

Abstract
Allergic contact dermatitis is a primarily T-cell-mediated inflammatory skin disease induced by exposure to small molecular-weight haptens, which covalently bind to proteins. The abundance of cutaneous T cells that recognize CD1a antigen-presenting molecules raises the possibility that MHC-independent antigen presentation may be relevant in some hapten-driven immune responses. Here we examine the ability of contact sensitizers to influence CD1-restricted immunity. Exposure of human antigen-presenting cells such as monocyte-derived dendritic cells and THP-1 cells to the prototypical contact sensitizer dinitrochlorobenzene potentiated the response of CD1a- and CD1d-autoreactive T cells, which released a vast array of cytokines in a CD1- and TCR-dependent manner. The potentiating effects of dinitrochlorobenzene depended upon newly synthesized CD1 molecules and the presence of endogenous stimulatory lipids. Further examination of a broad panel of contact sensitizers revealed 1,4-benzoquinone, resorcinol, isoeugenol and cinnamaldehyde to activate the same type of CD1-restricted responses. These findings provide a basis for the antigen-specific activation of skin-associated CD1-restricted T cells by small molecules and may have implications for contact sensitizer-induced inflammatory skin diseases.
Keywords: CD1, antigen-presentation, T cells, NKT cells, contact sensitivity
Introduction
Small molecular weight haptens are capable of inducing heterogenous T-cell-mediated responses manifested as cutaneous inflammation known as hapten-induced contact dermatitis in humans or contact hypersensitivity (CHS) in rodents [1, 2]. Haptens are unable to induce an immune response in their native state by virtue of their low molecular-weight and, therefore, must react with other molecules to become immunologically active [3]. Haptens may induce an immune response by several mechanisms, including a direct induction of innate immunity responses, and interaction with receptors activating adaptive immune response [4, 5]. Examples of clinically important haptens are electrophilic compounds that covalently bind to nucleophilic residues generating hapten-carrier complexes that represent novel T and B cell stimulatory compounds [6]. While the pathways through which contact sensitizers (CSs) act on MHC-restricted T cells are well described, very little is known of the potential roles of non MHC-restricted T cells within allergic contact dermatitis.
CD1 proteins are capable of binding a wide variety of microbial and self-lipid antigens and present them to T cells [7, 8]. In the context of skin-associated cell types, CD1d molecules are expressed by keratinocytes, CD1a by Langerhans cells, and all CD1 molecules by dermal dendritic cells (DCs). A significant proportion of skin-associated T cells are CD1a-restricted, with much smaller numbers recognizing antigens presented by CD1b, CD1c and CD1d [9]. The physiological relevance of these cells is largely unknown, while their role is some pathologies has been investigated in recent studies [10-13]. Translational research into the function of CD1-restricted T cells is hampered by the lack of expression of CD1a, b and c molecules in mice and rats [8]. Recently, human CD1a transgenic mice were instrumental in identifying the plant-derived lipid urushiol C15:2 as an antigen for CD1a-restricted T cells and in paving the way for studying such reactivities in patients with known sensitivity to this compound [14].
The presence of CD1d-restricted invariant Natural Killer T (iNKT) cells in skin lesions of patients with allergic contact dermatitis has been reported, together with an increased CD1d gene expression, at elicitation sites compared to normal skin [15]. Mouse studies also provided some evidence that iNKT cells may participate in the inflammatory response to haptens, typically via influencing the response of other immune cell types. iNKT cells may potentiate the inflammatory response to picryl chloride (2-chloro-1,3,5-trinitrobenzene, TNCB) [16] and oxazolone [17], may facilitate the activation of B1-cells involved in CHS initiation [18], and also the maturation of DCs contributing to CHS establishment [19]. A potential regulatory role of CD1d-restricted T cells in response to 2,4-dinitrochlorobenzene (DNCB) was also suggested [20], although the nature of involved cells and mechanisms remain unknown.
Within the present study we have investigated the effects of CSs on human CD1-restricted T-cell responses. We hypothesized that local exposure to small molecular-weight haptens may cause alterations in antigen-presentation to lipid-specific T cells. We focused on CD1d-restricted responses due to the existing knowledge about CSs and iNKT cells, and on CD1a-restricted T cells due to their known prevalence within the skin.
Results
DNCB-pulsed CD1+ human APCs activate CD1a- and CD1d-restricted T cells
To study the effects of CSs on CD1-restricted immunity, a panel of human T-cell clones was screened for reactivity to antigen-presenting cells (APCs) treated with the prototypical CS DNCB. DNCB treatment of monocyte-derived DCs (moDCs) prior to T-cell addition dose-dependently potentiated the response of the CD1a-restricted T-cell clone B13 (Fig. 1A). The B13 T-cell clone, was obtained from circulating T cells expressing CD4 and the CLA surface marker that identifies skin-homing T cells [21]. As the response of this T-cell clone was blocked by anti-CD1a mAbs and was observed only in the presence of CD1a-expressing APCs even in the absence of exogenously added lipids (Supporting Information Fig. 1A and B), the clone was considered bonafide CD1a-autoreactive. The CLA+ B13 cells released IL-22 upon stimulation (Supporting Information Fig. 1C), and expressed high levels of CCR4 expression (Supporting Information Fig. 1D), two additional hallmarks of skin-associated T cells. Incubation of APCs with DNCB at 6μM showed the highest T-cell stimulatory capacity, while exposure to concentrations above 6μM for 24 hours was toxic for the APCs (data not shown). In a new series of experiments we utilized as APCs CD1a-transfected THP-1 cells expressing stable levels of CD1a, as CD1 expression of DCs is variable between donors. Comparable results to those seen using moDC were observed with this type of APCs (Fig. 1B).
Figure 1.
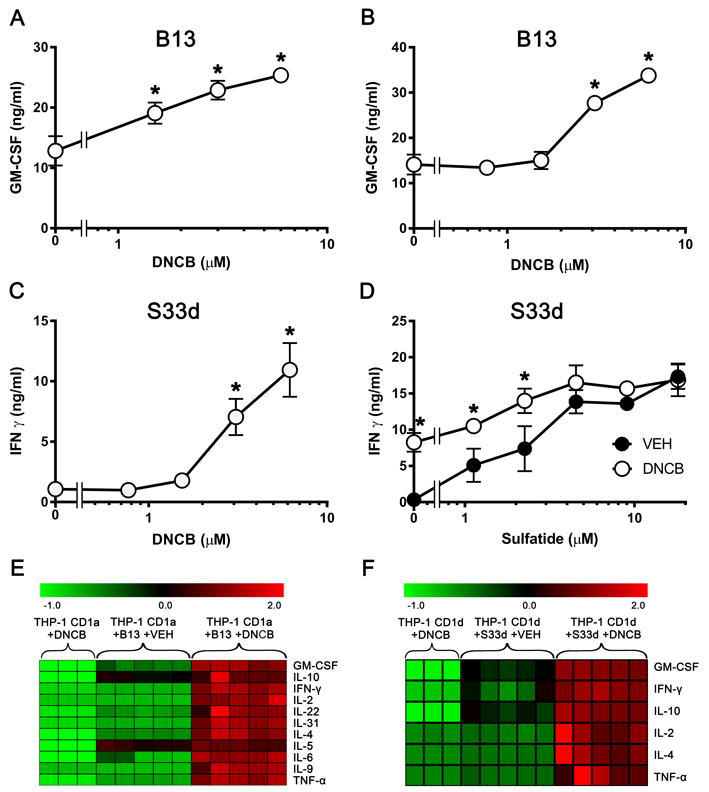
DNCB-pulsed CD1+APCs trigger the activation of CD1a- and CD1d-restricted clones. (A-D) B13 cells (A, B) and S33d cells (C, D) were stimulated with (A) DCs, (B) THP-1 CD1a, and (C) THP-1 CD1d cells by pulsing for 24 h with DNCB. (D) S33d cells were stimulated with sulfatide presented by THP-1 CD1d cells previously pulsed with DNCB (6 μM, open circles), or DMSO vehicle (VEH, closed circles). Production of (A, B) GM-CSF and (C, D) IFN-γ was measured by ELISA and shown as mean ± SD, n=3 for B13, n=4 for S33d cells. *p 0.01, t-test with Sidak multiple comparisons. Data shown are from single experiments representative of 3 independent experiments. (E) Heatmap of cytokines produced by B13 cells cultured with THP-1 CD1a cells previously pulsed with DNCB (6 μM) or VEH. Normalized data is expressed as the z-score. Absolute cytokine values are illustrated in Supporting Information Fig. 2. The following cytokines were tested, but were not released by the T-cell clone: IL-15, IL-17a/e/f, IL-21, IL-23, IL-27, IL-28, IL-33, TNF-β and MIP-1α. (F) Heatmap of cytokines produced by S33d cells cultured with THP-1 CD1d cells previously pulsed with DNCB (6 μM) or VEH. Normalized data are expressed as the z-score. Absolute cytokine values are illustrated in Supporting Information Fig. 3. The following cytokines were tested, but were not released by the T-cell clone: IL-5, IL-6, IL-9, IL-15, IL-17a/e/f, IL-21, IL-22, IL-23, IL-27, IL-28, IL-31, IL-33, TNF-β and MIP-1α. (E, F) Data in each column represent an individual replica of the indicated experimental condition.
We next screened for the existence of other types of CD1-restricted T cells positively influenced by APC pre-incubation with DNCB and identified the CD1d-restricted T-cell clone S33d (Fig. 1C). This clone is a NKT type II cell as it reacts to sulfatide, is CD1d-restricted, CD4+, and uses a non-invariant TCR [22].
As the antigen specificity of S33d cells were known, we could investigate the DNCB effect at different antigen doses. The potentiating effect of DNCB was significant at low and not at high sulfatide concentrations (≥4μM). DNCB seemed to act by increasing the potency and not the efficacy of sulfatide (Fig. 1D), thus suggesting a role in facilitating the formation of stimulatory CD1d-sulfatide complexes already at very low antigen doses. DNCB-treatment of THP-1 cells expressing the relevant CD1 molecules resulted in a broad potentiation of most cytokines, including IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-22, IL-31, GM-CSF, IFN-γ and TNFα, produced by B13 cells, as well as IL-2, IL-4, IL-10, GM-CFS, IFN-γ and TNFα, produced by S33d cells (Fig. 1E and F, absolute values Supporting Information Fig. 2 and 3).
In control experiments we asked whether DNCB had enhancing effect on other T-cell types. The CD1a-autoreactive K34B9.1 T cells (Supporting Information Fig. 4A), the CD1d-autoreactive JS63 iNKT cells (Supporting Information Fig. 4B), the α-galactosylceramide-responding VM-D5 iNKT cells (Supporting Information Fig. 4C), and the (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate-responding G2B2 TCR γδ cells (Supporting Information Fig. 4D) were not influenced by DNCB, thus excluding a generalized effect on T-cell responses.
The effect of DNCB on T-cell activation is CD1- and TCR-mediated
To evaluate whether the observed effect was mediated by cytokines released by APCs after exposure to DNCB, or if T-APC cognate interaction was required, S33d T-cell response was assessed separating DNCB-pulsed THP-1 CD1d cells in a transwell system (Supporting Information Fig. 5). The separation of S33d cells from APCs abrogated the activation, excluding a major role of soluble factors released by APCs and demonstrating the importance of cell-cell contact. Consistent with these data, the addition of anti-CD1 mAbs to DNCB-treated APCs prior to T-cell contact abrogated the T-cell response, demonstrating a CD1-dependent effect of the CS (Fig. 2A and B). To assess whether the observed effect was TCR-mediated, SKW-3 human T cells, which do not express endogenous TCR α, β, γ and δ genes, were transduced using lentiviruses containing the TCR α and β of the two DNCB-influenced T-cell clones, B13 (TRAV12-2*03, TRAJ5; TRBV14*01, TRBJ1-1) or S33d (TRAV14*01-TRAJ22; TRBV6-1*01-TRBJ2-2). The resulting SKW-B13 cells or SKW-S33d cells tested with DNCB-treated THP-1 CD1a or THP-1 CD1d cells, respectively, showed a dose-dependenT-cell surface upregulation of the CD69 activation marker, consistent with the TCR requirement to recognize the stimulatory CD1-antigen complex on DNCB-treated APCs (Fig. 2C and D). Collectively, these results suggest DNCB effects requiring specific TCRs and CD1-mediated antigen presentation.
Figure 2.
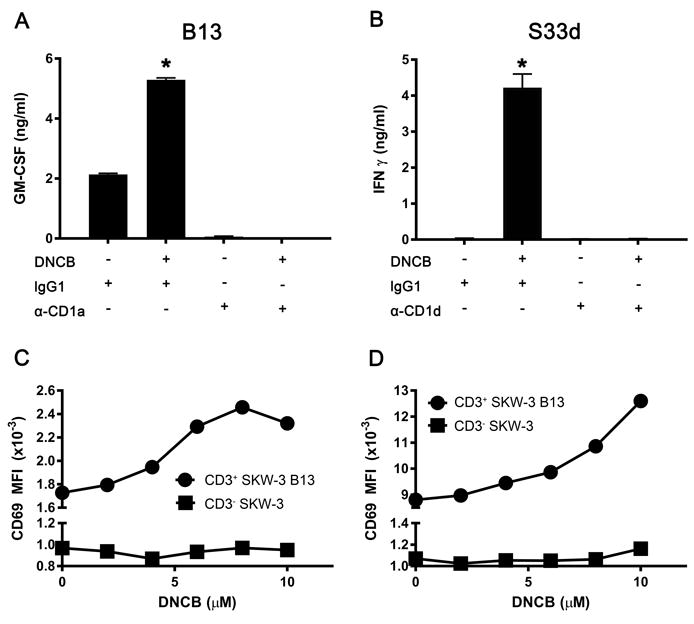
DNCB activity is mediated by CD1 and TCR. (A, B) Anti-CD1a (α-CD1a, A) and anti-CD1d (α-CD1d, B) or isotype-matched control mAbs (IgG1 and IgG2b, respectively) were added to (A) THP-1 CD1a cells and (B) THP-1 CD1d cells pulsed with DNCB (6 μM) or DMSO vehicle, before the incubation with (A) B13 and (B) S33d cells. Cytokines released by the T cells are expressed in ng/ml (mean + SD, n= 3-4) and results are from single experiments representative of 3 independent experiments. *p ≤0.001 (t-test with Sidak multiple comparisons). (C, D) Flow cytometry analysis of CD69 surface expression by (C) SKW-B13 and (D) SKW-S33d cells following incubation with (C) THP-1 CD1a and (D) THP-1 CD1d cells pretreated with DNCB. Control CD3-SKW-3 cells are also shown. Median fluorescence intensity (MFI) of CD3+ and of CD3- cells is plotted. Each plot represents data from a single experiment and is representative of 2 independent experiments.
Newly synthesized CD1 molecules are required for DNCB-mediated T-cell activation
In all experiments described above APCs were treated 24 hours with DNCB prior to extensive washing and addition of T cells. In a time-course experiment we determined that DNCB activity was detectable only after a minimum of 12 hours of incubation with APCs (Fig. 3A), suggesting a mechanism requiring prolonged APC exposure to DNCB. To investigate whether DNCB induces the generation of CD1-self antigen complexes, which accumulate over time, fixed THP-1 CD1d cells were used to stimulate S33d cells. THP-1 CD1d cells fixed before DNCB-treatment were unable to induce T-cell activation, while DNCB-treatment prior to fixation allowed increased activation compared to vehicle-treated controls (Fig. 3B). Thus, DNCB requires live APCs to exert its activation potential.
Figure 3.
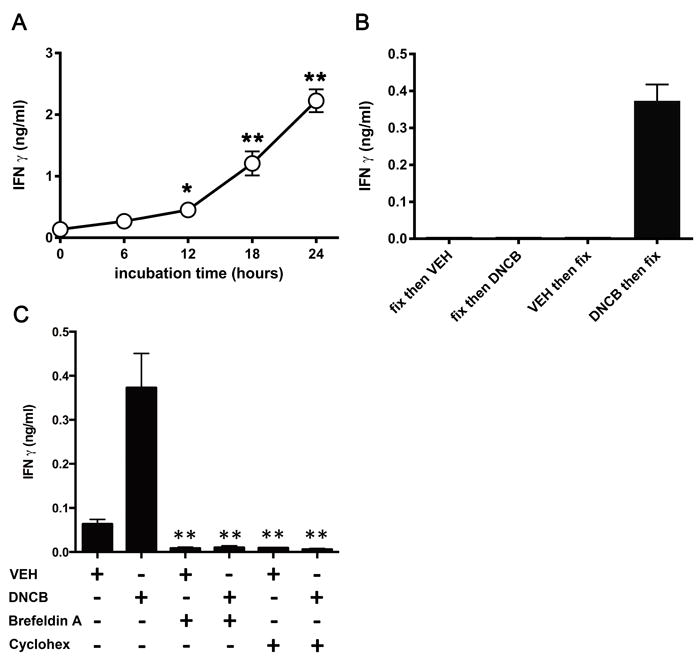
DNCB activity requires newly synthesized CD1 molecules. (A) Kinetic of THP-1 CD1d cell pulsing with DNCB (6 μM) before stimulation of S33d cells. IFN-γ release was measured by ELISA and data from single experiments representative of 3 independent experiments, are expressed as mean ± SD, n=3. (B) IFN-γ response of S33d cells to THP-1 CD1d cells fixed then pulsed with DNCB (6 μM) or pulsed first with DNCB (6 μM) and then fixed. Data are expressed as mean + SD, n=3, and are from a single experiment representative of 3 independent experiments. (C) IFN-γ response of S33d cells to DNCB (6 μM)- or DMSO vehicle (VEH)-pulsed THP-1 CD1d cells pre-treated with brefeldin A or cycloheximide (Cyclohex). Data are expressed as mean + SD, n=4, and are from a single experiment representative of 2-3 independent experiments. *p 0.05, **p 0.01 (t-test with Sidak multiple comparisons).
Taking into account the long time required by DNCB to exert its stimulatory effect, we treated APCs with cycloheximide and brefeldin A, drugs which block protein synthesis and egression from trans-Golgi network, respectively, to assess whether de novo synthesized CD1d complexes were involved. The stimulatory capacity of APCs was significantly reduced using both drugs, thus suggesting that newly synthesized CD1d-antigen complexes are the ones affected by DNCB (Fig. 3C).
Endogenous lipids are required for DNCB-mediated activation
To investigate whether the DNCB effect depended on the presence of endogenous lipid antigens, we used C1R cells expressing CD1d (C1R CD1d), which are able to present exogenous sulfatide in association with CD1d, but do not stimulate the S33d clone in the absence of exogenous antigen (Fig. 4A). Differently from what we observed with THP-1 CD1d cells, DNCB treatment of C1R CD1d cells did not result in the activation of S33d cells. Importantly, DNCB also did not change the response to the exogenously added sulfatide (Fig. 4A), suggesting that its effect depended on the endogenous lipids produced by some types of APCs.
Figure 4.
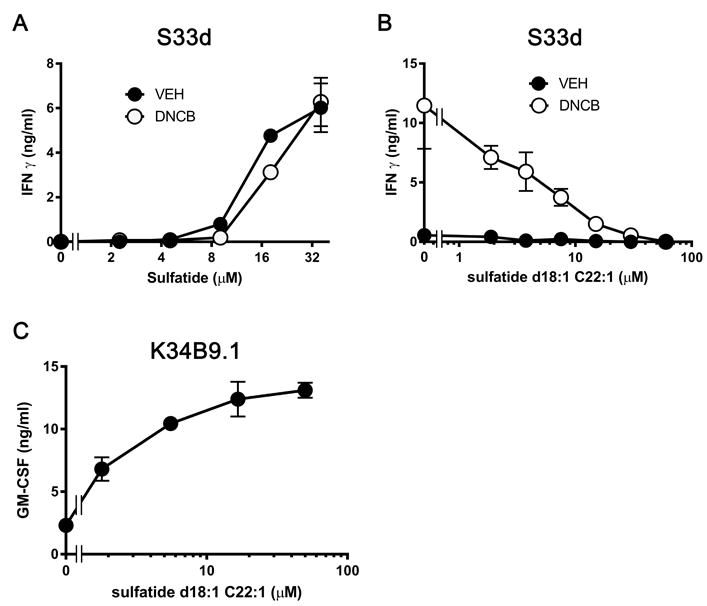
DNCB potentiates S33d cell activation through endogenous lipids. (A) IFN-γ response of S33d T cells to sulfatide presented by C1R CD1d cells pulsed with DNCB (6 μM, open circles) or DMSO vehicle (VEH, closed circles). (B) Non-stimulatory d18:1 C22:1 sulfatide was added to displace endogenous lipids from THP-1 CD1d cells previously pulsed with DNCB (6μM, open circles) or VEH (closed circles), before assessing S33d T-cell response. (C) Control response of the CD1a-restricted T-cell clone K34B9.1 to d18:1 C22:1 sulfatide presented by THP-1 CD1a cells. Data are expressed as mean ± SD, n=4,. *p≤0.05, t-test with Sidak multiple comparisons. Data are from single experiments representative of 2-3 independent experiments.
To further investigate this possibility, we performed a displacement experiment and incubated DNCB-treated THP-1 CD1d cells with synthetic d18:1-C22:1 sulfatide, a CD1d-binding sulfatide species non antigenic for S33d cells. This sulfatide attenuated, in a dose-dependent manner, the autoreactive response induced by DNCB with THP-1 CD1d cells, consistent with displacement of the endogenous stimulatory lipids (Fig. 4B). The observed attenuation was not due to toxic effects, as in control experiments the same synthetic lipid stimulated K34B9.1 cells, a second sulfatide-specific clone (Fig. 4C). These data suggested a key role for endogenous lipids permissive for DNCB-mediated activation of the CD1d-restricted S33d cells.
Resorcinol, isoeugenol and cinnamaldehyde trigger S33d T cells
To examine if the triggering of the S33d cell response was observed with other CSs, we screened more CSs, including two additional strong sensitizers, 1,4-benzoquinone and oxazolone, and four moderate sensitizers, para-phenylenediamine (PPD), cinnamaldehyde, resorcinol and isoeugenol. Incubation of THP-1 CD1d cells with resorcinol (Fig. 5A), isoeugenol (Fig. 5B) or cinnamaldehyde (Fig. 5C) resulted in a dose-dependent activation of S33d cells, and their effect was CD1d-mediated as it was blocked by the addition of anti-CD1d mAbs (Fig. 5D). The other compounds tested, 1,4-benzoquinone, oxazolone and PPD, did not exert any significant activity on S33d cell response (data not shown). 1,4-Benzoquinone, however, promoted activation of the B13 clone when it was used to treat THP-1 CD1a cells, and its effect was abrogated by the addition of anti-CD1a mAbs (Fig. 5E). These results show that several CSs have the capacity to elicit a response in CD1-restricted T cells.
Figure 5.
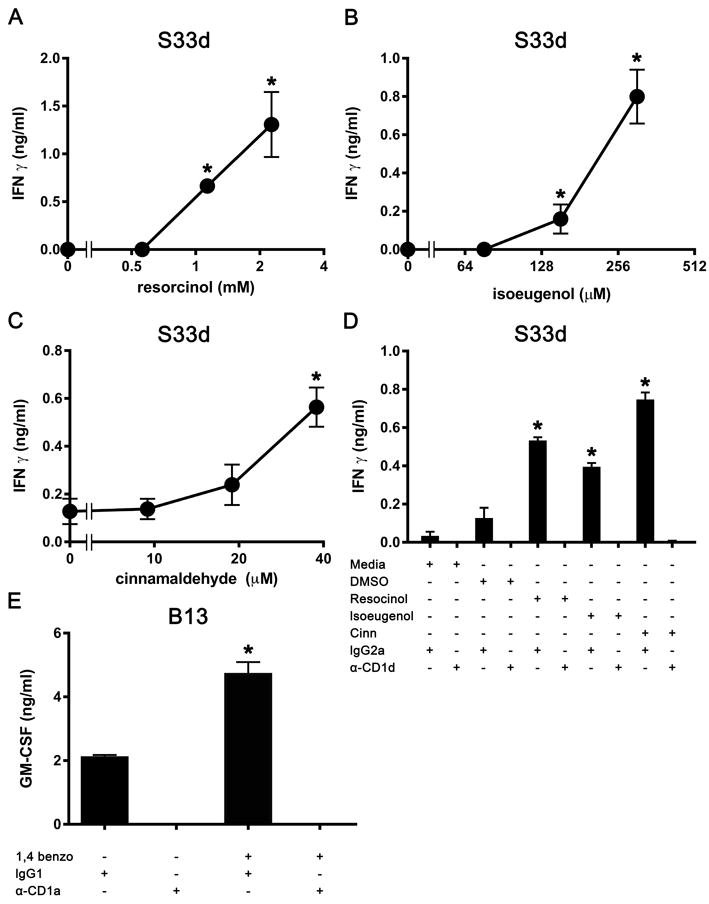
A wide range of contact sensitizers induce CD1-mediated T-cell activation. (A-C) The IFN-γ response of S33d cells to THP-1 CD1d cells pulsed with (A) resorcinol, (B) isoeugenol and (C) cinnamaldehyde. (D) S33d cell response in the presence of anti-CD1d (α-CD1d) or irrelevant (IgG2a) mAbs. Resorcinol was solubilized in medium and used at 2 mM, isoeugenol was solubilized in DMSO and used at 250 μM, cinnamaldehyde was solubilized in DMSO and used at 40 μM. (E) Response of B13 cells to THP-1 CD1a cells pulsed with 1,4 benzoquinone (10 μM in medium) and in the presence of anti-CD1a or irrelevant (IgG1) mAbs (α-CD1a). Data are expressed as mean ± SD, n=2-3, and are from single experiments representative of 2-3 independent experiments. *p 0.01 vs. the relevant vehicle controls, t-test with Sidak multiple comparisons.
Discussion
Modification of the interaction between the TCR and the antigen-presenting molecule by small compounds can induce strong, self-directed immune responses, manifesting clinically as inflammation of varying severity. The mechanisms by which a broad array of chemicals, including reactive haptens, drugs and metals act in the context of sensitization have been described for MHC class I and class II and the responding MHC-restricted T cells [2], whereas the ability of such small molecules to influence non-MHC-restricted T cells has mostly been uninvestigated. Within the current study we presented data indicating that small, low-molecular weight compounds are also able to influence CD1-mediated T-cell responses.
Upon incubation with a prototypic hapten and strong CS, DNCB, CD1-expressing APCs enhanced the response of self-reactive CD1a- and CD1d-restricted T cells. Two T-cell clones showed an important difference in their response. While the CD1a-restricted clone was CD1-autoreactive and its response was enhanced by DNCB, the CD1d-restricted clone showed CD1d-autoreactivity only in the presence of the compound, thus revealing that DNCB-sensitization may induce the appearance of otherwise silent self-reactivity. The DNCB effect was not observed with all tested T-cell clones. Indeed other CD1-restricted T cells, some of which were specific for endogenous antigens, were not affected by DNCB, suggesting the involvement of unique characteristics of their TCRs.
Our studies also addressed the mechanisms leading to DNCB-induced autoreactivity. First, only a prolonged DNCB-treatment of living APCs was effective. Secondly, DNCB effect required the presence of endogenous lipids that could be displaced by other CD1-binding lipids. Thirdly, not all CD1-expressing APCs were affected. Our data also excluded that the increased CD1 expression observed on APCs following treatment with DNCB at the relevant concentrations (Supporting Information Fig. 6 and 7), promoted autoreactivity. Moreover, a cytokine-mediated mechanism was excluded by the stimulatory capacity of DNCB-treated and fixed APCs. We interpret these findings with a direct effect of DNCB on the formation of CD1-self-antigen complexes, probably after increased synthesis of endogenous lipid-antigens in some cell types. This latter mechanism might explain why only some APCs (moDCs and THP-1 cells) were effective, while others (C1R and HeLa cells, not shown) were not, despite expressing high CD1 surface levels. A direct effect of DNCB on the formation of CD1-self-antigen complexes was further supported by two additional sets of data. First, autoreactivity following DNCB was reconstituted in TCR gene transduced cells, thus making unlikely the requirements of other unique T-cell surface molecules and confirming the precise antigen-specificity of autoreactivity. Secondly, DNCB effect was lost in the presence of exogenous lipids displacing the endogenous ones from CD1.
Intriguingly, DNCB effect was observed only after a prolonged exposure (>12 hours). This time is too long for a chemical modification induced by DNCB on mature and surface exposed CD1 molecules. In line with this observation, plate-bound assays using soluble recombinant CD1a and CD1d incubated with DNCB, did not stimulate the relevant T cells (data not shown). These results might reflect the requirements for continuous accumulation of newly synthesized CD1 molecules loaded with the stimulatory self-antigen. In agreement with this possibility, DNCB effects were prevented by drugs blocking protein synthesis and egression from the trans-Golgi. This mechanism is different from that applying to other contact sensitizers and MHC molecules, such as the unstable non-covalent interactions observed between sulfamethoxazole and nickel with MHC molecules [23, 24].
Importantly, we found that in addition to DNCB, other CSs induced the activation of the same T-cell clones, thus indicating that multiple CSs share this capability. The fact that they stimulate the same TCRs could be the consequence of a common mechanism of action. While the mechanism involving induced-self-antigens still remains plausible, an alternative one could be invoked by which the CS stably binds to nascent CD1 molecules and induces the formation of ternary complexes made by CD1, endogenous lipid-antigens and CS, in turn responsible for the stimulation of CD1-restricted T cells. This mechanism will thus resemble that of abacavir, the anti-HIV1 drug, which by binding to the HLA-B*57:01 binding cleft, alters its conformation and changes the self-peptide repertoire [25]. In the case of CSs, the formation of modified CD1-antigen complexes is probably the consequence of the high reactivity of the CSs, which were active in our model and are known to form adducts with cysteine and lysine residues, by reacting with aromatic nucleophilic substitution and forming covalent binding [26]. Although a direct structural proof of this ternary complex is missing, DNCB and the other CSs might alter the antigen-binding pocket of CD1 molecules, modify the positioning of endogenous antigens and induce a better response of certain T cells. Dedicated structural studies will be required to investigate the exact mechanism with each active CS.
The skin is home to approximately twice as many T cells as the blood [21], comprising of resident and migratory T cells located both within dermis and epidermis [27]. The close spatial association between T cells in the dermis or epidermis and skin cells expressing CD1 molecules raises the possibility of local interaction between T cells and APCs, and may account for the reactivity of cutaneous T cells to skin-associated lipids resulting in a basal level of lipid-mediated inflammation [10]. Considering the low dermal cellularity and the very high epidermal cellularity, direct interaction with keratinocytes is much more likely than interaction with fibroblasts. The large number of CD1a-restricted T cells within normal, non-inflammatory epidermis might promote their direct interaction with CD1a-expressing Langerhans cells. Thereby, exogenous small molecules, such as haptens, that can readily pass through the skin, would have ample opportunity to alter this immunological balance, resulting in potentiated local inflammation. In addition to CD1a-restricted T cells, we found that also CD1d-restricted T cells can be stimulated by CSs. Invariant NKT cells, while are largely absent in normal, non-inflammatory skin [28], infiltrate the epidermis during allergic contact dermatitis [15], and can kill keratinocytes in a CD1d-dependent manner [29]. Skin-infiltrating NKT cells, may thus become stimulated by APC that undergo prolonged interaction with CS.
Our demonstration that haptens can potentiate CD1-restricted T-cell activation raises broader questions concerning small molecules and skin-associated, lipid-specific T cells. To date most studies concerning type II drug adverse reactions have focused on MHC-dependent pathways, and while associations between HLA allotype and the risk of adverse reactions have been established, many seem allotype-unrelated and appear to be HLA-independent [4]. A recent publication by Kim and co-workers showed that urushiol binds to CD1a and induces skin inflammation in CD1a-transgenic mice by stimulating CD1a-restricted and urushiol-specific T cells [14]. Our findings differ from the ones above, as we found that several CSs stimulate T cells that are not CS-specific. Together with the work of Kim et al. (2016), our studies indicate that the activation of CD1-restricted T cells by a variety of CSs could be a novel mechanism in allergic contact dermatitis.
Self-reactive T cells restricted to CD1a, CD1c and CD1d molecules, with a polyclonal TCR repertoire and naïve/memory phenotype were detected in newborns and adults, with marked inter-individual variability [9, 30]. Our findings showed that despite the lack of functional polymorphism of human CD1 molecules would allow everybody to react to the allergens, only some T-cell clones were able to respond. In addition, not all CS-exposed CD1 expressing cell types induced such response. It is thus tempting to speculate that only the interaction of CSs with certain APCs producing unique sets of lipids and the concomitant activation of T cells with a matching TCR would contribute to the manifestation of contact dermatitis, in addition or in alternative to the classical CS-reactive MHC-restricted T-cell response. The requirement for such a combination would represent a natural limit to the number of individuals who develop lipid-specific T cells unleashed by CS presence.
The present identification of CD1-mediated activation of T-cell clones to some but not all CSs reflects the diversity of T-cell responses to these compounds, and underscores the complexity of immune responses to CSs. The involvement of the non-polymorphic CD1 molecules in the inflammatory response may prompt new therapies targeting CD1-restricted immune responses and may also allow new animal-free test strategies enabling skin sensitization safety assessments.
Materials and methods
Cells
Human monocytic THP-1 and B lymphoblast C1R cells expressing human CD1a, CD1b, CD1c or CD1d were previously described [31]. S33d, JS63, VM-D5 [22], K34B9.1 [32] and G2B2 [33] T-cell clones were maintained and re-stimulated in RPMI 1640 (Gibco, Thermo Fisher Scientific, Waltham, MA) supplemented with 5% AB+ human serum (Blood donation Centre, University Hospital Basel, Switzerland), 50 units/ml IL-2 [32]. To generate SKW-3 cells expressing the TCR from S33d or B13 cells, α and β cDNA were cloned into a vector with Lentiviral backbone (modified plenti-Blast, Addgene, Cambridge, MA) and transfected into HEK 293TLX cells using MetafectenePro (Biontex Laboratories, Munich, Germany) according to standard protocols. SKW-3 cells (Leibniz-Institut DSMZ, Braunschweig, Germany) were transduced with virus-containing supernatants supplemented with protamine sulfate (8μg/ml, Sigma-Aldrich, St Louis, MO) by spin-infection at 2000rpm for 45min at 37°C.
Isolation of B13 clone
Blood was obtained from healthy volunteers after informed consent and the study was approved by the Ethics Committee of the National University of Singapore (NUS-IRB 10-250). Peripheral blood mononuclear cells were separated by ficoll-paque gradient (GE Healthcare, Little Chalfont, UK) and enriched for those expressing cutaneous lymphocyte-associated antigen (CLA) using CLA-specific biotin-conjugated mAbs (HECA-542) and streptavidin-MACS beads (Miltenyi Biotec, Bergisch Gladbach, Germany). CLA-enriched cells were co-cultured with autologous moDCs in RPMI containing 5% autologous serum and 5 units/ml IL-2. After 12 days the cells were re-stimulated with autologous moDC in medium supplemented with 50 units/ml IL-2 and after another 12 days cloned by limiting dilution. Clones were screened with allogenic moDC in conjunction with mAbs specific for HLA-DR (L243), HLA-ABC (W6/32, both eBioscience, San Diego, CA), CD1a (OKT6), CD1b (BCD1b3.1), CD1c (F10/21A3.1 all purified in house) and CD1d (51.1, Biolegend, San Diego, CA).
Cellular assays
APCs were incubated in 10% FCS-supplemented RPMI 1640 with the following CSs: 2, 4-dinitrochlorobenzene (DNCB, solvent DMSO), cyclohexa-2,5-diene-1,4-dione (1,4-benzoquinone, solvent RPMI), 4-ethoxymethylene-2-phenyl-2-oxazolin-5-one (oxazolone, solvent DMSO), (2E)-3-phenylprop-2-enal (cinnamaldehyde, solvent DMSO), benzene-1,3-diol (resorcinol, solvent RPMI), 2-methoxy-4-(prop-1-en-1-yl)phenol (isoeugenol, all Sigma St Lois, MO, solvent DMSO), or 1,4-diaminobenzene (para-phenylenediamine, PPD, purity grade over 99.6%, L’Oreal laboratories, Paris, France, solvent RPMI) in the indicated solvents for 24h at 37°C, after which the APCs were washed and co-cultured with T cells at 1:2 ratio for 24-36h. For CD1-blocking experiments, APCs were incubated with the relevant antibodies for 2h prior to addition of T cells. For exogenous antigen presentation assays, α-galactosylceramide (KRN7000, Funakoshi, Tokyo, Japan) was solubilized in PBS 0.05% Tween20 and diluted in serum-free medium, sulfatides (3-O-sulfogalactosylceramides purified from bovine brain, Sigma-Fluka, St Louis, MO) and synthetic d18 C22:1 sulfatide (synthesized and purified according to standard methods, K.D. and A.R.H., unpublished) were solubilized in 2% methanol. For CS experiments, APCs were incubated with CS for 24h then washed prior to addition of the lipid. In some experiments, APCs were fixed with 0.05% glutaraldehyde [34] before or after the 24h incubation with CS, washed extensively and used with T cells at 1:1 ratio. Cycloheximide (50μg/ml, AbCam, Cambridge, UK) and brefeldin A (4μg/ml, eBioscience) were added to THP-1 cells 1h before DNCB and incubated for a further 24h, after which viability was confirmed with trypan blue, cells fixed with glutaraldehyde and used to stimulate T cells. Cytokine production was determined by ELISA (GM-CSF, IFN-γ, R&D systems, Minneapolis, MN) and Luminex assay (Merck Millipore, Billerica, MA). Luminex data is presented as heatmap based on the z-score, calculated as the individual cytokine data point value minus the mean value for the cytokine then divided by the standard deviation. This representation facilitates a comprehensive view of the significant changes of the T-cell response depending on APC treatment. Absolute values of cytokine released are illustrated in Supporting Information Fig. 2 and 3. For S33d clone, DNCB-treatment of THP-1 cells prior to stimulation did not change IL-6 production, while IL-15, IL-17a/e/f, IL-21, IL-22, IL-23, IL-27, IL-28, IL-31, IL-33, TNF-β and MIP-1α were not detected (data not shown). For B13 clone, DNCB-treatment of THP-1 cells did not change IL-13 production, while IL-15, IL-17a/e/f, IL-21, IL-23, IL-27, IL-28, IL-33, TNF-β and MIP-1α were not detected (data not shown). Activation of SKW-3 TCR transduced cells, was evaluated by flow cytometry as upregulation of CD69 on cells expressing CD3.
Flow cytometry
Human CD1 molecules were detected with mAbs specific for CD1a (HI149), CD1b (SN13), CD1c (L161) and CD1d (51.1, all eBioscience), following pre-incubation with Fc-block (BD Pharmingen). T-cell surface marker expression was evaluated using mAbs specific for CD3 (OKT3 or UCHT1), CD4 (OKT4), CD8 (SK1), CD69 (FN50), CLA (HECA-452) and CCR4 (TG6, all from Biolegend). THP-1 cell co-stimulatory molecules were examined with mAbs specific for CD80 (2D10), CD83 (HB15e), CD86 (IT2.2) and CD40 (5C3, all Biolegend). Samples were acquired on LSRII flow cytometer (Beckon-Dickinson, Franklin Lakes, NJ). Non-viable cells were excluded by FS/SS profile and DAPI (Life Technologies, Carlsbad, CA) or Pacific Orange (Biolegend) incorporation. Doublets were excluded by pulse-width parameter. Live cells were analyzed with FlowJo software (FlowJo LLC, Ashland, OR).
Statistical analysis
Data are expressed as mean ± SD and analyzed using Student’s t-test with Sidak multiple comparisons. P≤0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank O. Cexus, S.M. Ismail, R. Rozot, M.L. Tang, P. Wang, K.G. Srinivasan, F. Zolezzi and A. Gueniche for help and discussions, S. Koolarina d/o Suku, Z.X. Wong and M.H. Chong for assistance, M. Cavallari for T-cell clones, P. Savage, E. Padovan, A. Nardin, F. Ahmed, L. Breton, C. Bouez and M. Phong for advice. This work was supported by Singapore Immunology Network, L’Oreal Research and Innovation, the University of Basel and NIH RO1 GM087136 and U01 GM111849 grants to ARH.
Abbreviations
- APC
antigen-presenting cell
- CHS
contact hypersensitivity
- CS
contact sensitizer
- DC
dendritic cell
- DNCB
2,4-dinitrochlorobenzene
- HLA
human leukocyte antigen
- iNKT
invariant natural killer T
- MHC
major histocompatibility complex
- moDC
monocyte-derived dendritic cell
- NKT
natural killer T
- PPD
para-phenylenediamine
Footnotes
Conflict of interests:
All the authors declare no commercial or financial conflicts of interest. RJB, SM, ADB and SMT are employees and L’Oréal Research and Innovation.
References
- 1.Gell PG, Benacerraf B. Studies on hypersensitivity. IV. The relationship between contact and delayed sensitivity: a study of the specificity of cellular immune reactions. J Exp Med. 1961;113:571–585. doi: 10.1084/jem.113.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vocanson M, Hennino A, Rozieres A, Poyet G, Nicolas JF. Effector and regulatory mechanisms in allergic contact dermatitis. Allergy. 2009;64:1699–1714. doi: 10.1111/j.1398-9995.2009.02082.x. [DOI] [PubMed] [Google Scholar]
- 3.Pickard C, Smith AM, Cooper H, Strickland I, Jackson J, Healy E, Friedmann PS. Investigation of mechanisms underlying the T-cell response to the hapten 2,4-dinitrochlorobenzene. J Invest Dermatol. 2007;127:630–637. doi: 10.1038/sj.jid.5700581. [DOI] [PubMed] [Google Scholar]
- 4.Pichler WJ. The p-i Concept: Pharmacological Interaction of Drugs With Immune Receptors. World Allergy Organ J. 2008;1:96–102. doi: 10.1097/WOX.0b013e3181778282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan DH, Igyarto BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. 2012;12:114–124. doi: 10.1038/nri3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erkes DA, Selvan SR. Hapten-induced contact hypersensitivity, autoimmune reactions, and tumor regression: plausibility of mediating antitumor immunity. J Immunol Res. 2014;2014:175265. doi: 10.1155/2014/175265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Rhijn I, Godfrey DI, Rossjohn J, Moody DB. Lipid and small-molecule display by CD1 and MR1. Nat Rev Immunol. 2015;15:643–654. doi: 10.1038/nri3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mori L, Lepore M, De Libero G. The Immunology of CD1- and MR1-Restricted T Cells. Annu Rev Immunol. 2016;34:479–510. doi: 10.1146/annurev-immunol-032414-112008. [DOI] [PubMed] [Google Scholar]
- 9.de Jong A, Pena-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB. CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol. 2010;11:1102–1109. doi: 10.1038/ni.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, Van Rhijn I, Pena-Cruz V, Ruan DT, Altman JD, Rossjohn J, Moody DB. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol. 2014;15:177–185. doi: 10.1038/ni.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourgeois EA, Subramaniam S, Cheng TY, De Jong A, Layre E, Ly D, Salimi M, Legaspi A, Modlin RL, Salio M, Cerundolo V, Moody DB, Ogg G. Bee venom processes human skin lipids for presentation by CD1a. J Exp Med. 2015;212:149–163. doi: 10.1084/jem.20141505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subramaniam S, Aslam A, Misbah SA, Salio M, Cerundolo V, Moody DB, Ogg G. Elevated and cross-responsive CD1a-reactive T cells in bee and wasp venom allergic individuals. Eur J Immunol. 2016;46:242–252. doi: 10.1002/eji.201545869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, Hardman C, Xue L, Cerundolo V, Ogg G. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med. 2016;213:2399–2412. doi: 10.1084/jem.20160258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, Hu Y, Yongqing T, Kim J, Hughes VA, Le Nours J, Marquez EA, Purcell AW, Wan Q, Sugita M, Rossjohn J, Winau F. CD1a on Langerhans cells controls inflammatory skin disease. Nat Immunol. 2016;17:1159–1166. doi: 10.1038/ni.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gober MD, Fishelevich R, Zhao Y, Unutmaz D, Gaspari AA. Human natural killer T cells infiltrate into the skin at elicitation sites of allergic contact dermatitis. J Invest Dermatol. 2008;128:1460–1469. doi: 10.1038/sj.jid.5701199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campos RA, Szczepanik M, Itakura A, Akahira-Azuma M, Sidobre S, Kronenberg M, Askenase PW. Cutaneous immunization rapidly activates liver invariant Valpha14 NKT cells stimulating B-1 B cells to initiate T cell recruitment for elicitation of contact sensitivity. J Exp Med. 2003;198:1785–1796. doi: 10.1084/jem.20021562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nieuwenhuis EE, Gillessen S, Scheper RJ, Exley MA, Taniguchi M, Balk SP, Strominger JL, Dranoff G, Blumberg RS, Wilson SB. CD1d and CD1d-restricted iNKT cells play a pivotal role in contact hypersensitivity. Exp Dermatol. 2005;14:250–258. doi: 10.1111/j.0906-6705.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 18.Campos RA, Szczepanik M, Lisbonne M, Itakura A, Leite-de-Moraes M, Askenase PW. Invariant NKT cells rapidly activated via immunization with diverse contact antigens collaborate in vitro with B-1 cells to initiate contact sensitivity. J Immunol. 2006;177:3686–3694. doi: 10.4049/jimmunol.177.6.3686. [DOI] [PubMed] [Google Scholar]
- 19.Shimizuhira C, Otsuka A, Honda T, Kitoh A, Egawa G, Nakajima S, Nakashima C, Watarai H, Miyachi Y, Kabashima K. Natural killer T cells are essential for the development of contact hypersensitivity in BALB/c mice. J Invest Dermatol. 2014;134:2709–2718. doi: 10.1038/jid.2014.200. [DOI] [PubMed] [Google Scholar]
- 20.Fjelbye J, Antvorskov JC, Buschard K, Issazadeh-Navikas S, Engkilde K. CD1d knockout mice exhibit aggravated contact hypersensitivity responses due to reduced interleukin-10 production predominantly by regulatory B cells. Exp Dermatol. 2015;24:853–856. doi: 10.1111/exd.12792. [DOI] [PubMed] [Google Scholar]
- 21.Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, Kupper TS. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006;176:4431–4439. doi: 10.4049/jimmunol.176.7.4431. [DOI] [PubMed] [Google Scholar]
- 22.Facciotti F, Cavallari M, Angenieux C, Garcia-Alles LF, Signorino-Gelo F, Angman L, Gilleron M, Prandi J, Puzo G, Panza L, Xia C, Wang PG, Dellabona P, Casorati G, Porcelli SA, de la Salle H, Mori L, De Libero G. Fine tuning by human CD1e of lipid-specific immune responses. Proc Natl Acad Sci U S A. 2011;108:14228–14233. doi: 10.1073/pnas.1108809108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnyder B, Mauri-Hellweg D, Zanni M, Bettens F, Pichler WJ. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human alphabeta T cell clones. J Clin Invest. 1997;100:136–141. doi: 10.1172/JCI119505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gamerdinger K, Moulon C, Karp DR, Van Bergen J, Koning F, Wild D, Pflugfelder U, Weltzien HU. A new type of metal recognition by human T cells: contact residues for peptide-independent bridging of T cell receptor and major histocompatibility complex by nickel. J Exp Med. 2003;197:1345–1353. doi: 10.1084/jem.20030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, Miles JJ, Kjer-Nielsen L, Gras S, Williamson NA, Burrows SR, Purcell AW, Rossjohn J, McCluskey J. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012;486:554–558. doi: 10.1038/nature11147. [DOI] [PubMed] [Google Scholar]
- 26.Aleksic M, Pease CK, Basketter DA, Panico M, Morris HR, Dell A. Mass spectrometric identification of covalent adducts of the skin allergen 2,4-dinitro-1-chlorobenzene and model skin proteins. Toxicol In Vitro. 2008;22:1169–1176. doi: 10.1016/j.tiv.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. 2015;7:279ra239. doi: 10.1126/scitranslmed.3010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang XN, McGovern N, Gunawan M, Richardson C, Windebank M, Siah TW, Lim HY, Fink K, Li JL, Ng LG, Ginhoux F, Angeli V, Collin M, Haniffa M. A three-dimensional atlas of human dermal leukocytes, lymphatics, and blood vessels. J Invest Dermatol. 2014;134:965–974. doi: 10.1038/jid.2013.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balato A, Zhao Y, Harberts E, Groleau P, Liu J, Fishelevich R, Gaspari AA. CD1d-dependent, iNKT-cell cytotoxicity against keratinocytes in allergic contact dermatitis. Exp Dermatol. 2012;21:915–920. doi: 10.1111/exd.12036. [DOI] [PubMed] [Google Scholar]
- 30.de Lalla C, Lepore M, Piccolo FM, Rinaldi A, Scelfo A, Garavaglia C, Mori L, De Libero G, Dellabona P, Casorati G. High-frequency and adaptive-like dynamics of human CD1 self-reactive T cells. Eur J Immunol. 2011;41:602–610. doi: 10.1002/eji.201041211. [DOI] [PubMed] [Google Scholar]
- 31.de la Salle H, Mariotti S, Angenieux C, Gilleron M, Garcia-Alles LF, Malm D, Berg T, Paoletti S, Maitre B, Mourey L, Salamero J, Cazenave JP, Hanau D, Mori L, Puzo G, De Libero G. Assistance of microbial glycolipid antigen processing by CD1e. Science. 2005;310:1321–1324. doi: 10.1126/science.1115301. [DOI] [PubMed] [Google Scholar]
- 32.Shamshiev A, Gober HJ, Donda A, Mazorra Z, Mori L, De Libero G. Presentation of the same glycolipid by different CD1 molecules. J Exp Med. 2002;195:1013–1021. doi: 10.1084/jem.20011963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carena I, Shamshiev A, Donda A, Colonna M, Libero GD. Major histocompatibility complex class I molecules modulate activation threshold and early signaling of T cell antigen receptor-gamma/delta stimulated by nonpeptidic ligands. J Exp Med. 1997;186:1769–1774. doi: 10.1084/jem.186.10.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shamshiev A, Donda A, Prigozy TI, Mori L, Chigorno V, Benedict CA, Kappos L, Sonnino S, Kronenberg M, De Libero G. The alphabeta T cell response to self-glycolipids shows a novel mechanism of CD1b loading and a requirement for complex oligosaccharides. Immunity. 2000;13:255–264. doi: 10.1016/s1074-7613(00)00025-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.