

Abstract
Background.
Diffuse intrinsic pontine glioma (DIPG) is a high-grade brainstem glioma of children with dismal prognosis. There is no single unifying model about the cell of origin of DIPGs. Proliferating cells in the developing human and mouse pons, the site of DIPGs, express neural stem/progenitor cell (NPC) markers, including Sox2, nestin, vimentin, Olig2, and glial fibrillary acidic protein, in an overlapping and non-overlapping manner, suggesting progenitor cell heterogeneity in the pons. It is thought that during a restricted window of postnatal pons development, a differentiation block caused by genetic/epigenetic changes leads to unrestrained progenitor proliferation and DIPG development. Nearly 80% of DIPGs harbor a mutation in the H3F3A or the related HIST1H3B gene. Supporting the impaired differentiation model, NPCs derived from human induced pluripotent stem cells expressing the H3F3A mutation showed complete differentiation block. However, the mechanisms regulating an altered differentiation program in DIPG are unknown.
Methods.
We established syngeneic serum-dependent and independent primary DIPG lines, performed molecular characterization of DIPG lines in vitro and in an orthotopic xenograft model, and used small hairpin RNA to examine Olig2 function in DIPG.
Results.
The transcription factor Olig2 is highly expressed in 70%–80% of DIPGs. Here we report that Olig2 expression and DIPG differentiation are mutually exclusive events in vitro, and only DIPG cells that retained Olig2 in vitro formed robust Olig2-positive brainstem glioma with 100% penetrance in a xenograft model.
Conclusion.
Our results indicate Olig2 as an onco-requisite factor in DIPG and propose investigation of Olig2 target genes as novel candidates in DIPG therapy.
Keywords: differentiation, DIPG, Olig2
Diffuse intrinsic pontine gliomas (DIPGs) account for 10%–15% of brain tumors in children and are the leading cause of brain tumor–related deaths in children.1–3 DIPGs are infiltrative, highly aggressive tumors and involve ≥50% of the pons, resulting in death within one year from diagnosis.1–3 Tumor cells permeate the parenchyma, diffusely expanding through the pons, displacing neuronal cell bodies and axonal tracts, and sometimes through the white matter and granule cell layer of the cerebellum.1–4 Many patients demonstrate cerebral invasion, particularly of the frontal lobes and in the lateral subventricular zone, spinal metastases, or leptomeningeal spread at the time of recurrence or autopsy. The timing of DIPG development, which usually occurs during the first decade of life,5,6 strongly indicates a developmental window during which proliferation/differentiation of progenitor cells is affected during the normal development of the pons. Recent work demonstrates that the human pons, particularly the ventral pons (the site where DIPG is formed) expands dramatically during the first 7 months after birth.7,8 During this time, neural progenitor cells (NPCs) rapidly proliferate, particularly during the first month of life. NPC proliferation gradually declines between 2 and 7 months of age, following which only rare mitotic cells are observed through the first decade of life.7
Proliferating NPCs in the developing human and murine pons express the NPC intermediate filament nestin (neuroectodermal stem cell marker), the stem/progenitor cell transcription factors sex determining region Y–box 2 (Sox2), and the basic helix-loop-helix (bHLH) transcription factor Olig27,8 in overlapping and non-overlapping manner. For instance, only about half of nestin+ cells in the ventral human pons express Olig2, and in the murine ventral pons only some Olig2+ cells express Sox2. In the developing human pons, nestin/vimentin-expressing cells were found in the ventral pons but were mainly restricted to the lining of the fourth ventricle, while the majority of the proliferating cells in the pons parenchyma are Olig2+.7,8 A second wave of Olig2+ cells appears in the ventral pons during mid-childhood.8 In a mouse model, deletion of Ink4a‒ADP-ribosylation factor and overexpression of platelet derived growth factor (PDGF) in nestin+ cells gave rise to high-grade brainstem glioma.9 However, gene set enrichment analysis from fresh biopsy tissue revealed that DIPGs have a distinct expression profile compared with supratentorial and other brainstem high-grade glioma.10 Specifically, DIPGs overexpressed a set of bHLH transcription factors including Olig2, which was present in 70%–80% of cases.10 Olig2 expression also correlated with DIPG mitosis, aggressiveness, and shorter median overall survival.10,11
Rapid advances have been made in the last few years to understand the molecular basis of the disease12–16; however, the identity of cells that give rise to DIPG is unclear. We analyzed 5 DIPG lines, including one previously uncharacterized line established in our laboratory. We found that Olig2 expression is robust only in DIPG lines maintained in serum-free conditions and that Olig2 expression was important for in vivo engraftment and tumor formation. Isogenic lines maintained in serum from the beginning proliferated rapidly in vitro, expressed stem cell markers, including nestin, Musashi, and Nanog, but failed to form tumors in mice. Olig2-expressing serum-free lines resisted differentiation in vitro, and Olig2 silencing caused cell death in these lines. Immunohistochemistry (IHC) revealed robust Olig2 expression in DIPGs. Taken together, our results argue that Olig2 could be an onco-requisite factor during transformation of NPCs and evolution of DIPG in the human ventral pons.
Methods
Establishment of DIPG Cells in Culture from Autopsy Tissue
Tumor tissue was donated by a 5-year-old boy. Informed consent was received from the family and tissue was harvested in sterile conditions 16 hours after death in the autopsy suite following an institutional review board–approved institutional protocol (CCHMC Study No: 2013-1245; patient #PBTR3) and examined in the Pathology Core at CCHMC by expert pathologists. Cells were maintained in Dulbecco’s modified Eagle’s medium F/12 supplemented with heparin (10 ng/mL) and 20 μg/mL of each of epidermal growth factor (EGF), fibroblast growth factor (FGF)2, and PDGFα or with 10% primary serum (see Supplementary material and table for details). Cell proliferation/viability assay was done using CellTiter-Fluor Cell Viability Assay (Promega). Equal numbers of cells (usually 10000) were seeded in 96-well plates and viability was determined at indicated times. Absolute values from the plate reader were normalized (scaled down to 0–100/120) for simplification purposes and expressed as relative proliferation.
DIPG Differentiation, Immunocytochemistry, and Immunohistochemistry
DIPG spheres were differentiated for 5 days in medium containing 5% primary serum. An assay incorporating EdU (5-ethynyl-2'-deoxyuridine) was performed using Click-iT EdU Alexa Fluor 594 (Life Technologies). Immunocytochemistry and IHC were done on differentiated cells and formalin-fixed paraffin-embedded tissue obtained from the Pathology Core at CCHMC, respectively. The following primary antibodies were used: nestin, Olig2, O4 (all from Millipore), Tuj1 (Covance), Neurofilament (Sigma), Sox2, glial fibrillary acidic protein (GFAP), Musashi, PDGF receptor α (PDGFRα) (all from Cell Signaling Technology), EGF receptor (EGFR), and Nanog (both from Epitomics). For immunocytochemistry, appropriate Alexa-Fluor 488 or 594 secondary antibodies (Life Technologies) were used (see Supplementary data for detailed methods).
Orthotopic and flank xenograft
All animal procedures were carried out in accordance with the Institutional Animal Care and Use Committee–approved protocol of CCHMC. Animals were monitored daily by animal care personnel. Two microliters of medium containing 40000 luciferase+ cells was injected stereotactically into the fourth ventricle of cold-anesthetized P2 nonobese diabetic severe combined immunodeficient gamma (NSG) mice. The coordinates for injection were 3 mm posterior to lambda suture and 3 mm deep. Mice were monitored for neurological behaviors and imaged using the Xenogen IVIS Imaging System (STTARR). For flank xenograft, 1 × 106 cells were injected and mice were euthanized after 24 days.
Plasmids and virus
Lentiviral luciferase construct and doxycycline-inducible Olig2 construct were purchased from Addgene. Olig2 small hairpin (sh)RNA and nontarget shRNA clones in pLKO lentiviral transfer vector were purchased from the Lentiviral Core at CCHMC (originally TRC2.1 library, Sigma). Lentivirus was made using the 3 plasmid system following an Institutional Biosafety Committee–approved protocol.
Results
Growth Factor Responsiveness of a DIPG Line Established from Autopsy Tissue
Radiographic imaging showed a large tumor (Fig. 1A; Supplementary Fig. 1A, B) in the ventral pons measuring 6.5 × 4.5 × 4 cm in volume. Histopathology of autopsy tissue revealed glioblastoma (GBM; World Health Organization grade IV) with approximately 20% tumor necrosis (Supplementary Fig. 1C‒F). Tumor spread was observed in the midbrain, medulla, cerebellum, subthalamic cerebral cortex, oculomotor nerve, and dorsal cervical spinal cord (Supplementary Fig. 1G‒L). Sequencing results revealed H3K27M point mutation in H3.3 uniformly in primary pons, left and right anterior pons, right posterior pons, right basal ganglia, and leptomeningeal spread, while TP53 R273H mutation was found in all the above except primary pons (not shown).
Fig. 1.

Establishment of serum-free and serum-dependent DIPG lines. (A) Axial MRI scan showing location of the DIPG in the patient # PBTR3. The tumor is shown by the arrow. Bright field images of the serum-dependent line (B) and serum-free sphere line (C) established from autopsy tissue. Proliferation of serum-free line (D) and serum line (E) in the presence of combination of growth factors or serum. (F) Relative proliferation of serum-free DIPG line and normal human fetal brainstem neural progenitor cells (NPC) grown in identical conditions. (G) Proliferation of an adult GBM serum-free line in the presence or absence of growth factors. (H) Proliferation of the serum-free DIPG line in the presence or absence of growth factors.
To molecularly characterize the tumor cells, we established in vitro cultures by seeding one half of cells in 10% primary fetal bovine serum, and the remainder in growth factor supplemented serum-free medium. Adherent DIPG PBTR3 cells appeared within one week of seeding in the serum condition, and a confluent culture was established in one month (Fig. 1B). Small and large spheres also appeared within the first 2 weeks of seeding, and the isogenic serum-free line was also established in 4 weeks (Fig. 1C). Both lines retained the H3.3K27M mutation (not shown). To examine the growth factor(s) required for maximal viability of this DIPG line, we quantitated viable cells in cultures containing single or multiple growth factor combinations. EGF + FGF2 was sufficient to maintain a viable culture (Fig. 1D). Because FGF18 and its receptor FGFR2c are highly expressed in the developing mouse brainstem (Supplementary Fig. 2) much more than EGF or FGF2 (data retrieved from Allen’s mouse brain atlas), we tested whether FGF18 would further enhance growth of DIPGs. No single growth factor, including EGF, FGF2, PDGFα, FGF18, vascular endothelial growth factor α, or a combination of 4 or 5 growth factors could significantly improve viable cell numbers beyond what was achieved with EGF + FGF2, except for PDGFα, which in combination with EGF and FGF2 improved cell numbers by 15%–20% (Fig. 1D). Among the growth factors we tested, EGF alone was the most effective in being able to maintain about half maximal proliferation of this DIPG line. It is possible that EGF/FGF2-responsive cells were selected in our culture, and other growth factors like FGF18 might play an important role in vivo. In contrast to the serum-free line, no combination of growth factors matched the ability of serum to promote maximal proliferation and viability of the serum line (Fig. 1E), suggesting that additional serum factors stimulate DIPG proliferation. Cell-autonomous proliferation of serum-free DIPG cells was also higher compared with normal human fetal brainstem NPCs (Fig. 1F), and unlike adult GBM lines with constitutively active growth factor signaling (Fig. 1G), the serum-free DIPG line was completely growth-factor dependent for proliferation and survival (Fig. 1H). Collectively, our data show that DIPG cells are completely growth-factor dependent and that a combination of EGF, FGF2, and PDGFα is required for maximal viability of serum-free DIPG lines in vitro.
Olig2-expressing DIPG Cells Resist Differentiation In vitro
To molecularly characterize the DIPG lines, we performed immunocytochemistry. During proliferation in 10% serum, the serum line robustly expressed stem cell markers, including nestin, Sox2, Musashi, and Nanog, but did not express Olig2 (Fig. 2A‒E). About 25% of cells expressed the glial marker GFAP and about 20% of cells expressed the early neuronal marker Tuj1 (Fig. 2F, G). Consistent with others,17 EGFR was expressed by nearly all cells of our PBTR3 DIPG serum line; however, PDGFRα expression was heterogeneous (Fig. 2H, I). Surprisingly, ~40% of these cells were still able to incorporate EdU even in reduced serum condition (5%), but showed no Olig2 expression, indicating that these cells are able to proliferate in suboptimal conditions and in the absence of Olig2 (Fig. 2J).
Fig. 2 .

Immunophenotyping of PBTR3 serum line. Confocal immunofluorescence microscopy images (magnification 20×) of PBTR3 serum line showing marker expression: (A‒E) showing expression of stem cell/proliferation markers—nestin, Sox2, Musashi, Nanog, and Olig2. Note: the serum line expresses the typical stem cell/NPC markers except Olig2. Expression of the astrocytic marker (F) and the early neuronal marker Tuj1 (G) and that of 2 receptor tyrosine kinases, EGFR (H) and PDGFRα, (I) are shown. The arrows and asterisks in (H) and (I) shows heterogeneous expression of the receptor tyrosine kinases. (J) DNA synthesis (EdU incorporation) and Olig2 expression in reduced serum (5.0 serum %) condition. Nuclei were stained with 4′,6′-diamidino-2-phenylindole. Scale bars 2 µm.
In sharp contrast to the serum line, proliferating cells in serum-free spheres strongly expressed Olig2 in addition to nestin and Sox2 (Fig. 3A, B). Following 5 days of culture in differentiation medium, 90% of cells retained Sox2 and nestin (Fig. 3C, D). Some of these cells expressed either the glial intermediary filament GFAP or the neuronal filament Tuj1 (Fig. 3E). About 7% of cells aberrantly coexpressed GFAP and Tuj1 (Fig. 3E). Despite strong expression of the oligodendrocyte lineage transcription factor Olig2 by the proliferating cells in serum-free spheres (Fig. 3A), no significant differentiation into O4+ oligodendrocytes was observed (Fig. 3F). Instead, about 50% of cells continued to express Olig2 without differentiation into oligodendrocytes. A striking observation was the mutual exclusiveness of Olig2 with glial and neuronal differentiation markers. Ninety-eight percent of Olig2+ cells in the differentiation condition did not differentiate into GFAP astroglia or NF+ neurons (Fig. 3G, H, J, K). This observation in our PBTR3 serum-free line was recapitulated by 3 other lines: JHH-DIPG1 (Fig. 3I, L), SU-DIPG4, and SU-DIPG6 (not shown). To examine the fraction of Olig2+ cells that proliferated under this condition, we used EdU as before. In 3 independent sphere lines (SU-DIPG 4, SU-DIPG 6, and JHH-DIPG1), 30%–35% of Olig2+ cells also incorporated EdU, and about 15% EdU+ cells were Olig2− (Fig. 3M‒O). Therefore, proliferation of serum-free sphere lines in growth factor–free conditions occurred in the presence or absence of Olig2, although we cannot rule out that the Olig2− cells expressed undetectable levels of Olig2. Surprisingly, even prolonged exposure (4 wk) to serum-free conditions failed to restore Olig2 in the serum line. Together, these immunophenotyping results indicate that a serum-free condition allows retention of Olig2, while long-term exposure to serum represses Olig2 expression in DIPG cells.
Fig. 3 .

Mutually exclusive expression of Olig2 and differentiation markers during DIPG differentiation in vitro. (A‒H & J, K = PBTR3 serum-free; I, L = JHH-DIPG). Confocal immunofluorescence microscopy images showing marker expression during DIPG proliferation and differentiation: (A, B) expression of nestin and Olig2 in DIPG spheres in proliferation condition; (C‒L) marker expression during differentiation condition in the absence of growth factors. Note: DIPG cells under this condition continue to express stem cell markers Sox2 (C), nestin (D), and Olig2 (G–I); a fraction of cells also express the astrocytic marker GFAP and the early neuronal marker Tuj1 (E), and the mature neuronal marker neurofilament (H); very few cells expressed the oligodendrocyte marker O4 (F). Magnified images of G, H, and I showing mutual exclusivity between Olig2 and GFAP (J, L) and Olig2 and NF (K) in PBTR3 and JHH DIPG1. (M‒O) Edu incorporation assay, showing DNA synthesis in Olig2+ cells under differentiation condition. Nuclei were stained with 4′,6′-diamidino-2-phenylindole. Scale bars 2 µm (A‒I); 100 µm (M‒O).
Olig2 Expression Is High in DIPGs and Marks Tumor Cells in an Orthotopic DIPG Xenograft
The exclusivity of Olig2 expression in the serum-free PBTR3 line gave us an opportunity to test whether Olig2 expression defines the capacity of cell engraftment and tumor formation in vivo. We expressed luciferase through lentiviral transduction and transplanted equal number of isogenic PBTR3 cells to the fourth ventricle of postnatal day 2 NSG mice. All mice injected with the serum-free line (n = 5) developed neurological symptoms by 6 months, demonstrated strong luciferase signal (Fig. 4A), and were euthanized. Mice transplanted with the serum line (n = 6) remained tumor free for 12 months. Histological examination of mice injected with the serum-free line showed significantly enlarged pons (Fig. 4B) compared with mice injected with the isogenic serum line (Fig. 4C). Detailed examination showed hypercellular infiltration in the pons and white matter of the cerebellum (Fig. 4D, E) with histopathology similar to high-grade gliomas (Fig. 4F) seen in humans. Large numbers of diffusely infiltrating tumor cells were observed throughout the midbrain and cerebellum, interspersed with the granule cell neurons, and in the white matter tracts of every cerebellar folia (Fig. 4G). Leptomeningeal spread, a histopathological feature of DIPGs, was evident in our xenograft model (Fig. 4H). The overall histopathology of the pons xenograft was similar to that of DIPG with 2 noticeable exceptions: the patient tumor was significantly more vascular and displayed necrosis.
Fig. 4 .

Establishment of xenograft from PBTR3 serum-free line. (A) Imaging of luciferase-positive DIPG xenograft in the brainstem of NSG mice. (B, C) Hematoxylin and eosin (H&E) staining showing brainstem and cerebellum (magnification 1.6×) of mice injected with serum-free PBTR3 line (B) or isogenic serum line (C). Note: enlarged pons in (B) but not in (C). (D, E) Magnified images of pons and cerebellum of mice injected with the serum-free line showing high cellularity in both structures. (F, G) Magnified images of pons and cerebellum showing diffuse nature of the tumor in the pons (F) and infiltration of tumor cells in the cerebellar folia (G). (H) H&E image showing tumor cell spread across the meninges akin to leptomeningeal disease (arrows). (I‒K) IHC showing Olig2 positivity of nearly all tumor cells in the pons (I, J) and the cerebellum (K; inset magnification 63×) which shows infiltration of Olig2+ DIPG cells (red arrowhead) among granule cell neurons (blue arrowhead). (L, M, N) GFAP, nestin, and PDGFRα immunoreactivity, respectively. Scale bars 500 μm (D, E, I, K‒N); 200 μm (F, G,H, J).
To examine if Olig2 expression is retained in vivo during tumorigenesis, we performed IHC. Nearly all tumor cell nuclei were positive for Olig2 (Fig. 4I‒K). In the cerebellar granule cell layers, Olig2+ tumor cells (Fig. 4K, red arrowheads) were observed scattered among granule cell neurons (Fig. 4K, blue arrowheads). IHC also showed immunoreactivity of tumor cells for nestin, GFAP, and PDGFRα (Fig. 4L‒N). Analysis of the original patient tumor (PBTR3) showed strong Olig2 immunoreactivity in about 40% of tumor cells (Fig. 5A). In this tumor, GFAP expression was low (Fig. 5B), nestin expression was mainly restricted to endothelial cells of the neovasculature (Fig. 5C), and only a few tumor cells expressed nestin (Fig. 5C, inset). To verify whether Olig2 expression is common in DIPGs, we extended IHC to 3 additional tumors. All 3 tumors showed the presence of Olig2+ cells. DIPG #C13-12 was strongly positive for Olig2 (about 45% of cells) and weakly GFAP+, with few nestin+ cells (Fig. 5E‒G). DIPG #C13-54 was strong for Olig2 (about 40%) and highly positive for both GFAP and nestin (Fig. 5I‒K). DIPG #C13-69 had fewer Olig2+ cells (about 15%), many GFAP+ cells, and only a few nestin+ cells (Fig. 5M‒O). Thus, compared with nestin, Olig2 expression is abundant in DIPGs. Our IHC results also showed that all 4 tumors strongly expressed the PDGFα receptor (Fig. 5D‒P).
Fig. 5 .

Immunophenotyping of original PBTR3 and other DIPGs. IHC of original PBTR3 DIPG and 3 other DIPGs showing expression of Olig2 (A, E, I, M), GFAP (B, F, J, N), nestin (C, G, K, O), and PDGFRα (D, H, L, P). Magnifications 20× (A‒P); all insets equal area cropped 40×. Note that 3 out of 4 tumors express high Olig2 (A, D, G); GFAP was high in 2 tumors, DIPG # C13-54 (H) and C13-69 (K); only a few GFAP+ tumor cells were observed in PBTR3 and DIPG #C13-12 (B, E, inset); nestin expression was generally low except DIPG #C13-54 (I), while all tumors expressed high levels of PDGFRα. In the original PBTR3, nestin expression was primarily restricted to endothelial cells (C; inset in C shows a few nestin+ tumor cells). Scale bars 0.005 mm.
Consistent with the PBTR3 line, Olig2 was completely lost in JHH-DIPG1 upon exposure to 10% serum for 3 weeks (Supplementary Fig. 3A, B). To examine the mechanism of Olig2 suppression by serum, we performed chromatin immunoprecipitation analysis. Our results showed that while serum-free JHH-DIPG cells demonstrate a significant amount of active chromatin mark H3K4Me3 and diminished inactive chromatin mark H3K9Me3 in the Olig2 transcription start site, long-term serum-treated cells demonstrate the opposite (Supplementary Fig. 3C, D), indicating epigenetic regulation of Olig2 by serum factors. Unfortunately, this cell line did not reliably form tumors in vivo (not shown). In another line, CCHMC-DIPG1, serum only partially suppressed Olig2 expression (Supplementary Fig. 3E, F), suggesting additional mechanisms of Olig2 regulation in this line. Consistent with Olig2 retention, the serum-exposed CCHMC-DIPG1 line was able to form tumors in vivo. Although tumors grew slowly compared with the serum-free sphere line (Supplementary Fig. 3G, H), there was no significant difference in survival (not shown).
Olig2 Is Required for DIPG Proliferation
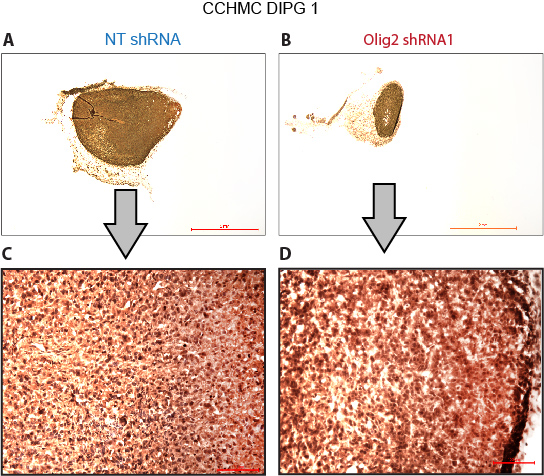
We examined whether Olig2 is specifically required for the proliferation of serum-free DIPGs. All serum-free lines expressed high levels of Olig2 (Fig. 6A; Supplementary Fig. 3D), while the PBTR3 serum line expressed very little Olig2. We validated Olig2 knockdown in the CCHMC-DIPG4 line (Fig. 6B). The PBTR3 serum line was resistant to Olig2 silencing, confirming the specificity of Olig2 shRNAs (Fig. 6C). In contrast, all serum-free lines were sensitive to Olig2 knockdown (Fig. 6D‒F). We transplanted CCHMC-DIPG1 expressing nontarget (NT) or Olig2 shRNA to the flank of Nu/Nu mice. While control (NT) cells formed robust tumors within 3 weeks, Olig2 shRNA tumors were significantly smaller (Fig. 6G, H). Olig2 shRNA tumors, however, showed Olig2 expression, suggesting inefficient silencing or de-repression of shRNA in vivo (Supplementary Fig. 4A‒D). When transplanted orthotopically, Olig2 knockdown significantly delayed tumorigenesis and enhanced survival of NSG mice (Fig. 6I, J). Our results suggest that Olig2 expression is retained at high levels in DIPG lines maintained in serum-free conditions, and together our in vitro and in vivo results suggest that the transcription factor Olig2 plays a key role in DIPG proliferation and could be a novel target in DIPG therapy.
Fig. 6.

Olig2 is required for DIPG proliferation. (A) Western blot showing Olig2 expression in serum-free DIPG4, JHH lines, and serum-dependent PBTR3 DIPG line. (B) Western blot showing Olig2 knockdown by Olig2 shRNA but not by control nontarget shRNA in DIPG4. (C‒F) Relative proliferation of PBTR3 serum line and serum-free SU-DIPG6, SU-DIPG4, and JHH-DIPG1 lines expressing nontarget or Olig2 shRNA. *P ≤ .005. Photomicrographs showing flank tumor formation in situ (G) or post resection (H) in Nu/Nu mice by CCHMC-DIPG1 serum-free line expressing nontarget shRNA or Olig2 shRNA. (I) NSG mice were imaged using the IVIS (Xenogen) system to monitor tumor growth at indicated days. (J) Kaplan‒Meier survival data of CCHMC-DIPG1 serum-free line expressing NT or Olig2 shRNA. *P = .0007.
Discussion
It is thought that an aborted cell differentiation program in NPCs of the developing pons leads to uncontrolled proliferation and development of DIPGs. In support of this premise, a combination of PDGFRα overexpression, TP53 knockdown, and H3.3K27M mutation in human embryonic stem cell‒derived NPCs blocked differentiation toward the astrocytic lineage.18 What factor(s) downstream of DIPG-specific mutations drive aberrant NPC proliferation is unclear. In this study, we report that the bHLH transcription factor Olig2, which is highly expressed in DIPGs, resists differentiation and is likely required for tumor formation in a xenograft model. We established 2 isogenic lines from an autopsy tissue (PBTR3): a serum-free line and a serum line. Both lines expressed the characteristic neural stem cell markers Sox2 and nestin, but only the serum-free line expressed Olig2. In vivo, only the Olig2-expressing serum-free lines were capable of forming diffuse, infiltrative tumors in the mouse pons with 100% penetrance, while the Olig2-negative serum line was incapable of forming tumors despite expressing other stem cell markers. Importantly, Olig2 silencing in a serum-free line significantly delayed tumorigenesis and enhanced survival in mice.
The serum-free line was completely growth-factor dependent for proliferation. It is important to note that both isogenic serum-dependent and serum-free PBTR3 lines expressed the stem cell markers Sox2 and nestin and serum-dependent PBTR3 incorporated EdU without expressing Olig2 in vitro, yet only the Olig2-expressing serum-free PBTR3 formed tumors in vivo. We suspect that chronic serum exposure suppressed Olig2 expression. The requirement of Olig2 for proliferation of serum-free DIPG lines but not serum-dependent PBTR3 is validated by our Olig2 shRNA experiments.
Olig2 is expressed along with Sox2 and nestin in mouse embryoid bodies, which upon differentiation along the neural lineage give rise to Olig2+ multipotent NPCs.19 Unlike nestin and Sox2, Olig2 is expressed exclusively in the CNS,20–23 and during development Olig2-expressing NPCs generate mainly oligodendrocytes and neuronal subtypes.24–26 However, Olig2 is also required for neural stem cell self-renewal and multipotency and is therefore expressed by transit-amplifying cells of the subventricular zone in the mammalian brain.27 Olig2 is induced by EGF and FGF2 in proliferating mammalian neural stem/progenitor cells and plays an obligate role in self-renewal and proliferation.28 While Olig2 is expressed by normal NPCs during proliferation, its expression is suppressed during in vitro differentiation and a subset of Olig2-expressing cells robustly differentiates into O4+ oligodendrocytes.29 Olig2 is not expressed in mature astroglia and accordingly overexpression of Olig2 in NPCs actively blocks astrocytic differentiation but induces oligodendrocyte differentiation. In light of these studies, our results show that DIPG cells maintained in EGF + FGF2–supplemented serum-free conditions express abundant Olig2 during proliferation but, in contrast to normal progenitor cells, retain Olig2 during in vitro differentiation. Importantly, nearly 100% of Olig2+ cells failed to differentiate into oligodendrocytes, neurons, or astrocytes, suggesting a differentiation block in Olig2+ cells. These results suggest that Olig2 regulation might occur perhaps at the epigenetic level. Indeed, compared with normal human NPCs, dramatic overexpression of Olig2 was observed in DIPG lines with the H3K27M mutation, and reduced H3K27Me3 trimethylation (indicative of de-repression) was found in the Olig2 locus.30
Despite antagonism with astrocytic differentiation, many adult astrocytic gliomas express Olig231 and nearly all proliferating cells including CD133+ stem cells in these tumors express Olig2. Indeed, Olig2 was required for glioma genesis in a mouse model of adult high-grade glioma,32 and Olig2+ oligodendrocyte precursors were defined as the cell of origin of murine glioma.33 In one study, 100% of proneural DIPGs and nearly 50% of mesenchymal DIPGs (which express GFAP and nestin) expressed Olig2, and median overall survival of children with Olig2+ DIPGs was significantly shorter than the non-Olig2 group (7.73 mo vs 12.37 mo).10 In another study, 22/24 DIPGs demonstrated Olig2 expression.11 In our analysis, 3 out of 4 cases showed robust Olig2 expression. Similar to other reports,3 we observed that nearly 100% of tumor cells in our xenograft model were Olig2+. Because we transplanted cells into the fourth ventricle, it is also possible that the presence of tumor cells on the ependymal surface of the fourth ventricle is due to tumor cell dissemination in situ and not a true leptomeningeal spread. The impact of Olig2 in DIPG is underscored by our result that Olig2 silencing significantly inhibited the viability of Olig2-expressing DIPG lines in vitro and in vivo.
Olig2+ cells are abundant in developing human and mouse pons7,8 (our data on mouse brain not shown). Our findings together with results from other labs indicate that some of these proliferating Olig2+ precursor cells could represent a potential cell of origin for DIPGs. The Monje group reported the presence of 2 groups of nestin+ cells in the developing human brainstem—one group present in the ventral pons and another lining the fourth ventricle.8 The first group of cells also expressed the radial glial marker vimentin but not the astrocytic intermediate filament GFAP, while distribution of nestin and vimentin was variable in the second group of cells. Nearly one-half of the nestin/vimentin double positive cells in the ventral pons also expressed Olig2. Interestingly, while similar to the results of Tate et al,7 cell proliferation was found to decline rapidly by the first 2 years after birth. Monje et al also observed appearance of a second wave of nestin+/vimentin+/Olig2+ cells around the age of 6 years, a time that parallels the occurrence of DIPG. Tate et al examined 123 autopsy samples of children between the ages of 3 days after birth through 18 years of age. In this study, Ki67+ (proliferating) cells in the pons did not express GFAP; however, 60% of Ki67+ cells colabeled with Olig2. Importantly, even when proliferation sharply declined at 7 months, the Olig2 population remained steady among the remaining Ki67+ cells, suggesting a potential regulatory role of this transcription factor in NPC proliferation in the ventral pons.
Further characterization of our serum-free PBTR3 line could not be done because unfortunately, the line was lost due to contamination. Nevertheless, key results obtained from our serum-free PBTR3 line, particularly retention of Olig2 during differentiation, were recapitulated by 3 other serum-free lines: JHH-DIPG1, SU-DIPG4, and SU-DIPG6.34,35 A critical unanswered question is whether (i) mutations occur in a preexisting Olig2+ population or (ii) epigenetic changes induced by the H3K27M mutation reprogram the epigenome of Olig2− cells to become Olig2+ and this regained Olig2 function plays a key role in DIPG pathogenesis. These possibilities may be mutually inclusive, since Olig2-expressing cells are likely to have a selective proliferative advantage due to Olig2-mediated suppression of P53 and P21.23,32 Indeed, murine cortical tumors initiated by dedifferentiation of astrocytes that originally lack Olig2, regain Olig2 during tumorigenesis,36 and following injury, reactive astrocytes acquire Olig2, which is required for the proliferation of these cells during glial scar formation.37 In summary, an Olig2-dependent transcription program could be critical for DIPG initiation and pathogenesis. Further work is needed to unravel the Olig2-dependent pathways in DIPGs for identification of novel targets downstream of Olig2.
Supplementary material
Supplementary data are available at Neuro-Oncology online.
Funding
This work was supported by CancerFreeKids and National Institutes of Health (1R01NS075291) to B.D.
Conflict of interest statement. The authors declare no conflict of interest.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are indebted to the family of the child who most generously agreed to donate tumor tissue. We thank Michelle Monje (Stanford University) for sharing primary DIPG lines (SU-DIPG4 and SU-DIPG6) and helpful comments on the manuscript, and Shabnam Pooya for help with the preparation and cutting of frozen DIPG sphere blocks. We also thank the Histology Core at CCHMC for providing the DIPG tissue sections.
References
- 1. Ostrom QT, Gittleman H, Liao P. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States 2007–2011. Neuro Oncol. 2014;16(4):1–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Front Oncol. 2012;2:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caretti V, Bugiani M, Freret M, et al. Subventricular spread of diffuse intrinsic pontine glioma. Acta Neuropathol. 2014;128(4):605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. 2014;128(4):573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher PG, Breiter SN, Carson BS, et al. A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocystic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer. 2000;89(7):1569–1576. [DOI] [PubMed] [Google Scholar]
- 6. Donaldson SS, Laningham F, Fisher PG. Advances toward an understanding of brainstem gliomas. J Clin Oncol. 2006;24(8):1266–1272. [DOI] [PubMed] [Google Scholar]
- 7. Tate MC, Lindquist RA, Nguyen T, et al. Postnatal growth of the human pons: a morphometric and immunohistochemical analysis. J Comp Neurol. 2015;523(3):449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Monje M, Mitra SS, Freret ME, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108(11):4453–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Becher OJ, Hambardzumyan D, Walker TR, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010;70(6):2548–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Puget S, Philippe C, Bax DA, et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS One. 2012;7(2):e30313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ballester LY, Wang Z, Shandilya S, et al. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am J Surg Pathol. 2013;37(9):1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bender S, Tang Y, Lindroth AM, et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell. 2013;24(5):660–672. [DOI] [PubMed] [Google Scholar]
- 13. Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. [DOI] [PubMed] [Google Scholar]
- 14. Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21(6):555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu G, Broniscer A, McEachron TA, et al. ; St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hashizume R, Andor N, Ihara Y, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat Med. 2014;20(12):1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zarghooni M, Bartels U, Lee E, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28(8):1337–1344. [DOI] [PubMed] [Google Scholar]
- 18. Okada Y, Shimazaki T, Sobue G, Okano H. Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Dev Biol. 2004;275(1):124–142. [DOI] [PubMed] [Google Scholar]
- 19. Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346(6216):1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. El-Helou V, Dupuis J, Proulx C, et al. Resident nestin+ neural-like cells and fibers are detected in normal and damaged rat myocardium. Hypertension. 2005;46(5):1219–1225. [DOI] [PubMed] [Google Scholar]
- 21. Yamada H, Takano T, Ito Y, et al. Expression of nestin mRNA is a differentiation marker in thyroid tumors. Cancer Lett. 2009;280(1):61–64. [DOI] [PubMed] [Google Scholar]
- 22. Strojnik T, Røsland GV, Sakariassen PO, Kavalar R, Lah T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: correlation of nestin with prognosis of patient survival. Surg Neurol. 2007;68(2):133–43; discussion 143. [DOI] [PubMed] [Google Scholar]
- 23. Mehta S, Huillard E, Kesari S, et al. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell. 2011;19(3):359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu QR, Sun T, Zhu Z, et al. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109(1):75–86. [DOI] [PubMed] [Google Scholar]
- 25. Takebayashi H, Yoshida S, Sugimori M, et al. Dynamic expression of basic helix-loop-helix Olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech Dev. 2000;99(1-2):143–148. [DOI] [PubMed] [Google Scholar]
- 26. Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109(1):61–73. [DOI] [PubMed] [Google Scholar]
- 27. Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, et al. PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron. 2006;51(2):187–199. [DOI] [PubMed] [Google Scholar]
- 28. Dromard C, Bartolami S, Deleyrolle L, et al. NG2 and Olig2 expression provides evidence for phenotypic deregulation of cultured central nervous system and peripheral nervous system neural precursor cells. Stem Cells. 2007;25(2):340–353. [DOI] [PubMed] [Google Scholar]
- 29. Dasgupta B, Gutmann DH. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci. 2005;25(23):5584–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan KM, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27(9):985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ligon KL, Alberta JA, Kho AT, et al. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol. 2004;63(5):499–509. [DOI] [PubMed] [Google Scholar]
- 32. Ligon KL, Huillard E, Mehta S, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53(4):503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu C, Sage JC, Miller MR, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor IC, Hütt-Cabezas M, Brandt WD, et al. Disrupting NOTCH slows diffuse intrinsic pontine glioma growth, enhances radiation sensitivity, and shows combinatorial efficacy with bromodomain inhibition. J Neuropathol Exp Neurol. 2015;74(8):778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Venkatesh HS, Johung TB, Caretti V, et al. Neuronal activity promotes glioma growth through Neuroligin-3 secretion. Cell. 2015;161(4):803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269–277. [DOI] [PubMed] [Google Scholar]
- 37. Chen Y, Miles DK, Hoang T, et al. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J Neurosci. 2008;28(43):10983–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.