

Abstract
Purpose
Second malignant neoplasms (SMNs) are severe late complications that occur in pediatric cancer survivors exposed to radiotherapy and other genotoxic treatments. To characterize the mutational landscape of treatment-induced sarcomas and to identify candidate SMN-predisposing variants we analyzed germline and SMN samples from pediatric cancer survivors.
Experimental Design
We performed whole exome sequencing (WES) and RNA sequencing on radiation-induced sarcomas arising from two pediatric cancer survivors. To assess the frequency of germline TP53 variants in SMNs, Sanger sequencing was performed to analyze germline TP53 in thirty-seven pediatric cancer survivors from the Childhood Cancer Survivor Study (CCSS) without history of a familial cancer predisposition syndrome but known to have developed SMNs.
Results
WES revealed TP53 mutations involving p53’s DNA binding domain in both index cases, one of which was also present in the germline. The germline and somatic TP53 mutant variants were enriched in the transcriptomes for both sarcomas. Analysis of TP53 coding exons in germline specimens from the CCSS survivor cohort identified a G215C variant encoding an R72P amino acid substitution in six patients and a synonymous single nucleotide polymorphism A639G in four others, resulting in ten out of 37 evaluable patients (27%) harboring a germline TP53 variant.
Conclusions
Currently, germline TP53 is not routinely assessed in pediatric cancer patients. These data support the concept that identifying germline TP53 variants at the time a primary cancer is diagnosed may identify patients at high risk for SMN development, who could benefit from modified therapeutic strategies and/or intensive post-treatment monitoring.
INTRODUCTION
Epidemiologic data indicate that there will be 500,000 survivors of pediatric cancers in the United States by 2020(2). Long term follow up of the expanding population of pediatric cancer survivors have identified substantial late toxicities resulting from intensive treatment regimens (3–7). Second malignant neoplasms (SMNs) arise after exposure to genotoxic therapies, which include radiotherapy and some chemotherapeutic agents. Sarcomas were the first histologic cancer type recognized after clinical radiotherapy (8), and continue to arise as SMNs in pediatric cancer survivors treated with modern regimens (7, 9). Compared to their de novo counterparts, SMNs are often clinically more aggressive, more difficult to treat, and associated with worse outcomes (4, 5, 10). This risk is elevated 35 years after treatment and appears to persist in patients treated in the modern era (11). Whereas epidemiologic studies have identified exposure to radiation and young age at time of therapy as major risk factors, the molecular basis for SMN development remains poorly understood.
To characterize the mutational landscape of treatment-induced sarcomas and to identify candidate predisposing germline and somatic variants, we performed whole exome sequencing (WES) of radiation-induced sarcoma SMNs and matched germline control DNA from two pediatric cancer survivors. RNA sequencing (RNA-Seq) validated WES-identified variants at the transcriptomic level. Both SMNs exhibited a similar pattern of base substitutions, and contained mutations in the tumor suppressor gene TP53, which was somatic in one patient and present in the germline of the other. Loss of normal TP53 transcripts and a corresponding enrichment of the variant TP53 transcripts occurred in both SMNs, highlighting the functional importance of these mutant p53 proteins. Analysis of a separate cohort of pediatric cancer survivors developing SMNs identified recurrent germline polymorphisms involving the DNA binding and proline rich domains of the p53 protein. These findings implicate exonic TP53 variants as a risk factor for SMN formation and suggest a strategy for identifying pediatric cancer survivors at high risk for developing SMNs.
MATERIALS AND METHODS
Human Research Protection
All work was performed under a research protocol approved by the UCSF Committee on Human Research (IRB protocol 11-07304).
Microscopy
Pathologic review was performed on hematoxylin and eosin-stained (H&E) sections by A.H. Photographs of histology were taken with an Olympus BX41 microscope, using Olympus UplanFl 10X/0.3 and 20X/0.5 objectives. An attached Olympus DP72 camera and Adobe Photoshop CS2 were used to capture the images.
Sample Preparations
The two SMNs analyzed by whole exome and transcriptome sequencing were isolated at UCSF. Genomic DNA was isolated from fresh frozen tumor samples and fresh peripheral blood samples using previously described techniques (12). RNA was isolated from fresh frozen tumor samples using the RNeasy RNA Isolation kit (Qiagen). H&E-stained sections of formalin fixed or fresh frozen tumor samples were reviewed with a pathologist (A.H.), who estimated greater than 90% tumor cell representation in the tumor samples.
Whole exome sequencing
Whole exome sequencing was performed using the NimbleGen Human exome v3.0 kit. Captured material was indexed and sequenced on the Illumina GAII and HiSeq2000 platform at the Institute for Human Genetics at UCSF. Successfully sequenced reads were then mapped to the human reference genome (GRCh37) using GATK best practices and somatic mutations were called using MuTect and Pindel as previously described (13). Annovar was used for variant annotation and to filter known human mutations to produce a cleaner list of variants (Supplementary Files 1 and 2). Mutation Assessor, SIFT, and PROVEAN were used to assign functional predictions. Validation of a subset of somatic variants by Sanger sequencing confirmed 92% of SNVs (23 of 25 tested).
RNA sequencing
cDNA was synthesized from RNA, followed by library preparation using the Illumina TruSeq Stranded kit with Ribodepletion-Gold (Illumina, San Diego, CA) according to the manufacturer’s instructions. 100bp paired end sequencing was performed on a HiSeq4000. Reads were aligned using Bowtie (14) and Tophat2-Fusion, (15) which included analysis for fusion transcripts. Only fusions with >100 paired-end reads supporting the mapping were considered. Those with <5 individual reads spanning the fusion, or those with >10 reads contradicting the fusion were also removed. Tophat-fusion-post was used to filter out false fusions due to highly similar sequences or pseudogenes. The 50 base pairs on the left side of a fusion and the 50 base pairs on the right side are combined, and then BLASTed (16) against the human blast database. If match length (range: 0 to 100) + identity percent (0 to 100) is greater than 160, the fusion was filtered out – which resulted in no predicted fusions being retained for either patient. Expression levels were estimated using Cufflinks. (17) TP53 expression for normal muscle control was obtained from the Genotype-Tissue Expression (GTEx) project (18).
Sanger Sequencing
Sanger sequencing was performed on exons 2 – 11 of TP53 (detailed in Supplementary File 4, with primer sequences listed in Supplementary File 5) in the germline DNA of CCSS samples.
Copy number analysis
Using the exome sequencing data we identified regions of copy number variation (CNV) in tumor DNAs relative to the matched normal using Control-FREEC with default parameters (19). Analysis was restricted to the region selected by the Human exome v3.0 capture kit (NimbleGen). CNVs were smoothed into 2.5 kb segments. Significance of Control-FREEC predictions were assessed by assigning Wilcoxon test and Kolmogorov-Smirnov test p-values in R.
Loss of heterozygosity analysis
Single nucleotide polymorphisms (SNPs) were called using UnifiedGenotyper (20). Only those that were heterozygous in the germline, present in dbSNP (build 132) and had 10 or more spanning reads were selected for analysis. Allelic frequency in the tumor, as represented by the proportion of reads supporting the A and B alleles, were plotted across chromosomes. Regions demonstrating loss of heterozygosity are represented by migration of the allele frequencies away from 0.5, which would represent approximately equal read depth of the maternal and paternal alleles.
Identification and classification of driver mutations
All somatic variants were compared against a list of potential driver genes (n=573) comprised of all genes identified in the COSMIC cancer gene census (October 2015)(21). Using the criteria outlined by Murugaesu et al. (22) we categorized variants as Category 1 (high confidence driver mutation), Category 2 (putative driver mutation), Category 3 (low confidence driver mutation) or Category 4 (representing a variant of unknown significance).
Statistical Analysis
Fisher’s Exact Test was used to determine whether the number of A639G substitutions in the SMN sample (4 mutations in 37 tissue samples for which chemical reactions succeeded out of 41 samples total) was higher than the frequencies observed in the 1000 Genomes Project (5 mutations in 1000 genomes sequenced). A one-sided p-value of 0.00015 was obtained by direct calculation using standard Microsoft Excel functions.
RESULTS
Clinical case histories
Patient 1 was an 11-year-old male with a pelvic embryonal rhabdomyosarcoma who was treated with chemotherapy and external beam radiotherapy as prescribed by Children’s Oncology Group protocol D9803(23). Radiotherapy to the bladder delivered 41.4 Gy in 23 fractions (Supplementary Fig. 1A). Nine years after his initial therapy, an MRI demonstrated a new 6 cm×6 cm mass in the right pubic component of the acetabulum (Supplementary Fig. 1B). A biopsy of the mass revealed pathology consistent with a high grade (3/3) chondroblastic osteosarcoma (Supplementary Fig. 1C).
Patient 2 was a 17-year-old male when first diagnosed with chondroblastic osteosarcoma of the pelvis and treated with neoadjuvant chemotherapy followed by resection. Adjuvant intensity-modulated radiotherapy delivered 60 Gy in 25 fractions to the right pubic ramus (Supplementary Fig. 1D). Seven years after the initial diagnosis, he developed a right lateral thigh mass within the previously irradiated volume (Supplementary Fig. 1E). Resection of this tumor revealed pathology consistent with a grade 3/3 deep pleomorphic sarcoma (Supplementary Fig. 1F).
DNA and RNA isolated from freshly frozen, unfixed tumor samples of both patients were analyzed by WES and RNA sequencing, respectively. Freshly collected peripheral blood from each patient served as germline control and was analyzed by WES. While tumor material received from Patient 1 was insufficient for intra-tumor heterogeneity to be investigated, Patient 2’s tumor sample was sufficiently large enough to enable sequencing of two physically separate regions (termed ‘a’ and ‘b’).
TP53 Mutations in SMN Specimens
TP53 is a tumor suppressor gene that defines the Li-Fraumeni tumor predisposition syndrome and also is commonly mutated in human malignancies (24, 25). WES of germline and tumor DNA from Patient 1 identified a somatic TP53 mutation (rs28934574, c.844C>T, p.Arg282Trp) (Fig. 1A). This mutation involves a recognized “hot spot” residue in p53’s DNA binding domain, and is among the most common TP53 mutations found in human cancers (26). Germline analysis of Patient 1 revealed no deleterious variants or mutations. In addition, there was no history of malignancy in Patient 1’s parents, sibling or grandparents, and thus no genetic testing was performed for his parents.
Figure 1. Somatic and germline mutations in TP53.

A. Sanger sequencing validation of a heterozygous somatic mutation identified by WES in the TP53 gene of Patient 1’s SMN. WES-derived read counts supporting each SNV are shown. B. Sanger sequencing validation of a heterozygous germline mutation identified by WES in the TP53 gene of Patient 2 that has been previously associated with de-novo Li-Fraumeni Syndrome (rs11540652, c.743G>A, p.Arg248Gln). WES-derived read counts supporting each SNV are shown. C/D. The top panels show genome wide copy number alterations for Patient 1 and Patient 2. Regions of normal copy number (ploidy = 2) are plotted in green, regions showing gain in red, and loss in blue. Segments of copy number change that were statistically significant from normal ploidy (Wilcoxon signed-rank test) are indicated by a line of the corresponding color (red indicates gain, blue indicates loss) either above or below the plot. Allelic ratio data of germline heterozygous loci are displayed in the lower panels. Variants in a normal genome would be centered at 0.5, which represents the presence of two alleles. Any deviation from 0.5 indicates allelic imbalance or loss of heterozygosity. Chromosomes are displayed consecutively, beginning with chromosome 1 at the far left. E. Copy number and allele frequency plots of chromosome 17 for Patient 1, highlighting the TP53 locus. F. Copy number and allele frequency plots of chromosome 17 for Patient 2, highlighting the TP53 locus.
By contrast, Patient 2 harbored a heterozygous germline mutation in the TP53 gene that has been previously associated with Li-Fraumeni Syndrome (rs11540652, c.743G>A, p.Arg248Gln) (Fig. 1B) (27, 28). This non-synonymous germline variant, which was also confirmed in a CLIA-approved laboratory, occurs in exon 7 and is classified as a Class I functional mutation (29) involving a residue that contacts the minor groove of the DNA helix. These types of pathogenic mutations are distinguished from Class II mutations, which destabilize the structural integrity of the DNA binding domain. Both exomes sequenced from Patient 2’s SMN (referred to as samples ‘a’ and ‘b’) showed enrichment of the germline TP53 mutation, as evidenced qualitatively by Sanger sequencing chromatograms and quantitatively by read count (Fig. 1B). Importantly, Patient 2 was not suspected of having a familial cancer predisposition syndrome at initial diagnosis and had not been referred for genetic evaluation before the SMN diagnosis. There was no history of malignancy in Patient 2’s parents or siblings, and no genetic testing was performed for his parents.
Somatic variants in radiation-induced SMNs
Deep WES was performed for SMNs (147-fold mean exome coverage, with 99% of the exome covered by ≥ 10 reads, and 90% covered by ≥ 50 reads in all samples - Supplementary Fig. 2). A total of 202 somatic single nucleotide variants (SNVs) were identified Patient 1’s sarcoma SMN (including 41 non-synonymous and 8 small indels – Supplementary File 1 and Supplementary Fig. 3). In Patient 2, the SMN demonstrated a total of 268 somatic SNVs (102 SNVs private to sample A, 55 SNVs private to sample B, and 111 SNVs present in both tumor samples). Sixteen indels were identified, including 8 and 5 that were unique to samples a and b, respectively, and two that were present in both (Supplementary File 2). SNVs were characterized by a predominance of C → T and G → A substitutions (Supplementary Fig. 3). Sixteen dinucleotide substitutions were identified across all tumor exomes, of which 10 were unique (Supplementary File 3).
Non-synonymous SNVs alter coding sequences and may have diverse consequences. Annovar software (30) was used annotate variants and estimate the functional impact of non-synonymous somatic variants arising in the sarcoma specimens. We categorized non-synonymous SNVs according to their likelihood of being a driver mutation as previously described (Materials and Methods and (22)). In addition to the high confidence TP53 driver mutations described above, Patient 2 also had a putative driver mutation (Category 2) in NCOA2, a gene encoding a transcriptional coactivator for nuclear receptors (31) known to be altered in sarcomas (32). Only one somatic variant from Patient 1's tumor was annotated to a gene in COSMIC (AFF3), and this variant was predicted to be functionally neutral.
Copy Number/Loss of Heterozygosity
Both SMN exomes were characterized by copy number variations (Fig. 1C/D and Supplementary Fig. 4). Each of the SMNs analyzed harbored large-scale (>25% of chromosome arm) alterations and focal chromosomal changes (Supplementary Files 1 and 2). Numerous copy number losses occurred on chromosomes 4, 5, 6, 7, 13, 16, and 17 in both sarcomas.
We also examined SMNs for loss of heterozygosity (LOH), defined as heterozygote allele frequencies deviating from 0.5. Large scale LOH (100 kb or greater) was present throughout the exomes of both patients, consistent with genomic instability (Fig. 1C/D and Supplementary Fig. 4). LOH frequently co-occurred with normal copy number profiles, indicating acquired uniparental disomy (UPD), also termed copy-neutral LOH. One such region encompasses the TP53 locus, located on the p arm of chromosome 17 (Fig. 1E/F). Interrogating the read depth at this position confirmed that in both patients the wildtype allele of TP53 had been lost and the variant allele duplicated.
We analyzed copy number changes involving genes described in the COSMIC cancer gene census (October 2015)(21), which likely contribute to tumorigenesis. Patient 1 had 216 genes affected by copy number change that were listed as associated with cancer in the COSMIC database. A large proportion of these (n = 53) were due to the gain of the entire length of chromosome 1. For Patient 2, there were 275 genes in the COSMIC database affected by copy number change, due in a large part to gains in chromosomes 1 (n = 39), 9 (n = 27), 17 (n = 27) and 19 (n = 28) (Supplementary Files 1 and 2).
Transcriptome analysis
Transcriptome sequencing revealed differential TP53 expression in both SMNs, with mutant TP53 highly overexpressed relative to normal skeletal muscle (Fig. 2A). Expression of mutant TP53 predominated in Patient 1’s SMN, with 96% of reads (105 out of 109 total reads) supporting the TP53 mutation. Similarly, in Patient 2’s SMN expression of the mutant germline TP53 dominated, supported by 95% of the reads covering this position (407 out of 430 total reads). We observed a significant preponderance of two transcript isoforms producing truncated p53 proteins with a shorter N-terminus region compared to the canonical tumor suppressor p53α, which is the most abundant isoform in normal tissues (33) (Fig. 2B/C). Patient 2’s SMN demonstrated increased expression of transcript isoform Δ40p53α (9.541 and 2.284 FPKM for Patients 2 and 1 respectively), whereas the Δ133p53α isoform was more abundant in Patient 1 (7.256 FPKM) compared to Patient 2 (2.943 FPKM) (Fig. 2B). Variant 1 of the Δ40p53α isoform is translated from the downstream in-frame start codon and lacks the first transactivation domain, impairing p53-mediated growth suppression (34) (GenBank accession: NM_001276760; Fig. 2C). Variant 5 of the Δ133p53α isoform is transcribed from an internal promoter and thus lacks all transactivation and part of DNA-binding domains. This variant stimulates angiogenesis and tumor progression in a p53α-dependent and independent manner when overexpressed (35, 36) (NM_001126115; Fig. 2C).
Figure 2. Mutant TP53 expression and isoforms.

A. TP53 gene expression levels quantified in FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) for Patient 1 (blue), Patient 2 (green), and normal skeletal tissue (grey, dbGAP accession number: phs000424.v6.p1; FPKM=4.3; n=148 samples). Each expression value is annotated with error bars reflecting the uncertainty in assigning reads to transcripts (estimated by Cufflinks statistical model), and indicating the lower and upper bound of the 95% confidence interval on the gene FPKM value. B. Expression levels of TP53 transcript isoforms between the two patients and normal skeletal muscle (ND, not detected): Δ40p53α (variants 1 and 8; GeneBank accession numbers NM_001276760 and NM_001126118 respectively), p53α (variant 2; NM_001126112), p53β (variant 3; NM_001126114), p53ɣ (variant 4; NM_001126113), Δ133p53α (variant 5; NM_001126115), Δ133p53β (variant 6; NM_001126116) and Δ133p53ɣ (variant 7; NM_001126117). C. Structural organization of the canonical p53 protein and its isoforms (from N- to C-termini: transactivation domains 1 and 2, proline-rich domain, DNA binding domain, nuclear localization domain, oligomerization domain and negative regulation domain).
Germline TP53 polymorphisms in pediatric cancer survivors developing SMNs
While the development of an SMN raises concern for a genetic susceptibility to tumorigenesis, most of these cancers arise in individuals without a known familial predisposition syndrome. To determine whether germline TP53 variants are present at higher frequencies in pediatric cancer survivors developing SMNs compared to the general population, we investigated a validation cohort of 37 pediatric cancer survivors registered in the Childhood Cancer Survivor Study (CCSS) known to have developed solid SMNs. These survivors had primary malignancies that included Hodgkin lymphoma, sarcomas, and CNS tumors and developed infiltrating ductal carcinoma of the breast and sarcomas as SMNs (Supplementary Table 1). None of these patients were reported to have a germline tumor predisposition syndrome (Li-Fraumeni, Neurofibromatosis I, Tuberous Sclerosis, Neurofibromatosis 2, or Ataxia Telangiectasia). Importantly, individuals in the validation cohort had no family history of cancer predispositions. These patients had both germline DNA and matched solid SMN tissue available. The SMN histologies were reviewed to confirm malignancy. Sanger sequencing was performed on exons 2 – 11 of TP53 in the germline DNA. Four out of 37 individuals (11%) harbored a germline variant A639G located in exon 6 (R213R in the protein) (Fig. 3A, Table 1), a synonymous single nucleotide polymorphism rs1800372 (37) with a minor allele frequency in the general population of 0.5% (p=0.00015 by Fisher’s exact test). This variant resides within the DNA binding domain of the p53 protein (Fig. 3B/C). A second germline variant identified in this cohort was G215C in exon 4 (R72P in the protein, rs1042522) (Supplementary Fig. 5, Table 1), which was present in 6 patients and mutually exclusive with the germline A639G variant, resulting in ten out of 37 evaluable patients (27%) harboring a germline TP53 variant. Exon 4 encodes the proline-rich domain, which is important for the apoptotic activity of p53 by nuclear exportation via MAPK (38). The R72P variant is not known to be deleterious, and in fact is present in 20–60% of the general population depending on ethnicity (39). Six out of 37 germline DNAs (16%) harbored this polymorphism, which is slightly below the low range for various ethnicities described in the literature and thus could not be considered enriched for any ethnicities. Table 1 shows the proportion of the various primary cancer groups who had a TP53 variant. Frequencies for all cancer types except HD were too small to analyze. The frequencies of each variant in HD were compared to the total of all other types combined. Proportions of HD with p53 variants did not differ from other types (p-value approximately 0.9). Most patients received both chemotherapy and radiotherapy (treatment exposures for each individual case shown in Supplementary Table 1). Given the low frequencies of patients receiving radiation only or chemotherapy only, this data does not permit testing for significant interactions between TP53 variants and specific therapies.
Figure 3. Germline TP53 variants.
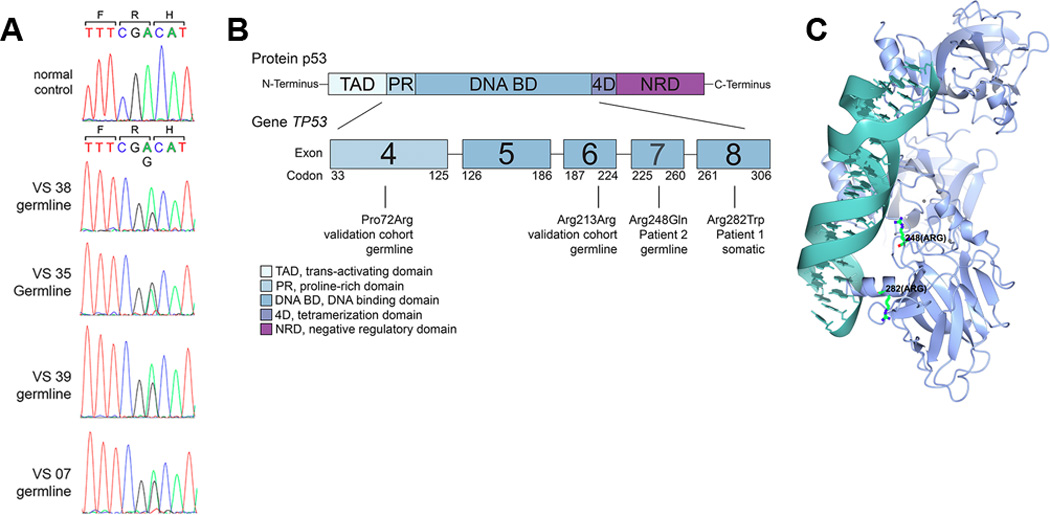
A. Sanger sequencing of exons 2–11 of TP53 was performed on the germline DNA of 37 pediatric cancer survivors who subsequently developed radiation-induced SMNs. Shown are chromatograms from the four individuals demonstrating the germline variant c.A639G (p.R213R), which is a synonymous single nucleotide polymorphism rs1800372. The trinucleotide codon is shown in color below the single letter amino acid symbol. B. Schematic of the p53 protein, indicating the locations of germline and somatic variants associated with SMNs. C. Three-dimensional structure of the p53 protein indicating the positions of the SMN-associated mutated amino acids.
Table 1. Malignancies in the Validation Cohort by TP53 Variant.
Table displaying the breakdown of genotyping for the two p53 variants (G215C, A639G). Thirty-nine total were available for analysis. The numbers of samples for each primary cancer diagnosis harboring G215C, A639G or neither are shown. “Sequencing failed” indicates samples for which we were unable to establish a genotype due to failure of sequencing reaction. Of note, no sample harbored both variants. Frequencies for all cancer types except HD were too small to analyze. The frequencies of each variant in HD were compared to the total of all other types combined. Proportions of HD with p53 variants did not differ from other types (p-value approximately 0.9). HD – Hodgkin Disease, AML – Acute myeloid Leukemia, ALL – Acute lymphoid leukemia, NHL – Non-Hodgkin Lymphoma
| Primary Cancer | No G215C Variant |
G215C present |
Sequencing failed |
No A639G Variant |
A639G present |
Sequencing failed |
|---|---|---|---|---|---|---|
| HD | 20 | 2 | 2 | 19 | 2 | 3 |
| Osteosarcoma | 3 | 0 | 0 | 3 | 0 | 0 |
| Wilms’ Tumor | 1 | 0 | 0 | 0 | 1 | 0 |
| Soft tissue sarcoma |
0 | 0 | 1 | 1 | 0 | 0 |
| Neuroblastoma | 1 | 0 | 0 | 1 | 0 | 0 |
| Astrocytoma | 1 | 0 | 0 | 1 | 0 | 0 |
| Ewing’s Sarcoma | 2 | 0 | 1 | 2 | 1 | 0 |
| AML | 1 | 2 | 0 | 2 | 0 | 1 |
| ALL | 0 | 0 | 1 | 1 | 0 | 0 |
| NHL | 0 | 1 | 0 | 1 | 0 | 0 |
| Total | 39 | 39 | ||||
DISCUSSION
Detecting somatic mutations in cancer specimens can inform prognosis and management, and defining germline mutations also informs individual cancer susceptibility. Pediatric cancers typically harbor many fewer somatic alterations than adult malignancies (1). Non-therapeutic mutagen exposure (e.g. tobacco or ultraviolet light) and aging also play little or no role in the pathogenesis of pediatric malignancies. By contrast, known inherited predispositions account for a much higher proportion of cancers arising during the first two decades of life. Thus, evaluating potential germline susceptibilities is particularly important in pediatric cancer patients, in whom a germline tumor suppressor gene mutation may underlie the primary cancer formation and potentially contribute to the risk of SMN (6, 40).
SMNs in pediatric cancer survivors are predominantly solid tumors, including soft tissue and bone sarcomas (9, 41), which represent approximately 15% of solid tumor SMNs (7). Our analysis of two radiation-induced sarcoma SMNs underscore the importance of TP53, a gene involved in DNA damage response and maintenance of genomic integrity (42). Using next-generation sequencing approaches this study identifies TP53 mutations in the exomes and transcriptomes of SMNs, which can arise as somatic alterations (as in the case of Patient 1) or germline mutations (as exemplified by Patient 2).
Coordinated WES and RNA-Seq also indicate how mutant TP53 can be represented in SMN transcriptomes. We found that in the presence of either somatic or germline TP53 mutations, wildtype TP53 is lost and mutant TP53 duplicated and overexpressed in the SMN. Copy number alterations were common in both SMNs, however in both cases, copy number neutral loss of heterozygosity occurred with TP53, suggesting a shared genetic mechanism distinguishable from the remainder of the exome.
The relationship of TP53 status to SMNs in pediatric cancer survivors without a known history of a cancer predisposition has been undefined. Prior analysis of an adult cancer patient with multiple primary tumors and a therapy-related acute myeloid leukemia identified a germline TP53 mutation, supporting the idea that cryptic variants in cancer susceptibility genes can define susceptibility to primary and secondary cancers (43). In our study, Patient 2 had Li-Fraumeni Syndrome but no first-degree relatives with malignancy, raising the possibility that TP53 variants are more common in pediatric survivors who develop SMNs as compared to the general population.
Individuals with Li-Fraumeni syndrome, who have a known germline TP53 mutation, are at high risk for developing a diverse array of malignancies, including sarcomas, which can present in early childhood (44). To manage this life-long risk, biochemical and imaging surveillance of individuals with Li-Fraumeni syndrome have been devised and shown to improve survival (45). Our findings in radiotherapy-induced sarcomas strongly argue that these individuals are also susceptible to SMNs. Using next-generation sequencing of both SMN exomes and transcriptomes, this work presents molecular evidence establishing that the germline TP53 variant is exclusively represented in the transcriptome in specific mRNA isoforms.
This analysis identified the recurrent SNP rs1800372 in a validation cohort of pediatric cancer survivors who subsequently developed SMNs. Although rs1800372 is a synonymous variant, synonymous variants are not necessarily silent or neutral in cancer evolution, as TP53 synonymous mutations can inactivate function (46). Indeed, rs1800372 is associated with poor outcome in primary breast cancer (47), and synonymous variants in the mouse Trp53 gene have been shown to influence binding of the Trp53 transcript to MDM2 and subsequent p53 function (48). Similarly, the R72P p53 variant (rs1042522), which is not classified as a deleterious mutation, has also been implicated in cancer susceptibility in humans (49). To our knowledge, this analysis is the first multi-omics analysis of SMNs and determination of germline TP53 variants in survivors of pediatric malignancies.
Similar to Patient 2, the CCSS validation cohort that we analyzed also did not have histories of familial tumor predisposition syndromes. However, clinical histories can be inadequate for identifying individuals with germline mutations in cancer predisposition genes, and germline mutational analysis may play an important role when estimating SMN risk. Our data suggest that analyzing germline TP53 exons at the time of diagnosis will identify patients at high risk for SMN development. These individuals may then benefit from dedicated surveillance protocols to screen for SMN formation (Fig. 4). The precise value of germline TP53 sequencing of all new sarcomas must be assessed by the analysis of a larger number of tumors than presented in this work.
Figure 4. Potential Clinical Implications.

Assessing SMN susceptibility may be integrated into the work up for an initial cancer diagnosis. Germline sequencing in patients may differentiate individuals with elevated risk. These individuals might be offered adjusted therapies and more focused surveillance.
Patient 1 did not harbor a germline mutation in a tumor suppressor gene, but epidemiologic data suggest that children diagnosed with rhabdomyosarcoma (embryonal and pleomorphic subtypes) have heightened risk of developing SMNs (50). Clearly the molecular basis for SMN risk remains to be defined for many pediatric cancer survivors.
The National Comprehensive Cancer Network (NCCN) guidelines for Adolescent and Young Adult (AYA) Oncology (51) defines second cancer screening recommendations based on dose delivered to at-risk anatomy. These guidelines address breast cancer, thyroid cancer, and colorectal cancer. Germline screening-based risk stratification is not integrated. Recommendations are radiation dose-dependent, with cancer screening recommended for patients receiving higher radiation doses (51). The Children’s Oncology Group (COG) has also outlined comprehensive recommendations for the long-term follow-up of pediatric cancer survivors (52). Similar to NCCN guidelines, COG’s guidelines also highlight higher radiation dose as a risk factor for second malignancies. In addition, for specific types of second malignancies, germline mutations in TP53, RB1 and NF1 are listed as risk factors. However, germline mutational analysis is not routinely performed in pediatric cancer patients. Furthermore, it is likely that germline-mediated SMN risk varies between different germline variants. Additional sequencing analyses of SMNs from pediatric cancer survivors may lead to additional tiers of recommendations based on the presence of specific germline variants. Insights into the biologic basis for SMN formation, such as germline-based risk factors, may similarly permit rational strategies to manage or mitigate this risk in cancer survivors (Fig. 4).
This study is limited by the small size of the analyzed cohort, which reflects the challenges of obtaining high quality SMN tissue for analyses, given the long latency of SMN development. Analysis of a larger cohort of pediatric cancer survivors developing SMNs may identify additional genetic variants that significantly correlate with second cancer development. Another limitation is that the corresponding SMNs for the validation cohort were not analyzed by whole exome or transcriptome sequencing.
New approaches for mitigating SMN induction are needed. Appropriately applying such approaches requires accurately identifying which individuals are at greatest risk for SMN development, and our findings strongly suggest that analyzing the TP53 gene for variants, both synonymous and non-synonymous, may be a viable strategy for identifying individuals at greatest risk of SMN formation. Identifying at-risk individuals is important because advances in radiotherapy delivery alone will not eliminate SMNs in future survivors. Notably, both patients received conformal radiotherapy, which is representative of modern radiotherapy and is employed to limit radiation to normal tissues and reduce late toxicities. Proton-based radiotherapy, which produces conformal dose deposition due to the unique physical characteristics of heavy particles, has been proposed as an approach that will reduce SMN risk in pediatric cancer patients. However, SMNs caused by neutrons produced during proton radiotherapy has been raised as a concern (53). The clinical experience to date are inconclusive and indicate no significant difference in SMN rate between protons and photon radiotherapy, although with longer follow up differences may be detected (53).
As oncologic care continues to improve survival, considerations of late toxicity become increasingly important, and our work suggests that the identification of germline-mediated risk should be an integral component of strategies to reduce SMN formation.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Most pediatric cancer survivors who develop SMNs have no known tumor predisposition syndrome, and accurately identifying which individuals are at greatest risk for SMN development is currently challenging. This work presents next-generation sequencing of exomes and transcriptomes of SMNs to resolve the role of TP53 in SMNs at a level of molecular detail not previously shown. We identify somatic and germline TP53 mutations in sarcoma SMNs, and in a cohort of pediatric cancer survivors known to develop SMNs a TP53 polymorphism is significantly more frequent than the estimated frequency in the general population. These findings strongly suggest that analyzing the TP53 gene for variants, both synonymous and non-synonymous, may be a viable strategy for identifying individuals at greatest risk of SMN formation. Our work suggests the utility of germline analysis at the time of diagnosis to actualize personalized medicine and offer stratified cancer survivorship care.
Acknowledgments
J. Nakamura was supported by grants from Alex’s Lemonade Stand Foundation, St. Baldrick’s Scholar Award, and the Hagar Family Foundation. The Childhood Cancer Survivor Study is supported by U24 CA55727 (G.T. Armstrong, Principal Investigator). The NIH/NCI award U54CA196519 supported J. Nakamura, V. Lavergne, A. Horvai, M. Loh, K. Shannon, and S. Bhatia.
Footnotes
Conflicts of interest: none
REFERENCES
- 1.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: life-long risks and responsibilities. Nat Rev Cancer. 2014;14:61–70. doi: 10.1038/nrc3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neglia JP, Meadows AT, Robison LL, Kim TH, Newton WA, Ruymann FB, et al. Second neoplasms after acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;325:1330–1336. doi: 10.1056/NEJM199111073251902. [DOI] [PubMed] [Google Scholar]
- 4.Ng AK, Bernardo MV, Weller E, Backstrand K, Silver B, Marcus KC, et al. Second malignancy after Hodgkin disease treated with radiation therapy with or without chemotherapy: long-term risks and risk factors. Blood. 2002;100:1989–1996. doi: 10.1182/blood-2002-02-0634. [DOI] [PubMed] [Google Scholar]
- 5.Travis LB, Hill DA, Dores GM, Gospodarowicz M, van Leeuwen FE, Holowaty E, et al. Breast cancer following radiotherapy and chemotherapy among young women with Hodgkin disease. Jama. 2003;290:465–475. doi: 10.1001/jama.290.4.465. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia S. Genetic variation as a modifier of association between therapeutic exposure and subsequent malignant neoplasms in cancer survivors. Cancer. 2015;121:648–663. doi: 10.1002/cncr.29096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman DL, Whitton J, Leisenring W, Mertens AC, Hammond S, Stovall M, et al. Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2010;102:1083–1095. doi: 10.1093/jnci/djq238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahan WG, Woodard HQ, et al. Sarcoma arising in irradiated bone; report of 11 cases. Cancer. 1948;1:3–29. doi: 10.1002/1097-0142(194805)1:1<3::aid-cncr2820010103>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Reulen RC, Frobisher C, Winter DL, Kelly J, Lancashire ER, Stiller CA, et al. Long-term risks of subsequent primary neoplasms among survivors of childhood cancer. JAMA. 2011;305:2311–2319. doi: 10.1001/jama.2011.747. [DOI] [PubMed] [Google Scholar]
- 10.Travis LB, Hill D, Dores GM, Gospodarowicz M, van Leeuwen FE, Holowaty E, et al. Cumulative absolute breast cancer risk for young women treated for Hodgkin lymphoma. J Natl Cancer Inst. 2005;97:1428–1437. doi: 10.1093/jnci/dji290. [DOI] [PubMed] [Google Scholar]
- 11.Schaapveld M, Aleman BM, van Eggermond AM, Janus CP, Krol AD, van der Maazen RW, et al. Second Cancer Risk Up to 40 Years after Treatment for Hodgkin's Lymphoma. N Engl J Med. 2015;373:2499–2511. doi: 10.1056/NEJMoa1505949. [DOI] [PubMed] [Google Scholar]
- 12.Sherborne AL, Davidson PR, Yu K, Nakamura AO, Rashid M, Nakamura JL. Mutational Analysis of Ionizing Radiation Induced Neoplasms. Cell Rep. 2015;12:1915–1926. doi: 10.1016/j.celrep.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343:189–193. doi: 10.1126/science.1239947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh S, Chan CK. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol Biol. 2016;1374:339–361. doi: 10.1007/978-1-4939-3167-5_18. [DOI] [PubMed] [Google Scholar]
- 18.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–425. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murugaesu N, Wilson GA, Birkbak NJ, Watkins TB, McGranahan N, Kumar S, et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer discovery. 2015;5:821–831. doi: 10.1158/2159-8290.CD-15-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arndt CA, Stoner JA, Hawkins DS, Rodeberg DA, Hayes-Jordan AA, Paidas CN, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J Clin Oncol. 2009;27:5182–5188. doi: 10.1200/JCO.2009.22.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saha T, Kar RK, Sa G. Structural and sequential context of p53: A review of experimental and theoretical evidence. Prog Biophys Mol Biol. 2015;117:250–263. doi: 10.1016/j.pbiomolbio.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Muller PA, Vousden KH. p53 mutations in cancer. Nature cell biology. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 26.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez KD, Buzin CH, Noltner KA, Gu D, Li W, Malkin D, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet. 2009;46:689–693. doi: 10.1136/jmg.2008.058958. [DOI] [PubMed] [Google Scholar]
- 28.Toguchida J, Yamaguchi T, Dayton SH, Beauchamp RL, Herrera GE, Ishizaki K, et al. Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. The New England journal of medicine. 1992;326:1301–1308. doi: 10.1056/NEJM199205143262001. [DOI] [PubMed] [Google Scholar]
- 29.Prives C. How loops, beta sheets, and alpha helices help us to understand p53. Cell. 1994;78:543–546. doi: 10.1016/0092-8674(94)90519-3. [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong H, Kohli K, Garabedian MJ, Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol. 1997;17:2735–2744. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakayama R, Miura Y, Ogino J, Susa M, Watanabe I, Horiuchi K, et al. Detection of HEY1-NCOA2 fusion by fluorescence in-situ hybridization in formalin-fixed paraffin-embedded tissues as a possible diagnostic tool for mesenchymal chondrosarcoma. Pathol Int. 2012;62:823–826. doi: 10.1111/pin.12022. [DOI] [PubMed] [Google Scholar]
- 33.Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57–68. doi: 10.2147/OTT.S53876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, et al. DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene. 2002;21:6722–6728. doi: 10.1038/sj.onc.1205874. [DOI] [PubMed] [Google Scholar]
- 35.Bernard H, Garmy-Susini B, Ainaoui N, Van Den Berghe L, Peurichard A, Javerzat S, et al. The p53 isoform, Delta133p53alpha, stimulates angiogenesis and tumour progression. Oncogene. 2013;32:2150–2160. doi: 10.1038/onc.2012.242. [DOI] [PubMed] [Google Scholar]
- 36.Marcel V, Petit I, Murray-Zmijewski F, Goullet de Rugy T, Fernandes K, Meuray V, et al. Diverse p63 and p73 isoforms regulate Delta133p53 expression through modulation of the internal TP53 promoter activity. Cell Death Differ. 2012;19:816–826. doi: 10.1038/cdd.2011.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.NCBI. Reference SNP Cluster Report: rs1800372. http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=1800372.
- 38.Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 1998;17:4668–4679. doi: 10.1093/emboj/17.16.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sjalander A, Birgander R, Saha N, Beckman L, Beckman G. p53 polymorphisms and haplotypes show distinct differences between major ethnic groups. Hum Hered. 1996;46:41–48. doi: 10.1159/000154324. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med. 2015 doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatia S, Sklar C. Second cancers in survivors of childhood cancer. Nat Rev Cancer. 2002;2:124–132. doi: 10.1038/nrc722. [DOI] [PubMed] [Google Scholar]
- 42.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Link DC, Schuettpelz LG, Shen D, Wang J, Walter MJ, Kulkarni S, et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA. 2011;305:1568–1576. doi: 10.1001/jama.2011.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li FP, Fraumeni JF., Jr Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 45.Villani A, Tabori U, Schiffman J, Shlien A, Beyene J, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12:559–567. doi: 10.1016/S1470-2045(11)70119-X. [DOI] [PubMed] [Google Scholar]
- 46.Supek F, Minana B, Valcarcel J, Gabaldon T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 47.Berns EM, van Staveren IL, Look MP, Smid M, Klijn JG, Foekens JA. Mutations in residues of TP53 that directly contact DNA predict poor outcome in human primary breast cancer. British journal of cancer. 1998;77:1130–1136. doi: 10.1038/bjc.1998.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, et al. P53 mRNA controls p53 activity by managing Mdm2 functions. Nature cell biology. 2008;10:1098–1105. doi: 10.1038/ncb1770. [DOI] [PubMed] [Google Scholar]
- 49.Yadav P, Masroor M, Tanwer K, Mir R, Javid J, Ahmad I, et al. Clinical significance of TP53 (R72P) and MDM2 (T309G) polymorphisms in breast cancer patients. Clin Transl Oncol. 2015 doi: 10.1007/s12094-015-1425-5. [DOI] [PubMed] [Google Scholar]
- 50.Archer NM, Amorim RP, Naves R, Hettmer S, Diller LR, Ribeiro KB, et al. An Increased Risk of Second Malignant Neoplasms After Rhabdomyosarcoma: Population-Based Evidence for a Cancer Predisposition Syndrome? Pediatr Blood Cancer. 2016;63:196–201. doi: 10.1002/pbc.25678. [DOI] [PubMed] [Google Scholar]
- 51.NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Adolescent and Young Adult (AYA) Oncology. 2016 https://www.nccn.org/professionals/physician_gls/f_guidelines.asp. [Google Scholar]
- 52.Children's Oncology Group (COG) Long-Term Follow-Up Guidelines. 2014 http://www.survivorshipguidelines.org/ [Google Scholar]
- 53.Chung CS, Yock TI, Nelson K, Xu Y, Keating NL, Tarbell NJ. Incidence of second malignancies among patients treated with proton versus photon radiation. Int J Radiat Oncol Biol Phys. 2013;87:46–52. doi: 10.1016/j.ijrobp.2013.04.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.