an integral part of the ZMM group of proteins. HFM1/Mer3 has been shown to be required for the completion of synapsis during meiosis, and deletion of Hfm1 closely resembles the phenotype of a Brg1 knockdown (43). While the details of meiotic regulation are unlikely to be exactly the same, it is interesting to note that the Swi/Snf complex has retained a role in regulating meiosis through evolution. Finally, global changes in splicing have been reported during male meiosis in mammals (44), so understanding the mechanism underlying this regulation and determining if Snf2 contributes to it are of particular interest.
This work reveals multiple mechanisms by which Swi/Snf acts as a nexus point in the regulation of meiosis in S. cerevisiae. All or a subset of these mechanisms may also be crucial in the gene regulation response to other physiological conditions that yeast might experience, which we are currently exploring.
ACCESSION NUMBERS
Data generated in this study is available under GEO accession number GSE94404.
Supplementary Material
ACKNOWLEDGEMENTS
We would also like to thank Dr Jerry Workman for the kind gift of the Snf2 K1493,1497R strain and the isogenic WT. We would also like to thank Lauren Neves and other members of the lab for critical reading of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences [GM-085474]; Whitcome Molecular Biology Pre-doctoral Fellowship (to S.V.); Whitcome Summer Research Fellowship (to A.R.G.). Funding for open access charge: National Institute of General Medical Sciences [GM-085474].
Conflict of interest statement. None declared.
REFERENCES
- 1. Naftelberg S., Schor I.E., Ast G., Kornblihtt A.R.. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 2015; 84:165–198. [DOI] [PubMed] [Google Scholar]
- 2. Pleiss J.A., Whitworth G.B., Bergkessel M., Guthrie C.. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol. Cell. 2007; 27:928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bergkessel M., Whitworth G.B., Guthrie C.. Diverse environmental stresses elicit distinct responses at the level of pre-mRNA processing in yeast. RNA. 2011; 17:1461–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson T.L., Vilardell J.. Regulated pre-mRNA splicing: the ghostwriter of the eukaryotic genome. Biochim. Biophys. Acta. 2012; 1819:538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Munding E.M., Igel A.H., Shiue L., Dorighi K.M., Trevino L.R., Ares M. Jr. Integration of a splicing regulatory network within the meiotic gene expression program of Saccharomyces cerevisiae. Genes Dev. 2010; 24:2693–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spingola M., Ares M. Jr. A yeast intronic splicing enhancer and Nam8p are required for Mer1p-activated splicing. Mol. Cell. 2000; 6:329–338. [DOI] [PubMed] [Google Scholar]
- 7. Qiu Z.R., Schwer B., Shuman S.. Determinants of Nam8-dependent splicing of meiotic pre-mRNAs. Nucleic Acids Res. 2011; 39:3427–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kadosh D., Struhl K.. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell. 1997; 89:365–371. [DOI] [PubMed] [Google Scholar]
- 9. Munding E.M., Shiue L., Katzman S., Donohue J.P., Ares M. Jr. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol. Cell. 2013; 51:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baumgartner B.L., Bennett M.R., Ferry M., Johnson T.L., Tsimring L.S., Hasty J.. Antagonistic gene transcripts regulate adaptation to new growth environments. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:21087–21092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Berg M.G., Singh L.N., Younis I., Liu Q., Pinto A.M., Kaida D., Zhang Z., Cho S., Sherrill-Mix S., Wan L. et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell. 2012; 150:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Merkhofer E.C., Johnson T.L.. U1 snRNA rewrites the "script. Cell. 2012; 150:9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dutta A., Gogol M., Kim J.H., Smolle M., Venkatesh S., Gilmore J., Florens L., Washburn M.P., Workman J.L.. Swi/Snf dynamics on stress-responsive genes is governed by competitive bromodomain interactions. Genes Dev. 2014; 28:2314–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grate L., Ares M. Jr. Searching yeast intron data at Ares lab web site. Methods Enzymol. 2002; 350:380–392. [DOI] [PubMed] [Google Scholar]
- 15. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Livak K.J., Schmittgen T.D.. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408. [DOI] [PubMed] [Google Scholar]
- 17. Deutschbauer A.M., Williams R.M., Chu A.M., Davis R.W.. Parallel phenotypic analysis of sporulation and postgermination growth in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:15530–15535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Padmore R., Cao L., Kleckner N.. Temporal comparison of recombination and synaptonemal complex formation during meiosis in S. cerevisiae. Cell. 1991; 66:1239–1256. [DOI] [PubMed] [Google Scholar]
- 19. Rockmill B., Lambie E.J., Roeder G.S.. Spore enrichment. Methods Enzymol. 1991; 194:146–149. [DOI] [PubMed] [Google Scholar]
- 20. Khare A.K., Singh B., Singh J.. A fast and inexpensive method for random spore analysis in Schizosaccharomyces pombe. Yeast. 2011; 28:527–533. [DOI] [PubMed] [Google Scholar]
- 21. Gunderson F.Q., Johnson T.L.. Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 2009; 5:e1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gunderson F.Q., Merkhofer E.C., Johnson T.L.. Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:2004–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knight B., Kubik S., Ghosh B., Bruzzone M.J., Geertz M., Martin V., Denervaud N., Jacquet P., Ozkan B., Rougemont J. et al. Two distinct promoter architectures centered on dynamic nucleosomes control ribosomal protein gene transcription. Genes Dev. 2014; 28:1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mallory M.J., Cooper K.F., Strich R.. Meiosis-specific destruction of the Ume6p repressor by the Cdc20-directed APC/C. Mol. Cell. 2007; 27:951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martin D.E., Soulard A., Hall M.N.. TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell. 2004; 119:969–979. [DOI] [PubMed] [Google Scholar]
- 26. Brar G.A., Yassour M., Friedman N., Regev A., Ingolia N.T., Weissman J.S.. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012; 335:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nachman I., Regev A., Ramanathan S.. Dissecting timing variability in yeast meiosis. Cell. 2007; 131:544–556. [DOI] [PubMed] [Google Scholar]
- 28. Engebrecht J., Roeder G.S.. MER1, a yeast gene required for chromosome pairing and genetic recombination, is induced in meiosis. Mol. Cell. Biol. 1990; 10:2379–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Awad S., Hassan A.H.. The Swi2/Snf2 bromodomain is important for the full binding and remodeling activity of the Swi/Snf complex on H3- and H4-acetylated nucleosomes. Ann. N.Y. Acad. Sci. 2008; 1138:366–375. [DOI] [PubMed] [Google Scholar]
- 30. Pnueli L., Edry I., Cohen M., Kassir Y.. Glucose and nitrogen regulate the switch from histone deacetylation to acetylation for expression of early meiosis-specific genes in budding yeast. Mol. Cell. Biol. 2004; 24:5197–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hooks K.B., Delneri D., Griffiths-Jones S.. Intron evolution in Saccharomycetaceae. Genome Biol. Evol. 2014; 6:2543–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inai T., Yukawa M., Tsuchiya E.. Interplay between chromatin and trans-acting factors on the IME2 promoter upon induction of the gene at the onset of meiosis. Mol. Cell. Biol. 2007; 27:1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim J.H., Saraf A., Florens L., Washburn M., Workman J.L.. Gcn5 regulates the dissociation of Swi/Snf from chromatin by acetylation of Swi2/Snf2. Genes Dev. 2010; 24:2766–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dutta A., Sardiu M., Gogol M., Gilmore J., Zhang D., Florens L., Abmayr S.M., Washburn M.P., Workman J.L.. Composition and function of mutant Swi/Snf complexes. Cell Rep. 2017; 18:2124–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sen P., Luo J., Hada A., Hailu S.G., Dechassa M.L., Persinger J., Brahma S., Paul S., Ranish J., Bartholomew B.. Loss of Snf5 induces formation of an aberrant Swi/Snf complex. Cell Rep. 2017; 18:2135–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kleene K.C. Patterns, mechanisms, and functions of translation regulation in mammalian spermatogenic cells. Cytogenet. Genome Res. 2003; 103:217–224. [DOI] [PubMed] [Google Scholar]
- 37. Patrick K.L., Ryan C.J., Xu J., Lipp J.J., Nissen K.E., Roguev A., Shales M., Krogan N.J., Guthrie C.. Genetic interaction mapping reveals a role for the Swi/Snf nucleosome remodeler in spliceosome activation in fission yeast. PLoS Genet. 2015; 11:e1005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monahan B.J., Villen J., Marguerat S., Bahler J., Gygi S.P., Winston F.. Fission yeast Swi/Snf and RSC complexes show compositional and functional differences from budding yeast. Nat. Struct. Mol. Biol. 2008; 15:873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Batsche E., Yaniv M., Muchardt C.. The human Swi/Snf subunit Brm is a regulator of alternative splicing. Nat. Struct. Mol. Biol. 2006; 13:22–29. [DOI] [PubMed] [Google Scholar]
- 40. Tyagi A., Ryme J., Brodin D., Ostlund Farrants A.K., Visa N.. Swi/Snf associates with nascent pre-mRNPs and regulates alternative pre-mRNA processing. PLoS Genet. 2009; 5:e1000470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim Y., Fedoriw A.M., Magnuson T.. An essential role for a mammalian Swi/Snf chromatin-remodeling complex during male meiosis. Development. 2012; 139:1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J., Gu H., Lin H., Chi T.. Essential roles of the chromatin remodeling factor BRG1 in spermatogenesis in mice. Biol. Reprod. 2012; 86:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guiraldelli M.F., Eyster C., Wilkerson J.L., Dresser M.E., Pezza R.J.. Mouse HFM1/Mer3 is required for crossover formation and complete synapsis of homologous chromosomes during meiosis. PLoS Genet. 2013; 9:e1003383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmid R., Grellscheid S.N., Ehrmann I., Dalgliesh C., Danilenko M., Paronetto M.P., Pedrotti S., Grellscheid D., Dixon R.J., Sette C. et al. The splicing landscape is globally reprogrammed during male meiosis. Nucleic Acids Res. 2013; 41:10170–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.