

Abstract
LL-37 is an immune peptide that regulates innate and adaptive immune responses in the upper airways. Elevated levels of LL-37 have been linked to cell death and inflammatory diseases, such as chronic rhinosinusitis (CRS). Glycosaminoglycans (GAGs) are polysaccharides that are found on respiratory epithelial cells and serve important roles in mucosal surface repair. Recent findings suggest that a synthetic glycosaminoglycan (GM-0111) can protect against LL-37-induced sinonasal mucosal inflammation and cell death in a murine model of acute RS. Herein, we elucidated the mechanisms by which LL-37 causes sinonasal inflammation and how GM-0111 can prevent these mechanisms. When challenged with LL-37, human nasal epithelial cells (HNEpCs) and mouse macrophages (J774.2) demonstrated increased release of adenosine triphosphate (ATP) and interleukin (IL)-6 and -8, as well as cell death and lysis. These cellular responses were all blocked dose-dependently by pre-treatment with GM-0111. We identified that LL-37-induced cell death is associated with caspase-1 and -8 activation, but not activation of caspase-3/7. These responses were again blocked by GM-0111. Our data suggest that LL-37 causes cellular death of HNEpCs and macrophages through the pro-inflammatory necrotic and/or pyroptotic pathways rather than apoptosis, and that a GM-0111 is capable of inhibiting these pro-inflammatory cellular events.
Introduction
Chronic rhinosinusitis (CRS) is a debilitating condition of sinonasal mucosal inflammation that affects up to 49 million Americans.[1,2,3,4,5] Patients with CRS experience significant declines in quality of life more disabling than other chronic conditions such as coronary heart disease and Parkinson’s Disease.[6,7,8,9,10,11] Despite its large impact on society, the pathogenesis of this condition remains unclear, as CRS is complex with multiple etiologies (e.g., allergen, bacterial, viral, or fungal exposure).[12,13,14,15] Regardless of etiology, a seemingly unchecked state of persistent inflammation is common in most patients. Immunological hallmarks of CRS include increased permeability and damage to the sinonasal epithelial cell barrier induced by epithelial cell death and the infiltration of innate and adaptive immune cells. Current treatments of CRS primarily target nasal symptom relief and sinonasal inflammation with intranasal steroid sprays.[4] However, approximately 20% of patients are unresponsive to available medical therapies and turn to endoscopic sinus surgery as an alternative,[2,16,17] underscoring the need for improved anti-inflammatory therapeutics.
We previously developed a murine model of sinonasal mucosal inflammation to mirror the underlying inflammatory state of rhinosinusitis through intranasal instillation of the inflammatory-modulating peptide LL-37. This model was also developed for testing potential anti-inflammatory therapeutics capable of preventing and reducing such inflammatory effects. LL-37 has cytotoxic effects on bronchial and alveolar epithelial cells and has been shown to enhance mucus production in the airways, contributing to the pathogenesis of chronic obstructive pulmonary disorder.[18,19] LL-37 is expressed in many different cell types, including upper airway epithelial cells, immune cells, and cells comprising the sinonasal mucosa [20,21,22,23] and is upregulated in the nasal secretions and sinonasal mucosa of CRS patients.[24,25] In this model, we found robust responses consistent with RS-related changes, including histologic evidence of inflammation of the mucosa and increased inflammatory cell infiltration and cell death.[26] We further reported that a synthetic glycosaminoglycan (GAG; GM-0111) can prevent these inflammatory changes by blocking cell death and immune cell infiltration into the sinonasal epithelium and mucosa.[27] GAGs are polysaccharides normally found on respiratory epithelial cells and mucosal glands which are known to play important and diverse roles in the upper airways, including fluid homeostasis, inflammation, and mucosal tissue repair and remodeling.[28,29,30,31,32]
In the present study, we elucidated the molecular mechanisms through which LL-37 might be inducing sinonasal mucosal inflammation and how GM-0111 can block these mechanisms. The specific death pathways being activated and inhibited were examined on the single cell level using human primary nasal epithelial cells (HNEpCs) and mouse monocyte macrophage cell line J744.2 as an in vitro model of sinonasal mucosal inflammation. Using this model, secreted factors indicative of cellular stress (adenosine triphosphate (ATP)) and cytotoxicity (interleukin (IL)-6 and IL-8) were quantitated, whereas cell morphological changes were qualitatively interpreted within the context of sinonasal mucosal inflammation.
Materials and methods
Reagents
LL-37 is a C-terminal peptide fragment from human cathelicidin with a sequence of LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES. LL-37 was obtained from the DNA/Peptide Synthesis Core Facility at the University of Utah (Salt Lake City, UT) at >95% purity. GM-0111 was supplied by GlycoMira Therapeutics (Salt Lake City, UT).[33] Compounds were dissolved in NanoPure double-distilled water (ddH2O) or phosphate buffered saline (PBS; pH 7.4) and filtered through a sterile 0.22 μm filter before use.
Cell culture
HNEpCs and recommended cell culture supplies were obtained from Celprogen (Torrance, CA). J774.2 cells, a BALB/C mouse monocyte macrophage cell line, were obtained from Sigma Aldrich (St. Louis, MO); the recommended cell culture supplies for J774.2 cells were obtained from ThermoFisher Scientific (Grand Island, NY). Cells were maintained at 37°C and 5% CO2. All expanding, freezing, and culturing protocols were performed according to the suppliers’ instructions.
ATP release and death quantitation of HNEpCs and J774.2 cells
For analyses of LL-37-induced ATP release and cell death, HNEpCs and J774.2 cells were first detached from culture flasks using Accutase (Innovative Cell Technologies; San Diego, CA), delivered to complete medium, pelleted by centrifugation, and then resuspended in 1 mL of complete medium. Cells were counted using a hemocytometer, examined for viability with trypan blue (0.4% solution, Thermo Fisher Scientific; Hampton, NH), and only used when the population was >90% viable. For ATP, cell death, and caspase assays the HNEpCs and J774.2 cells were plated into 24-well plates at a density of 500,000 cells/well. For ELISA assays HNEpCs were plated in 96-well plates at a density of 10,000 cells/well. Cells were maintained overnight at 37°C and 5% CO2 before use in experiments.
HNEpCs and J774.2 cells were then washed with sterile PBS (3 x 500 μL) and incubated in serum-free medium or GM-0111 (0, 30, 100, or 300 μg/mL) diluted in serum-free medium, for 1 h (37°C, 5% CO2). LL-37 (10 μM), or the LL-37 diluent only (controls), was then added to each well for 15 min. Supernatant (120 μL) was then collected, centrifuged, and subjected to ATP quantification under sterile conditions using an ENLITEN®ATP Assay System kit (Promega; Madison, WI) following the manufacturer’s instructions, and analyzed with a Tecan Infinite®200 PRO plate reader (Männedorf, Switzerland) in luminescence mode.
Fifteen minutes after the addition of LL-37 (10 μM), cells were then detached using Accutase and added to the remaining volume of their respective supernatant, and centrifuged. Cells were washed with PBS, centrifuged, and resuspended in 100 μL of PBS containing FITC-Annexin V (BioLegend; San Diego, CA) and 7-AAD (BioLegend; San Diego, CA) (10:2:1 PBS/FITC Annexin V/7-AAD) for 30 min at 37°C. The reaction was quenched with PBS. The cells were then centrifuged, resuspended in PBS, and analyzed using a Guava EasyCyte HT8 (Millipore; Billerica, MA) flow cytometer. These assays were performed in quadruplicate for each condition (n = 4).
Morphologic change imaging of HNEpCs and J774.2 cells
HNEpCs and J774.2 cells were plated in μ-Slide 8 well glass bottom plates (Ibidi USA, Inc., Fitchburg, WI) and mLexamined for cell morphological changes under a Nikon TMS T009 (Nikon Inc., Melville, NY) microscope at 40x magnification before (time 0) and 60 min after the addition of vehicle (saline), LL-37 (10 μM), or GM-0111 (100 μg/mL) + LL-37 (10 μM).
Cytokine quantitation by ELISA of HNEpCs
ELISA MAX™ Deluxe Sets were used for quantification of Human IL-6 and IL-8 protein (BioLegend; Sand Diego, CA). The ELISA plates were prepared the day before the assay per manufacturer instructions by addition of 100 μL of the appropriate Capture Antibody solution to each well, then sealing and incubating the plate overnight at 4°C. HNEpCs were plated at a density of 10,000 cells/well in 96-well plates and grown overnight (37°C, 5% CO2). The cells were then washed with sterile PBS (3 x 500 μL) and incubated in GM-0111 (30, 100, 300, or 1,000 μg/mL) or serum-free medium alone for 1 h (37°C, 5% CO2). Next, LL-37 (60 or 30 μM) or medium (controls) was delivered to each well. All conditions were performed in quadruplicate (n = 4). After 1 h incubation with LL-37, the supernatant from each sample was gently aspirated and analyzed by enzyme-linked immunosorbent assay (ELISA) according to manufacturer instructions. Absorbance was measured at 450 and 570 nm (570 nm subtracted) using a Tecan Infinite®200 PRO plate reader (Austria). A standard curve relating sample absorbance at 450 nm to concentration (pg/mL) of IL-6 or -8 was calculated with strong fit (r = 0.999) based on diluted standards of known concentration and used to calculate concentration of the samples.
Caspase activation assay of HNEpCs and J774.2 cells
HNEpCs and J774.2 cells were plated and treated as described for death assays with GM-0111 (0, 1, 10, or 100 μg/mL) added in serum-free medium for 1 h (37°C, 5% CO2), followed by LL-37 (0 or 10 μM) for 15 min. FAM-FLICA™ assays (ImmunoChemistry Technologies; Bloomington, MN) specific to the detection of caspase-1, -3/7, and -8 activation were performed according to the manufacturer’s instructions and analyzed using a Guava EasyCyte HT8 flow cytometer (FITC filter). Cells were simultaneously labeled with 7-AAD (near red filter) as an indicator of terminal cell death. Cells with co-labeling of both active caspase and 7-AAD were selected for comparison between treatment groups. All conditions were performed in triplicate to quintuplicate (n = 3 to 5).
Statistics
A normal Gaussian distribution was assumed to satisfy the requirements of the parametric one-way ANOVA test. However, formal nests of normality (Shapiro-Wilk and the D'Agostino & Pearson omnibus normality tests) could not be performed due to the small sample size per treatment group (n = 3 or 4). Pair-wise comparisons were made by one-way ANOVA, followed by Tukey’s post-test to adjust for multiple comparisons (P-value ≤ 0.05 indicates a statistically significant difference). For dose response curves a post-test for linear trend was also applied to the one-way ANOVA to determine significance of the apparent dose-response relationship. For the purpose of graphical representation and statistical analysis, ELISA data were transformed by addition of the absolute value of the most negative value in the data set (-3.0474 for IL-6 and -3.0727 for IL-8) as negative values of low or no signal groups resulted from prediction based on the curve of fit, though are physically nonsensical. Statistical tests and graphing were performed using Prism 6 for Windows (GraphPad Software; La Jolla, CA).
Results
LL-37 causes increased ATP secretion from HNEpCs and J774.2 cells, which is dose-dependently blocked by GM-0111
The release of ATP from HNEpCs and J774.2 cells was measured in response to treatment with LL-37 ranging from 0 to 30 μM for 15 min. LL-37 caused a dose-dependent increase in ATP release from both HNEpCs and J774.2 cells (Fig 1A). A dose of 10 μM LL-37 was chosen for subsequent experiments, as this dose resulted in a pronounced cytotoxic effect while also maintaining a viable population (approximate median effective dose (ED50).
Fig 1. LL-37 causes a dose-dependent increase in ATP release from both HNEpC and J774.2 cells (A), which is blocked by GM-0111 in a dose-dependent manner (B).

Each column represents the mean ± SD (n = 4). ****P ≤ 0.0001, ***P ≤ 0.001, *P ≤ 0.05, ns (not significant) P > 0.05.
HNEpCs and J774.2 cells were then pre-treated with GM-0111 (0, 1, 10, or 100 μg/mL), followed by incubation with 10 μM LL-37. GM-0111 was found to significantly reduce LL-37-induced ATP release compared to cells treated only with LL-37 (Fig 1B) in a dose-dependent manner with a significant linear trend for this effect for both HNEpCs (P < 0.0001) and J774.2 cells (P < 0.001). For HNEpCs, mean ATP levels were increased from 9.8 nM ± 0.94 with no treatment to 112.6 nM ± 2.96 with 10 μM LL-37 (P < 0.0001), and levels were decreased to 12.44 ± 1.43 when pre-treated with 100 μg/mL GM-0111 in addition to the 10 μM LL-37 (P < 0.0001 for comparison to LL-37 alone). For J774.2 cells, mean ATP levels were increased from 2.27 nM ± 0.28 with no treatment to 9.15 nM ± 3.13 with 10 μM LL-37 (P < 0.001), and levels were decreased to 2.79 ± 0.72 when pre-treated with 100 μg/mL GM-0111 (P < 0.001 for comparison to LL-37 alone).
LL-37 causes increased IL-6 and IL-8 release from HNEpCs, which is dose-dependently blocked by GM-0111
HNEpCs were incubated with a dose ranging from 0 to 60 μM LL-37 for 15 or 60 min to determine the response of IL-6 and IL-8 cytokine levels. A robust increase in levels of both cytokines was seen at higher doses, but remained below the limit of detection at ≤ 3 μM (Fig 2A). Similar cytokine levels were observed with both 15 and 60 min treatment durations; 60 min incubation was chosen for subsequent experiments. A dose-dependent effect of LL-37 concentration on IL-6 and IL-8 levels, with a significant linear trend (P < 0.0001), was observed over the dose range of 10, 30, and 60 μM.
Fig 2.

Treatment of HNEpCs with LL-37 for 60 min results in a dose-dependent increase in cytokines IL-6 and -8 (A). Pre-treatment with GM-0111 causes significant dose-dependent reductions in LL-37-induced cytokine release (B). There is no significant effect of GM-0111 alone on IL-6 or -8 levels. ****P ≤ 0.0001.
We then determined the effect of GM-0111 treatment on LL-37-induced IL-6 and IL-8 release from HNEpCs. Cells were pre-treated with GM-0111 (0, 30, 100, 300, or 1,000 μg/mL), then treated with 30 μM LL-37. This higher dose (relative to ATP experiments) was chosen for these experiments based on preliminary dose response curves demonstrating only a modest increase in cytokine levels with 10 μM. GM-0111 pre-treatment resulted in a dose-dependent decrease in LL-37-induced IL-6 and IL-8 levels (Fig 2B), with a significant linear trend (P < 0.0001). No significant effect on the levels of these cytokines was observed with GM-0111 treatment alone up to 1,000 μg/mL. Mean levels of IL-6 and IL-8, respectively, were increased from 0.47 pg/mL ± 0.33 and 0.61 pg/mL ± 0.80 in controls, to 34.59 pg/mL ± 9.21 and 28.47 pg/mL ± 2.72 with 30 μM LL-37 (P < 0.0001). When pre-treated with 100 μg/mL GM-0111 followed by 30 μM LL-37, cytokine levels were respectively decreased to 4.27 pg/mL ± 5.69 and 1.41 pg/mL ± 0.14 (P < 0.001 for comparison to LL-37 alone).
LL-37 causes gross morphologic changes in HNEpCs and J774.2 cells, which are prevented with GM-0111
The effect of LL-37 and GM-0111 pre-treatment on cell death was assayed based on morphologic characteristics. Using light microscopy we obtained still images (Fig 3) following treatment of HNEpCs and J774.2 cells with either phosphate buffered saline (PBS; vehicle control), LL-37 (10 μM), or both GM-0111 (100 μg/mL) and LL-37 (10 μM). Representative images are shown at time points of 0 and 60 min after treatment (Fig 3). When treated with LL-37, gross morphologic changes consistent with cell death were observed, including cell swelling and rupture over 15 min (Fig 3, column 2). When pre-treated with GM-0111 (Fig 3, column 3), these morphologic changes were not observed; the cells were grossly indistinguishable from controls.
Fig 3. LL-37 induces morphologic change in HNEpCs (upper two rows) and J774.2 cells (bottom two rows), which are prevented with GM-0111 treatment.

All images are at 40x magnification.
LL-37 causes increased death of HNEpCs and J774.2 cells, which is dose-dependently blocked by GM-0111
Cell death was quantitated by flow cytometric detection of respective early and late cell death markers, Annexin V and 7-aminoactinomycin D (7-AAD).[34,35] HNEpCs and J774.2 cells were treated first with a range of LL-37 doses for 15 min, to determine a dose that induced pronounced cytotoxic effect while maintaining a viable population. The data demonstrated a clear dose-dependent increase in the number of Annexin V- and 7-AAD-postive cells, with near complete cell death at 30 μM, and approximately 55% (approximate ED50) of the total Annexin V- and 7-AAD-positive cell population when treated with 10 μM LL-37 for 15 min (Fig 4A, HNEpCs). These findings were consistent with our previous data; and 10 μM was used for subsequent studies.
Fig 4. LL-37 increases cell death, which is reduced by GM-0111.

A. Treatment with LL-37 alone results in a dose-dependent increase in late cell death (Annexin V+/7-AAD+) in HNEpCs. B. The percentage of total HNEpCs and J774.2 cells that are viable (Annexin V-/7-AAD-) is significantly reduced with LL-37 treatment, and this is dose-dependently reversed with GM-0111. C. The percentage of cells undergoing early death (Annexin V+/7-AAD-) is significantly increased in HNEpCs treated with LL-37, and the effect is reversed with GM-0111. D. The percentage of HNEpCs and J774.2 in late cell death (Annexin V+/7-AAD+) is also significantly increased with LL-37 treatment and dose-dependently reversed with GM-0111. ****P ≤ 0.0001, ***P ≤ 0.001, **P ≤ 0.01, *P ≤ 0.05, ns (not significant) P > 0.05. The data represent the means ± SD (n = 4).
HNEpCs and J774.2 cells were pre-treated with GM-0111 (0, 30, 100, or 300 μg/mL), followed by incubation with 10 μM LL-37, labeled for Annexin V and 7-AAD, and then subjected to flow cytometry. GM-0111 reduced LL-37-induced cell death in a dose-dependent manner when compared to cells treated only with LL-37 (Fig 4B–4D). The percentage of cells negative for both Annexin V and 7-AAD (considered viable) was significantly reduced from 68.96% ± 4.72 and 69.04% ± 4.89 with no treatment, to 35.91% ± 15.30 (P < 0.001) and 26.04% ± 3.02 (P < 0.0001) with 10 μM LL-37, for HNEpC and J774.2 cells, respectively (Fig 4B). This was reversed in a dose-dependent manner with GM-0111 pre-treatment (P < 0.0001; linear trend). Interestingly, at the highest dose of GM-0111 pre-treatment (300 μg/mL GM-0111 with 10 μM LL-37), cellular viability exceeded even that of the controls, indicating that GM-0111 may possess cytoprotective effects; 68.96% ± 4.72 and 69.04% ± 4.89 viable controls compared to 85.68% ± 6.73 and 82.65% ± 4.83 (P < 0.01) for HNEpC and J774.2 cells, respectively, though this did not reach statistical significance for HNEpCs.
The percentage of cells demonstrating late cell death changes (Annexin V and 7-AAD positive) increased significantly after treatment with 10 μM LL-37 in HNEpCs (P < 0.01) and J774.2s (P < 0.0001); a dose-dependent decrease in late cell death occurred with GM-0111 pre-treatment (Fig 4D) (P < 0.0001; linear trend). In HNEpCs, early death (Annexin V-positive, 7-AAD-negative) followed the same trend as observed with late cell death; there was a significant increase in the percent of cells with early cell death changes after treatment with 10 μM LL-37 (P < 0.05) and a dose-dependent decrease with GM-0111 pre-treatment (P < 0.0001; linear trend). However, in the J774.2 cells there was a significant decrease in early cell death with 10 μM LL-37 (P < 0.01) which was reversed with GM-0111 pre-treatment (P < 0.01 for 10 μM LL-37 alone compared to pre-treatment of 10 μM LL-37 followed by 30 μg/mL GM-0111); there was no apparent dose-dependent relationship of GM-0111 and the percentage of cells in early cell death as the percentage remained insignificantly different from controls at doses of 30 μg/mL GM-0111 and higher (Fig 4C).
LL-37 induces caspase-1 and -8 activation in HNEpCs and JJ74.2 cells, and GM-0111 protects against activation
Caspases are enzymes that have unique roles in regulating the phenotype of cellular death.[36] After J774.2 cells were incubated with 10 μM LL-37, the percentage of total cells that were both dead (7-AAD positive) and containing active caspase-1 increased from 5.72% ± 2.24 to 22.51% ± 2.76 (P < 0.0001) in HNEpCs, and from 9.82% ± 1.27 to 41.35% ± 4.53 (P < 0.0001). For both cell types, caspase-1 activation was inhibited dose-dependently by GM-0111 pre-treatment (P < 0.0001; linear trend), with the percentage of cells both dead and expressing active caspase-1 reduced to similar levels as controls when treated with both 10 μM LL-37 and 30 μg/mL GM-0111 (Fig 5A).
Fig 5. In cells committed to terminal cell death, caspase-1 and -8 activity is increased with LL-37 treatment and reduced with GM-0111.
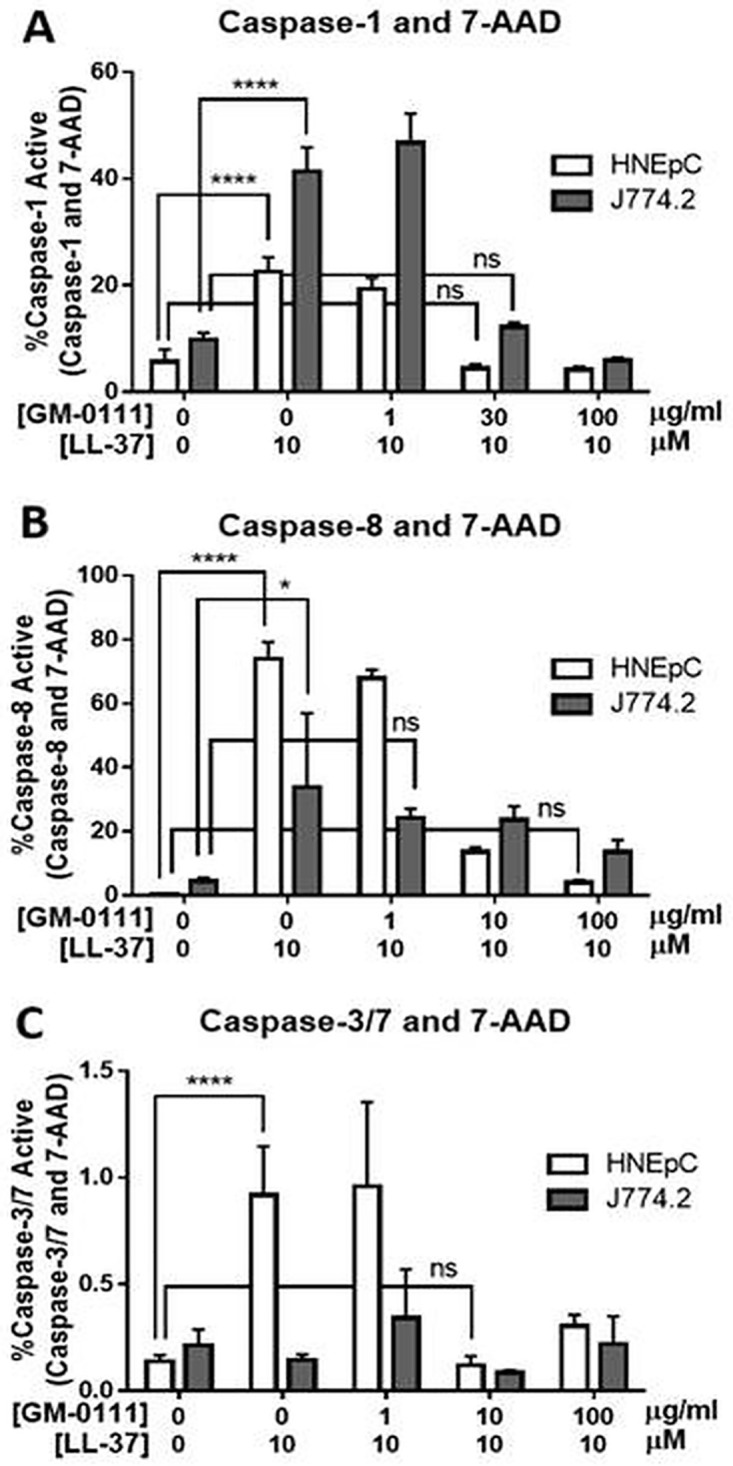
A. The percentage of total HNEpCs and J774.2 cells with active caspase-1 and 7-AAD (late cell death marker) is significantly increased with LL-37 treatment, and this is dose-dependently reversed with GM-0111 treatment. B. The percentage of total HNEpCs and J774.2 cells with active caspase-8 and 7-AAD is significantly increased with LL-37 treatment and dose-dependently decreased with GM-0111. ****P ≤ 0.0001, *P ≤ 0.05, ns (not significant) P > 0.05. The data represent the means ± SD (n = 2–5). Note the differences in y-axis scale (panel B y-axis maximum only 1.5%), adjusted to allow visual resolution but heights of bars are not comparable between panels. C. There is a statistically significant though minimal (<1%) increase in caspase-3 or -7 activity in HNEpCs also positive for 7-AAD after LL-37 treatment, and the effect is reversed with GM-0111. There is no significant change in caspase-3 or -7 activity with LL-37 or GM-0111 in J774.2 cells.
The percentage of the total cells that were both dead and expressing active caspase-8 increased significantly with 10 μM LL-37 treatment, from 0.42% ± 0.09 to 73.95% ± 5.25 (P < 0.0001) in HNEpCs, and from 4.38% ± 1.19 to 33.77% ± 23.19 (P < 0.05) in J774.2 cells. Caspase-8 activation was dose-dependently blocked by GM-0111 pre-treatment for both cell types (HNEpC: P < 0.0001; J744.2: P < 0.05; linear trends) (Fig 5B).
LL-37 does not induce caspase-3 or -7 activation in HNEpCs or J774.2 cells
For J774.2 cells, no significant effect of LL-37 treatment on the percentage of total cells that were 7-AAD positive and demonstrated caspase-3 or -7 activity was observed, despite indications of cell death and toxicity (i.e., morphologic changes, ATP release, and Annexin V/7-AAD labeling). For HNEpCs, a statistically significant, but biologically negligible increase in 7-AAD/caspase-3 and -7-positive cells was observed (0.14% ± 0.03 to 0.92% ± 0.23) after 10 μM LL-37 treatment. This small increase in caspase-3 and -7 activity decreased in a dose-dependent fashion with GM-0111 pre-treatment (P < 0.0001; linear trend).
Discussion
The pathogenesis of RS-associated sinonasal mucosal inflammation is complex and poorly understood despite extensive research into its pathophysiology. The heterogeneity of the disease has made it challenging to determine specific molecular mechanisms and inflammatory pathways resulting in the end phenotype of sinonasal inflammation, as well as to develop new therapies to prevent and treat this condition. The objectives of this work were to determine (1) the molecular mechanisms by which the inflammatory-modulating peptide LL-37 might contribute to sinonasal mucosal inflammation and cell death, and (2) how a non-steroidal, synthetic GAG (GM-0111) prevents this.
Our data demonstrate, that LL-37 treatment results in pro-inflammatory signaling, with increased levels of ATP, IL-6, and IL-8, and that these effects are dose-dependently blocked by GM-0111. In response to stress and damage, cells secrete ATP, stimulating pro-inflammatory mediators through purinergic receptor activation.[37] Increased ATP release from human intestinal epithelial cells and human urothelial cells has been linked to cell death within the context of inflammatory gut and bladder disease, respectively.[35,38] Similarly, increased levels of the pro-inflammatory cytokines IL-6 and IL-8 have been demonstrated from airway cells after stimulation by bacteria,[39] viral infection,[40] and in nasal epithelial cells after challenge with an antigen.[41]
Significantly increased levels of IL-6 have also been demonstrated in patients with CRS compared to controls,[42,43,44,45,46,47] and IL-8 levels have been previously shown to correlate with nasal symptoms in acute respiratory infections and in patients with CRS.[48,49] IL-8 is also increased in the sinonasal mucosal tissue and nasal discharge of patients with CRS relative to controls [47,50] and positively correlated with radiographic evidence of CRS-specific disease severity as well.[51] Taken together, our findings are consistent with what has been previously identified in CRS.
Our data show that LL-37 induces marked cellular morphology changes that are consistent with cell death and that GM-0111 protects against these changes. Necrosis is morphologically distinct from programmed cell death (apoptosis). Necrosis is characterized by cytoplasmic swelling (oncosis), moderate chromatin condensation, and rupture of the plasma membrane; in contrast to the cell rounding, nuclear fragmentation, and blebbing of the plasma membrane that leads to phagocytes of apoptotic bodies.[52,53] Observation of both HNEpCs and J774.2 cells after treatment with LL-37 revealed morphologic changes of cellular membrane rupture and extrusion of intracellular contents, but also cellular swelling as seen with the process of oncosis (Fig 3). These morphologic changes of cell death were prevented with GM-0111. These particular morphologic findings appear to be consistent with the cell death processes of necrosis and/or pyroptosis, within the context of our other enzymological data. Pyroptosis has been described morphologically as cell rupture, followed by membrane “re-sealing,” and cell swelling with nuclear condensation.[54]
Similarly, cells externalize phosphatidylserine (PS) on an intact cell membrane during oncosis in early necrosis, as well as when undergoing pyroptosis.[55,56,57,58] Pyroptotic cells also undergo PS-dependent phagocytosis.[58] Observed changes in the percentage of cells positive for Annexin V without 7-AAD (Fig 4C) therefore are interpreted to indicate early cell death without inferring a specific cell death process, as Annexin V binds PS when exposed by externalization on an intact membrane such as in early apoptosis or after loss of cell membrane integrity. HNEpCs were found to have a significant increase in early cell death with LL-37 that was abolished with GM-0111, whereas J774.2 cells did not demonstrate significant changes with treatment, demonstrating a difference in the time course of cell death. Interestingly, a higher dose of GM-0111 reduced cell death below that observed with controls, suggesting a mechanism of cytoprotection beyond simple sequestration of LL-37 by charge neutralization, which would be expected to prevent the adverse effects of LL-37 without impact on basal levels of viability.
Examining the specific death pathways activated by LL-37 and blocked by GM-0111, we assayed which caspases were involved. We observed that LL-37 caused robust increases in caspase-1 and -8, but not in caspases-3/7 in HNEpCs and J774.2 cells. All caspase activity was dose-dependently blocked with GM-0111 pre-treatment. Caspase-3, -7, and -8 serve roles in apoptotic cell death, whereas Caspase-1 has a distinct role in pro-inflammatory cell death.[59,60] Caspase-3 is specifically required for characteristic features of apoptotic cell death such as DNA fragmentation.[61,62,63] Here, caspase activity was measured only in cells also positive for 7-AAD for selection of cells terminally committed to death. These data suggest that cell death is not occurring through classical apoptosis.
Caspase-1 is associated with the pro-inflammatory death mechanism of pyroptosis,[64] as demonstrated in macrophages and dendritic cells.[65,66] This cell death pathway is initiated when Nod-like receptors (NLRs) respond to a variety of cellular danger signals, including extracellular ATP, forming the multiprotein inflammasome complex. The inflammasome activates caspase-1, which in turn initiates pyroptotic cell death and pro-inflammatory cytokines.[66,67] Increased caspase-1 activity in response to LL-37 is therefore consistent with a pro-inflammatory model of pyroptotic cell death and increased inflammatory signaling, as opposed to an anti-inflammatory apoptotic cell death mediated by caspase-3 and -7.
While active caspase-8 is classically known to activate caspase-3 in apoptotic cell death,[64] there is evidence for more diverse roles of caspase-8 in inflammatory signaling and caspase-1-mediated cell death by pyroptosis.[68,69,70,71] Our findings thus support a mechanism of LL-37-induced pyroptotic cell death in HNEpCs and J774.2 cells, though observed rapid morphologic changes also suggest the possibility of some necrotic/non-programmed cell death (Fig 6).
Fig 6. Diagram of the proposed mechanism of LL-37-induced cell death and protection from GM-0111.

Nasal epithelial cells subjected to LL-37 demonstrate a pro-inflammatory response, characterized by increased ATP, IL-6, and -8 production and pyroptosis and/or necrosis via caspase-1 and -8 but not caspase-3 or -7 activation. These changes are prevented by GM-0111 treatment. IL-6 and -8 promote an inflammatory response in vivo through the recruitment of neutrophils and further inflammatory signaling in a positive feedback loop. The process of pro-inflammatory cell death is propagated to nearby cells due to these changes in the local environment, resulting in an unchecked cytotoxic response initiated by LL-37.
A limitation to this study is the difficulty in validating LL-37-induced inflammation as an accurate in vitro model of RS-associated sinonasal mucosal inflammation. Reported endogenous levels of LL-37 within the context of airway inflammation range from 2 to 8 nM.[18,72,73] In this study, the supraphysiological concentrations of LL-37 employed fell within the range of previous in vitro functional studies using LL-37 to induce different biological outcomes [74,75,76] and were optimized to produce a robust response that could be reproducibly measured. Findings from the present study and previous work support the plausibility of this model by similarities to clinical findings of patients with RS. Our prior animal model demonstrated histologic changes of inflammation and a similar inflammatory infiltrate and cell death to that seen in RS-associated sinonasal disease.[26] Molecular changes from the present study are more difficult to relate to in vivo findings from patients, due to the heterogeneity of the disease process and likely multiple mechanisms that may result in a similar disease phenotype. Perpetual pro-inflammatory processes initiated by the inflammasome and caspase-1, as well as the downstream pro-inflammatory effects of pyroptotic and/or necrotic cell death is consistent with the seemingly uncontrolled sinonasal inflammation observed in RS.
In summary, our findings suggest that LL-37-induced cell death of HNEpCs and J774.2 cells occurs via the mechanism of pyroptosis or a combination of pyroptosis/necrosis. We demonstrate that a synthetic GAG (GM-0111) dose-dependently prevents increased inflammatory mediator production and apparent pyroptotic and/or necrotic cell death induced by LL-37, supporting its potential utility as a novel therapeutic for upper airway inflammatory conditions. Further work is needed to assess the effects of other therapeutics in this inflammatory model, as well as to apply GM-0111 in other models and to further validate its utility in different contexts of sinonasal mucosal inflammation (e.g., allergic and/or infectious in vivo models).
Acknowledgments
The authors thank Drs. Won Yong Lee and Justin Savage at GlycoMira Therapeutics, Inc. for their helpful scientific discussions when interpreting the results of this work.
Data Availability
All data and calculations contributing to the final data are within the paper.
Funding Statement
This work was supported by grant numbers KL2TR001065 (JAA) and R43 AI126987-01 (AP, JAA) from the University of Utah Program in Personalized Health and the National Center for Advancing Translational Sciences and National Institute on Allergy and Infectious Diseases (NIAID), respectively. The funders had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript. J.A.A. is a consultant for Spirox (Menlo Park, CA; USA) and Medtronic, Inc. (Jacksonville, FL; USA), which are not affiliated with this investigation. AP and JAA declare financial interest and/or other relationships with GlycoMira Therapeutics, Inc. (Salt Lake City, UT; USA). GlycoMira Therapeutics, Inc. provided support in the form of salary for AP and reagents/supplies, but did not have additional roles in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
References
- 1.Bachert C, Gevaert P, Holtappels G, Johansson SG, van Cauwenberge P (2001) Total and specific IgE in nasal polyps is related to local eosinophilic inflammation. The Journal of allergy and clinical immunology 107: 607–614. doi: 10.1067/mai.2001.112374 [DOI] [PubMed] [Google Scholar]
- 2.Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, et al. (2012) EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology 50: 1–12 [DOI] [PubMed] [Google Scholar]
- 3.Meltzer EO, Hamilos DL, Hadley JA, Lanza DC, Marple BF, Nicklas RA, et al. (2004) Rhinosinusitis: establishing definitions for clinical research and patient care. The Journal of allergy and clinical immunology 114: 155–212. doi: 10.1016/j.jaci.2004.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orlandi RR, Kingdom TT, Hwang PH (2016) International Consensus Statement on Allergy and Rhinology: Rhinosinusitis Executive Summary. International forum of allergy & rhinology 6 Suppl 1: S3–21. [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeld RM, Piccirillo JF, Chandrasekhar SS, Brook I, Ashok Kumar K, Kramper M, et al. (2015) Clinical practice guideline (update): adult sinusitis. Otolaryngology—head and neck surgery: official journal of American Academy of Otolaryngology-Head and Neck Surgery 152: S1–S39. [DOI] [PubMed] [Google Scholar]
- 6.Alt JA, Mace JC, Buniel MC, Soler ZM, Smith TL (2014) Predictors of olfactory dysfunction in rhinosinusitis using the brief smell identification test. The Laryngoscope. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alt JA, Smith TL (2013) Chronic rhinosinusitis and sleep: a contemporary review. International forum of allergy & rhinology 3: 941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alt JA, Smith TL, Mace JC, Soler ZM (2013) Sleep quality and disease severity in patients with chronic rhinosinusitis. The Laryngoscope 123: 2364–2370. doi: 10.1002/lary.24040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krueger JM (2008) The role of cytokines in sleep regulation. Current pharmaceutical design 14: 3408–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litvack JR, Mace JC, Smith TL (2009) Olfactory function and disease severity in chronic rhinosinusitis. Am J Rhinol Allergy 23: 139–144. doi: 10.2500/ajra.2009.23.3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soler ZM, Wittenberg E, Schlosser RJ, Mace JC, Smith TL (2011) Health state utility values in patients undergoing endoscopic sinus surgery. The Laryngoscope 121: 2672–2678. doi: 10.1002/lary.21847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akdis CA, Bachert C, Cingi C, Dykewicz MS, Hellings PW, Naclerio RM, et al. (2013) Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. The Journal of allergy and clinical immunology 131: 1479–1490. doi: 10.1016/j.jaci.2013.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy JL, Borish L (2013) Chronic sinusitis pathophysiology: the role of allergy. Am J Rhinol Allergy 27: 367–371. doi: 10.2500/ajra.2013.27.3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Lane AP (2011) Chronic rhinosinusitis as a multifactorial inflammatory disorder. Current infectious disease reports 13: 159–168. doi: 10.1007/s11908-011-0166-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan BK, Kern RC, Schleimer RP, Schwartz BS (2013) Chronic rhinosinusitis: the unrecognized epidemic. American journal of respiratory and critical care medicine 188: 1275–1277. doi: 10.1164/rccm.201308-1500ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharyya N (2007) Progress in surgical management of chronic rhinosinusitis and nasal polyposis. Current allergy and asthma reports 7: 216–220. [DOI] [PubMed] [Google Scholar]
- 17.Smith TL, Litvack JR, Hwang PH, Loehrl TA, Mace JC, Fong KJ, et al. (2010) Determinants of outcomes of sinus surgery: a multi-institutional prospective cohort study. Otolaryngol Head Neck Surg 142: 55–63. doi: 10.1016/j.otohns.2009.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang YY, Xiao W, Zhu MX, Yang ZH, Pan XJ, Zhang Y, et al. (2012) The effect of human antibacterial peptide LL-37 in the pathogenesis of chronic obstructive pulmonary disease. Respir Med 106: 1680–1689. doi: 10.1016/j.rmed.2012.08.018 [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Jiang Y, Sun C, Wang Q, Yang Z, Pan X, et al. (2014) The human cathelicidin LL-37 enhances airway mucus production in chronic obstructive pulmonary disease. Biochem Biophys Res Commun 443: 103–109. doi: 10.1016/j.bbrc.2013.11.074 [DOI] [PubMed] [Google Scholar]
- 20.Chen PH, Fang SY (2004) The expression of human antimicrobial peptide LL-37 in the human nasal mucosa. Am J Rhinol 18: 381–385. [PubMed] [Google Scholar]
- 21.Lau YE, Bowdish DM, Cosseau C, Hancock RE, Davidson DJ (2006) Apoptosis of airway epithelial cells: human serum sensitive induction by the cathelicidin LL-37. American journal of respiratory cell and molecular biology 34: 399–409. doi: 10.1165/rcmb.2005-0170OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mader JS, Mookherjee N, Hancock RE, Bleackley RC (2009) The human host defense peptide LL-37 induces apoptosis in a calpain- and apoptosis-inducing factor-dependent manner involving Bax activity. Molecular cancer research: MCR 7: 689–702. doi: 10.1158/1541-7786.MCR-08-0274 [DOI] [PubMed] [Google Scholar]
- 23.Nell MJ, Tjabringa GS, Vonk MJ, Hiemstra PS, Grote JJ (2004) Bacterial products increase expression of the human cathelicidin hCAP-18/LL-37 in cultured human sinus epithelial cells. FEMS Immunol Med Microbiol 42: 225–231. doi: 10.1016/j.femsim.2004.05.013 [DOI] [PubMed] [Google Scholar]
- 24.Ooi EH, Wormald PJ, Carney AS, James CL, Tan LW (2007) Human cathelicidin antimicrobial peptide is up-regulated in the eosinophilic mucus subgroup of chronic rhinosinusitis patients. American journal of rhinology 21: 395–401. [DOI] [PubMed] [Google Scholar]
- 25.Thienhaus ML, Wohlers J, Podschun R, Hedderich J, Ambrosch P, Laudien M (2011) Antimicrobial peptides in nasal secretion and mucosa with respect to Staphylococcus aureus colonization in chronic rhinosinusitis with nasal polyps. Rhinology 49: 554–561 [DOI] [PubMed] [Google Scholar]
- 26.Alt JA, Qin X, Pulsipher A, Orb Q, Orlandi RR, Zhang J, et al. (2015) Topical cathelicidin (LL-37) an innate immune peptide induces acute olfactory epithelium inflammation in a mouse model. International forum of allergy & rhinology 5: 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pulsipher A, Qin X, Thomas AJ, Prestwich GD, Oottamasathien S, Alt JA (2016) Prevention of sinonasal inflammation by a synthetic glycosaminoglycan. International forum of allergy & rhinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turino GM, Cantor JO (2003) Hyaluronan in respiratory injury and repair. American journal of respiratory and critical care medicine 167: 1169–1175. doi: 10.1164/rccm.200205-449PP [DOI] [PubMed] [Google Scholar]
- 29.Manuele C, Paola V, Antonio M, Giuseppe O, Lorenzo S, Valentina G, et al. (2016) Hyaluronic acid and upper airway inflammation in pediatric population: A systematic review. International journal of pediatric otorhinolaryngology 85: 22–26. doi: 10.1016/j.ijporl.2016.03.015 [DOI] [PubMed] [Google Scholar]
- 30.Gerdin B, Hallgren R (1997) Dynamic role of hyaluronan (HYA) in connective tissue activation and inflammation. Journal of internal medicine 242: 49–55. [DOI] [PubMed] [Google Scholar]
- 31.Tammi MI, Day AJ, Turley EA (2002) Hyaluronan and homeostasis: a balancing act. J Biol Chem 277: 4581–4584. doi: 10.1074/jbc.R100037200 [DOI] [PubMed] [Google Scholar]
- 32.Negrini D, Passi A, Moriondo A (2008) The role of proteoglycans in pulmonary edema development. Intensive care medicine 34: 610–618. doi: 10.1007/s00134-007-0962-y [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Xu X, Rao NV, Argyle B, McCoard L, Rusho WJ, et al. (2011) Novel sulfated polysaccharides disrupt cathelicidins, inhibit RAGE and reduce cutaneous inflammation in a mouse model of rosacea. PloS one 6: e16658 doi: 10.1371/journal.pone.0016658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee WY, Savage JR, Zhang J, Jia W, Oottamasathien S, Prestwich GD (2013) Prevention of anti-microbial peptide LL-37-induced apoptosis and ATP release in the urinary bladder by a modified glycosaminoglycan. PloS one 8: e77854 doi: 10.1371/journal.pone.0077854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C (1995) A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. Journal of immunological methods 184: 39–51. [DOI] [PubMed] [Google Scholar]
- 36.Riedl SJ, Shi Y (2004) Molecular mechanisms of caspase regulation during apoptosis. Nature reviews Molecular cell biology 5: 897–907. doi: 10.1038/nrm1496 [DOI] [PubMed] [Google Scholar]
- 37.la Sala A, Ferrari D, Di Virgilio F, Idzko M, Norgauer J, Girolomoni G (2003) Alerting and tuning the immune response by extracellular nucleotides. Journal of leukocyte biology 73: 339–343. [DOI] [PubMed] [Google Scholar]
- 38.Souza CO, Santoro GF, Figliuolo VR, Nanini HF, de Souza HS, Castelo-Branco MT, et al. (2012) Extracellular ATP induces cell death in human intestinal epithelial cells. Biochimica et biophysica acta 1820: 1867–1878. doi: 10.1016/j.bbagen.2012.08.013 [DOI] [PubMed] [Google Scholar]
- 39.Larsson BM, Larsson K, Malmberg P, Palmberg L (1999) Gram positive bacteria induce IL-6 and IL-8 production in human alveolar macrophages and epithelial cells. Inflammation 23: 217–230. [DOI] [PubMed] [Google Scholar]
- 40.Becker S, Quay J, Soukup J (1991) Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. Journal of immunology 147: 4307–4312. [PubMed] [Google Scholar]
- 41.Ohkubo K, Ikeda M, Pawankar R, Gotoh M, Yagi T, Okuda M (1998) Mechanisms of IL-6, IL-8, and GM-CSF release in nasal secretions of allergic patients after nasal challenge. Rhinology 36: 156–161. [PubMed] [Google Scholar]
- 42.Ghaffar O, Lavigne F, Kamil A, Renzi P, Hamid Q (1998) Interleukin-6 expression in chronic sinusitis: colocalization of gene transcripts to eosinophils, macrophages, T lymphocytes, and mast cells. Otolaryngology—head and neck surgery: official journal of American Academy of Otolaryngology-Head and Neck Surgery 118: 504–511. [DOI] [PubMed] [Google Scholar]
- 43.Min YG, Lee KS (2000) The role of cytokines in rhinosinusitis. Journal of Korean medical science 15: 255–259. doi: 10.3346/jkms.2000.15.3.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peters AT, Kato A, Zhang N, Conley DB, Suh L, Tancowny B, et al. (2010) Evidence for altered activity of the IL-6 pathway in chronic rhinosinusitis with nasal polyps. The Journal of allergy and clinical immunology 125: 397–403 e310. doi: 10.1016/j.jaci.2009.10.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riechelmann H, Deutschle T, Rozsasi A, Keck T, Polzehl D, Burner H (2005) Nasal biomarker profiles in acute and chronic rhinosinusitis. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology 35: 1186–1191. [DOI] [PubMed] [Google Scholar]
- 46.Sejima T, Holtappels G, Kikuchi H, Imayoshi S, Ichimura K, Bachert C (2012) Cytokine profiles in Japanese patients with chronic rhinosinusitis. Allergology international: official journal of the Japanese Society of Allergology 61: 115–122. [DOI] [PubMed] [Google Scholar]
- 47.Min YG, Lee CH, Rhee CS, Hong SK, Kwon SH (1999) Increased expression of IL-4, IL-5, IFN-gamma, IL-6, IL-8, and TGF-beta mRNAs in maxillary mucosa of patients with chronic sinusitis. American journal of rhinology 13: 339–343. [DOI] [PubMed] [Google Scholar]
- 48.Henriquez KM, Hayney MS, Xie Y, Zhang Z, Barrett B (2015) Association of interleukin-8 and neutrophils with nasal symptom severity during acute respiratory infection. Journal of medical virology 87: 330–337. doi: 10.1002/jmv.24042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rhyoo C, Sanders SP, Leopold DA, Proud D (1999) Sinus mucosal IL-8 gene expression in chronic rhinosinusitis. The Journal of allergy and clinical immunology 103: 395–400. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki H, Takahashi Y, Wataya H, Ikeda K, Nakabayashi S, Shimomura A, et al. (1996) Mechanism of neutrophil recruitment induced by IL-8 in chronic sinusitis. The Journal of allergy and clinical immunology 98: 659–670. [DOI] [PubMed] [Google Scholar]
- 51.Lund VJ, Mackay IS (1993) Staging in rhinosinusitus. Rhinology 31: 183–184. [PubMed] [Google Scholar]
- 52.Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer 26: 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell death and differentiation 16: 3–11. doi: 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Labbe K, Saleh M (2008) Cell death in the host response to infection. Cell death and differentiation 15: 1339–1349. doi: 10.1038/cdd.2008.91 [DOI] [PubMed] [Google Scholar]
- 55.Lecoeur H, Prevost MC, Gougeon ML (2001) Oncosis is associated with exposure of phosphatidylserine residues on the outside layer of the plasma membrane: a reconsideration of the specificity of the annexin V/propidium iodide assay. Cytometry 44: 65–72. [DOI] [PubMed] [Google Scholar]
- 56.Krysko O, De Ridder L, Cornelissen M (2004) Phosphatidylserine exposure during early primary necrosis (oncosis) in JB6 cells as evidenced by immunogold labeling technique. Apoptosis: an international journal on programmed cell death 9: 495–500. [DOI] [PubMed] [Google Scholar]
- 57.Waring P, Lambert D, Sjaarda A, Hurne A, Beaver J (1999) Increased cell surface exposure of phosphatidylserine on propidium iodide negative thymocytes undergoing death by necrosis. Cell death and differentiation 6: 624–637. doi: 10.1038/sj.cdd.4400540 [DOI] [PubMed] [Google Scholar]
- 58.Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T (2013) Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. International immunology 25: 363–372. doi: 10.1093/intimm/dxs161 [DOI] [PubMed] [Google Scholar]
- 59.Martinon F, Tschopp J (2007) Inflammatory caspases and inflammasomes: master switches of inflammation. Cell death and differentiation 14: 10–22. doi: 10.1038/sj.cdd.4402038 [DOI] [PubMed] [Google Scholar]
- 60.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665. doi: 10.1038/nature15514 [DOI] [PubMed] [Google Scholar]
- 61.Janicke RU, Sprengart ML, Wati MR, Porter AG (1998) Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 273: 9357–9360. [DOI] [PubMed] [Google Scholar]
- 62.Lamkanfi M, Kanneganti TD (2010) Caspase-7: a protease involved in apoptosis and inflammation. The international journal of biochemistry & cell biology 42: 21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walsh JG, Cullen SP, Sheridan C, Luthi AU, Gerner C, Martin SJ (2008) Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proceedings of the National Academy of Sciences of the United States of America 105: 12815–12819. doi: 10.1073/pnas.0707715105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siegel RM (2006) Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol 6: 308–317. doi: 10.1038/nri1809 [DOI] [PubMed] [Google Scholar]
- 65.Miao EA, Rajan JV, Aderem A (2011) Caspase-1-induced pyroptotic cell death. Immunological reviews 243: 206–214. doi: 10.1111/j.1600-065X.2011.01044.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brennan MA, Cookson BT (2000) Salmonella induces macrophage death by caspase-1-dependent necrosis. Molecular microbiology 38: 31–40. [DOI] [PubMed] [Google Scholar]
- 67.Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR (2005) Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-kappaB-driven protein synthesis. Journal of immunology 175: 7611–7622. [DOI] [PubMed] [Google Scholar]
- 68.Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. (2015) Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem 290: 20167–20184. doi: 10.1074/jbc.M115.652321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monie TP, Bryant CE (2015) Caspase-8 functions as a key mediator of inflammation and pro-IL-1beta processing via both canonical and non-canonical pathways. Immunological reviews 265: 181–193. doi: 10.1111/imr.12284 [DOI] [PubMed] [Google Scholar]
- 70.Philip NH, Dillon CP, Snyder AG, Fitzgerald P, Wynosky-Dolfi MA, Zwack EE, et al. (2014) Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. Proceedings of the National Academy of Sciences of the United States of America 111: 7385–7390. doi: 10.1073/pnas.1403252111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, et al. (2014) Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proceedings of the National Academy of Sciences of the United States of America 111: 7391–7396. doi: 10.1073/pnas.1403477111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elenius V, Palomares O, Waris M, Turunen R, Puhakka T, Ruckert B, et al. (2017) The relationship of serum vitamins A, D, E and LL-37 levels with allergic status, tonsillar virus detection and immune response. PLoS One 12: e0172350 doi: 10.1371/journal.pone.0172350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xie D, Guo Y, Wu D, Xie D (2010) [Expressions of LL-37 and IL-8 in chronic sinusitis with nasal polyps]. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 24: 337–340. [PubMed] [Google Scholar]
- 74.Alagarasu K, Patil PS, Shil P, Seervi M, Kakade MB, Tillu H, et al. (2017) In-vitro effect of human cathelicidin antimicrobial peptide LL-37 on dengue virus type 2. Peptides 92: 23–30. doi: 10.1016/j.peptides.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 75.Edfeldt K, Agerberth B, Rottenberg ME, Gudmundsson GH, Wang XB, Mandal K, et al. (2006) Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler Thromb Vasc Biol 26: 1551–1557. doi: 10.1161/01.ATV.0000223901.08459.57 [DOI] [PubMed] [Google Scholar]
- 76.Torres-Juarez F, Cardenas-Vargas A, Montoya-Rosales A, Gonzalez-Curiel I, Garcia-Hernandez MH, Enciso-Moreno JA, et al. (2015) LL-37 immunomodulatory activity during Mycobacterium tuberculosis infection in macrophages. Infect Immun 83: 4495–4503. doi: 10.1128/IAI.00936-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and calculations contributing to the final data are within the paper.