

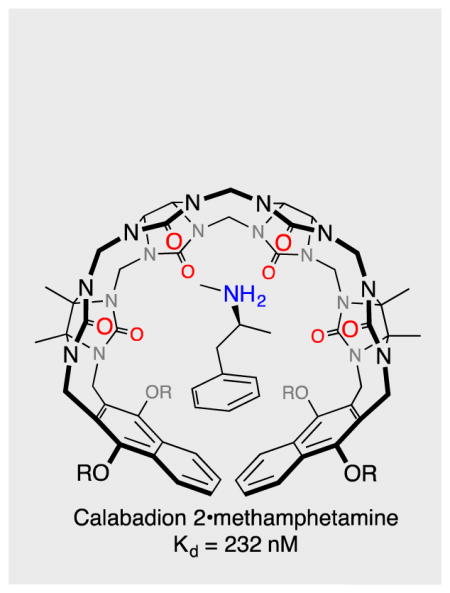
Abstract
We measured the affinity of five molecular container compounds (Calabadion 1 and 2, CB[7], sulfocalix[4]arene and HP-β-CD) toward seven drugs of abuse in homogenous aqueous solution at physiological pH by various (1H NMR, UV/Vis, ITC) binding assays and find that they span from less than 102 to over 108 M−1. X-ray crystal structures of CB[7]•methamphetamine and 1•methamphetamine are reported. We find that 2, but not CB[7], is able to ameliorate the hyperlocomotive activity of rats treated with methamphetamine.
Keywords: molecular container, cucurbituril, reversal agent, drugs of abuse, methamphetamine
Be My Guest
We measured the binding affinity of five molecular container compounds toward 7 drugs of abuse and found that the Calabadions display broad spectrum affinity. X-ray crystal structures of 1•methamphetamine and CB[7]•methamphetamine elucidate the geometry of the complexes. Administration of Calabadion 2 to rats treated with methamphetamine ameliorates the hyperlocomotive activity of the drug.
The recreational use of drugs of abuse is a major problem worldwide and overdoses and the associated mortality are common.[1] For example, the Substance Abuse and Mental Health Services Administration (SAMHSA) estimated that 10.2% of the US population 12 and older (27 million) had used an illicit drug in the past month.[2] Illicit drugs include methamphetamine, cocaine, marijuana, heroin, hallucinogens (ketamine, phencyclidine), inhalants, or prescription-type psychotherapeutics (pain relievers, tranquilizers, stimulants, and sedatives) when used non-medically (Scheme 1). The heathcare costs of emergency room visits related to illicit drugs was ≈ $11 billion in 2011 and overall costs associated with crime and lost work productivity was ≈ $193 billion.[3] Accordingly, the development of treatments for illicit drug overdose and addiction is an important biomedical problem with significant socio-economic consequences.
Scheme 1.

Chemical structures of the: a) molecular containers, and b) drugs of abuse used in this study.
Current treatments for drug overdose and addiction are based on either pharmacodynamic (PD) or pharmacokinetic (PK) strategies. PD strategies aim to the disrupt the effects of the illicit drug at its site(s) of action (e.g. drug receptors) in the body. The PD approach is currently used clinically for treating opioid overdose and addiction (e.g. naloxone, naltrexone).[4] While this treatment approach has been successful for opioids, there are currently no pharmacotherapies approved by the US FDA to treat cocaine, methamphetamine, or cannabis use disorders.[5] In contrast, the PK strategy does not require a precise molecular level knowledge of the drug•receptor interaction needed in the PD strategy, but only the ability to decrease drug concentration below its effective concentration.[6] For example, enzyme based therapeutics (e.g. human butyrylcholine esterase) are being explored that can hydrolyze cocaine into its inactive metabolites (ecgonine methyl ester and benzoic acid), thereby creating a negative concentration gradient between the brain and blood which results in a reduced concentration of the drug at its receptor in the brain.[4, 7] Immunotherapeutics are also being developed for cocaine, methamphetamine, and fentanyl, most notably by Janda,[8] which act as peripheral blockers by binding and sequestering the “antigenic” drug and preventing it from crossing the blood brain barrier.[9]
The PK strategy has also been implemented using synthetic molecular container compounds (Scheme 1) to directly compete with biological molecular recognition events. For example, the β-cyclodextrin derivative sugammadex has been shown to bind with high affinity (≈ 107 M−1) toward the neuromuscular blocking agents rocuronium and vecuronium in vitro and to act as an reversal agent for neuromuscular block in vivo.[10] Sugammadex is marketed worldwide by Merck under the trade name Bridion™. Since the turn of the millenium, much attention has been paid to cucurbit[n]uril (CB[n]) family of molecular containers because of their outstanding recognition properties toward hydrophobic cations in aqueous solution (e.g. high affinity, high selectivity, and stimuli responsiveness).[11, 12] Recently, Isaacs and Eikermann have shown that acyclic CB[n]-type molecular containers (Calabadion 1 and 2) bind strongly to rocuronium, vecuronium and cisatracurium in vitro and function as in vivo reversal agents in rats.[13, 14, 15] Given that CB[n]-type molecular containers generally possess a high affinity toward hydrophobic cations and that many drugs of abuse fall into this class of compounds, we decided to evaluate the potential of molecular containers as agents for the in vivo reversal of the biological effects of drugs of abuse by the PK strategy. In particular, the use of molecular containers to treat intoxication with drugs of abuse – as opposed to addiction where long circulation times would be needed – seems most promising.
Initially, we choose to screen the affinity of the five molecular containers (Calabadion 1 and 2, CB[7], 4, and HP-β-CD) toward a panel of drugs of abuse (6 – 12) in order to inform and prioritize subsequent in vivo work. The molecular containers used in this study were selected for several reasons. First, all of these classes of containers are known to bind to hydrophobic cations (e.g. for sensing, drug delivery, and reversal applications)[10, 12, 16] which is a common structural element in drugs 6 – 12. Second, these molecular containers possess good levels of aqueous solubility (≥ 10 mM) which is required for efficient formulation. Third, all of these containers are easily synthesized on large scale by efficient procedures which makes them viable scaffolds for further medicinal chemical optimization. Finally, all of these containers display good biocompatibility (in vitro and in vivo) and one (HP-β-CD) has even been used to formulate drugs administered to humans (e.g. Janssen’s formulation of Itraconazole (Sporonox™)). The disparate molecular structures of the five containers will result in different binding modes and types of interactions (ion-dipole, ion-ion, hydrophobic, π–π, etc.) between container and drug; an important goal of our study was to determine which class of molecular containers serves as the best lead scaffold for further medicinal chemistry optimization in the future. The drug panel includes stimulants (6, 8), hallucinogens (9, 10), and prescription-type psychotherapeutics used for pain relief (7, 11, 12). All these drugs contain the amino functional group which is protonated at physiological pH. First, we qualititatively studied the binding of containers 1 – 5 toward the drugs of abuse by 1H NMR spectroscopy. For containers 1 – 4 whose cavity constitutes an NMR shielding region we typically observe upfield shifts in the 1H NMR resonances of included drugs 6 – 12, although the kinetics of exchange is generally fast on the chemical shift timescale (Supporting Information) for 1 – 4. For example, Figure 1a–d shows the 1H NMR spectra recorded for methamphetamine (6), Calabadion 1, a 1:1 mixture of 1 and 6, and a 1:2 mixture of 1 and 6. The 1H NMR resonances for the aromatic ring of 6 (He – Hg) shift significantly upfield within 1•6 (Figure 1c) and appear as sharp resonances at 1:1 stoichiometry. When 2 equivalents of 6 are present (Figure 1d) the resonances for He – Hg shift back toward the chemical shifts for free 6 due to the fast kinetics of guest exchange on the chemical shift timescale. The resonances for the aromatic wall of 1 (Hu) appears as a pair of doublets due to the chiral nature of the guest which renders these protons diastereotopic (u, u′) in the complex. For HP-β-CD, 1H NMR was not a useful analytical tool because only very minor changes in guest chemical shift are typically observed upon HP-β-CD•guest binding. The 1:1 nature of the various host•guest complexes was established by either Job plots, x-ray crystallography, 1H NMR, isothermal titration calorimetry (ITC), or literature precedent.
Figure 1.

1H NMR spectra recorded (400 MHz, RT, 20 mM sodium phosphate buffered D2O, pH 7.4) for: a) 6 (2 mM), b) 1 (1 mM), c) a mixture of 6 (1 mM) and 1 (1 mM), and d) a mixture of 6 (2 mM) and 1 (1 mM).
We were fortunate to obtain single crystals of CB[7]•6 and 1•6 that were suitable for structure determination by x-ray crystallography.[17] Figure 2 shows stereoscopic representations of individual structures of the CB[7]•6 and 1•6 complexes in the crystal. In accord with the NMR studies, the aromatic ring of 6 is bound within the cavity of 1 featuring π–π stacking (Figure 2a). The ammonium ion is H-bonded to one of the ureidyl C=O groups (H•••O=C, 2.093 Å; N-H•••O angle, 140.0˚) and also benefits from ion-dipole interactions with the carbonyl portal. The C-shaped backbone of 1 undergoes a helical distortion within the 1•6 complex as observed previously with different guests.[18–20] The crystal structure of CB[7]•6 (Figure 2b) also shows a deep penetration of the aromatic ring of 6 into the cavity of CB[7] along with ion-dipole and H-bonding interactions at the ureidyl C=O portal. Interestingly, four CB[7]•methamphetamine complexes pack into a square in the crystal driven byinteractions between the C-H groups on the convex face of CB[7] and the C=O portal on an adjacent CB[7].
Figure 2.
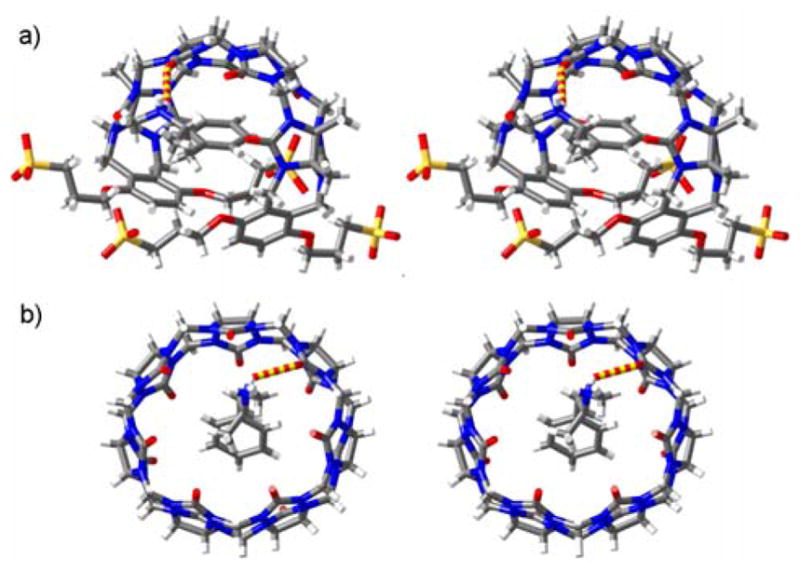
Cross-eyed stereoviews of individual complexes of: a) 1•6, and b) CB[7]•6 in the crystal. Color code: C, grey; H, white; N, blue; O, red; S, yellow; H-bond, red-yellow striped.
After having qualitatively investigated the binding between 1 – 5 and 6 – 12 by 1H NMR and confirming the binding mode for 6 toward CB[7] and 1 by x-ray crystallography we set out to measure the Ka values of the various complexes initially using NMR and UV/Vis methods and later incorporating isothermal titration calorimetry (ITC) when this instrument became available to us in the later stages of our study. For this purpose, we exployed various binding assays as appropriate. For example, for the weaker complexes (Ka < 5000 M−1) formed between hosts 3 and 4 whose cavities constitute NMR shielding regions and drugs 6 and 8 – 11 we used direct 1H NMR titrations of guest with host and fitted the change in chemical shift to a standard 1:1 binding model (Table 1, Supporting Information). Based on our extensive experience measuring CB[n]•guest binding constants, we chose a 1H NMR competition assay[22] to determine Ka = 1.2 × 108 M−1 for CB[7]•methamphetamine. For stronger complexes involving 1 and 2 we employed UV/Vis competition assays developed by us previously.[23] For this purpose, we form the 1•rhodamine 6G or 2•rhodamine 6G complexes which show different UV/Vis spectra than free rhodamine 6G and then titrate those complexes with drug which induces the reverse UV/Vis change. Based on a knowledge of the total concentrations of container, rhodamine 6G and the 1•rhodamine 6G and 2•rhodamine 6G binding constants we fitted the change in UV/Vis absorbance versus [drug] to a competition binding model[23] to extract the unknown container•drug complex Ka value (Table 1). For example, Figure 3a shows the UV/Vis spectra recorded when a solution of 2 (4.97 μM) and rhodamine 6G (4.97 μM) was titrated with a solution of methamphetamine (0 – 219 μM) whereas Figure 3b shows the fitting of the data to the competition binding model to extract Ka = 4.3 × 106 M−1 for 2•methamphetamine. The remaining complexes – mainly HP-β-CD•drug but also container•fentanyl and container•hydromorphone – were measured by ITC when the instrumentation became available in the lab (Table 1, Supporting Information). For example, Figure 3c,d shows the thermogram recorded for the titration of 2 (87 μM) with hydromorphone 12 (0 – 200 μM) which was fitted with the MicroCal PEAQ-ITC analysis software to deliver Ka = (6.8 ± 0.1) × 105 M−1 and ΔH = −2.1 ± 0.3 kcal mol−1 for the 2•12 complex. The other ITC derived thermodynamic parameters given in Table 1 were carried out analogously. Previously, we have shown that the results of UV/Vis competition assays and direct ITC titrations involving Calabadions 1 and 2 and guests give comparable values which gives us confidence in comparisons between Ka values measured by the different techniques.[15, 24]
Table 1.
Binding constants (Ka, M−1) determined for the various container•drug complexes.
| Guest | Calabadion 1 | Calabadion 2 | 3 (CB[7]) | 4 | 5 (HP-β-CD) |
|---|---|---|---|---|---|
| 6 methamphetamine | (7.5 ± 2.9) × 106 a) | (4.3 ± 1.0) × 106 a) | 1.2 × 108 c) | (3.8 ± 0.6) × 104 b) | (1.9 ± 0.3) × 102 d) |
| 7 fentanyl | (1.1 ± 0.0) × 107 d) | (7.6 ± 0.5) × 106 d) | 1.8 × 107 d) | (1.1 ± 0.1) × 104 d) | (3.0 ± 0.5) × 103 d) |
| 8 cocaine | (6.6 ± 0.4) × 105 a) | (1.0 ± 0.1) × 106 a) | (2.3 ± 0.2) ×103 b) | (4.3 ± 1.5) × 104 b) | (1.7 ± 0.3) × 102 d) |
| 9 ketamine | (1.1 ± 0.1) × 104 a),e) | (2.1 ± 0.1) × 105 a),e) | (6.4 ± 1.2) ×102 b) | (2.0 ± 0.2) × 104 d) | (1.1 ± 0.2) × 102 d) |
| 10 phencyclidine | (4.7 ± 0.5) × 104 a) | (2.1 ± 0.1) × 105 a) | (4.4 ± 0.9) ×103 b) | (4.5 ± 0.3) × 104 d) | (1.0 ± 0.0) × 103 d) |
| 11 morphine | (5.3 ± 0.3) × 105 a) | (5.3 ± 0.4) × 105 a),f) | n.b. | (4.9 ± 0.3) × 104 b) | n.b. |
| 12 hydromorphone | (1.8 ± 0.0) × 105 d) | (6.8 ± 0.1) × 105 d) | n.b. | (1.0 ± 0.0) × 104 d) | (2.8 ± 0.5) × 102 d) |
Figure 3.

a) UV/Vis spectra recorded during the titration of a mixture of 2 (4.97 μM) and rhodamine 6G (4.97 μM) with methamphetamine (6, 0 – 219 μM) in 20 mM NaH2PO4 buffered water (pH 7.4). b) Plot of absorbance versus [2] fitted to a competitive binding model with Ka = (4.3 ± 1.0) × 106 M−1. c) ITC thermogram recorded during the titration of 2 (87 μM) in the cell with 12 (0.93 mM) in the syringe. d) Fitting of the data to a 1:1 binding model with Ka = (6.8 ± 0.1) × 105 M−1.
An examination of the Ka values given in Table 1 reveals that they span a wide dynamic range from no binding to over 108 M−1. HP-β-CD, which is well known as a promiscuous and low affinity host, unsurprisingly displays modest Ka values in the 0 – 103 M−1 range. The lack of charge complementarity between neutral HP-β-CD and cationic drugs as well as the absence of typical high affinity binding epitopes (e.g. adamantyl residue) on the drugs is presumably responsible for the weaker binding. In contrast, the tetraanionic calix[4]arene derivative 4 displays larger Ka values but they cluster in the narrow range of 1.0 × 104 – 4.9 × 104 M−1. The lack of selectivity in this case may be due to the relative importance of the electrostatic contributions to the binding free energy. Most interesting, and encouraging as lead compounds for the further development of reversal agents for in vivo intoxication with drugs of abuse, are the three CB[n]-type molecular containers Calabadion 1 and 2 and CB[7]. These three containers bind to selected drugs of abuse with Ka values that fall in the 104 – 108 M−1 range. Given the presence of phenethylammonium binding epitopes it is not surprising that CB[7] displays high affinity toward both methamphetamine (Ka = 1.2 × 108 M−1) and fentanyl (Ka = 1.8 × 107 M−1). Furthermore, CB[7] discriminates against the more sterically encumbered drugs (Ka < 4400 M−1) due to unfavorable steric interactions in the CB[7]•guest complexes. In contrast, the acyclic CB[n]-type containers Calabadion 1 and Calabadion 2 have a more flexible cavity which can flex to accommodate a range of guest sizes and display both good levels of affinity (Ka from 104 – 107 M−1) and some selectivity toward the panel of drugs of abuse. The complexes between the tetra-anionic Calabadions and cationic drugs benefit from electrostatic interactions and π–π interactions between guest aromatic rings and host aromatic walls. Similar to CB[7], Calabadion 1 and Calabadion 2 display highest affinity toward methamphetamine and fentanyl with Ka values in the 106 – 107 M−1 range. Among Calabadions 1 and 2 and CB[7], CB[7] displays the highest levels of affinity and selectivity toward the drugs of abuse panel. However, the affinity and selectivity of 1 and 2 seen herein and also documented in the context of neuromuscular block reversal,[15] is substantial and suggests that 1 and 2 could serve as core scaffolds for further structural optimization. We are mindful of the fact that metabolites of amino acids, especially phenylalanine, will likely display similar binding affinity toward Calabadion 2 as methamphetamine does which could compromise in vivo efficacy. Accordingly, and given the fact that Calabadion•guest complexes are eliminated in the urine over several hours,[14, 21] we do not envision the use of Calabadion 2 for long term treatment of methamphetamine addiction. However, such consideration of circulation time and discrimination against related metabolites are less relevant in the context of treatment of drug overdose and intoxication where dose can simply be adjusted to prevent mortality. Accordingly, we decided to test the ability of an acyclic CB[n]-type receptor and unfunctionalized CB[7] as an in vivo reversal agent for methamphetamine intoxication. We choose Calabadion 2 as the acyclic CB[n] test compound because of its documented high biocompatibility (e.g. low in vitro cytotoxicity, high in vivo maximum tolerated dose, passed Ames test, no hERG ion channel inhibition)[21] and because of its generally higher binding affinity toward its guests.[13, 19]
Next, we set out to determine whether the high in vitro binding affinities of Calabadion 2 and CB[7] toward methamphetamine could prevent or reverse its in vivo biological effects. For our assay, we took advantage of the known increase of locomotor activity (e.g. movement) of rats upon treatment with methamphetamine.[25] A total of 21 male Sprague-Dawley rats (weight, mean ± SD: 288 ± 14 g) were tested repeatedly in a randomized controlled crossover setting. A single bolus of methamphetamine (0.3 mg kg−1) or placebo was injected intravenously via a tail vein catheter to assess baseline locomotor activity. Two types of studies were performed. In the treatment studies, Calabadion 2 (65 or 130 mg kg−1) or CB[7] (45 or 90 mg kg−1) was administered 30 seconds after methamphetamine whereas in the prevention studies, rats were pretreated with Calabadion 2 (Figure 5) 30 seconds before methamphetamine treatment via tail vein catheter. The catheter was flushed with 1 mL normal saline in between injections to ensure that methamphetamine was not simply complexed by Calabadion 2 inside the catheter. The animals were placed in the center of a 100 × 100 × 40 cm open field and their locomotor activity was video recorded for 20 minutes. The location of the rat and the total distance traveled was analyzed using the ANY-maze (version 4.5, Stoelting, Wood Dale, IL, USA) software. Values are given in percent of the placebo + methamphetamine activity level and reported as mean ± standard error of the mean. As expected, the rats only treated with placebo in addition to methamphetamine exhibit enhanced levels of locomotor activity relative to their baseline: (n = 21; 26.5 ± 4.9) m vs. (5.8 ± 1.5) m; p<0.001. Encouragingly, the preventive use of Calabadion 2 (65 or 130 mg kg−1) significantly decreased the traveled distance by (64.1 ± 8.4)% (p=0.001) and (74.4 ± 3.8)% (p<0.001), respectively, compared to placebo + methamphetamine (Figure 4). Similarly, the treatment study with Calabadion 2 (130 mg/kg) significantly decreased the traveled distance by (43.8 ± 11.2)% compared to placebo + methamphetamine (p=0.004), whereas no significant reduction of methamphetamine induced hyperlocomotion was observed when treated with 65 mg kg−1 of 2 (p=0.625). As a control, we determined that a bolus of Calabadion 2 alone (130 mg kg−1) without methamphetamine did not affect the locomotion compared to baseline (33.5 ± 17.7% vs. 32.9 ± 15.0%; p = 0.982). A smaller dose of Calabadion 2 was required in a prevention setting compared to a treatment setting to reverse methamphetamine-induced hyperlocomotion (interaction effect of indication × Calabadion 2 dose for locomotor activity: p<0.001), indicating effect modification. In our previous studies of the use of Calabadion 1 and 2 to reverse neuromuscular block, we found that the majority of the drug was eliminated in the urine of the rats as the non-metabolized Calabadion•drug complex within two hours.[14, 21] We posit that Calabadion 2 functions in a similar way to modulate the hyperlocomotive effect induced by methamphetamine described herein. The substantially higher doses of Calabadion 2 required for methamphetamine reversal than for neuromuscular block reversal likely reflects the tighter binding of methamphetamine to its biological targets and/or kinetic issues of intravenous sequestration of methamphetamine once it has crossed the blood-brain barrier. We expect that the Calabadion framework will enable a medicinal chemistry structural optimization to enhance both binding affinity and selectivity.
Figure 4.

Results from behavioural tests in the open field. a) Bar graph showing distance traveled in percent of the placebo + methamphetamine activity level within 20 minutes; error bars represent the standard error of the mean. *p<0.01 compared to methamphetamine + placebo. **p<0.001 compared to methamphetamine + placebo. #interaction term indication × Calabadion 2 dose: p<0.001, indicating effect modification by time of administration of Calabadion 2. Tracking plots illustrating the traveled distance of one rat within 20 minutes in the open field: b) Baseline, no methamphetamine given, c) following methamphetamine (0.3 mg kg−1) + placebo, d) following methamphetamine (0.3 mg kg −1) + calabadion 2 (65 mg kg−1), and e) following methamphetamine (0.3 mg kg−1) + calabadion 2 (130 mg kg−1).
In contrast, treatment with CB[7] (45 mg kg−1 or 90 mg kg−1) did not affect the distance traveled compared to placebo + methamphetamine: (87.0 ± 42.6)%, p=0.773 and (119.5 ± 55.4)%, p=0.739, respectively). As a control, we verified that injection of CB[7] alone had no significant effect on locomotion compared to baseline (19.3 ± 6.0% vs. 21.6 ± 6.9%; p =0.546).
In summary, we have screened the binding affinity of molecular containers 1 – 5 toward seven drugs of abuse by a combination of direct and competitive 1H NMR, UV/Vis, and ITC binding assays. The structures of the 1•methamphetamine and CB[7]•methamphetamine complexes were solved by single crystal x-ray diffraction. Calabadion 1 and Calabadion 2, in particular, display very good levels of affinity across the full range of drugs and display highest affinity toward methamphetamine and fentanyl. We find that the hyperlocomotive activity of rats given methamphetamine could be both prevented and treated by Calabadion 2 (65 or 130 mg kg−1). The documented biocompatibility of the Calabadions[18, 21, 26] and their convergent building block synthesis[19] suggest fruitful pathways toward their further structural optimization for development as reversal agents for intoxication with non-opioid drugs of abuse for which no treatments are currently available.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (CA168365 to L.I.), University of Maryland (Graduate Deans Dissertation Fellowship to S. G.), Merck Inc. (to M.E.), and Jeff and Judy Buzen (philanthropic donation to M.E.) for financial support.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Facing Addition in America. [accessed April 6, 2017];The Surgeon General’s Report on Alcohol, Drugs, and Health. 2016 https://www.surgeongeneral.gov/library/2016alcoholdrugshealth/index.html. [PubMed]
- 2.Behavioral Health Trends in the United States. https://www.samhsa.gov/data/sites/default/files/NSDUH-FRR1-2014/NSDUH-FRR1-2014.pdf (accessed.
- 3. [accessed April 6, 2017];National Drug Threat Assessment. 2011 http://www.justice.gov/archive/ndic/pubs44/44849/44849p.pdf.
- 4.Gorelick DA. Future Med Chem. 2012;4:227–243. doi: 10.4155/fmc.11.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skolnick P. Trends in Pharmacological Sciences. 2015;36:628–635. doi: 10.1016/j.tips.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montoya ID. Adicciones. 2012;24:95–103. [PubMed] [Google Scholar]; Zheng F, Zhan C-G. Future Med Chem. 2012;4:125–128. doi: 10.4155/fmc.11.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhan C-G, Zheng F, Landry DW. J Am Chem Soc. 2003;125:2462–2474. doi: 10.1021/ja020850+. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zheng F, Yang W, Ko M-C, Liu J, Cho H, Gao D, Tong M, Tai H-H, Woods JH, Zhan C-G. J Am Chem Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shram MJ, Cohen-Barak O, Chakraborty B, Bassan M, Schoedel KA, Hallak H, Eyal E, Weiss S, Gilgun Y, Sellers EM, Faulknor J, Spiegelstein O. J Clin Psychopharmacol. 2015;35:396–405. doi: 10.1097/JCP.0000000000000347. [DOI] [PubMed] [Google Scholar]
- 8.Bremer PT, Kimishima A, Schlosburg JE, Zhou B, Collins KC, Janda KD. Angew Chem, Int Ed. 2016;55:3772–3775. doi: 10.1002/anie.201511654. [DOI] [PMC free article] [PubMed] [Google Scholar]; Collins KC, Schlosburg JE, Bremer PT, Janda KD. J Med Chem. 2016;59:3878–3885. doi: 10.1021/acs.jmedchem.6b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kimishima A, Wenthur CJ, Eubanks LM, Sato S, Janda KD. Mol Pharmaceutics. 2016;13:3884–3890. doi: 10.1021/acs.molpharmaceut.6b00682. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gooyit M, Miranda PO, Wenthur CJ, Ducime A, Janda KD. ACS Chem Neurosci. 2017;8:468–472. doi: 10.1021/acschemneuro.6b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosten TR, Domingo CB, Shorter D, Orson F, Green C, Somoza E, Sekerka R, Levin FR, Mariani JJ, Stitzer M, Tompkins DA, Rotrosen J, Thakkar V, Smoak B, Kampman K. Drug Alcohol Depend. 2014;140:42–47. doi: 10.1016/j.drugalcdep.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Stevens MW, Henry RL, Owens SM, Schutz R, Gentry WB. MAbs. 2014;6:1649–1656. doi: 10.4161/19420862.2014.976431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bom A, Bradley M, Cameron K, Clark JK, Van Egmond J, Feilden H, MacLean EJ, Muir AW, Palin R, Rees DC, Zhang M-Q. Angew Chem, Int Ed. 2002;41:265–270. doi: 10.1002/1521-3773(20020118)41:2<265::aid-anie265>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 11.Isaacs L. Acc Chem Res. 2014;47:2052–2062. doi: 10.1021/ar500075g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ko YH, Kim E, Hwang I, Kim K. Chem Commun. 2007:1305–1315. doi: 10.1039/b615103e. [DOI] [PubMed] [Google Scholar]; Shetty D, Khedkar JK, Park KM, Kim K. Chem Soc Rev. 2015;44:8747–8761. doi: 10.1039/c5cs00631g. [DOI] [PubMed] [Google Scholar]; Del Barrio J, Horton P, Lairez D, Lloyd G, Toprakcioglu C, Scherman O. J Am Chem Soc. 2013;135:11760–11763. doi: 10.1021/ja406556h. [DOI] [PubMed] [Google Scholar]; Barrow SJ, Kasera S, Rowland MJ, del Barrio J, Scherman OA. Chem Rev. 2015;115:12320–12406. doi: 10.1021/acs.chemrev.5b00341. [DOI] [PubMed] [Google Scholar]; Masson E, Ling X, Joseph R, Kyeremeh-Mensah L, Lu X. RSC Adv. 2012;2:1213–1247. [Google Scholar]
- 12.Ghale G, Nau WM. Acc Chem Res. 2014;47:2150–2159. doi: 10.1021/ar500116d. [DOI] [PubMed] [Google Scholar]
- 13.Ma D, Zhang B, Hoffmann U, Sundrup MG, Eikermann M, Isaacs L. Angew Chem, Int Ed. 2012;51:11358–11362. doi: 10.1002/anie.201206031. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann U, Grosse-Sundrup M, Eikermann-Haerter K, Zaremba S, Ayata C, Zhang B, Ma D, Isaacs L, Eikermann M. Anesthesiology. 2013;119:317–325. doi: 10.1097/ALN.0b013e3182910213. [DOI] [PubMed] [Google Scholar]; Haerter F, Simons JCP, Foerster U, Duarte IM, Diaz-Gil D, Eikermann-Haerter K, Ayata C, Ganapati S, Zhang B, Blobner M, Isaacs L, Eikermann M. Anesthesiology. 2015;123:1337–1349. doi: 10.1097/ALN.0000000000000868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ganapati S, Zavalij PY, Eikermann M, Isaacs L. Org Biomol Chem. 2016;14:1277–1287. doi: 10.1039/c5ob02356d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinalli R, Dalcanale E. Acc Chem Res. 2013;46:399–411. doi: 10.1021/ar300178m. [DOI] [PubMed] [Google Scholar]; Biavardi E, Federici S, Tudisco C, Menozzi D, Massera C, Sottini A, Condorelli GG, Bergese P, Dalcanale E. Angew Chem, Int Ed. 2014;53:9183–9188. doi: 10.1002/anie.201404774. [DOI] [PubMed] [Google Scholar]; Guo D-S, Liu Y. Acc Chem Res. 2014;47:1925–1934. doi: 10.1021/ar500009g. [DOI] [PubMed] [Google Scholar]
- 17.CCDC 1544715 (CB[7]•6) and CCDC 1544716 (1•6) contain the supplementary crystallographic data for this paperThese data can be obtained free of charge from The Cambridge Crystallographic Data Centrevia http://www.ccdc.cam.ac.uk/data_request/cif.
- 18.Ma D, Hettiarachchi G, Nguyen D, Zhang B, Wittenberg JB, Zavalij PY, Briken V, Isaacs L. Nat Chem. 2012;4:503–510. doi: 10.1038/nchem.1326. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B, Isaacs L. J Med Chem. 2014;57:9554–9563. doi: 10.1021/jm501276u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B, Zavalij PY, Isaacs L. Org Biomol Chem. 2014;12:2413–2422. doi: 10.1039/c3ob42603c. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gilberg L, Zhang B, Zavalij PY, Sindelar V, Isaacs L. Org Biomol Chem. 2015;13:4041–4050. doi: 10.1039/c5ob00184f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz-Gil D, Haerter F, Falcinelli S, Ganapati S, Hettiarachchi GK, Simons JCP, Zhang B, Grabitz SD, Moreno Duarte I, Cotten JF, Eikermann-Haerter K, Deng H, Chamberlin NL, Isaacs L, Briken V, Eikermann M. Anesthesiology. 2016;125:333–345. doi: 10.1097/ALN.0000000000001199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J Am Chem Soc. 2005;127:15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]; Cao L, Isaacs L. Supramol Chem. 2014;26:251–258. [Google Scholar]; Cao L, Šekutor M, Zavalij PY, Mlinarić-Majerski K, Glaser R, Isaacs L. Angew Chem, Int Ed. 2014;53:988–993. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]
- 23.Ma D, Zavalij PY, Isaacs L. J Org Chem. 2010;75:4786–4795. doi: 10.1021/jo100760g. [DOI] [PubMed] [Google Scholar]
- 24.Isaacs L, Eikermann M. The binding constants reported in reference 15 were reproduced by an independent contract research organization sponsored by Calabash Bioscience 2016 unpublished work. [Google Scholar]
- 25.Gentry WB, Ghafoor AU, Wessinger WD, Laurenzana EM, Hendrickson HP, Owens SM. Pharmacol Biochem Behav. 2004;79:751–760. doi: 10.1016/j.pbb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Hettiarachchi G, Samanta S, Falcinelli S, Zhang B, Moncelet D, Isaacs L, Briken V. Mol Pharmaceut. 2016;13:809–818. doi: 10.1021/acs.molpharmaceut.5b00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.