

ABSTRACT
The mature microbiome is a stable ecosystem that resists perturbation despite constant host exposure to exogenous microbes. However, the microbial mechanisms determining microbiome development and composition are poorly understood. We recently demonstrated that a non-toxigenic B. fragilis (NTBF) strain restricts enteric colonization by an enterotoxigenic (ETBF) strain dependent on a type VI secretion system (T6SS). We show here that a second enterotoxigenic strain is competent to colonize, dependent on the Bacteroides fragilis pathogenicity island (BFPAI). Additional data showing complex environmental regulation of the Bacteroides fragilis toxin (BFT) suggest that virulence factors may be adapted to modify the colonic niche to provide a strain-specific colonization advantage. We conclude that more complex models of host-microbe-microbiome interactions are needed to investigate this hypothesis.
keywords: bacteroides, competition, pathogenesis, secretion, toxin
Introduction
The identity and function of specific factors that determine composition of the commensal gut microbiota is an enduring question in medicine and microbiology. Enteric microbial development follows a predictable course in which the sterile neonate is progressively colonized by the environmental contaminant flavibacteria, then lactobacilli, followed by facultative anaerobic coliforms, and finally the strictly anaerobic Bacteroidales.1 Despite this apparently ordered process, prediction of which specific organisms constitute the fully mature flora has proved challenging. Microbial exposure is necessary but not sufficient: culture independent surveys of the indoor environment have revealed all manner of commensal and pathogenic microbes absent in individual microbiota despite persistent exposure.2,3 Mouse models have demonstrated that host genotype is also insufficient to predict the steady-state microbiota,4 confirmed in human twin studies that show an observable, but minor, contribution of genotype to microbial inheritance.5 An understanding of how microbial determinants influence microbiota assembly is lacking.
The mature, adult microbiome is relatively static, with minimal strain replacement over time.6 A molecular understanding of complex microbial colonization factors may permit targeted interventions that minimally disrupt the enteric ecosystem while eliminating specific commensal microbes, called pathobionts,7 that can underpin disease. While antimicrobial therapies may achieve the same end point of pathobiont elimination, their effect is not targeted and, as such, contributes to unwanted elimination of beneficial microbes and fueling of antimicrobial resistance. Importantly, time-limited antimicrobial therapy cannot preclude re-colonization with the same pathobiont.
Many human diseases are associated with commensal microbes or an inappropriate immune response to commensal microbes. The chronic colitis associated with Crohn's disease and celiac disease is a prominent example of an inflammatory reaction against enteric flora. Commensal microbes may also mediate acute episodes of colitis, including many cases of childhood and antibiotic-associated diarrhea where no pathogen sensu strictu is detectable.8 One interpretation is that the disease state results from a dysregulated interaction between the host and a “healthy” flora. Many factors likely contribute to this miscommunication, including the particular configuration of the microbial community, host genotype, ontogeny, diet, exposure to antibiotics, and other environmental variables. In this view, colonization resistance and virulence are not isolated properties of the host or microbe, but an emergent property of the interaction between host, microbe, and microbiome.
Animal modeling of complex colonization events with toxigenic organisms are required to investigate this hypothesis. Toxins have long been studied in models of pathogenesis and understood exclusively as virulence factors. However, Bacteroides fragilis toxin (BFT) does not fit neatly within this paradigm. BFT is a metalloprotease encoded on a transposable element denoted B. fragilis pathogenicity island (BFPAI). BFT has been most extensively characterized in models of pathogenesis as its expression in enterotoxigenic B. fragilis (ETBF) is associated with childhood diarrhea and colon cancer.9,10 In vitro, intestinal epithelial cell intoxication with BFT leads to E-cadherin cleavage, loss of cell adhesion, and secretion of inflammatory signaling molecules.11,12 Murine ETBF monocolonization stimulates a BFT-dependent activation of Stat3 in immune and epithelial cells, followed by infiltration of T cells expressing IL-17.13 In response to this injury, neutrophil recruitment controls microbial translocation, while Stat3 activation leads to IL-22-driven epithelial regeneration, antimicrobial peptide secretion, and mucus glycosylation. B. fragilis may be able to withstand this pro-inflammatory environment by the action of LpxF, a phosphatase of lipid A14 that provides immunity to mucus-bound antimicrobial peptides (AMPs) and allows for evasion of the phagocytic TLR4 response elicited by LPS.15
Whether host signals induced by expression of BFT confer an advantage to the bacterium during colonic niche establishment has not been investigated. Yet, studies of clinical isolates suggest that 20% of individuals colonized with B. fragilis are carrying ETBF.16 Most of these individuals do not display evidence of toxin-mediated disease. As BFT is not known to be required for B. fragilis commensalism, and indeed many non-toxigenic strains colonize the human population, we consider ETBF to be a uniquely tractable model for testing the role of a bacterial toxin in commensalism. We propose that while BFT is not essential for survival in the mammalian gut, the toxin may confer advantages to the bacterium in the competitive microbial ecosystem of the host colon.
We recently described type VI secretion (T6S) as a determinant of in vivo competition between an established and secondary challenge strain of B. fragilis.17 Mice colonized with a non-toxigenic B. fragilis (NTBF) strain were protected against colonization with ETBF and rescued from enteric disease. In contrast, an isogenic NTBF mutant lacking a critical component of the type VI secretion system (T6SS) permitted ETBF colonization, demonstrating the necessity of T6S in predicting the outcome of strain co-colonization. We now demonstrate that the BFPAI locus also contributes to the outcome of strain competition. Thus, canonical ‘virulence factors’ may be adapted to modify the colonic environment to provide a strain-specific advantage during competitive colonization. This finding underlines the importance of understanding microbiota composition in modeling enteric disease and highlights the need for development and interrogation of more sophisticated animal models to understand multifactorial host-microbe-microbiome interactions.
Methods
Bacterial strains, culture conditions, and plasmids. B. fragilis strains TM4000 (NTBF), ATCC 43858 (ETBF) and ATCC 43859 (ETBF) were used for this study. All strains were grown in Brain Heart Infusion (BHI) broth anaerobically at 37°C with a gas mix of 5% H2, 10% CO2 and 85% N2. BHI was supplemented with 0.0005% hemin and 0.5 μg/mL vitamin K1 for optimal growth (BHIS). Plasmids pFD340 and pAH2 were conjugated into B. fragilis strains TM4000 and ATCC 43859 from E. coli as described previously.17 Carbohydrates for toxin suppression testing were used at final concentration of 0.5% (weight/volume). For testing of toxin production during various growth phases of ATCC 43858, stationary phase overnight cultures were diluted 1:50 into fresh BHIS. Samples were removed from the culture each hour to measure optical density until stationary phase was reached.
In-frame deletion of bfpai was generated through allelic exchange using a protocol modified from previous studies.18 1 kb upstream and 1 kb downstream of bfpai was amplified from ATCC 43859 and fused via overlap PCR. This construct was cloned into pKNOCK and conjugated into ATCC 43859 as described previously.18 Single clones resistant to clindamycin, indicating genomic integration, were passaged (1:100) daily without antibiotics. After 5–10 passages, single clones were patched onto selective (clindamycin) and non-selective plates. Sensitive colonies were PCR screened for loss of bfpai.
B. fragilis pellet and supernatant fraction preparation. For detection of BFT in the cell pellet or supernatant fraction, the samples were prepared as follows. 1 mL of ATCC 43858 culture at the indicated time point was pelleted at 5,000 g for 5 minutes at room temperature and separated into cell pellet and supernatant fractions. The cell pellet was resuspended in 2X Laemmli sample buffer and heated to 95°C for 10 minutes. The supernatant was precipitated in a final concentration of 10% TCA, incubated for 1 hour on ice, and the precipitate was pelleted by 1 hour centrifugation on benchtop centrifuge at 15,000 g. The supernatant of this spin was removed and washed with 100% acetone to resuspend the precipitate. The washed precipitate was then spun for 10 minutes at maximum speed, followed by removal of the supernatant, a repeat wash and spin. After removal of the final wash, the pellet was air-dried for 30 minutes and resuspended in 2X Laemmli sample buffer for gel electrophoresis.
Immunoblots. For all immunoblots, samples were suspended in 2X Laemmli sample buffer run on 12% SDS-PAGE gels and transferred onto activated PVDF membrane. Membranes were subsequently blocked with 5% skim milk in TBS buffer supplemented with 0.1% Tween-20 (TBST), incubated with primary antibody for one hour, and washed thrice in TBST for 5 minutes. Secondary antibody was incubated with the membrane for one hour, proceeded by 3 TBST washes. The membrane was subsequently imaged on a Li-Cor Odyssey system. Rabbit anti-BFT antibody was generated as previously reported,19 and used at 1:2000 dilution in TBST. Goat anti-rabbit Alexa Fluor 680 (Life Technologies) was used as the secondary antibody at a 1:10,000 concentration in TBST.
Quantitative Reverse Transcription PCR. To test the mRNA levels of bft, quantitative reverse transcription PCR (qRT-PCR) was used. RNA was collected from ATCC 43858 cell culture using the RNeasy kit and RNA protect (Qiagen), according to manufacturer's instructions. RNase-free DNase (Fisher) was used to digest contaminating genomic DNA in the samples. First strand cDNA synthesis was performed with iScript cDNA synthesis kit (Bio-Rad) and qPCR was performed with SYBR Green (Bio-Rad) on a Bio-Rad CFX96 machine. BFT transcript was quantified with BFT-specific primers and normalized to B. fragilis 16s rRNA as described previously.17 Statistical analysis was performed using GraphPad Prism software. Pairwise comparisons were performed using the unpaired, parametric, 2-tailed Student's t-test.
Mouse modeling. All animal studies were conducted in accord with ethical regulations under protocols approved by the University of Chicago Animal Care and Use Committee and Institutional Biosafety Committee. SPF C57BL/6 mice were bred in-house from mice originally purchased from Jackson Laboratory. There was no investigator blinding in animal experimentation, and no animals were excluded from analysis. Mice were pre-treated with 100 mg/L clindamycin in drinking water for one day prior to and throughout the course of infection. Sequential colonization was performed with a modified protocol as described previously17 and summarized as follows. Primary colonization was achieved by inoculation of 108 CFU of TM4000 encoding pFD340 via oral gavage. After colonization for 7 days, secondary challenge was performed via oral gavage with 108 CFU of ATCC 43859 WT or Δbfpai encoding pAH2. To analyze fecal CFU following oral inoculation, serial 10-fold dilutions of fecal slurry in PBS were plated on BHIS agar containing 200 μg/mL gentamicin and 5 μg/mL clindamycin plus either 10 μg/mL tetracycline, to monitor ATCC 43859 recovery, or 20 μg/mL rifampicin, to determine TM4000 recovery. CFU/g feces for each clone was calculated, log10 transformed and plotted over time. Limit of detection is dependent upon the weight of each fecal pellet, indicated based on average fecal pellet weight at ∼103.5. Statistical analysis was performed using GraphPad Prism software. Pairwise comparisons were performed using the unpaired, parametric, 2-tailed Student's t-test.
Results
The Bacteroides fragilis pathogenicity island is a putative locus for enhanced secondary colonization
B. fragilis shows self-exclusion in a mouse model of competitive colonization in which a challenge inoculum fails to colonize the gut after initial engraftment of the same strain, dependent on the commensal colonization factors (ccf) locus.20 Competition between strains shows a more complex pattern of exclusion.17 In particular, we have demonstrated that ETBF strain ATCC 43859 was alone in successfully colonizing the murine gut in the context of initial engraftment with NTBF strains that are otherwise highly exclusionary. This led us to conclude that ATCC 43859 possesses a genetically encoded phenotype we term ‘enhanced secondary colonization fitness’. This phenotype provided an opportunity to test the hypothesis that bfpai is advantageous to colonization. Indeed, specific deletion of the bfpai locus (Δbfpai) results in a 5-log reduction of fecal CFU relative to wild-type (WT) ATCC 43859 after primary colonization with NTBF strain TM4000 (Fig. 1a, b). As the distinction between the WT and mutant ETBF clones emerges over time, and indeed is absent one day post-challenge, the defect is a function of the dynamic characteristics of the colonic niche.
Figure 1.
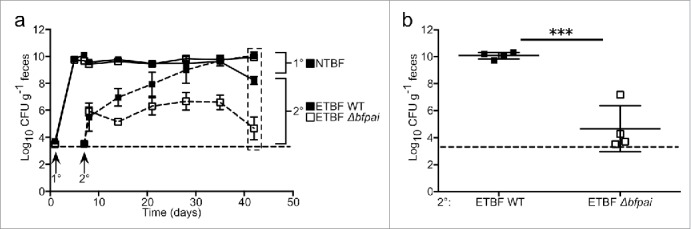
bfpai locus deletion impairs fitness during competitive secondary colonization. (A-B) Mice were primarily colonized with NTBF strain TM4000 638R and subsequently challenged with either ETBF strain ATCC 43859 or an isogenic mutant with bfpai deleted (Δbfpai). Clindamycin (100 mg/L) was maintained in drinking water throughout the experiment. Fecal CFU was monitored for 5 weeks post-challenge. N = 4 mice per group. Figures are representative of 3 independent experiments. Error bars: mean +/− s.e.m (A), mean +/− s.d. (B). Dashed lines indicate the limit of detection. n.s., not significant.
Importantly, bfpai is neither necessary nor sufficient for enhanced secondary colonization by ATCC 43859. We found the phenotype to be dependent on the primary colonizing strain, as Δbfpai was not impaired in colonizing hosts carrying NTBF strains NCTC 9343 or YCH46 (not shown). Moreover, a second ETBF strain (ATCC 43858) is prevented from invading the microbiota of mice colonized with TM4000 despite an intact bfpai.17 The high degree of strain specificity in sequential colonization illustrates that differential encoding of genetic elements in the secondary strain can have an impact on colonization outcomes. Future work will be required to determine which elements in the pathogenicity island are required for enhanced secondary colonization and if these factors directly impact on the competing organism or have an indirect effect by modifying the colonic environment through host signaling.
The pathogenicity island-encoded Bacteroides fragilis toxin is regulated by environmental cues
As we found bfpai to be advantageous for secondary colonization in vivo, we examined conditions affecting BFT regulation in vitro to understand the host niche for which it may be adapted. Toxin production can be broken down into 3 phases: transcription of the full-length toxin mRNA, translation of the pro-toxin and cleavage of pro-toxin into its active secreted form by the cysteine protease Fragipain (Fpn).19 Superimposed on these phases is toxin release into the environment, which occurs by an as yet unidentified mechanism. Glucose is known to suppress BFT production.21 We performed a time course with and without supplemental glucose, serially collecting supernatant, cell pellet, and RNA fractions. This experiment revealed several key pieces of information: first, glucose suppressed toxin expression at every time point as measured by western blot analysis for full-length toxin (Fig. 2a, FLBFT), the active toxin moiety (Fig. 2a, BFT*), and quantitative analysis of bft RNA (Fig. 2b), indicating that this phenotype is not sensitive to population density. Second, toxin expression was minimal in all 3 fractions before early stationary phase, despite the large majority of the total cell growth already having occurred. Third, without glucose supplementation, BFTproduction rapidly increased during early stationary phase in all fractions, with the majority of expression occurring within a 1–2 hour period, thus confirming and extending data from previous literature.21 Importantly, glucose supplementation did not alter bacterial growth kinetics (Fig. 2c).
Figure 2.
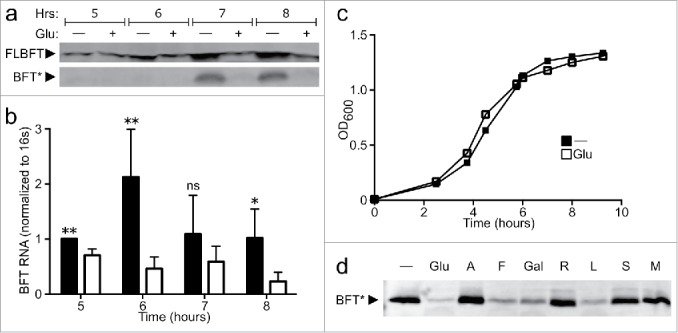
BFT is transcriptionally regulated by fermentable carbohydrates. (A) Full-length protoxin (FLBFT) in cell pellet and cleaved active BFT (BFT*) in culture supernatant was probed by western blot during log (5, 6 h) or early stationary (7, 8 h) phase growth from cultures with or without 0.5% glucose. (B) RNA from cell pellet in (a) was assayed by qPCR at various time points during log (5, 6 h) or early stationary (7, 8 h) phase growth from cultures with (filled bars) or without (empty bars) 0.5% glucose. *p<0.05, ** p<0.01; ns, not significant. (C) B. fragilis cultures show log phase growth in BHIS (filled squares) with early stationary phase beginning around 7–8 hours. The addition of 0.5% glucose to BHIS (open squares) does not affect growth kinetics. (D) ETBF was grown overnight in BHIS supplemented with PBS (-) or 0.5% of various carbohydrates (Glu = glucose, A = arabinose, F = fructose, Gal = galactose, R = rhamnose, L = lactose, S = sorbitol, M = manitol) and cleaved active BFT (BFT*) was probed by western blot from culture supernatants.
Free glucose is not found in the mammalian colon.22 Therefore, we sought to determine if suppression of BFT is specific to glucose or extends to other carbohydrates found in the gut. To accomplish this, ATCC 43858 was grown overnight with a series of supplemental carbohydrates and the supernatant screened for toxin production through anti-BFT immunoblot. Comparison of active toxin in the supernatant reveals that, in addition to glucose, multiple carbohydrates suppress toxin output relative to the PBS control, while others have no effect (Fig. 2d). This phenotype is associated with fermentation, as B. fragilis can metabolize only the suppressive carbohydrates, indicating that toxin is co-regulated with simple carbohydrate metabolism.23 Together, these data suggest that toxin production in vitro is dependent upon available fermentable carbohydrates and growth phase, regulated through a transcriptional mechanism. Several of these carbohydrates are constituents of mucus and may be important to BFT regulation in vivo.24
BFT is expressed as a pro-toxin and requires processing dependent on Fpn for its activity.19 Fpn is most active at pH 6.5–819, consistent with the classical mechanism of action for cysteine proteases. Since B. fragilis cultures acidify to pH 5.6 during stationary phase,21 we reasoned that the pH of the environment might affect the levels of active BFT seen during in vitro culture. Production of both BFT transcript and protein occurs during late stationary phase (Fig. 2a), with degradation of mRNA within an hour of production, and levels of cleaved BFT in culture supernatants remains stable for up to 24 hours. As predicted, transfer of stationary phase bacteria into fresh media at a pH 7.4 allows for increased production of cleaved BFT relative to transfer into fresh media adjusted to pH 5.6, indicating that environmental pH can affect BFT maturation (Fig. 3a). Interestingly, pH is variable across the colonic mucosa, increasing from 5.6 in pre-formed mucus secreted from the goblet cell to 6.8–7.2 in the fully stratified mucus coating the epithelium of the distal colon.25 These data indicate that BFT activity may be tied via pH to a spatially restricted niche.
Figure 3.
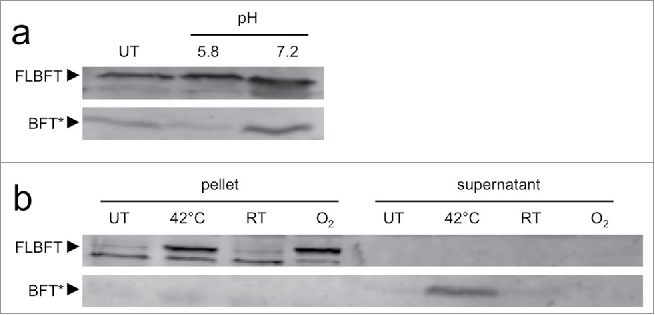
Complex protein-level regulation of BFT by environmental cues. (A) Bacteria from stationary phase cultures of ETBF strain ATCC4 3858 were sedimented, resuspended in spent media from normal growth conditions (UT) or fresh BHIS at pH 5.6 and pH 7.4, incubated for 1 hour at 37°C, and separated into pellet and supernatant fractions. BFT in the supernatant fraction was analyzed by Western blot for full length protoxin (FLBFT, upper band) and cleaved active toxin (BFT*). (B) Heat and oxidative stress upregulate BFT production. ETBF strain ATCC 43858 was grown to late-log phase, a growth phase during which BFT expression is not normally detected. Cultures were then exposed to different conditions for 1 hour: normal growth conditions (UT), 42°C, room temperature (RT), or room air (O2). Cell pellet and supernatant fractions were probed for full length protoxin (FLBFT, upper band) and cleaved active toxin (BFT*). The lower band is nonspecific.
We also observed that exposure of ATCC 43858 to either oxidative stress through aeration or heat stress increased BFT production (Fig. 3b). Room air increased levels of full-length protoxin, while heat increased levels of both pro-toxin and active cleaved BFT (Fig. 3b). Aerobicity is significant at the epithelial surface in vivo but rapidly decreases with distance. Thus, oxygen tension is a defining element of the colonic niche. Enteric microbes have been described to modify this variable with virulence factors for the selective benefit of their growth.26 Together with a bfpai-dependent advantage during competitive colonization, these data revealing environmental regulation of BFT indicate B. fragilis may fit within such a paradigm.
Conclusion
Deletion of bfpai in ETBF ATCC 43859 eliminates this strain's ability to successfully compete in vivo against an initial colonizing NTBF strain TM4000. BFT, the toxin encoded on bfpai, responds to several environmental cues. Maximum expression of bft in vitro occurs during early stationary phase and can be inhibited by free glucose and other fermentable carbohydrates, including galactose, which is among the most common glycosylations of colonic mucus. BFT activity at the protein level is regulated by Fpn, a cysteine protease that operates efficiently at pH 6.5 to 8.0, the range found in human feces and mature colonic mucus. Lastly, we show that heat and oxygen upregulate BFT.
Together, these data suggest a genetic link between virulence and niche competition through regulation of toxin over both time and space (Fig. 4). We propose that competition during enteric microbial development in early life drives ETBF into the host mucus layer, a niche inaccessible to many other microbes due to the presence of cationic antimicrobial peptides that are ineffective against Bacteroides.14 BFT expression during this period activates host immune signals that modify the composition of mucus to include carbohydrate constituents most beneficial for ETBF fermentation, leading to nutritional exclusion of competing strains from the mucus niche. This host-microbe interaction becomes homeostatic during development as the colon becomes anaerobic, downregulating BFT expression. Concurrently, the mucus layer matures, becoming structured such that pH-dependent Fpn activity is maximized at a distance from the epithelium where oxygen is scarce, fermentable carbohydrates from the diet and mucus are plentiful, and injury from BFT activation is minimized.
Figure 4.
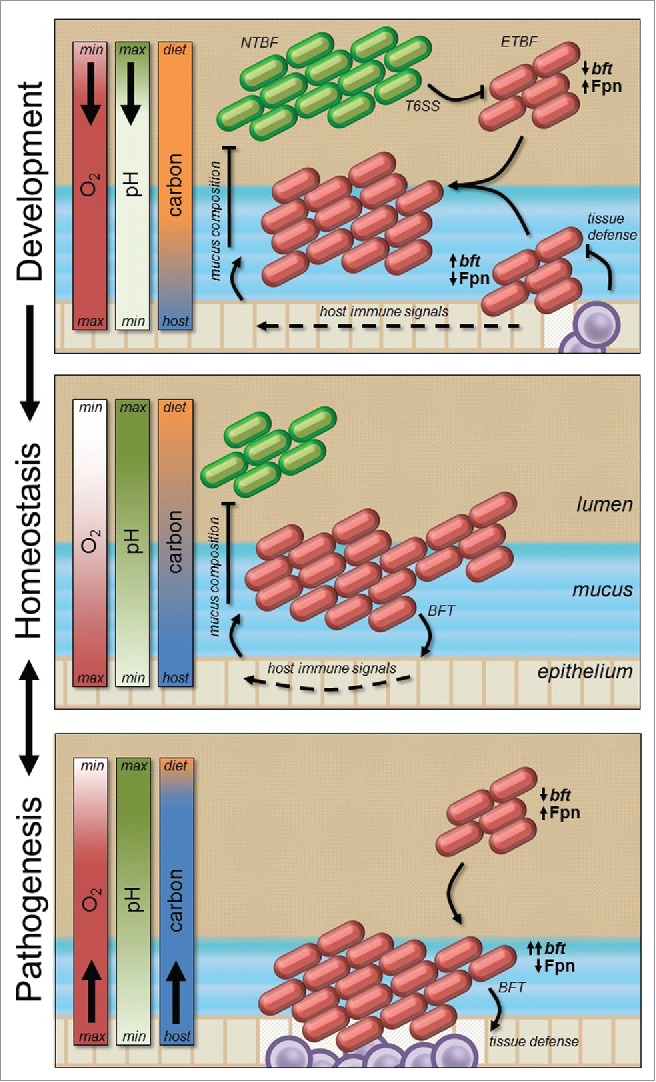
Spatiotemporal regulation of BFT predicts the outcome of virulence and competition. Bacterial competition during host development drives ETBF into colonic mucus where BFT activity manipulates the carbohydrate composition for its advantage by stimulating host immunity (top panel). This interaction becomes homeostatic during mucus layer maturation as Fragipain deactivates in immature acidic mucus most proximate to the epithelium (middle panel). Depletion of fermentable carbohydrates due to depletion of the mucus layer, or insufficient dietary fiber intake, drives ETBF further into the mucosa, where host-microbe interaction is further dysregulated by aerobic upregulation of BFT (bottom). In this case Fpn deactivation is compensated for by the number of organisms, volume of bft transcription, or host proteases.
Environmental conditions that alter the set points for Fpn activity or bft expression, for example elevated mucosal pH or a diet low in fermentable substrates, may predispose to chronic subacute disease.27,28 Exogenous perturbations may upset this balance further to induce acute disease pathogenesis. Such perturbations include withdrawal of dietary fiber (e.g. fasting during acute illness), transient hyperoxia due to enteric tissue reperfusion injury after surgery, or mucus layer depletion during microbial dysbiosis. Dynamic regulation by Fpn and these environmental cues warrants further investigation as potentially important genetic determinants of virulence and strain competition.
Virulence as a dimension of enteric microbial ecology
We propose that BFT, previously only considered as a virulence factor, is an adaptation for colonization in a competitive enteric environment. By manipulating mucosal immunity, BFT empowers ETBF to construct a colonic niche defined by particular resources and other conditions, such as mucus mobility, pH, and aerobicity, most advantageous to ETBF. Once constructed, niche access is regulated by bacterial adaptations for occupancy, such as mucus adhesion factors29 and polysaccharide utilization loci.20 Occupancy may not be exclusive to ETBF, as closely related strains could express many of the same adaptations. Thus, our model predicts that colonization is dependent on 2 layers of ecological priority after initial niche construction by ETBF. First, niche access is dictated by relatedness of accessory genes to the original colonizing strain. Second, niche occupancy is dictated by type VI contact-dependent compatibility between the established and invading strain.
This ecological view of virulence and colonization offers several important predictions. If this view is biologically relevant, we predict that co-colonizing strains in the mature human microbiome will be either highly related with compatible type VI secretion systems occupying the same niche, or so unrelated that they are adapted for different niches; highly related strains with incompatible type VI secretion systems should not co-occur. Serial competitive replacement of B. fragilis strains during development means that the mature ecosystem is expected to reach an optimum that is highly stable and homeostatic, with strain replacement occurring only rarely when a novel strain is encountered.30 Moreover, the initial colonist that constructs the niche may be replaced during host development given substantial changes in anatomy and physiology.6 The ability of strains to persist throughout this period reflects competitive fitness in environmental conditions that are unpredictable during initial enteric colonization, including changes in diet and exposure to other enteric microbes in childhood and beyond.31 Finally, because individual strains make sequential, potentially unique, modifications to the niche, early colonizing strains may influence the nature of the niche and its final occupants even after their elimination. Early life colonization with ETBF may therefore be a key event in shaping the adult microbiome, and represents an untapped opportunity for probiotic therapy.
Genetic determinants of virulence and competition may depend on experimental model
Testing these predictions requires novel animals models of colonization and competition. Although B. fragilis acquisition by humans is likely to occur during development and in the context of myriad other microbes, existing murine models have relied on gnotobiotic systems or antibiotic control to enable B. fragilis colonization. These models do not account for either microbial acquisition in a complex colonic ecosystem or native microbial acquisition during perinatal development. Murine models are needed that facilitate a detailed examination of early neonatal colonization in a developing and complex microbial environment, including potentially important events that govern durable colonization, such as birth, suckling, and weaning. Revealing microbial adaptations for strain competition and virulence may require animal models that account for these features of host development.
Several barriers exist to the development of suitable models. Stable B. fragilis colonization of the specific pathogen-free (SPF) mouse requires antibiotic decolonization of the gut, a treatment program that necessarily prevents analysis of complex host-microbe-microbiome interactions.20 Moreover, quantification of B. fragilis colonization over time may prove challenging if B. fragilis cannot be readily distinguished from the SPF fecal flora, possibly requiring culture-independent methods of quantification.
Moreover, to test the hypothesis that ecological perturbations underlie host-commensal driven disease, animals lacking genes required for structuring the enteric ecosystem will also be critical. Several such models exist, including mice deficient in the secreted colonic mucin Muc2, the fucosyltransferase Fut2 that attaches fucose modifications to mucopolysaccharides, and intestinal core 1- and core 3-derived O-glycans.32 These animals show increased disease susceptibility in several colitis models,32-34 however niche occupancy by B. fragilis has not been tested in these host variants. ETBF is a common enteric commensal in the developed world35 and must only rarely induce acute disease pathogenesis. Accounting for this distinctive feature of B. fragilis is likely to uncover a broader understanding of how genetic determinants of virulence and competition shape the microbiome and confer resistance or susceptibility to disease.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Department of Pediatrics at the University of Chicago and a Pilot and Feasibility Award from the Digestive Diseases Research Core Center at the University of Chicago (NIDDK P30DK42086). J.B.W. is a recipient of a Burroughs Wellcome Foundation Investigators in the Pathogenesis of Infectious Disease Fellowship. B.W.C., A.L.H., and V.M.C. are trainees of the National Institutes of Health Medical Scientist Training Program at the University of Chicago (GM007281).
References
- [1].Schaedler RW, Dubos R, Costello R. The development of the bacterial flora in the gastrointestinal tract of mice. J Exp Med 1965; 122:59-66; PMID:14325473; http://dx.doi.org/ 10.1084/jem.122.1.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, Gibbons SM, Larsen P, Shogan BD, Weiss S, et al.. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 2014; 345:1048-52; PMID:25170151; http://dx.doi.org/ 10.1126/science.1254529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gibbons SM, Schwartz T, Fouquier J, Mitchell M, Sangwan N, Gilbert JA, Kelley ST. Ecological succession and viability of human-associated microbiota on restroom surfaces. Appl Environ Microbiol 2015; 81:765-73; PMID:25398865; http://dx.doi.org/ 10.1128/AEM.03117-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005; 102:11070-5; PMID:16033867; http://dx.doi.org/ 10.1073/pnas.0504978102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al.. A core gut microbiome in obese and lean twins. Nature 2009; 457:480-4; PMID:19043404; http://dx.doi.org/ 10.1038/nature07540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science 2009; 326:1694-7; PMID:19892944;http://dx.doi.org/ 10.1126/science.1177486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chow J, Mazmanian SK. A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 2010; 7:265-76; PMID:20413095;http://dx.doi.org/ 10.1016/j.chom.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bartlett JG. Antibiotic-associated diarrhea. N Engl J Med 2002; 346:334-9; PMID:11821511; http://dx.doi.org/ 10.1056/NEJMcp011603 [DOI] [PubMed] [Google Scholar]
- [9].Sears CL, Islam S, Saha A, Arjumand M, Alam NH, Faruque AS, Salam MA, Shin J, Hecht D, Weintraub A, et al.. Association of enterotoxigenic Bacteroides fragilis infection with inflammatory diarrhea. Clin Infect Dis Off Publ Infect Dis Soc Am 2008; 47:797-803; http://dx.doi.org/ 10.1086/591130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al.. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009; 15:1016-22; PMID:19701202; http://dx.doi.org/ 10.1038/nm.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim JM, Jung HY, Lee JY, Youn J, Lee CH, Kim KH. Mitogen-activated protein kinase and activator protein-1 dependent signals are essential for Bacteroides fragilis enterotoxin-induced enteritis. Eur J Immunol 2005; 35:2648-57; PMID:16114110; http://dx.doi.org/ 10.1002/eji.200526321 [DOI] [PubMed] [Google Scholar]
- [12].Wu S, Powell J, Mathioudakis N, Kane S, Fernandez E, Sears CL. Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-kappaB pathway. Infect Immun 2004; 72:5832-9; PMID:15385484; http://dx.doi.org/ 10.1128/IAI.72.10.5832-5839.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wick EC, Rabizadeh S, Albesiano E, Wu X, Wu S, Chan J, Rhee KJ, Ortega G, Huso DL, Pardoll D, et al.. Stat3 activation in murine colitis induced by enterotoxigenic Bacteroides fragilis. Inflamm Bowel Dis 2014; 20:821-34; PMID:24704822; http://dx.doi.org/ 10.1097/MIB.0000000000000019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cullen TW, Schofield WB, Barry NA, Putnam EE, Rundell EA, Trent MS, Degnan PH, Booth CJ, Yu H, Goodman AL. Gut microbiota. Antimicrobial peptide resistance mediates resilience of prominent gut commensals during inflammation. Science 2015; 347:170-5; PMID:25574022; http://dx.doi.org/ 10.1126/science.1260580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tan Y, Zanoni I, Cullen TW, Goodman AL, Kagan JC. Mechanisms of toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity 2015; 43:909-22; PMID:26546281; http://dx.doi.org/ 10.1016/j.immuni.2015.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].San Joaquin VH, Griffis JC, Lee C, Sears CL. Association of Bacteroides fragilis with childhood diarrhea. Scand J Infect Dis 1995; 27:211-5; PMID:8539543; http://dx.doi.org/ 10.3109/00365549509019011 [DOI] [PubMed] [Google Scholar]
- [17].Hecht AL, Casterline BW, Earley ZM, Goo YA, Goodlett DR, Bubeck Wardenburg J, et al.. Strain competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Rep 2016; 17:1281-91; PMID:27432285; http://dx.doi.org/ 10.15252/embr.201642282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Comstock LE, Coyne MJ, Tzianabos AO, Pantosti A, Onderdonk AB, Kasper DL. Analysis of a capsular polysaccharide biosynthesis locus of Bacteroides fragilis. Infect Immun 1999; 67:3525-32; PMID:10377135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Choi VM, Herrou J, Hecht AL, Teoh WP, Turner JR, Crosson S, Bubeck Wardenburg J. Activation of Bacteroides fragilis toxin by a novel bacterial protease contributes to anaerobic sepsis in mice. Nat Med 2016; 22:563-7; PMID:27089515; http://dx.doi.org/ 10.1038/nm.4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, Mazmanian SK. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 2013; 501:426-9; PMID:23955152; http://dx.doi.org/ 10.1038/nature12447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Van Tassell RL, Lyerly DM, Wilkins TD. Purification and characterization of an enterotoxin from Bacteroides fragilis. Infect Immun 1992; 60:1343-50; PMID:1548060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gastrointestinal Physiology - 8th Edition Available at: https://www.elsevier.com/books/gastrointestinal-physiology/johnson/978-0-323-10085-4. (Accessed: 9thJanuary2017) [Google Scholar]
- [23].Cato EP, Johnson JL. Reinstatement of Species Rank for Bacteroides fragilis, B. ovatus,B. distasonis,B. thetaiotaomicron, and B. vulgatus: Designation of Neotype Strains for Bacteroides fragilis (Veillon and Zuber) Castellani and Chalmers and Bacteroides thetaiotaomicron (Distaso) Castellani and Chalmers. Int J Syst Evol Microbiol 1976; 26:230-7 [Google Scholar]
- [24].Koropatkin NM, Cameron EA, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat Rev Microbiol 2012; 10:323-35; PMID:22491358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ambort D, Johansson ME, Gustafsson JK, Nilsson HE, Ermund A, Johansson BR, Koeck PJ, Hebert H, Hansson GC. Calcium and pH-dependent packing and release of the gel-forming MUC2 mucin. Proc Natl Acad Sci U S A 2012; 109:5645-50; PMID:22451922; http://dx.doi.org/ 10.1073/pnas.1120269109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lopez CA, Miller BM, Rivera-Chávez F, Velazquez EM, Byndloss MX, Chávez-Arroyo A, Lokken KL, Tsolis RM, Winter SE, Bäumler AJ. Virulence factors enhance Citrobacter rodentium expansion through aerobic respiration. Science 2016; 353:1249-53; PMID:27634526; http://dx.doi.org/ 10.1126/science.aag3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, Sonnenburg JL. Quantitative Imaging of Gut Microbiota Spatial Organization. Cell Host Microbe 2015; 18:478-88; PMID:26439864; http://dx.doi.org/ 10.1016/j.chom.2015.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al.. A Dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 2016; 167:1339-53; PMID:27863247; http://dx.doi.org/ 10.1016/j.cell.2016.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huang JY, Lee SM, Mazmanian SK. The human commensal Bacteroides fragilis binds intestinal mucin. Anaerobe 2011; 17:137-41; PMID:21664470; http://dx.doi.org/ 10.1016/j.anaerobe.2011.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Oh J, Byrd AL, Park M; NISC Comparative Sequencing Program, Kong HH, Segre JA. Temporal stability of the human skin microbiome. Cell 2016; 165:854-66; PMID:27153496;http://dx.doi.org/ 10.1016/j.cell.2016.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Griffin NW, Ahern PP, Cheng J, Heath AC, Ilkayeva O, Newgard CB, Fontana L, Gordon JI. Prior dietary practices and connections to a human gut microbial metacommunity alter responses to diet interventions. Cell Host Microbe 2016; 21(1):84-96; PMID:28041931; http://dx.doi.org/ 10.1016/j.chom.2016.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bergstrom K, Fu J, Johansson ME, Liu X, Gao N, Wu Q, Song J, McDaniel JM, McGee S, Chen W, et al.. Core 1- and 3-derived O-glycans collectively maintain the colonic mucus barrier and protect against spontaneous colitis in mice. MucosalImmunol 2016; 10(1):91-103; PMID:27143302; http://dx.doi.org/ 10.1038/mi.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, et al.. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006; 131:117-29; PMID:16831596; http://dx.doi.org/ 10.1053/j.gastro.2006.04.020 [DOI] [PubMed] [Google Scholar]
- [34].Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, Bogatyrev SR, Ismagilov RF, Pamer EG, Turnbaugh PJ, et al.. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 2014; 514:638-41;PMID:25274297; http://dx.doi.org/ 10.1038/nature13823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Newton RJ, McLellan SL, Dila DK, Vineis JH, Morrison HG, Eren AM, Sogin ML. Sewage reflects the microbiomes of human populations. mBio 2015; 6:e02574; PMID:25714718; http://dx.doi.org/ 10.1128/mBio.02574-14 [DOI] [PMC free article] [PubMed] [Google Scholar]