

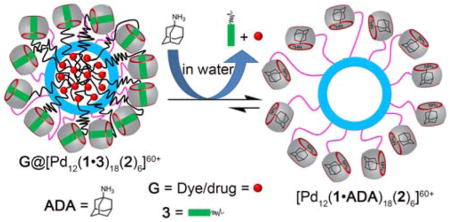
Abstract
Mixed self-assembly of ligand 1, 2, 1,6-hexanediamine (HDA) and Pd(NO3)2 afforded Fujita-type metal organic polyhedron MOP1 (diameter ≈ 8.2 nm) which is covalently functionalized with an average of 18 cucurbit[7]uril (CB[7]) units as evidenced by 1H NMR, diffusion ordered spectroscopy NMR and transmission electron microscopy measurements. By virtue of the host-guest properties of CB[7], the inner cavity of MOP can be rendered hydrophobic by using octadecyl HDA (3) as guest during the self-assembly process. The hydrophobic cavity was successfully utilized to trap the hydrophobic dye Nile Red (NR) and the anticancer drug Doxorubicin (DOX). The stimuli responsive release of encapsulated NR or DOX occurs: 1) upon addition of a competitive binder (e.g. adamantane ammonium (ADA)) for CB[7], 2) by a dual pH-chemical stimulus involving the protonation state change of adamantane carboxylate at pH 5.8, and 3) a dual pH-photochemical stimulus involving photoisomerization of trans-6 to cis-6 at pH 5.8. NR is released from NR@MOP2 within HeLa cancer cells. This body of work suggests that the covalent attachment of cucurbit[n]uril to metal organic polyhedra constitutes a promising vehicle for the development of both diagnostic and therapeutic nanoparticles.
Graphical Abstract
Introduction
The cucurbit[n]urils (CB[n]) – which are pumpkin shaped molecular containers – have received significant attention over the past decade because their unique aqueous host-guest chemistry provides a means to create a wide range of functional molecular and supramolecular systems.1 Advantageously, such CB[n] derived systems often exhibit large changes in constitution upon photochemical or electrochemical stimulation, upon addition of competive binders, and upon changes in the pH of the medium due to the accompanying large changes in CB[n]•guest binding affinity.1g,2 Within the CB[n] family, CB[7] is perhaps the most attractive member due to its very good water solubility (~20 mM),1e and its exceptionally high binding affinity (Ka routinely in the 106 – 1012 M−1 range) and selectivity towards cationic guests.1b,3 In addition, CB[n]-type receptors exhibit good biocompatibility, and have therefore been used to formulate, protect, and deliver drugs, as reversal agents to mitigate drug (side) effects, and for various sensing applications.4 Earlier, the excellent recognition properties of CB[n] were combined with nanoparticles for biological and material applications.5 For example, the Zink, Stoddart, and Yang groups have created mesoporous silica nanoparticles that are gated by host-guest recognition processes.6 In recent years, the availability of (mono)functionalized CB[n] compounds1d,7 has extended the scope of functional CB[n] derived systems8 to include membrane protein isolation,8c–e vesicle fusion,8f enhancement of biopharmaceuticals by supramolecular PEGylation,8a affinity purification of proteins featuring N-terminal Phe, and cellular imaging.8b In this work, we describe the self-assembly of monofunctionalized CB[7] derivative 1 to deliver a metal organic polyhedron (MOP) and investigation of its potential as a stimuli responsive theranostic nanoparticle.
A major goal in anti-cancer chemotherapy is to achieve highly selective delivery to the tumor to ameliorate the adverse effects of these highly potent drugs on healthy tissue. It is known that drug delivery systems based on large nanoparticles (e.g. > 5 nm diameter) benefit from the enhanced permeability and retention (EPR) effect,9 which results in selective accumulation of nanoparticles in the tumor compartment. Accordingly, various stimuli–responsive (e.g. temperature changes, magnetic fields, ultrasound, light, electric field, pH)6c,10 nanoparticle drug delivery systems have been explored for cancer therapy and imaging. Despite these advances, the delivery of effective doses of anti-cancer drugs with high spatio-temporal control is still a major challenge. For example, even the blockbuster drug Doxil™ suffers from the slow release of doxorubicin (< 5% in 24 h) which hampers its efficacy.11 Therefore, the development of new drug delivery vehicles equipped for targeted delivery and stimuli responsive release is urgently needed.
Over the years, supramolecular chemists have mastered the preparation and functionalization of monodisperse metal-organic cage structures.12 For example, the Fujita group has prepared an elegant family of spherical MOPs (PdnL2n composition) with diameters in the 3 – 9 nm13 range by simply mixing Pd2+ and angular bispyridine ligands (L), demonstrated exohedral and endohedral functionalization for molecular recognition, and even controlled the hydrophobicity of the MOP cavity.14 Despite the substantial progress in the field of nanoparticle-based systems for theranostic application mentioned above, the use of MOPs for this purpose was not explored until recently.15 For example, Lippard reported that a self-assembled metal-organic octahedron could enhance the delivery of adamantane functionalized cis-platin prodrugs.15a,16 Stang showed that Pt-based supramolecular coordination complexes can be utilized as theranostic supramolecular nanoparticles.17 Nitschke and Mascarenas demonstrated that host•guest binding of an Fe-cage to an oligoarginine peptides triggered the cellular uptake of the peptide.18 Recently, we reported a Fujita-type cubooctahedral MOP studded with 24 methyl viologen groups which non-covalently recruited CB[8] and doxorubicin prodrugs by heteroternary complexation and its ability to deliver doxorubicin to HeLa cells.19 These MOPs which are non-covalently functionalized with CB[n] are less robust and less attractive for in vivo application due to their potential for dissociation which lead us to consider more robust analogues based on a non-covalent but mechanically interlocked architecture.20 In this paper, we explore a covalently functionalized architecture featuring a cubooctahedral MOP functionalized with CB[7] units. Complexation of the CB[7] units with alkanediammonium ions functionalized with hydrophobic tails creates a hydrophobic nanoenvironment that is capable of taking up Nile Red (NR) or Doxorubicin (DOX). The inherent stimuli responsiveness of CB[7]•guest complexes enables the multi-stimuli triggered destruction of the nanoenvironment which causes concomitant release of DOX or NR.
Results and Discussion
This results and discussion section is subdivided into sections on the synthesis and characterization of ligand 1 and MOP1, followed by demonstration of the formation of a hydrophobic inner nanospace by non-covalent complexation and uptake of NR and DOX, and finally the stimulus responsive release of NR and DOX upon pH, light, and/or chemical changes. Finally, cellular uptake of MOP and the release of dye is described.
Ligand Design and Synthesis
To covalently decorate a cubooctahedral Pd12L24 assembly with CB[7] units, we needed to prepare a bis(pyridine) functionalized CB[7] derivative to use in the self-assembly process. Given the ready availability of azidobutyl-CB[7],7b we decided to prepare a propargylated bis(pyridine) ligand to unite them by click chemistry. Alkylation of the known alcohol 2 with propargyl bromide under basic conditions delivered 2a in 80% yield (Scheme 1). Subsequently, 2a was allowed to react with azidobutyl-CB[7] using Pericàs catalyst21 in a mixed solvent system (H2O:DMSO, 1:1) at 80 °C for 4 days to afford compound 1 in 55% yield. The synthesis of guests 3 – 5 and the characterization of all new compounds are presented in the Supporting Information.
Scheme 1.

A) Synthesis of ligand 1. Conditions: a) Propargyl bromide, NaH, THF-DMF, 0 °C to room temperature, 3 h, 80%, b) Azidobutyl-CB[7], Pericàs Catalyst, H2O-DMSO, 80 °C, 4 days, 55%. B) Chemical structures of compounds used in this study.
Synthesis and Characterization of CB[7] Functionalized MOP
We first examined the formation of a CB[7]-functionalized cubooctahedral MOP by self-assembly of ligand 1 and Pd(NO3)2 but did not observe the formation of an assembly by 1H NMR. Experiments with added HDA – designed to block the CB[7] cavity from complexing its own pyridine ligand – were also unsuccessful. Subsequently, we decided to perform the co-assembly of 1 and less bulky bis(pyridine) ligand 2 in a 3:1 molar ratio.14a,14b,22 When the mixture of 1, 2, HDA, and Pd(NO3)2 was heated in DMSO at 70 °C for 72 h we observed the quantitative formation of the cubooctahedral M12L24 spherical complex MOP1 (Figure 1) in accord with the precedent of Fujita on related bis(pyridine) ligands. Although we depict MOP1 as being composed of an 18:6 ratio of ligands 1 and 2, we believe that the stoichiometry represents a statistical distribution around the 18:6 ratio in accord with the results on two component assemblies previously prepared by Fujita.14b Analogous experiments conducted in the absence of HDA were unsuccessful which indicates an important role for HDA in blocking the recognition properties of CB[7] toward guest inclusion and or metal (e.g. Pd2+) complexation at its electrostatically negative ureidyl C=O portals.23
Figure 1.

(a) Self-assembly of MOP1 and MOP2 in DMSO. 2D DOSY spectra recorded (600 MHz, D2O) for: b) MOP1, c) 1, and d) MOP2. 1H NMR spectra recorded (600 MHz, RT) for: e) MOP2 (in D2O), f) MOP1 (in D2O), g) 1 (in D2O), h) MOP1 (in DMSO-d6), and i) MOP2 (in DMSO-d6). TEM images of: j and k) MOP2 (120 μM), and l) MOP1 (100 μM).
MOP1 was characterized by 1H NMR and diffusion-ordered spectroscopy (DOSY). We observe a single broadened set of resonances in the 1H NMR spectrum arising from the pyridyl groups which is consistent with a statistical distribution of ligands throughout the structure.14b The 1H NMR spectrum of MOP1 in DMSO (Figure 2h & S19) shows the typical down-field shifting of pyridine protons (Ha and Hb) upon self-assembly (Ha: to 9.49 ppm; Hb: to 8.42 ppm). The broadening of the signals reflect their slower tumbling motion on NMR timescale due to the large size of the complexes. The diffusion coefficient of MOP1 was measured by DOSY in DMSO as D = 2.90 × 10−11 m2/s (Figure S21). Application of the Stokes–Einstein equation allowed us to estimate the diameter of MOP1 as 7.6 nm which is in agreement with estimates based on the MMFF optimized geometry of MOP1 (Figure S59 and Table S1). MOP1 is highly cationic (60+) due to its twelve Pd2+ ions and 18 dicationic HDA guests bound to CB[7] which promotes its aqueous solubility. Accordingly, a DMSO-d6 solution of MOP1 was dialyzed (MWCO = 3500) against D2O for 48 h to afford an aqueous solution of MOP1. The 1H and DOSY spectra (Figures 2b, 2f & S20) recorded for MOP1 in D2O are analogous to those measured in DMSO. Based on the DOSY results, the diameter was calculated to be 7.8 nm, which establishes both the size and stability of MOP1 in water. We were also successful in visualizing MOP1 by transmission electron microscopy (TEM) by deposition of an aqueous solution of MOP1 on a carbon-coated Cu grid (Figure 2l & S55). The TEM results provide an independent measure of the size (≈ 7.0 nm in diameter) and spherical shape of the individual MOP1 assemblies. We also performed energy dispersive spectroscopy by SEM to confirm the presence of elements arising from both the metal (Pd) and ligand (1 and 2: C, N, O) components of MOP1 and MOP2 (vide infra, Figure S57 and S58).
Figure 2.

a) Schematic representation of the chemical-responsive release of NR or DOX from the hydrophobic cavity of MOP2 by chemical stimulus with ADA. b & c) Naked eye detection of hydrophobic guest encapsulation and chemical-responsive release (b, DOX; c, NR). d) UV/Vis spectrum (in water) of I) NR (1.0 μM), II) NR@MOP2 ([MOP2] = 6 μM), III) NR@MOP2 ([MOP2] = 6 μM) with 24 equiv. of ADA. e) UV/Vis spectrum (in water) of I) hydrophobic DOX (13 μM), II) DOX@MOP2 ([MOP2] = 75 μM) and III) DOX@MOP2 ([MOP2] = 75 μM) after treatment with 24 equiv. ADA.
Next, we decided to confirm the ability to perform guest exchange processes on MOP1. From the literature it is known that CB[7] binds with moderate affinity to HDA (Ka = 8.97 × 107 M−1 toward CB[7]3b) but that much higher affinities are achieved with adamantane derived guests (e.g. adamantane amine (ADA), Ka = 4.23 × 1012 M−1).3b Accordingly, we expected that ADA would replace HDA bound to MOP1 by a competitive binding process (Figure S39). Experimentally, when we added ADA (24 equiv.) to a solution of MOP1, we observed a large upfield shift of the adamantane protons to 1.23–1.36 ppm in the 1H NMR spectrum (Supporting Information, Figure S22) which indicates that ADA is bound inside CB[7]. Additionally, resonances for uncomplexed HDA now appear in the spectrum (3.0 ppm). We used DOSY NMR to determine the diffusion coefficient of nanocage [Pd12(1•ADA)18(2)6](NO3)42 (D = 6.5 × 10−11 m2/s, Figure S23) which is comparable to that of MOP1 (D = 6.31 × 10−11 m2/s); this establishes that [Pd12(1•ADA)18(2)6](NO3)42 and MOP1 have similar hydrodynamic diameters (Table S1).
Cage Stuffed with Hydrocarbon
After successfully preparing MOP1 and confirming the guest exchange process on MOP1, we turned our attention towards the central nanospace of MOP1 for guest encapsulation and release. Previously, Fujita reported that endohedral covalent alkyl functionalization of a cubooctahedral cage promoted encapsulation of hydrophobic guest molecules.14d,24 We envisioned that we could use CB[7]•guest non-covalent interactions to recruit hydrophobic moieties to the MOP1 assembly. By virtue of the six square and eight triangular apertures of this cubooctahedral MOP, alkyl chains that are non-covalently complexed with the exohedral CB[7] units of the MOP are able to fold back and penetrate the cage to create a self-assembled hydrophobic phase inside the MOP. The non-covalent nature of the CB[7]•guest complexation enables the creation, modification, and destruction of the nanoenvironment inside the MOP using the stimuli responsiveness of CB[7].
Accordingly, we designed and synthesized guest 3 which features a hexanediammonium ion binding domain for CB[7] and a pendant hydrophobic C18H37 chain. MOP2 was prepared by the self-assembly of a mixture of 1, 2, Pd(NO3)2 and 3 (18:6:24:18 molar ratio) in DMSO at 70 °C (Figure 2). The formation of MOP2 was confirmed by 1H and DOSY NMR both in DMSO and in D2O (Figure S24–26). The 1H NMR spectrum of MOP2 clearly exhibited the characteristic down-field shifted and broadened pyridine resonances upon coordination to the Pd2+ metal center (Figure 1e & 1i). Comparison of the diffusion coefficients measured for MOP1 (6.31 × 10−11 m2/s) and MOP2 (7.94 × 10−11 m2/s, Figure 2d) shows that MOP2 diffuses faster than MOP1. The diameter of MOP2 was calculated as ≈ 6.2 nm based on the Stokes-Einstein equation, which is approximately 1.5 nm smaller than MOP1 (Table S1). We believe that this size contraction reflects the accumulation of the C18 alkyl chain inside MOP2 which pulls the CB[7] units toward the surface of the MOP. The total volumes of the cubooctahedral MOP framework as defined by the innermost atoms on the central phenylene units of the ligand is estimated to be 8.2 nm3 (Supporting Information). Considering Rebek’s 55% solution for the packing coefficient in liquids to be an upper limit,25 the available empty volume within MOP2 for hydrocarbon is ≈ 4.5 nm3. Assuming an equal contribution from each alkyl chain, we estimate that 7 C-atoms of each C18 alkyl chain is able to penetrate the MOP cavity to create a densely packed hydrophobic cavity within MOP2. The TEM image of MOP2 shows spherical nanoparticles with an average particle size of ≈ 6.5 nm diameter (Figure S56) which further confirms the size contraction.
Encapsulation and Triggered Release
The hydrophobic cavity created within MOP2 was then utilized to encapsulate the hydrophobic dye nile red (NR).14d,26 Addition of an excess of solid NR to an aqueous solution MOP2 (6.0 μM, 4.0 mL) led to the encapsulation of NR (supporting information) within the hydrophobic cavity of MOP2 as visually observed by the intense color of the solution (Figure 2a,c,d). As a control experiment, we attempted to solubilize NR under identical conditions in the absence of MOP2 but the solution remained nearly colorless. UV/Vis spectroscopy confirmed these visual observations, in particular the strong absorption at 570 nm for NR@MOP2. The bathochromic change in the UV/Vis λmax value for NR inside MOP2 (λmax = 594 nm for uncomplexed NR in water) reflects the reduced polarity of the nanoenvironment inside MOP2 and is in accord with the known solvatochromism of NR.14d Encapsulated NR is expelled from the cavity into bulk solvent upon the reduction of solvent polarity by dilution with CH3CN (Figure S44). UV/Vis spectroscopy of the aq. CH3CN solutions shows an absorption band at 541 which is consistent with that of free NR. The molar extinction coefficient of free NR (38000 M−1 cm−1) in this solvent allowed us to calculate that ≈ 7–8 molecules of NR are encapsulated within each MOP2 assembly (Figure S43 & Table S2).
We next sought to demonstrate the triggered release of the encapsulated NR cargo. For this purpose, we added ADA (24 equiv.) which forms an ultratight complex with CB[7]3b to a solution of NR@MOP2 with the expectation that ADA would outcompete 3 for CB[7] binding, disrupting the non-covalent interactions that promoted the formation of the hydrophobic nanoenvironment inside MOP2 and thereby cause its destruction and the release of NR (Figure 2a). Figure 2c shows pictures of the solution of NR@MOP2 and of the precipitate observed immediately after addition of ADA. The UV/Vis spectrum (Figure 2d) showed a sudden drop of absorption intensity at λ = 580 nm upon addition of ADA to an aqueous solution of NR@MOP2. These results confirm that addition of ADA displaced amphiphilic guest 3 from CB[7] and triggered the cascade that resulted in the release of encapsulated NR dye. We wanted to be sure that the solubilization of NR was not due to the formation of micelles of amphiphile 3 on its own. Accordingly, we measured the critical micellar concentration (CMC) of amphiphile 3 (CMC = 1.5 mM, Figure S54) which is far above the 110 μM concentrations of guest 3 used to create MOP2 in the triggered release experiments described above. The ability of 3 to promote solubilization of NR is clearly tied to its non-covalent complexation to the CB[7] units on the surface of MOP2.
The encapsulation of NR dye inside MOP2 and its release in chemical-responsive fashion, encouraged us to study the encapsulation of the anticancer drug doxorubicin (DOX). To explore the use of MOP2 for the stimuli responsive spatiotemporal controlled release of drug, DOX free base was loaded inside MOP2 following the protocol used for NR encapsulation (Supporting Information).27 Both naked-eye and UV/Vis spectroscopic detection confirmed the encapsulation of DOX within the hydrophobic cavity of MOP2 (Figure 2b,e). DOX@MOP2 ([MOP2] = 75 μM) displayed an intense UV/Vis absorption band at 487 nm whereas hydrophobic DOX in water gave only very weak absorption band (Figure 2e). Addition of a nonpolar solvent like acetonitrile caused the expulsion of DOX from the hydrophobic nanoenvironment to the bulk solution (Figure S48). The UV/Vis absorbance intensity allowed us to calculate that ≈ 8–9 molecules of DOX are encapsulated within MOP2 (Figure S47 & Table S2). Similarly addition of the competitive guest ADA (24 equiv.) triggered the release of DOX from the hydrophobic cavity of DOX@MOP2 as observed by the dramatic decrease in UV/Vis absorbance at 477 nm and naked-eye detection (Figure 2b and 2e). Addition of ADA to concentrated solutions of DOX@MOP2 leads to the precipitation of hydrophobic DOX from water.
After successful encapsulation and triggered release of NR and DOX, we set out to investigate the release kinetics of cargo molecules from the cavity of MOP2. In order to develop a novel nanocarrier for biomedical applications, premature drug releases must be avoided as it is detrimental and can lead to side effects. Accordingly we measured the fluorescence intensity of freshly prepared samples of NR@MOP2 and DOX@MOP2 over 12 h (Figure S50). We did not observe significant changes in fluorescence intensity over time (≤10% release of DOX observed) for both samples (NR@MOP2 and DOX@MOP2) which suggests premature release from MOP2 will not be problematic (Figure 3b,d). However, guest release can be promoted by addition of ADA. The fluorescence emission intensity (at 650 nm) of NR@MOP2 ([MOP2] = 0.6 μM) was quenched upon titration with ADA (14.5 μM) reflecting the chemical responsive release of NR from the hydrophobic cavity of MOP2 (Figure 3a,b). Approximately 90% of NR dye was released from upon addition of 24 equivalents of ADA with respect to 18 equiv. CB[7] in MOP2. Similar results were obtained for DOX@MOP2. The fluorescence emission intensity at 590 nm of an aqueous solution of DOX@MOP2 ([MOP2] = 2.5 μM) decreases significantly upon addition of ADA (60 μM) indicating the release of DOX from the hydrophobic cavity to the aqueous solution (Figure 3c,d). It is calculated that approximately 85% of DOX is released to the bulk solution upon addition of 24 equiv. of ADA. It should be noted that the fluorescence intensity does not change significantly after 18 equiv. of ADA (Figure 3b and 3d) which indicates that an excess of ADA relative to CB[7] is not required.
Figure 3.
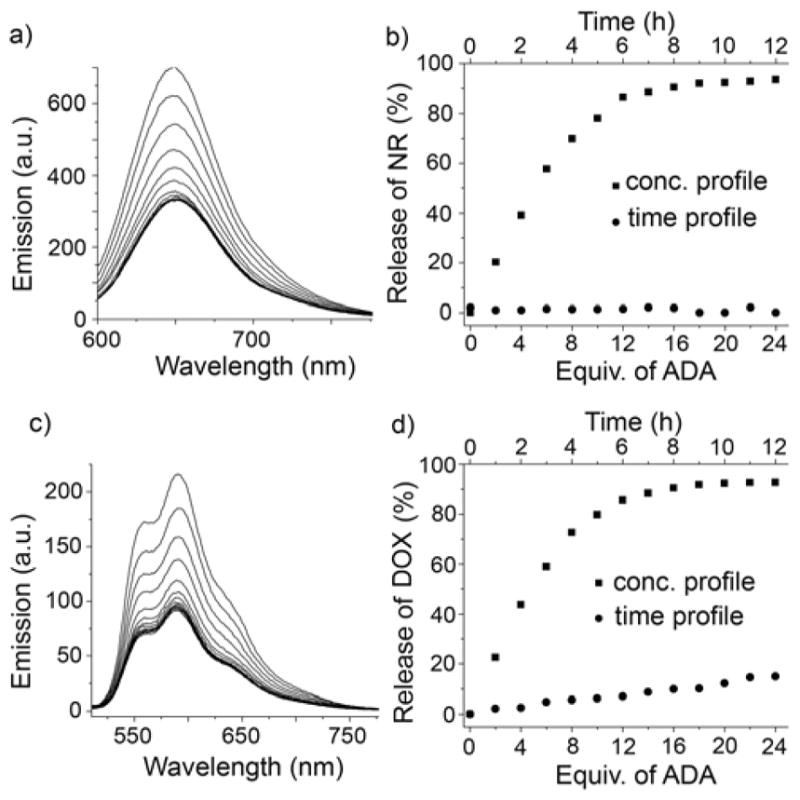
a) Fluorescence titration of NR@MOP2 ([NR] = 5 μM) with ADA (λex = 580 nm) in water. b) Plot of release of NR (%) based on emission intensity at 650 nm for NR@MOP2 in the absence of ADA versus time and separately with increasing concentration of ADA. c) Fluorescence titration of DOX@MOP2 ([DOX] = 20 μM) with ADA (λex = 488 nm) in water. d) Plot of release of DOX (%) based on emission intensity at 590 nm for DOX@MOP2 in the absence of ADA versus time and separately with increasing concentration of ADA.
pH-Chemical and pH-Photochemical Triggered Release
After we had successfully developed the chemical stimuli responsive cargo release from MOP2, we turned our attention toward incorporating pH and photochemical responsiveness release. The slightly acidic environment (pH ≈ 6.0)28 of cancer cells compared to normal cells (pH = 7.4) is often found to be beneficial for designing smart drug delivery vehicles.10a,28b,29 Similarly, light has previously been used for non-invasive cancer photodynamic therapy with spatiotemporal control.10a,10f,30 Accordingly, we thought it would be interesting to demonstrate the triggered release of cargo from the hydrophobic cavity of a MOP by pH-photochemical stimulus. As proof-of-concept the triggered release of NR dye by pH-chemical and pH-photochemical stimuli were investigated (Figure 4).
Figure 4.

Schematic representation of pH-chemical and pH-photochemical responsive release of cargo from the hydrophobic inner phase of MOP3 and MOP4, respectively. Please note the MOP3 and MOP4 differ from MOP2 in that octadecyl butanediammonium 5 is used to create MOP3 and octadecyl pentanediammonium ion 4 is used to create MOP4. The different affinities of 3 – 5 toward CB[7] provide the appropriate thermodynamics to create the pH-chemical and pH-photochemical responsive release.
The design of the pH-chemical and pH-photochemically triggerable systems is based on the large changes in Ka values that are common for CB[n]•guest complexes upon application of appropriate stimuli.3b Model studies (Supporting Information, Figure S34) show that the CB[7]•BDA complex is not perturbed by the presence of adamantane carboxylic acid (ADAc) at pH 7.4 in sodium phosphate buffer (10 mM) because ADAc is present in its anionic carboxylate form.3b,3c However, at pH 5.8, neutral ADAc is able to displace BDA from CB[7] cavity to form the stronger CB[7]•ADAc complex (Figure S33). The longer diammonium ions HDA (Figure S37) or PDA (Figure S35) bind more strongly to CB[7] and cannot be displaced by ADAc at pH 5.8. Accordingly we designed and synthesized guest 5 where BDA is functionalized with a C18 alkyl chain (Scheme 1). Following the reaction protocol used for MOP2, a mixture of 1, 2, Pd(NO3)2, and 5 in 18:6:24:18 ratio afforded MOP3 (Figure 4) which was characterized by 1H NMR and DOSY NMR (Figure S28 & S29). The diffusion coefficient of MOP3 (D = 8.12 × 10−11 m2/s) was similar to that measured for MOP2 (D = 7.94 × 10−11 m2/s) which establishes that they are comparable in diameter (Table S1). The hydrophobic cavity within MOP3 was loaded with NR dye; the loading capacity of MOP3 was ≈ 8–9 NR molecules per molecule of MOP3 (Figure S45 & Table S2) which is comparable to that of MOP2 as expected. The pH-triggered release of NR was monitored by fluorescence spectroscopy (Figure 5b). As a control (Figure S51), an aqueous solution of NR@MOP3 in 10 mM sodium phosphate buffer at pH 7.4 was titrated with ADAc (0 – 36 equiv.). Only a small decrease in fluorescence emission intensity was observed indicating ≤10% NR release from the hydrophobic cavity of MOP3 at pH 7.4 (Figure 5b). However, titration of NR@MOP3 solution with ADAc at pH 5.8 results in a decrease in fluorescence emission intensity at 650 nm indicating the release of NR from the cavity of MOP3 to aqueous solution (Figure 5a). We calculate that 75% of the NR dye is released by the dual pH-chemical stimulus (Figure 5b). Control experiments (Figure 5b & S51) indicated that ≤ 5% encapsulated NR was released from the cavity of MOP3 in the absence of ADAc at pH 5.8 which rules out the possibility of dye leakage and establishes the stability of MOP3 at slightly acidic pH.
Figure 5.
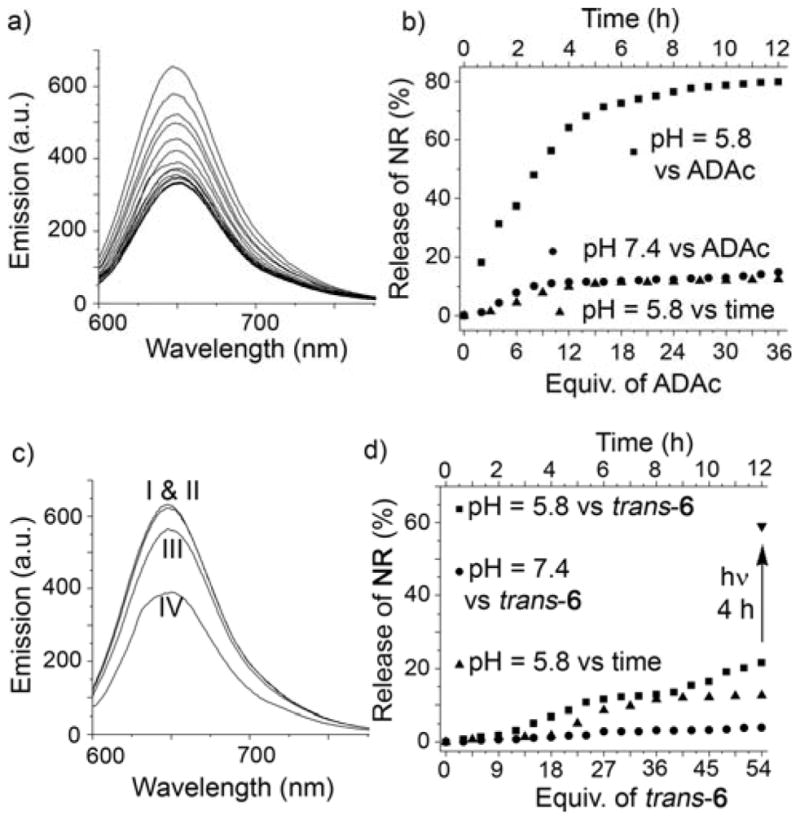
a) Fluorescence titration of NR@MOP3 ([NR] = 5 μM) with ADAc at pH 5.8 (λex = 580 nm) in aqueous acetate buffer (10 mM). b) Plot of NR release from NR@MOP3 ([NR] = 5 μM) versus increasing equiv. of ADAc at pH 5.8 and pH 7.4 and separately versus time at pH 5.8 in the absence of ADAc. c) Fluorescence emission intensity (λex = 580 nm) of NR@MOP4 ([NR] = 5 μM) (I) with trans-6 (54 equiv.) at pH 7.4 (II) or pH 5.8 (III) and after photoirradiation at 350 nm at pH 5.8 for 4 hours (IV). d) Plot of NR release from NR@MOP4 ([NR] = 5 μM) versus trans-6 in pH 7.4 in sodium phosphate buffer (10 mM), in the presence of increasing equiv. of trans-6 at pH 5.8 in sodium acetate (10 mM) buffer, and separately versus time at pH 5.8 in the absence of trans-6.
To promote the release photochemically, we used the well known photoresponsive ligand trans-4,4′-diaminostilbene (trans-6). Kim and co-workers previously demonstrated that photoirradiation converts CB[7]•trans-6 into the tighter CB[7]•cis-6 complex (Figure S37).31 At pH 7.4, both cis-6 and trans-6 are poorly water soluble and do not exhibit any binding toward CB[7]. In model studies (Figure S38), we found that the CB[7]•BDA complex at pH 5.8 fully dissociates upon addition of trans-6 to yield CB[7]•trans-6. In contrast, the CB[7]•HDA complex does not dissociate in the presence of either trans-6 or cis-6 (Figure S41 & S42). We found that PDA which has intermediate affinity for CB[7] was ideal in this case. A 1H NMR model study showed that only 30% of PDA is displaced from CB[7]•PDA by the addition of 3.0 equiv. of trans-6 (Supporting Information Figure S40). Photoirradiation of this mixture at 350 nm for 3h resulted in trans-to-cis isomerization which lead to the dissociation of >90% of the CB[7]•PDA complex (Figure S41) and the concomitant formation of the stronger CB[7]•cis-6 complex. After developing the model system, we designed and synthesized guest 4 (Scheme 1) which features a pentanediammonium ion unit connected to a hydrophobic C18 alkyl chain. The self-assembly of mixture of 1, 2, Pd(NO3)2 and 4 in 18:6:24:18 ratio afforded MOP4 as characterized by 1H and DOSY NMR (D = 7.51 × 10−11 m2/s) (Figure S30 & S31). Similar to MOP2 and MOP3, MOP4 solubilizes NR within its hydrophobic inner phase. We used fluorescence spectroscopy to study the release of NR. As expected based on the model studies, at pH 7.4 (10 mM sodium phosphate buffer) neither trans-6 in the absence of light nor cis-6 under photoirradiation were able to trigger the release of NR from hydrophobic cavity of MOP4 (Figure 5c, 5d & S52). Addition of a large excess of (54 equiv.) of trans-6 to an aqueous solution of NR@MOP4 in 10 mM sodium acetate buffer at pH 5.8 triggered the release of only 15% of NR (Figure 5d & S53). However, after photoirradiation of the mixture at 350 nm for 4 h at pH 5.8, the fluorescence intensity decreases significantly indicating ≈ 60% release of NR from the hydrophobic cavity of MOP4 (Figure 5c,d). In combination, these results demonstrate that the innate chemical, pH, and photochemical responsiveness of the CB[7]•guest complexes can be successfully transferred to far more complex architectures like MOP2 – MOP4 which augurs well for their use as a switch in advanced applications.
In Vitro Assessment of toxicity, uptake and release
After successfully developing stimuli-responsive MOPs for triggered guest release, we next sought to determine the suitability of the MOPs for delivery to eukaryotic cells. First, we aimed to evaluate the toxicity of MOP2. HeLa cells were incubated with increasing concentrations of MOP2 for 24 h (Figure 6a). Afterward a standard MTS assay was used to determine cell viability. MOP2 demonstrated increased toxicity to HeLa cells at concentrations higher than 3.125 μM which is consistent with the reported toxicities of Pd containing molecules against eukaryotic cells. We believe that in addition to delivery of anti-cancer drugs, the sequestration of MOP2 in tumors due to the EPR effect along with its inherent toxicity will increase the effectiveness of the nanocarrier toward cancer cells. Next, we determined the uptake characteristics of MOP2. For this purpose, we treated THP-1 monocytes with NR@MOP2 (6 μM) for 20 min followed by analysis of uptake by flow cytometry (Figure 6b). THP-1 cells treated with equimolar amounts of NR served as a positive control. Incubation of THP-1 cells with NR@MOP2 resulted in increased red fluorescence compared to untreated cells indicating significant uptake of NR@MOP2. THP-1 cells treated with NR showed increased red fluorescence compared to NR@MOP2. This is not surprising given that NR is freely diffusible across eukaryotic membranes whereas NR@MOP2 requires active uptake by the cell. Finally, we analyzed the release of NR from NR@MOP2 after uptake by HeLa cells. Cells were treated with either NR@MOP2 or an equimolar amount of NR for 1 h followed by three washes with phosphate buffered saline and finally counter stained with DAPI to visualize the cell nuclei. Stained cells were then visualized by fluorescence microscopy (Figure 6c,d). Red fluorescence staining patterns in both samples showed punctate staining of lipid bodies in the cytosol as well as diffuse staining of the cellular membrane. The staining patterns seen with NR and NR@MOP2 were identical which indicates that NR undergoes passive release from NR@MOP2 after cellular uptake. Unfortunately, this passive release process prevents us from studying the triggered release of NR intracellularly.
Figure 6.
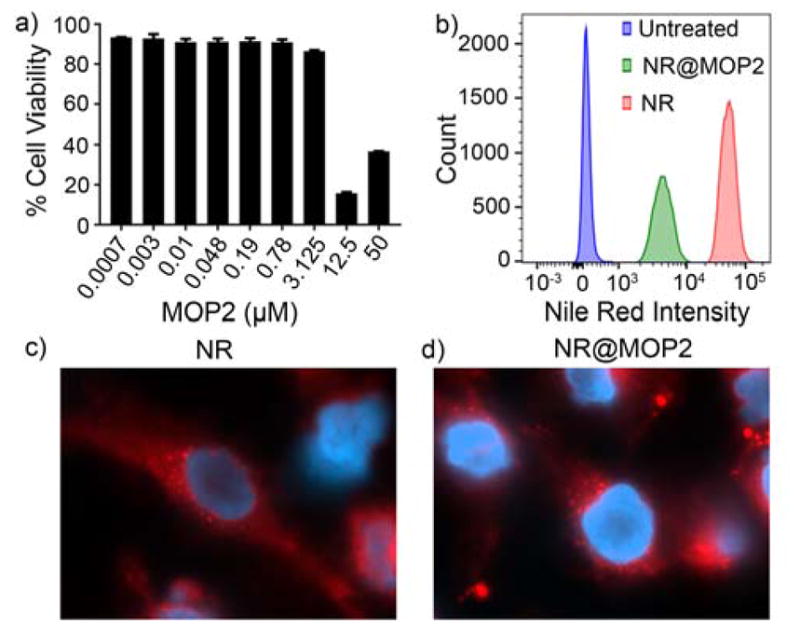
a) Cytotoxicity analysis (MTS assay) of HeLa cells treated with increasing concentration of MOP2 for 24 h. b) Flow cytometry analysis of the uptake of NR@MOP2 by THP-1 cells. The figure is representative of two independent experiments. c) Fluorescence microscopic analysis of HeLa cells stained with NR (6 μM) for 1 hour and counter stained with DAPI (blue). d) Fluorescence microscopy analysis of HeLa cells stained with NR@MOP2 (6 μM) for 1 h and counter stained with DAPI (blue).
Conclusion
In summary, we have presented the use of clickable monofunctionalized CB[7] to prepare bis(pyridine)-CB[7] ligand 1 which undergoes self-assembly with 2 and Pd(NO3)2 to yield MOP1 which bears 18 covalently attached CB[7] groups (diameter ≈ 7.8 nm). Complexation of MOP1 with amphiphilic HDA derivative 3 gives MOP2 whose hydrophobic side chains sequester themselves inside the MOP cavity which results in a size contraction (diameter ≈ 6.2 nm) as characterized by 1H NMR, DOSY and TEM. The hydrophobic inner phase is capable of taking up multiple NR and DOX molecules. The fact that the inner hydrophobic phase of MOP2 is built up by CB[7]•guest interactions renders the entire assembly stimulus responsive. For example, MOP2 was shown to respond to the presence of ADA as a competitive chemical stimulus that results in the dissociation of the CB[7]•3 complex, the destruction of the hydrophobic inner phase and the release of the encapsulated NR or DOX. MOP3 and MOP4 were shown to respond to dual pH-chemical and photo-chemical-chemical stimuli. Finally, we showed that the NR@MOP2 assembly is taken up by HeLa cancer cells and that NR is passively released intracellularly. Overall, the work exemplifies the high architectural complexity that can be achieved using clickable monofunctionalized CB[7] derivatives and the transferability of the stimuli responsiveness of individual CB[7]•guest complexes to create functional uptake and release systems. Looking forward, the multiplicity of CB[7] units on the surface of the MOP1 suggests a variety of plug-and-play non-covalent functionalization schemes to enable diagnostic and therapeutic applications.
Supplementary Material
Acknowledgments
We thank the National Cancer Institute of the National Institutes of Health (CA168365) for financial support. B.V. thanks the University of Maryland for a Millard and Lee Alexander Fellowship.
Footnotes
Supporting Information. Experimental procedures, 1H NMR, 13C NMR, and DOSY NMR spectra, ESI-MS, UV/Vis and fluorescence spectra, TEM images, photographic images, details of the in vitro study, and molecular modeling results.
References
- 1.a) Lagona J, Mukhopadhyay P, Chakrabarti S, Isaacs L. Angew Chem, Int Ed. 2005;44:4844–4870. doi: 10.1002/anie.200460675. [DOI] [PubMed] [Google Scholar]; b) Liu W, Samanta SK, Smith BD, Isaacs L. Chem Soc Rev. 2017;46:2391–2403. doi: 10.1039/c7cs00011a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mock WL. Top Curr Chem. 1995;175:1–24. [Google Scholar]; d) Kim K, Selvapalam N, Ko YH, Park KM, Kim D, Kim J. Chem Soc Rev. 2007;36:267–279. doi: 10.1039/b603088m. [DOI] [PubMed] [Google Scholar]; e) Lee JW, Samal S, Selvapalam N, Kim HJ, Kim K. Acc Chem Res. 2003;36:621–630. doi: 10.1021/ar020254k. [DOI] [PubMed] [Google Scholar]; f) Masson E, Ling X, Joseph R, Kyeremeh-Mensah L, Lu X. RSC Adv. 2012;2:1213–1247. [Google Scholar]; g) Barrow SJ, Kasera S, Rowland MJ, del Barrio J, Scherman OA. Chem Rev. 2015;115:12320–12406. doi: 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]; h) Assaf KI, Nau WM. Chem Soc Rev. 2015;44:394–418. doi: 10.1039/c4cs00273c. [DOI] [PubMed] [Google Scholar]; i) Kaifer AE. Acc Chem Res. 2014;47:2160–2167. doi: 10.1021/ar5001204. [DOI] [PubMed] [Google Scholar]
- 2.a) Ghale G, Nau WM. Acc Chem Res. 2014;47:2150–2159. doi: 10.1021/ar500116d. [DOI] [PubMed] [Google Scholar]; b) Ko YH, Kim E, Hwang I, Kim K. Chem Commun. 2007:1305–1315. doi: 10.1039/b615103e. [DOI] [PubMed] [Google Scholar]; c) Del Barrio J, Horton P, Lairez D, Lloyd G, Toprakcioglu C, Scherman O. J Am Chem Soc. 2013;135:11760–11763. doi: 10.1021/ja406556h. [DOI] [PubMed] [Google Scholar]; d) Benyettou F, Nchimi-Nono K, Jouiad M, Lalatonne Y, Milosevic I, Motte L, Olsen J-C, Saleh Ni, Trabolsi A. Chem - Eur J. 2015;21:4607–4613. doi: 10.1002/chem.201405774. [DOI] [PubMed] [Google Scholar]; e) Chinai JM, Taylor AB, Ryno LM, Hargreaves ND, Morris CA, Hart PJ, Urbach AR. J Am Chem Soc. 2011;133:8810–8813. doi: 10.1021/ja201581x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Shetty D, Khedkar JK, Park KM, Kim K. Chem Soc Rev. 2015;44:8747–8761. doi: 10.1039/c5cs00631g. [DOI] [PubMed] [Google Scholar]; b) Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J Am Chem Soc. 2005;127:15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]; c) Jeon WS, Moon K, Park SH, Chun H, Ko YH, Lee JY, Lee ES, Samal S, Selvapalam N, Rekharsky MV, Sindelar V, Sobransingh D, Inoue Y, Kaifer AE, Kim K. J Am Chem Soc. 2005;127:12984–12989. doi: 10.1021/ja052912c. [DOI] [PubMed] [Google Scholar]; d) Rekharsky MV, Mori T, Yang C, Ko YH, Selvapalam N, Kim H, Sobransingh D, Kaifer AE, Liu S, Isaacs L, Chen W, Moghaddam S, Gilson MK, Kim K, Inoue Y. Proc Natl Acad Sci U S A. 2007;104:20737–20742. doi: 10.1073/pnas.0706407105. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Cao L, Sekutor M, Zavalij PY, Mlinaric-Majerski K, Glaser R, Isaacs L. Angew Chem, Int Ed. 2014;53:988–993. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]
- 4.a) Ma D, Zhang B, Hoffmann U, Sundrup MG, Eikermann M, Isaacs L. Angew Chem, Int Ed. 2012;51:11358–11362. doi: 10.1002/anie.201206031. [DOI] [PubMed] [Google Scholar]; b) Ma D, Hettiarachchi G, Nguyen D, Zhang B, Wittenberg JB, Zavalij PY, Briken V, Isaacs L. Nat Chem. 2012;4:503–510. doi: 10.1038/nchem.1326. [DOI] [PubMed] [Google Scholar]; c) Dong N, Xue SF, Zhu QJ, Tao Z, Zhao Y, Yang LX. Supramol Chem. 2008;20:659–665. [Google Scholar]; d) Zhao Y, Buck DP, Morris DL, Pourgholami MH, Day AI, Collins JG. Org Biomol Chem. 2008;6:4509–4515. doi: 10.1039/b813759e. [DOI] [PubMed] [Google Scholar]; e) Walker S, Oun R, McInnes FJ, Wheate NJ. Isr J Chem. 2011;51:616–624. [Google Scholar]; f) Chen H, Y-W, CJ, Li S, Liu JJ, Wyman I, Lee SM-Y, Macartney DH, Wang R. RSC Adv. 2015;5:63745–63752. [Google Scholar]
- 5.a) Kim C, Agasti SS, Zhu Z, Isaacs L, Rotello VM. Nat Chem. 2010;2:962–966. doi: 10.1038/nchem.858. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tonga GY, Jeong Y, Duncan B, Mizuhara T, Mout R, Das R, Kim ST, Yeh YC, Yan B, Hou S, Rotello VM. Nat Chem. 2015;7:597–603. doi: 10.1038/nchem.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Wu Z, Song N, Menz R, Pingali B, Yang YW, Zheng Y. Nanomedicine. 2015;10:1493–1514. doi: 10.2217/nnm.15.1. [DOI] [PubMed] [Google Scholar]; b) Ambrogio MW, Thomas CR, Zhao YL, Zink JI, Stoddart JF. Acc Chem Res. 2011;44:903–913. doi: 10.1021/ar200018x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Thomas CR, Ferris DP, Lee JH, Choi E, Cho MH, Kim ES, Stoddart JF, Shin JS, Cheon J, Zink JI. J Am Chem Soc. 2010;132:10623–10625. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]
- 7.a) Jon SY, Selvapalam N, Oh DH, Kang JK, Kim SY, Jeon YJ, Lee JW, Kim K. J Am Chem Soc. 2003;125:10186–10187. doi: 10.1021/ja036536c. [DOI] [PubMed] [Google Scholar]; b) Vinciguerra B, Cao L, Cannon JR, Zavalij PY, Fenselau C, Isaacs L. J Am Chem Soc. 2012;134:13133–13140. doi: 10.1021/ja3058502. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ahn Y, Jang Y, Selvapalam N, Yun G, Kim K. Angew Chem, Int Ed. 2013;52:3140–3144. doi: 10.1002/anie.201209382. [DOI] [PubMed] [Google Scholar]
- 8.a) Webber MJ, Appel EA, Vinciguerra B, Cortinas AB, Thapa LS, Jhunjhunwala S, Isaacs L, Langer R, Anderson DG. Proc Natl Acad Sci U S A. 2016;113:14189–14194. doi: 10.1073/pnas.1616639113. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bockus AT, Smith LC, Grice AG, Ali OA, Young CC, Mobley W, Leek A, Roberts JL, Vinciguerra B, Isaacs L, Urbach AR. J Am Chem Soc. 2016;138:16549–16552. doi: 10.1021/jacs.6b11140. [DOI] [PubMed] [Google Scholar]; c) Li W, Bockus AT, Vinciguerra B, Isaacs L, Urbach AR. Chem Commun. 2016;52:8537–8540. doi: 10.1039/c6cc03193e. [DOI] [PubMed] [Google Scholar]; d) Lee DW, Park KM, Banerjee M, Ha SH, Lee T, Suh K, Paul S, Jung H, Kim J, Selvapalam N, Ryu SH, Kim K. Nat Chem. 2011;3:154–159. doi: 10.1038/nchem.928. [DOI] [PubMed] [Google Scholar]; e) Murray J, Sim J, Oh K, Sung G, Lee A, Shrinidhi A, Thirunarayanan A, Shetty D, Kim K. Angew Chem, Int Ed. 2017;56:2395–2398. doi: 10.1002/anie.201611894. [DOI] [PubMed] [Google Scholar]; f) Gong B, Choi BK, Kim JY, Shetty D, Ko YH, Selvapalam N, Lee NK, Kim K. J Am Chem Soc. 2015;137:8908–8911. doi: 10.1021/jacs.5b05385. [DOI] [PubMed] [Google Scholar]
- 9.a) Fang J, Nakamura H, Maeda H. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]; b) Longmire M, Choyke PL, Kobayashi H. Nanomedicine. 2008;3:703–717. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Mura S, Nicolas J, Couvreur P. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]; b) Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Science. 1978;202:1290. doi: 10.1126/science.364652. [DOI] [PubMed] [Google Scholar]; c) Chen KJ, Liang HF, Chen HL, Wang Y, Cheng PY, Liu HL, Xia Y, Sung HW. ACS Nano. 2013;7:438–446. doi: 10.1021/nn304474j. [DOI] [PubMed] [Google Scholar]; d) Ruiz-Hernández E, Baeza A, Vallet-Regí M. ACS Nano. 2011;5:1259–1266. doi: 10.1021/nn1029229. [DOI] [PubMed] [Google Scholar]; e) Rapoport NY, Kennedy AM, Shea JE, Scaife CL, Nam KH. J Control Release. 2009;138:268–276. doi: 10.1016/j.jconrel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Schroeder A, Goldberg MS, Kastrup C, Wang Y, Jiang S, Joseph BJ, Levins CG, Kannan ST, Langer R, Anderson DG. Nano Lett. 2012;12:2685–2689. doi: 10.1021/nl2036047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 12.a) McConnell AJ, Wood CS, Neelakandan PP, Nitschke JR. Chem Rev. 2015;115:7729–7793. doi: 10.1021/cr500632f. [DOI] [PubMed] [Google Scholar]; b) Tranchemontagne DJ, Mendoza-Cortes JL, O’Keeffe M, Yaghi OM. Chem Soc Rev. 2009;38:1257–1283. doi: 10.1039/b817735j. [DOI] [PubMed] [Google Scholar]; c) Kumari H, Deakyne CA, Atwood JL. Acc Chem Res. 2014;47:3080–3088. doi: 10.1021/ar500222w. [DOI] [PubMed] [Google Scholar]; d) Brown CJ, Toste FD, Bergman RG, Raymond KN. Chem Rev. 2015;115:3012–3035. doi: 10.1021/cr4001226. [DOI] [PubMed] [Google Scholar]; e) Harris K, Fujita D, Fujita M. Chem Commun. 2013;49:6703–6712. doi: 10.1039/c3cc43191f. [DOI] [PubMed] [Google Scholar]; f) Lehn JM. Chem Soc Rev. 2007;36:151–160. doi: 10.1039/b616752g. [DOI] [PubMed] [Google Scholar]; g) Samanta SK, Rana A, Schmittel M. Angew Chem, Int Ed. 2016;55:2267–2272. doi: 10.1002/anie.201509108. [DOI] [PubMed] [Google Scholar]; h) Clever GH, Tashiro S, Shionoya M. J Am Chem Soc. 2010;132:9973–9975. doi: 10.1021/ja103620z. [DOI] [PubMed] [Google Scholar]; i) Holloway LR, McGarraugh HH, Young MC, Sontising W, Beran GJO, Hooley RJ. Chem Sci. 2016;7:4423–4427. doi: 10.1039/c6sc01038e. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Wang W, Wang YX, Yang HB. Chem Soc Rev. 2016;45:2656–2693. doi: 10.1039/c5cs00301f. [DOI] [PubMed] [Google Scholar]; k) Kishi N, Li Z, Yoza K, Akita M, Yoshizawa M. J Am Chem Soc. 2011;133:11438–11441. doi: 10.1021/ja2037029. [DOI] [PubMed] [Google Scholar]; l) Yazaki K, Noda S, Tanaka Y, Sei Y, Akita M, Yoshizawa M. Angew Chem Int Ed. 2016;55:15031–15034. doi: 10.1002/anie.201608350. [DOI] [PubMed] [Google Scholar]; m) Johnson AM, Wiley CA, Young MC, Zhang X, Lyon Y, Julian RR, Hooley RJ. Angew Chem, Int Ed. 2015;54:5641–5645. doi: 10.1002/anie.201500400. [DOI] [PubMed] [Google Scholar]; n) Carnes ME, Collins MS, Johnson DW. Chem Soc Rev. 2014;43:1825–1834. doi: 10.1039/c3cs60349k. [DOI] [PubMed] [Google Scholar]
- 13.a) Fujita D, Ueda Y, Sato S, Mizuno N, Kumasaka T, Fujita M. Nature. 2016;540:563–566. doi: 10.1038/nature20771. [DOI] [PubMed] [Google Scholar]; b) Sun QF, Iwasa J, Ogawa D, Ishido Y, Sato S, Ozeki T, Sei Y, Yamaguchi K, Fujita M. Science. 2010;328:1144–1147. doi: 10.1126/science.1188605. [DOI] [PubMed] [Google Scholar]; c) Tominaga M, Suzuki K, Kawano M, Kusukawa T, Ozeki T, Shakamoto S, Yamaguchi K, Fujita M. Angew Chem, Int Ed. 2004;43:5621–5625. doi: 10.1002/anie.200461422. [DOI] [PubMed] [Google Scholar]
- 14.a) Bruns CJ, Fujita D, Hoshino M, Sato S, Stoddart JF, Fujita M. J Am Chem Soc. 2014;136:12027–12034. doi: 10.1021/ja505296e. [DOI] [PubMed] [Google Scholar]; b) Sato S, Ikemi M, Kikuchi T, Matsumura S, Shiba K, Fujita M. J Am Chem Soc. 2015;137:12890–12896. doi: 10.1021/jacs.5b06184. [DOI] [PubMed] [Google Scholar]; c) Sato S, Iida J, Suzuki K, Kawano M, Ozeki T, Fujita M. Science. 2006;313:1273–1276. doi: 10.1126/science.1129830. [DOI] [PubMed] [Google Scholar]; d) Suzuki K, Iida J, Sato S, Kawano M, Fujita M. Angew Chem, Int Ed. 2008;47:5780–5782. doi: 10.1002/anie.200801700. [DOI] [PubMed] [Google Scholar]
- 15.a) Zheng YR, Suntharalingam K, Johnstone TC, Lippard SJ. Chem Sci. 2015;6:1189–1193. doi: 10.1039/c4sc01892c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yi JW, Barry NPE, Furrer MA, Zava O, Dyson PJ, Therrien B, Kim BH. Bioconjugate Chem. 2012;23:461–471. doi: 10.1021/bc200472n. [DOI] [PubMed] [Google Scholar]; c) He C, Lu K, Liu D, Lin W. J Am Chem Soc. 2014;136:5181–5184. doi: 10.1021/ja4098862. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cook TR, Vajpayee V, Lee MH, Stang PJ, Chi KW. Acc Chem Res. 2013;46:2464–2474. doi: 10.1021/ar400010v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitt F, Freudenreich J, Barry NPE, Juillerat-Jeanneret L, Suss-Fink G, Therrien B. J Am Chem Soc. 2012;134:754–757. doi: 10.1021/ja207784t. [DOI] [PubMed] [Google Scholar]
- 17.Zhang M, Li S, Yan X, Zhou Z, Saha ML, Wang YC, Stang PJ. Proc Natl Acad Sci U S A. 2016;113:11100–11105. doi: 10.1073/pnas.1612898113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez J, Mosquera J, Couceiro JR, Nitschke JR, Vazquez ME, Mascarenas JL. J Am Chem Soc. 2017;139:55–58. doi: 10.1021/jacs.6b11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samanta SK, Moncelet D, Briken V, Isaacs L. J Am Chem Soc. 2016;138:14488–14496. doi: 10.1021/jacs.6b09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samanta SK, Brady KG, Isaacs L. Chem Commun. 2017;53:2756–2759. doi: 10.1039/c6cc10328f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Özçubukçu S, Ozkal E, Jimeno C, Pericàs MA. Org Lett. 2009;11:4680–4683. doi: 10.1021/ol9018776. [DOI] [PubMed] [Google Scholar]
- 22.Gramage-Doria R, Hessels J, Leenders SHAM, Troeppner O, Duerr M, Ivanovic-Burmazovic I, Reek JNH. Angew Chem, Int Ed. 2014;53:13380–13384. doi: 10.1002/anie.201406415. [DOI] [PubMed] [Google Scholar]
- 23.Ni XL, Xiao X, Cong H, Liang LL, Cheng K, Cheng XJ, Ji NN, Zhu QJ, Xue SF, Tao Z. Chem Soc Rev. 2013;42:9480–9508. doi: 10.1039/c3cs60261c. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki K, Takao K, Sato S, Fujita M. J Am Chem Soc. 2010;132:2544–2545. doi: 10.1021/ja910836h. [DOI] [PubMed] [Google Scholar]
- 25.Mecozzi S, Rebek JJ. Chem Eur J. 1998;4:1016–1022. [Google Scholar]
- 26.Gillies ER, Jonsson TB, Fréchet JMJ. J Am Chem Soc. 2004;126:11936–11943. doi: 10.1021/ja0463738. [DOI] [PubMed] [Google Scholar]
- 27.Chi X, Yu G, Shao L, Chen J, Huang F. J Am Chem Soc. 2016;138:3168–3174. doi: 10.1021/jacs.5b13173. [DOI] [PubMed] [Google Scholar]
- 28.a) Gatenby RA, Gillies RJ. Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]; b) Mizuhara T, Saha K, Moyano DF, Kim CS, Yan B, Kim YK, Rotello VM. Angew Chem, Int Ed. 2015;54:6567–6570. doi: 10.1002/anie.201411615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helmlinger G, Yuan F, Dellian M, Jain RK. Nature Medicine. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 30.Lerch MM, Hansen MJ, van Dam GM, Szymanski W, Feringa BL. Angew Chem, Int Ed. 2016;55:10978–10999. doi: 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]
- 31.Choi S, Park SH, Ziganshina AY, Ko YH, Lee JW, Kim K. Chem Commun. 2003:2176–2177. doi: 10.1039/b306832c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.