

Abstract
Lipotoxicity induced by saturated fatty acids (SFAs) plays a central role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD); however, the exact mechanism(s) remain to be fully elucidated. SIRT3 is an NAD+-dependent deacetylase primarily located inside mitochondria. In this study, we demonstrated that a SFAs-rich high-fat diet (HFD) was more detrimental to the liver than an isocaloric unsaturated FAs-rich HFD. Unexpectedly, SIRT3 expression/activity were significantly elevated in the livers of mice exposed to the SFAs-rich HFD. Using cultured HepG2 and AML-12 hepatocytes, we demonstrated that unlike monounsaturated FAs, SFAs upregulates SIRT3 expression/activity. SIRT3 overexpression renders both the liver and hepatocytes susceptible to palmitate-induced cell death, which can be alleviated by SIRT3 siRNA transfection. In contrast, SIRT3 suppression protects hepatocytes from palmitate cytotoxicity. Further studies revealed that SIRT3 acts as a negative regulator of autophagy, whereby enhancing the susceptibility of hepatocytes to SFAs-induced cytotoxicity. Mechanistic investigations elucidate that SIRT3 overexpression causes manganese superoxide dismutase (MnSOD) deacetylation/activation, which depleted intracellular superoxide contents, leading to AMP-activated protein kinase (AMPK) inhibition and mTORC1 activation, resulting in autophagy suppression. In contrast, SIRT3 siRNA gene silencing enhances autophagy flux. The similar result was observed in the liver tissue from SIRT3 knockout mice.
Conclusion
our data identified SIRT3 to be a novel negative regulator of autophagy, whose activation by SFAs contributes to lipotoxicity in hepatocytes and suggest that restraining SIRT3 overactivation can be a potential therapeutic choice for the treatment of NAFLD as well as other metabolic disorders, with lipotoxicity being the principal pathomechanism.
Keywords: Fatty acids, AMPK, Mitochondria, MnSOD, Acetylation
Introduction
Lipotoxicity refers to cellular dysfunction and/or cell death derived from excess exposure to elevated levels of fatty acids and lipid accumulation in non-adipose tissues, including liver, heart, skeletal muscle, and pancreas.(1) Lipotoxicity accounts for many manifestations of the metabolic syndrome, including non-alcoholic fatty liver disease (NAFLD).(2) Hepatocyte cell death resulting from lipotoxicity, also called lipoapoptosis, is a hallmark of NAFLD.(3) Overall, lipotoxicity is most commonly induced by saturated fatty acids (SFAs), in specific palmitic acid (16:0, PA), the most abundant SFA in the diet and in the circulation.(4) The cellular mechanisms underlying SFAs-induced lipotoxicity in hepatocytes remain to be fully understood; however, it is well documented that endoplasmic reticulum (ER) stress and oxidative stress are the major intracellular pathways mechanistically involved in lipoapoptosis.(5, 6) Protecting hepatocytes from SFAs-induced hepatotoxicity provides a conceivable strategy for preventing the development of NAFLD.
Autophagy is an evolutionarily conserved physiological process that represents a system of bulk protein degradation aimed at removal and breakdown of cellular components (organelles and proteins) during starvation, thereby redistributing nutrients for maintenance of cellular energetic balance.(7) It also plays a critical role in eliminating damaged proteins and organelles.(8) We previously reported that autophagy activation protected hepatocytes against SFAs-induced lipotoxicity.(9) Similarly, autophagy activation confers protection against palmitate-induced cytotoxicity in pancreatic beta-cells.(10)
Sirtuins (SIRTs) are highly conserved nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases that regulate a wide variety of biological functions.(11–14) Of the seven mammalian sirtuins, SIRT3, SIRT4, and SIRT5 are predominantly localized inside mitochondria, with SIRT3 being considered as the primary modulator of mitochondrial protein acetylation.(15) SIRT3 is highly expressed in metabolically active tissues, such as the liver, and is a key regulator of mitochondrial functions, including metabolism, energy production, and modulation of the response to oxidative stress.(16–21) The function of SIRT3 in cellular response is stimulus- and cell type-specific,(22) with both anti-apoptotic and pro-apoptotic activities being reported.(23–25)
The present study aims to investigate the role of SIRT3 in SFAs-induced hepatotoxicity. Unexpectedly, we demonstrate that the sensitivity of hepatocytes to SFAs-induced lipotoxicity is positively associated with SIRT3 activity. Our detailed mechanistic investigations provide evidence for a novel role of SIRT3 in the regulation of autophagy induction via an AMP-activated protein kinase (AMPK)-dependent mechanism.
Materials and Methods
Animal Model and Experimental Protocol
All animal studies were performed in accordance with the animal care committee at the University of Illinois at Chicago. All mice were fed ad libitum, and housed on a 12:12 h light: dark cycle at 25°C. Male SIRT3−/− mice and WT 129Sv mice (8-week of age) were purchased from Jackson Laboratory (Bar Harbor, ME), and maintained on a standard chow diet. Male C57BL/6 mice (8-week old, Jackson Laboratory, Bar Harbor, ME) were fed with normal (n = 8) or high-fat diet using either palm oil (n = 8) or corn oil (n = 8) as fat source (Supplementary data, Table. 1, Bio-Serv, Flemington, NJ). For liver-specific SIRT3 overexpression, the animals were injected in the lateral tail vein with recombinant adeno-associated viral (AAV) serotype 8 gene transfer vectors bearing a liver-specific promoter combination (albumin promoter) with mouse SIRT3 sequence. The detailed protocols were shown in the online supplementary data. Mice were sacrificed 8 weeks later.
Establishment of stable human SIRT3 over-expression cell lines
The eukaryotic expression vector plasmid pcDNA3.1+/hSIRT3 containing the human SIRT3 gene sequence was kindly provided by Dr. Yu Wang (University of Hong Kong).(26) HepG2 cells were transfected with 0.8 μg/well of the expression vector pcDNA3.1+/hSIRT3 or empty vector control pcDNA3.1+ (Invitrogen, Grand Island, NY) in 24-well plate using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY) following the manufacturer’s guidelines. After 48 hours transfection, cells were trypsinized into 24-well plates at a ratio of 1:100 for monoclonal cell selection using 800 μg/ml G418. The stable cell line was established and confirmed by both western blot analysis and Real Time PCR.
MnSOD Overexpression
Recombinant lentivirus vector (pLV[Exp]-EGFP:T2A:Neo) containing human MnSOD (NM_001024465.1) under the control of EF-1alpha promoter was generated by Cyagen Biosciences Inc. (Guangzhou, China). A noncoding vector carrying the EF1alpha promoter was used to produce vector control particles. The optimal virus titer used for cell transfection was screened according to the manufacturer’s instructions. HepG2 cells were transfected with either pLV[Exp]-EGFP:T2A:Neo-EF1alpha-hMnSOD to overexpress MnSOD or pLV[Exp]-EGFP:T2A:Neo-EF1alpha-null as a vector control.
Analysis of autophagic flux
The autophagic flux was measured as described previously.(27) Briefly, the cells were pretreated with chloroquine (CQ), an inhibitor of lysosome acidification, after SIRT3 overexpression or knockdown. The autophagic flux was determined by detecting GFP-LC3 puncta and LC3-II expression using laser scanning confocal microscope (Nikon A1R, Japan) and Western blot, respectively. Additionally, cells were transiently transfected with recombinant adenovirus mRFP-GFP-LC3 (Hanbio Biotechnology Co. Ltd., Shanghai, China). Yellow or red puncta was detected by confocal microscope from at least 50 cells for each individual experiment after different treatment.
Statistical Analysis
All data are expressed as mean ± SD of at least three independent experiments. Statistical analysis was performed using a one-way ANOVA and analyzed by post-hoc test with Fisher’s least significant difference (LSD). Differences between treatments were considered to be statistically significant at p < 0.05.
Results
SIRT3 Negatively Regulates Autophagy in Hepatocytes
To directly test the effect of SIRT3 on autophagy in hepatocytes, a stable SIRT3-overexpressing HepG2 cell line was established (Supplementary data, Fig. 1). SIRT3 overexpression strongly decreased intracellular autophagosome formation and microtubule-associated protein 1 (MAP1) light chain 3 (LC3)-II conversion, the well-established markers of autophagy induction, while increased accumulation of p62, a protein specifically degraded by autophagy, was observed (Fig. 1A). Moreover, SIRT3 overexpression blunted rapamycin-induced autophagy activation (Supplementary data, Fig. 2) and attenuated autophagic flux, which was evidenced by reduced autophagosome formation and LC3-II conversion in the presence of CQ, and decreased autophagosomes (yellow puncta) and autolysosomes (red puncta) number (Fig. 1A). Conversely, SIRT3 knockdown via siRNA transfection led to increased LC3-II conversion, decreased p62 accumulation (Fig. 1B; Supplementary data, Fig. 3), and increased autophagic flux (Fig. 1B). Furthermore, SIRT3−/− and the liver-specific SIRT3 overexpression mice were collected. As shown in Fig. 1C, SIRT3−/− mice manifested enhanced hepatic LC3-II conversion and decreased accumulation of p62 in both fasting and fed states (Fig. 1C), while the mice with liver-specific SIRT3 overexpression manifested decreased LC3-II conversion and increased accumulation of p62 in the liver (Fig. 1D). These data altogether suggest that SIRT3 is a negative regulator of autophagy.
Figure 1.
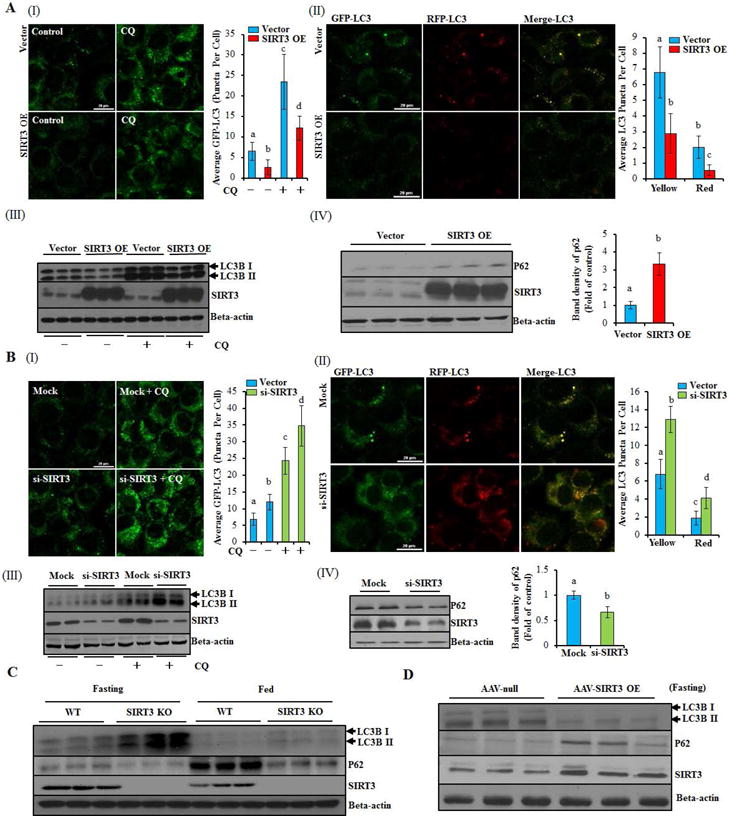
SIRT3 negatively regulates autophagy in hepatocytes. (A) SIRT3-overexpressing (SIRT3 OE) or vector control HepG2 cells were transfected with recombinant adenovirus GFP-LC3 or mRFP-GFP-LC3, and treated with or without CQ (20 μM) for 12 h. Green puncta was counted (I); Yellow and red puncta was counted (II); Immunoblotting assay for SIRT3 and LC3-II (III), SIRT3 and p62 (IV). (B) HepG2 cells were transfected with siSIRT3 or scramble siRNA. Recombinant adenovirus GFP-LC3 or mRFP-GFP-LC3 was transfected into cells and followed by CQ treatment as indicated. Green puncta was counted (I); Yellow and red puncta was counted (II); Immunoblotting assay for SIRT3 and LC3-II (III) and p62 (IV). (C) LC3-II, p62, and SIRT3 expressions were detected in SIRT3−/− or wild type mice liver with or without 12 h fasting, respectively (n = 5). (D) LC3-II, p62, and SIRT3 expressions were detected in liver specific SIRT3 OE or control mice liver after 12 h fasting. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly, p ˂ 0.05.
FFAs Differentially Regulate SIRT3 Expression in the Liver/Hepatocytes
SFAs-induced hepatocyte cell death contributes to the pathogenies of NAFLD(9). We showed that both PA and SA induced significant cell death (Supplementary data, Fig. 4A–F), while oleic acid (OA) showed no effect on cell viability (Supplementary data, Fig. 4G & H). In our in vivo investigations, male C57BL/6 mice were exposed to either a SFAs-rich (palm oil) high-fat diet (HFD) or an isocaloric unsaturated fatty acids (unSFAs)-rich (corn oil) HFD for 8 weeks. We demonstrated here that the SFAs-rich HFD more prominent hepatic pathological alterations than the isocaloric unSFAs-rich HFD (Fig. 2A; Supplementary data, Fig. 5). Intriguingly, hepatic SIRT3 expression was significantly elevated in the setting of SFAs-rich HFD feeding (Fig. 2B; Supplementary data, Fig. 6A). Correspondingly, mitochondrial protein acetylation status in the liver was significantly reduced in mice fed the SFAs-rich HFD (Fig. 2C).
Figure 2.
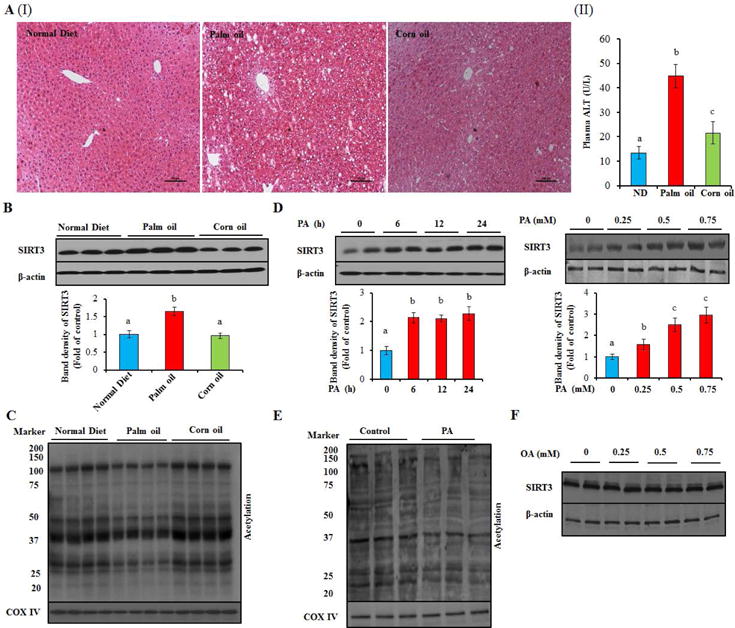
SFAs upregulates SIRT3 expression and activity in the liver and HepG2 cells. Male C57BL/6 mice were fed with normal or high-fat diet using either palm oil or corn oil as fat source. Plasma and liver were collected for analysis. HepG2 cells were treated with 0.5 mM PA or OA for 12 h (or treated with the indicated dose and duration in the figure). Total cellular lysates from hepatocytes and liver tissues (n = 8) were subjected to immunoblotting assay for SIRT3. Mitochondrial proteins were extracted from both hepatocytes and liver tissues using a commercial Mitochondria Isolation kit (Beyotime, China) for the measurement of acetylated proteins abundance. (A) Liver H&E stain (I); Plasma ALT (II). (B) SIRT3 protein expression. (C) Mitochondrial protein acetylation. (D) SIRT3 protein expression. (E) Mitochondrial protein acetylation. (F) SIRT3 protein expression. Each in vitro test was performed at least three times (n = 8 for mice), and a representative blot was shown. All values are denoted as means ± SD. Bars with different characters differ significantly, p ˂ 0.05.
To directly determine the effect of different types of fatty acids on SIRT3 expression and activity, cultured hepatocytes were exposed to either PA or OA. PA exposure increased SIRT3 expression in a dose- and time-dependent manner in both human (HepG2) and mouse (AML-12, a non-transformed mouse hepatocyte cell line) hepatocytes (Fig. 2D; Supplementary data, Fig. 7A & B) without affecting its stability (Supplementary data, Fig. 6B & C). Accordingly, acetylated mitochondrial protein abundance was reduced by PA exposure (Fig. 2E; Supplementary data, Fig. 7C). OA exposure had no effect on SIRT3 expression (Fig. 2F).
SIRT3 Controls Sensitivity of Hepatocyte/Liver to SFAs-induced Lipotoxicity
SIRT3 upregulation by PA, but not OA, spurred us to investigate the role of SIRT3 activity in SFA-induced lipotoxicity. Somewhat unexpectedly, SIRT3 overexpression increased cellular susceptibility to PA-induced cell death (Fig. 3A). In contrast, SIRT3 inhibition, with either genetic (siRNA transfection) or pharmacologic (nicotinamide, NAM) approach, alleviated PA-induced cell death in both control and SIRT3-overexpressing cells (Fig. 3B–D; Supplementary data, Fig. 8 & 9). Similar results were observed when SA was applied (Supplementary data, Fig. 10). These results suggest that SIRT3 activity modulates the sensitivity of hepatocyte to SFAs-induced lipotoxicity. This notion was corroborated by our in vivo investigation using the liver-specific SIRT3 overexpression mice. As shown in Fig. 3E and Supplementary data, Fig. 11, liver-specific SIRT3 overexpression aggravated liver damage in response to an 8-week palm oil-rich HFD feeding.
Figure 3.
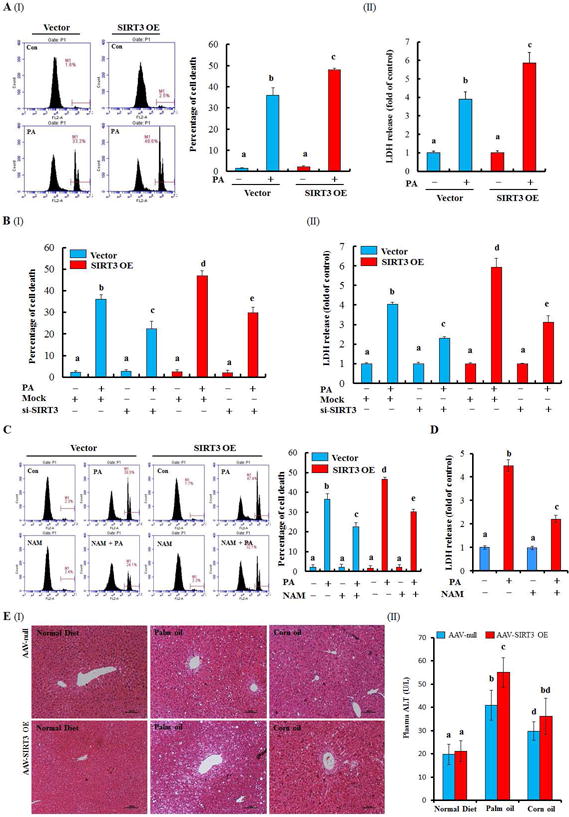
SIRT3-overexpressing increases the susceptibility of PA/palm oil-induced injury in HepG2 cells and liver. Vector control or SIRT3 OE HepG2 cells were suffered to PA (0.5 mM) exposure for 12 h. For SIRT3 inhibition, HepG2 cells were either transfected siSIRT3 or pretreated with NAM (5 mM), a pharmacologic inhibitor of SIRT3 for 1 h. Cell death was detected by the measurements of LDH release and PI staining. Liver specific SIRT3 OE mice model was established and fed with normal or high-fat diet using either palm oil or corn oil as described in the Methods. Plasma and liver were collected for analysis (n = 8). (A)-(D) Cell death was detected by the measurements of PI staining or LDH release after the indicated treatments. (E) Liver H&E stain (I); Plasma ALT (II). All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly, p ˂ 0.05.
SIRT3 Sensitizes Hepatocytes to Lipotoxicity via Negatively Mediating Autophagy
In keeping with previous studies, including ours,(9) we showed here that autophagy induction, via several well-established approaches, including rapamycin supplementation, amino acid and serum-depleted medium (EBSS), as well as serum-free medium, provided strong protection against PA-induced cell death (Supplementary data, Fig. 12A & B). Conversely, the autophagy inhibitors, bafilomycin A1 (Baf) and CQ, aggravated PA hepatotoxicity (Supplementary data, Fig. 12C & D). Similar results were observed when SA was used (Supplementary data, Fig. 12E & F). However, OA supplementation with or without autophagy inhibitors had no effect on cell death (Supplementary data, Fig. 12G & H).
To determine whether the negative regulation of SIRT3 on autophagy is involved in PA-induced lipotoxicity, rapamycin was exploited to activate autophagy in SIRT3-overexpressing cells before PA exposure. As shown in Fig. 4A, rapamycin pretreatment blocked PA-induced lipotoxicity in SIRT3-overexpressing cells. Conversely, autophagy suppression via either pharmacological (CQ) or genetic (siRNA for Atg5, Supplementary data, Fig. 13) approach blunted the protective effect of SIRT3 siRNA transfection on PA-induced cell death (Fig. 4B & C; Supplementary data, Fig. 14). These results suggest that SIRT3 controls the sensitivity of hepatocytes to PA-induced lipotoxicity via negatively regulating autophagy.
Figure 4.
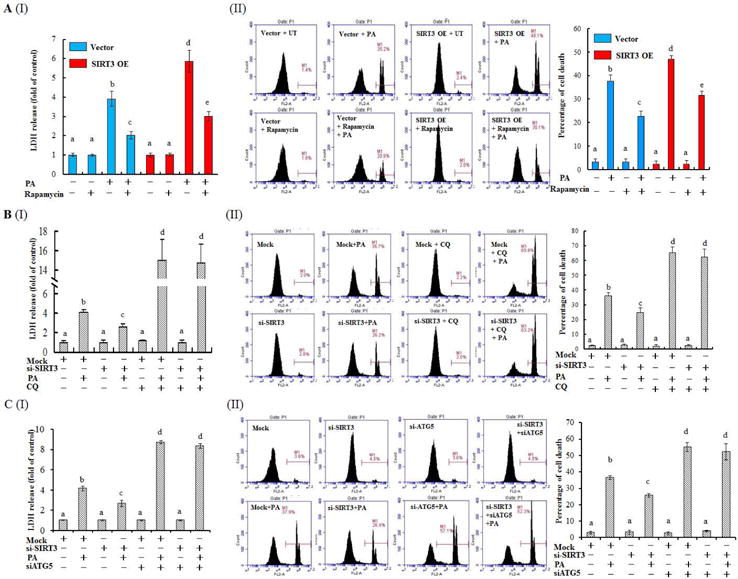
Autophagy alteration contributes to SIRT3-regulated lipotoxicity in HepG2 cells. (A) SIRT3 OE and vector control HepG2 cells were treated with 0.5 mM PA for 12 h with or without 1 h pre-incubation with 50 nM rapamycin. Cell death was detected by the measurements of LDH release and PI staining, respectively. (B) HepG2 cells were transfected with either scramble siRNA or SIRT3 siRNA for overnight, followed with 0.5 mM PA exposure for 12 h. Autophagy inhibitor CQ (20 μM) was added 1 h ahead of PA treatment. Cell death was detected. (C) HepG2 cells were co-transfected with siRNA for SIRT3 and Atg5 siRNA, followed with 0.5 mM PA exposure for 12 h. Cell death was detected. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly, p ˂ 0.05.
SIRT3 Negatively Regulates Autophagy via Suppressing AMPK
AMPK activation induces autophagy by suppressing mammalian target of rapamycin C1 (mTORC1).(28) To determine the role of AMPK in SIRT3-regulated autophagy suppression, we first examined the effect of SIRT3 on AMPK activation. As shown in Fig. 5A, SIRT3 overexpression was associated with suppressed AMPK activation, evidenced by decreased protein phosphorylation of AMPK and ACC, an AMPK target protein, as well as increased phospho-p70S6k1, a well-known downstream target of mTORC1 activation. Moreover, SIRT3 overexpression blunted AICAR-instigated AMPK activation (Supplementary data, Fig. 15). Conversely, SIRT3 siRNA silencing led to a robust enhancement of AMPK activation and p70S6k1 inhibition (Fig. 5A; Supplementary data, Fig. 16). Accordingly, the livers from SIRT3−/− mice demonstrated upregulated p-AMPK and p-ACC levels, as well as decreased phospho-p70S6k1 abundance in comparison to wild type mice (Fig. 5B; Supplementary data, Fig. 17A), while the opposite changes of these proteins were observed in the livers from hepatocyte-specific SIRT3 overexpressing mice (Fig. 5C).
Figure 5.
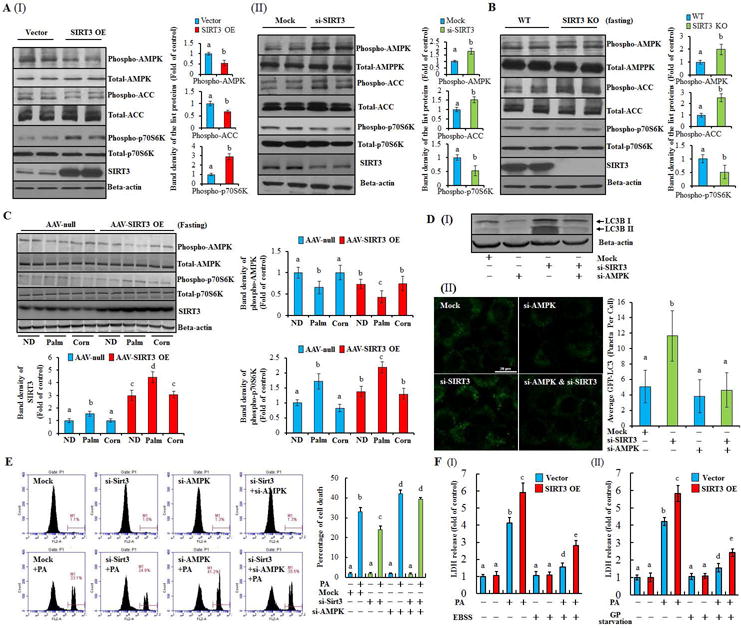
AMPK is mechanistically involved in SIRT3-regulated autophagy and lipotoxicity in hepatocytes. (A) Total cellular lysates were obtained from SIRT3 OE or SIRT3 knocking-down HepG2 cells. Immunoblotting assay was performed for SIRT3, phospho-AMPK, -ACC, -p70s6k, and SIRT3. (B) Immunoblotting assay was performed for phospho-AMPK, -ACC, -p70s6k, and SIRT3 expressions in the liver form SIRT3−/− or wild type mice after 12 h fasting (n = 5). (C) Immunoblotting assay was performed for phospho-AMPK, p-70s6k, and SIRT3 expressions in the liver form liver specific SIRT3 OE mice. (D) HepG2 cells were co-transfected with siRNAs for SIRT3 and AMPK. The expression of LC3-II was detected (I); HepG2 cells were transfected with recombinant adenovirus GFP-LC3, and subsequently re-seeded into the chamber (BD Falcon, CA) and co-transfected with siRNAs for SIRT3 and AMPK. Green puncta was counted (II). (E) HepG2 cells were treated with PA after co-transfecting with siRNAs for SIRT3 and AMPK. Cell death was detected. (F) SIRT3 OE or vector control HepG2 cells were treated with PA in either EBSS (amino acid and serum-depleted) or GP starvation (glucose, L-glutamine, pyruvate, and serum-depleted) medium, respectively. LDH release was detected. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly, p ˂ 0.05.
Inasmuch as the existence of a reverse association between SIRT3 and AMPK activity, we next sought to determine if AMPK is mechanistically implicated in the effect of SIRT3 on autophagy. AMPK siRNA silencing prevented LC3-II conversion and LC3 puncta accumulation resulting from SIRT3 suppression (Fig. 5D), indicating that AMPK is the main downstream target of SIRT3 in regulating autophagy. Although both ERK1/2 and Akt were reported to regulate autophagy and be potentially impacted by SIRT3 activation,(29) their involvement was not supported by our investigations (Supplementary data, Fig. 17).
PA inhibits AMPK activation in hepatocytes.(30) In accordance, we observed that PA strongly inhibited AMPK activation both in vitro and in vivo, whereas OA activated AMPK (Supplementary data, Fig. 18). Importantly, we showed that silencing AMPK blunted the protection against PA-induced cell death in SIRT3 siRNA- transfected HepG2 cells (Fig. 5E; Supplementary data, Fig. 19). In contrast, either EBSS (amino acid and serum-depleted) or GP starvation (glucose, L-glutamine, pyruvate, and serum-depleted) medium protected against PA-induced cell death in both control and SIRT3-overexpressing cells, both being associated with the strong activation of AMPK and autophagy (Fig. 5F; Supplementary data, Fig. 20 & 21). These results altogether suggest that AMPK activation contributes to SIRT3-regulated autophagy and sensitivity to PA-induced lipotoxicity.
Altered Intracellular Redox Status Contributes to the Regulatory Role of SIRT3 on AMPK
Other than altered AMP/ATP ratio, intracellular redox status modulates AMPK activation.(31) To gain insight on mechanism(s) involved in SIRT3-regulated AMPK activation, we first examined the effect of SIRT3 on intracellular redox status. As shown in Fig. 6A, SIRT3 overexpression markedly decreased intracellular O2•− levels, while intracellular H2O2 content was significantly increased. In contrast, siRNA silencing of SIRT3 significantly enhanced intracellular O2•− levels but reduced H2O2 content (Fig. 6B; Supplementary data, Fig. 22). These observations directed us to subsequently analyze the effect of O2•− on AMPK activity and autophagy by treating HepG2 with rotenone, a well-established O2•− inducer that acts via inhibition of mitochondrial complex I. As expected, rotenone exposure led to a significant increase of intracellular O2•− production (Supplementary data, Fig. 23A). Interestingly, cellular p-AMPK level was also markedly increased in response to rotenone exposure (Supplementary data, Fig. 23B). Importantly, both rotenone-induced O2•− overproduction and AMPK activation were associated with autophagy induction, as shown by increased LC3-II conversion and LC3 puncta accumulation (Supplementary data, Fig. 23B & C). Reducing intracellular O2•− by N-acetylcysteine (NAC), a O2•− cleaner (Supplementary data, Fig. 24A), markedly reduced p-AMPK abundance, and inhibited autophagy (Supplementary data, Fig. 24B & C). Furthermore, we showed that AMPK siRNA silencing abolished rotenone-triggered LC3-II conversion, while AICAR-induced LC3-II conversion was not affected by NAC supplementation (Fig. 6C). These data altogether suggest that decreased O2•− generation contributes to SIRT3 overexpression-induced AMPK inhibition and resultant autophagy suppression in hepatocytes.
Figure 6.
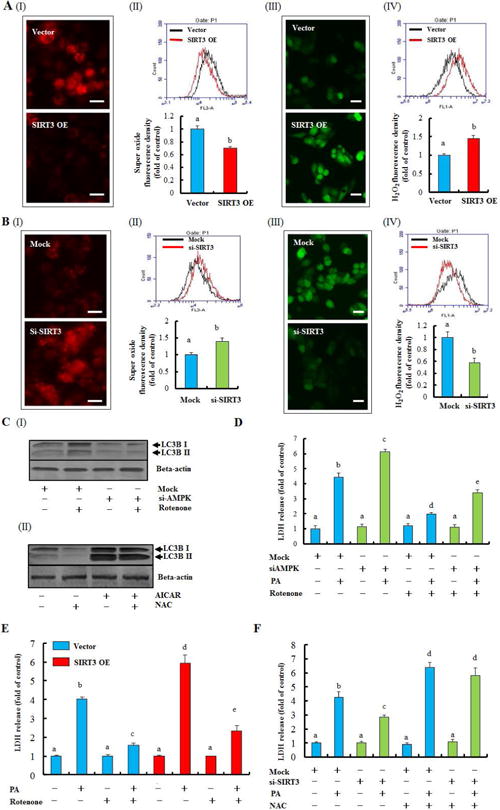
Redox-mediated AMPK activity contributes to SIRT3-regulated autophagy and lipotoxicity. Intracellular O2•− and H2O2 were measured in SIRT3 OE and vector control HepG2 cells, and also in HepG2 cells with SIRT3 siRNA transfection by fluorescence microscope and flow cytometry. (A) O2•− level (I & II); H2O2 level (III & IV). (B) O2•− level (I & II); H2O2 level (III & IV). (C) HepG2 cells were incubated with rotenone (10 nM) for 12 h after AMPK siRNA transfection. LC3-II was detected (I); AICAR (2.5 mM) was added 1 h ahead of NAC (5 mM, 12 h) treatment. LC3-II was detected (II). (D) HepG2 cells were transfected with AMPK siRNA and exposed to 0.5 mM PA for 12 h. Rotenone (10 nM) was added 1 h before PA treatment. LDH release was detected. (E) SIRT3 OE and vector control cells were incubated with 0.5 mM PA with or without 1 h rotenone (10 nM) pre-treatment. LDH release was detected. (F) HepG2 cells were transfected with SIRT3 siRNA and exposed to 0.5 mM PA for 12 h. NAC (5 mM) was added 1 h before PA treatment. LDH release was detected. All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly, p ˂ 0.05.
Our data support that SIRT3 overexpression-induced autophagy suppression involves AMPK inhibition via depleting cellular O2•− levels. We next asked whether the alteration of intracellular redox status is able to affect PA-induced lipotoxicity. As shown in Supplementary data, Fig. 25A, increasing intracellular O2•− by rotenone markedly alleviated PA-induced cell death, which was abolished by both siRNA silencing of AMPK and autophagy inhibition (Fig. 6D; Supplementary data, Fig. 25B). In addition, rotenone also alleviated PA-induced cell death in SIRT3 overexpressing cells (Fig. 6E). Conversely, depleting cellular O2•− content by NAC aggravated PA-induced lipotoxicity (Supplementary data, Fig. 26). Accordingly, NAC blunted the protection conferred by SIRT3 siRNA silencing against PA-induced cell death (Fig. 6F).
SIRT3 Mediated Intracellular O2•− Homeostasis via Regulating Manganese Superoxide Dismutase (MnSOD) Activity
SIRT3-mediated deacetylation reaction enhances MnSOD activity,(20) a mitochondrial antioxidant enzyme catalyzing the conversion of O2•− to H2O2. In line with previous reports, our result showed that whereas SIRT3 overexpression robustly decreased acetylated MnSOD protein abundance, SIRT3 silencing by siRNA led to increased MnSOD protein acetylation (Fig. 7A & B). Of note, treatment of HepG2 cells with 2-methoxyestradiol (2-ME), a commonly used MnSOD inhibitor, mimicked the effect of SIRT3 siRNA knockdown on intracellular ROS production (Fig, 7C; Supplementary data, Fig. 27A & B), whereas MnSOD overexpression in HepG2 cells mimicked the effect of SIRT3 overexpression (Fig. 7D). Correspondingly, 2-ME prevented PA-induced cell death in both normal and SIRT3-overexpressing cells (Fig. 7E; Supplementary data, Fig. 27C), which, as expected, was blunted by autophagy suppression (Supplementary data, Fig. 27D), while MnSOD overexpression suppressed autophagy and aggravated PA-induced cell death (Fig. 7F; Supplementary data, Fig. 28 & 29).
Figure 7.
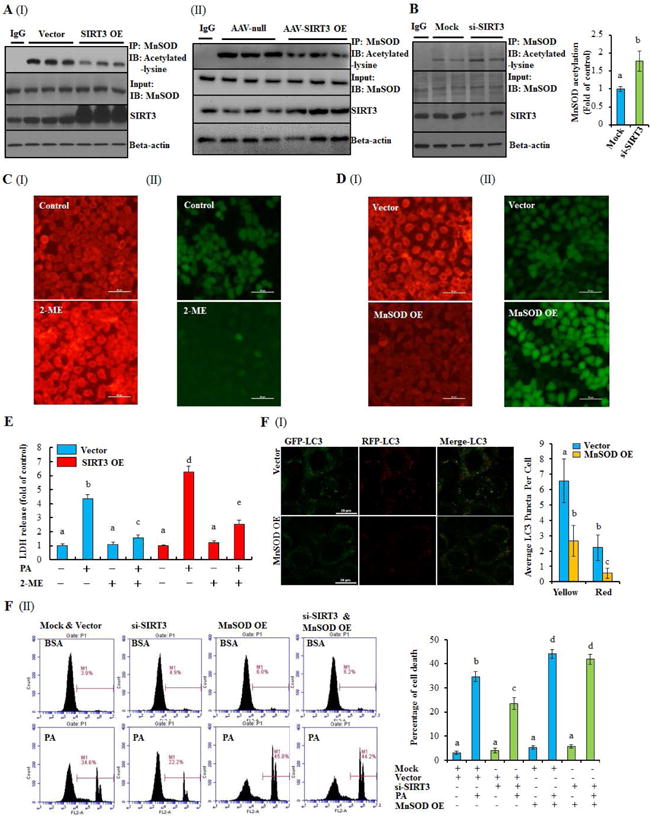
SIRT3 aggravates lipotoxicity via de-acetylating MnSOD through mediating intracellular O2•− homeostasis. (A) & (B) Altered MnSOD acetylation was detected in SIRT3 OE HepG2 cells, AAV-SIRT3 OE livers, or SIRT3 silencing HepG2 cells. (C) HepG2 cells were treated with MnSOD inhibitor, 2-ME (100 μM), for 12 h. O2•− (I) and H2O2 level (II) were detected. (D) HepG2 cells were transfected with recombinant lentivirus containing human MnSOD or empty vector as control. O2•− (I) and H2O2 level (II) were detected. (E) SIRT3 OE and control HepG2 cells were incubated with 0.5 mM PA with or without 1 h pretreatment of 2-ME (100 μM). LDH release was detected. (F) MnSOD OE or vector control HepG2 cells were transfected with recombinant adenovirus mRFP-GFP-LC3. Yellow and red puncta was counted (I). SIRT3 siRNA was transfected into MnSOD OE or vector control HepG2 cells, and followed with 0.5 mM PA treatment. Cell death was detected by flow cytometry (II). All values are denoted as means ± SD from three or more independent batches of cells. Bars with different characters differ significantly, p ˂ 0.05.
Palmitate Exposure is Associated with MnSOD Deacetylation and Reduced O2•− Generation
Finally, the effects of PA exposure on MnSOD protein acetylation status and intracellular O2•− homeostasis were determined. In harmony with its positive effect on SIRT3, a 12-hour PA exposure (0.5 mM) evidently decreased MnSOD protein acetylation (Fig. 8A). In accordance, PA exposure reduced intracellular O2•− levels and attenuated rotenone-triggered intracellular O2•− increase (Fig. 8B & C).
Figure 8.
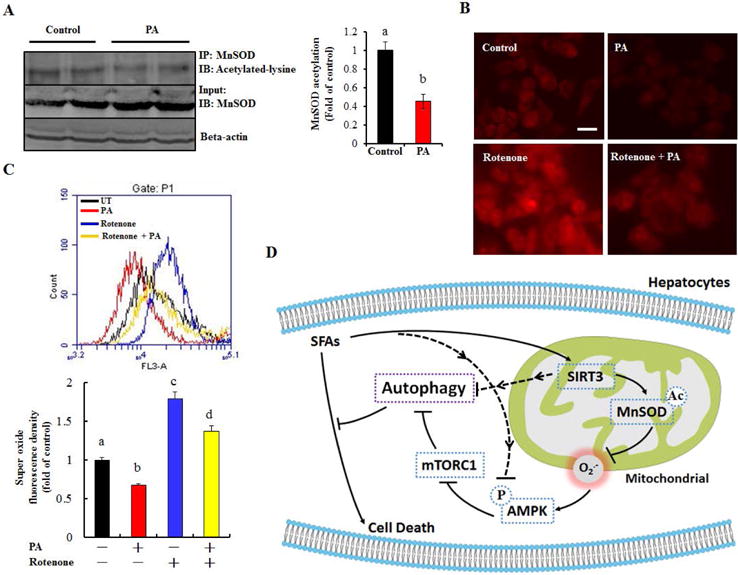
Palmitate-reduced intracellular O2•− is associated with MnSOD deacetylation. (A) HepG2 cells were treated with 0.5 mM PA for 12 h. Acetylated-MnSOD was detected. (B) & (C) HepG2 cells were treated with 0.5 mM PA for 12 h. Rotenone (10 nM) was added 1 h before PA treatment. Intracellular O2•− level was detected. All values are denoted as means ± SD. Bars with different characters differ significantly, p ˂ 0.05. (D) The proposed model of SIRT3-mediated hepatocyte susceptibility to lipotoxicity. The dotted lines denote the potential alternative mechanisms which were not investigated by the current study.
Discussion
Lipotoxicity plays a pathological role in the development of NAFLD.(32) However, the exact mechanism(s) underlying SFAs-triggered hepatocyte cell death remains to be fully elucidated. In the present study, we provide evidence for a novel SIRT3-dependent mechanism exacerbating SFAs-induced lipotoxicity in hepatocytes. We demonstrate that hepatic SIRT3 overexpression renders hepatocytes more susceptible to SFAs-induced cell death and aggravates NAFLD progression in mice in the setting of SFAs-rich HFD feeding through negatively regulating autophagy induction. Our results reveal that AMPK inhibition, resulting from MnSOD activation-derived O2•− depletion, is mechanistically involved in SIRT3-induced autophagy suppression and aggravated lipotoxicity in hepatocytes (see proposed model in Fig. 8D).
SIRT3 plays a pivotal role in maintaining mitochondrial protein acylation homeostasis, and thereby metabolism, by deacetylaing critical enzymes in a variety of metabolic pathways.(16–21, 25) SIRT3 activation is one of the major mechanisms accounting for many calorie restriction-derived benefits.(20, 33) Similarly, the beneficial effects of SIRT3 activation have been reported by numerous studies in a variety of experimental and clinical settings.(34, 35) In this work, we demonstrate that SFAs and unsaturated FAs differentially regulate SIRT3 expression in hepatocytes. Unexpectedly, our data suggest that SIRT3 activation is associated with increased susceptibility of hepatocytes to PA-induced cell death. These observations are in direct contrast with a previous report that HFD feeding downregulated hepatic SIRT3 expression and that SIRT3 activation conferred beneficial effect on PA-induced hepatotoxicity by preventing oxidative stress derived from mitochondrial dysfunction.(36) These discrepancies are difficult to be fully clarified at present. However, one possible explanation can be the difference in the SIRT3 overexpression protocol employed. In comparison to the transient SIRT3 overexpression used by Bao et al.,(36) our stable overexpression system mimics in vivo experimental conditions and is therefore more clinically relevant. Also noteworthy is that, although oxidative stress has been mechanistically implicated in PA-induced lipotoxicity in several cell types, including hepatocytes, our previous study documented that PA exposure was associated with increased intracellular glutathione concentration in HepG2 cells,(37) suggestive of the existence of oxidative stress-independent mechanism(s) in PA-induced hepatotoxicity. Furthermore, exacerbated PA-induced hepatocyte cell death by the antioxidant NAC implied that a certain amount of ROS could in fact be required to protect against lipotoxicity.(37)
Our studies point to a critical role of SIRT3 as a negative regulator of autophagy in hepatocytes, leading to exacerbated SFAs-induced lipotoxicity. Our data revealed that SIRT3 gene silencing/knockout is associated with AMPK activation in hepatocyte/the liver. These observations are consistent with a previous report which demonstrated that the reduced SIRT3 expression represents a fundamental mechanism in metformin-induced AMPK activation in hepatocytes.(38) Conversely, SIRT3 overexpression in HepG2 cells led to a significant inhibition of AMPK activation. This observation disagree with a previous report showing that SIRT3 overexpression in HepG2 cells was associated with AMPK activation.(39) The transient SIRT3 overexpression system employed in that study might potentially account for the discrepancy. AMPK activation induces autophagy by inhibiting mTORC1, a strong inhibitor of autophagy.(28) Given the well-documented positive role of AMPK activity in the regulation of autophagy, we therefore postulate that AMPK may represent a mechanistic link between SIRT3 and autophagy. This notion is supported by our data that genetic inhibition of AMPK alleviated autophagy induction in SIRT3 siRNA-transfected hepatocytes and abolished SIRT3 silencing-conferred protection against lipotoxicity, while activating AMPK significantly reduced PA-induced cell death in SIRT3-overexpressed hepatocytes. Nevertheless, under regular diet, mice with liver AAV-SIRT3 OE intriguingly failed to recapitulate the effects of SIRT3 on AMPK and mTORC1 as observed in SIRT3 OE hepatocytes, in spite of apparent autophagy induction. This observation implied the existence of the AMPK/mTORC1 signaling pathway-independent mechanism in the regulation of SIRT3 OE-instigated autophagy suppression in the in vivo system. On the other hand, we observed that, under high palm oil-HFD feeding, AAV-SIRT3 OE mice manifested suppressed hepatic AMPK activation and increased mTORC1 activation (p-S6K1) in comparison to that in AAV-null mice (Fig. 5C), suggesting that the effect of SIRT3 on AMPK/mTOR signaling pathway is context-dependent, which may require environmental/dietary cues.
AMPK is a positive regulator of autophagy. It was previously reported that SIRT3−/− mice manifested a significantly reduced hepatic ATP level in comparison to wild type animals.(16) Moreover, AMPK activation by metformin was associated with SIRT3 downregulation and the subsequent reduction of cellular ATP levels in both hepatocytes and the liver, which was recovered by SIRT3 overexpression.(39) These observations provided the first line of evidence supporting the existence of a negative relationship between SIRT3 and AMPK. Other than altered AMP/ATP ratio, it was also reported that O2•− plays an important role in the activation of AMPK and autophagy.(31, 40) To gain further insight on the mechanism(s) involved in SIRT3-triggered AMPK suppression, we investigates the effect of SIRT3 on ROS production and their role in AMPK activation. Our studies provide initial evidence suggesting that O2•− depletion plays a mechanistic role in SIRT3 overexpression-induced AMPK suppression. This notion is supported by several findings: 1. SIRT3-overexpressing HepG2 cells have significantly low levels of O2•−, whereas hydrogen peroxide was significantly increased. Conversely, SIRT3 gene silencing was associated with markedly increased O2•− levels, while hydrogen peroxide levels were significantly reduced; 2. Rotenone, a potent O2•− inducer via suppressing mitochondrial complex I, increased cellular O2•− production, activated AMPK, induced autophagy, and protected HepG2 cells against lipotoxicity; 3. NAC, a potent ROS scavenger, inhibited AMPK and autophagy activation, and potentiated PA-induced hepatotoxicity; 4. MnSOD negatively regulates O2•− production and AMPK activation. Overall, our studies support the notion that SIRT3-mediated O2•− depletion mechanistically links SIRT3 activation to AMPK suppression.
MnSOD, the mitochondrial form of SOD converting O2•− into H2O2, is crucial for the maintenance of mitochondria-generated O2•− homeostasis. Interestingly, MnSOD is a target of SIRT3, whose deacetylation by SIRT3 results in enhanced enzymatic activity.(20) In line with previous reports, we found that SIRT3 overexpression robustly decreased MnSOD protein acetylation, whereas its silencing by siRNA led to increased acetylated MnSOD. Moreover, we found that MnSOD inhibition mimics SIRT3 gene silencing in terms of the ROS generation pattern, whereas MnSOD overexpression mimics SIRT3 overexpression phenotype. Importantly, similar to SIRT3 siRNA transfection, MnSOD inhibition prevented PA-induced cell death in both normal and SIRT3-overexpressing HepG2 cells. These data suggest that SIRT3 mediates mitochondria-derived O2•− protection via affecting MnSOD acetylation status.
The negative effect of palmitate on AMPK was previously reported in several cell types, including hepatocytes, the exact mechanism(s) remain unclear. The ceramide-dependent PP2A activation seemed to play an important role in palmitate-induced AMPK dephosphorylation/suppression.(41, 42) Moreover, palmitate-induced PPAR-α suppression was also reported to contribute to AMPK inhibition.(43) The cell culture experiments in the present study provided strong evidence that SIRT3 upregulation is mechanistically involved in palmitate-elicited AMPK suppression. At the same time, we also recognized that our in vitro observations were not able to be completely recapitulated in our in vivo investigations. Specifically, our data showed that long-term high palm oil-HFD feeding exerted much profound effect on AMPK/mTORC1 signaling pathway than AAV-SIRT3 overexpression did. Therefore, the role of SIRT3-independent pathways in the regulation of palmitate-induced AMPK inhibition cannot be excluded and warrant further investigations.
In summary, this study provides initial experimental evidence that SFAs exposure of hepatocytes leads to SIRT3 upregulation and activation both in vitro and in vivo, which in turn exacerbates SFAs-triggered hepatotoxicity by negatively regulating autophagy induction. Intriguingly, our studies uncover that AMPK activation due to MnSOD activation-dependent O2•− depletion plays a mechanistic role in SIRT3-mediated autophagy suppression in hepatocytes. Given that the both muscle- and liver-specific SIRT3 knockout mice do not manifest any overt metabolic phenotype under either chow or high-fat diet conditions(44) and that SIRT3 knockout mice are protected from acetaminophen-induced hepatotoxicity,(45) our findings suggest that curbing SIRT3 overactivation can be a potential therapeutic choice for the treatment of NAFLD as well as other metabolic disorders, with lipotoxicity being the principal pathomechanism.
Supplementary Material
Acknowledgments
We thank Dr. Giamila Fantuzzi (University of Illinois at Chicago) for her technical support and great assistance during manuscript preparation.
Financial Support: This study was supported by grants from NIH-NIAAAR01 AA017442 (Z Song), Natural Science Foundation of China (81470845, Z Song; 81573132, S Li; and 81473393 X Dou), Natural Science Foundation of Heilongjiang Province (LC2015030, S Li), and National Basic Program Research of China (973 program, 2014 CB543001, X Dou).
Abbreviation
- NAFLD
non-alcoholic fatty liver disease
- SA
stearic acid
- PA
palmitic acid
- SFAs
saturated fatty acids
- ER
endoplasmic reticulum
- SIRTs
Sirtuins
- NAD
nicotinamide adenine dinucleotide
- AMPK
AMP-activated protein kinase
- OA
oleic acid
- BSA
bovine serum albumin
- ALT
alanine aminotransferase
- DMEM
Dulbecco’s Modified Eagle Medium
- LDH
lactate dehydrogenase
- PI
propidium iodide
- TEM
Transmission electron microscopy
- CQ
chloroquine
- IP
immunoprecipitation
- ROS
Reactive oxygen species
- O2•−
intracellular superoxide
- H2O2
hydrogen peroxide
- MAP1
microtubule-associated protein 1
- LC3
microtubule-associated protein 1 light chain 3
- EBSS
Earle’s Balanced Salt Solution
- SCD-1
stearoyl-CoA desaturase-1
- NAM
nicotinamide
- Baf
bafilomycin A1
- mTORC1
mammalian target of rapamycin C1
- AICAR
5-Aminoimidazole-4-carboxamide1-β-D-ribofuranoside
- NAC
N-acetylcysteine, MnSOD, manganese superoxide dismutase
- 2-ME
2-methoxyestradiol
Footnotes
Conflicts of Interest
None.
References
- 1.Schaffer JE. Lipotoxicity: when tissues overeat. Curr Opin Lipidol. 2003;14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Angulo P. Nonalcoholic fatty liver disease. The New England journal of medicine. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 3.Ibrahim SH, Kohli R, Gores GJ. Mechanisms of lipotoxicity in NAFLD and clinical implications. J Pediatr Gastroenterol Nutr. 2011;53:131–140. doi: 10.1097/MPG.0b013e31822578db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baylin A, Kabagambe EK, Siles X, Campos H. Adipose tissue biomarkers of fatty acid intake. The American journal of clinical nutrition. 2002;76:750–757. doi: 10.1093/ajcn/76.4.750. [DOI] [PubMed] [Google Scholar]
- 5.Lu J, Wang Q, Huang L, Dong H, Lin L, Lin N, Zheng F, et al. Palmitate causes endoplasmic reticulum stress and apoptosis in human mesenchymal stem cells: prevention by AMPK activator. Endocrinology. 2012;153:5275–5284. doi: 10.1210/en.2012-1418. [DOI] [PubMed] [Google Scholar]
- 6.Muller C, Gardemann A, Keilhoff G, Peter D, Wiswedel I, Schild L. Prevention of free fatty acid-induced lipid accumulation, oxidative stress, and cell death in primary hepatocyte cultures by a Gynostemma pentaphyllum extract. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2012;19:395–401. doi: 10.1016/j.phymed.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell death and differentiation. 2005;12(Suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Li J, Shen C, Zhang X, Sun S, Cho M, Sun C, et al. tert-Butylhydroquinone (tBHQ) protects hepatocytes against lipotoxicity via inducing autophagy independently of Nrf2 activation. Biochim Biophys Acta. 2014;1841:22–33. doi: 10.1016/j.bbalip.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ, Lee JH, Kim Y, et al. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology. 2009;150:126–134. doi: 10.1210/en.2008-0483. [DOI] [PubMed] [Google Scholar]
- 11.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 13.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Cimen H, Han MJ, Shi T, Deng JH, Koc H, Palacios OM, et al. NAD+-dependent deacetylase SIRT3 regulates mitochondrial protein synthesis by deacetylation of the ribosomal protein MRPL10. J Biol Chem. 2010;285:7417–7429. doi: 10.1074/jbc.M109.053421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 20.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 21.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lombard DB, Tishkoff DX, Bao J. Mitochondrial sirtuins in the regulation of mitochondrial activity and metabolic adaptation. Handb Exp Pharmacol. 2011;206:163–188. doi: 10.1007/978-3-642-21631-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allison SJ, Milner J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle. 2007;6:2669–2677. doi: 10.4161/cc.6.21.4866. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Fu LL, Wen X, Wang XY, Liu J, Cheng Y, Huang J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giralt A, Villarroya F. SIRT3, a pivotal actor in mitochondrial functions: metabolism, cell death and aging. Biochem J. 2012;444:1–10. doi: 10.1042/BJ20120030. [DOI] [PubMed] [Google Scholar]
- 26.Law IK, Liu L, Xu A, Lam KS, Vanhoutte PM, Che CM, Leung PT, et al. Identification and characterization of proteins interacting with SIRT1 and SIRT3: implications in the anti-aging and metabolic effects of sirtuins. Proteomics. 2009;9:2444–2456. doi: 10.1002/pmic.200800738. [DOI] [PubMed] [Google Scholar]
- 27.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy B, Pattanaik AK, Das J, Bhutia SK, Behera B, Singh P, Maiti TK. Role of PI3K/Akt/mTOR and MEK/ERK pathway in Concanavalin A induced autophagy in HeLa cells. Chem Biol Interact. 2014;210:96–102. doi: 10.1016/j.cbi.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Lee J, Hong SW, Chae SW, Kim DH, Choi JH, Bae JC, Park SE, et al. Exendin-4 improves steatohepatitis by increasing Sirt1 expression in high-fat diet-induced obese C57BL/6J mice. PLoS One. 2012;7:e31394. doi: 10.1371/journal.pone.0031394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han Y, Wang Q, Song P, Zhu Y, Zou MH. Redox regulation of the AMP-activated protein kinase. PLoS One. 2010;5:e15420. doi: 10.1371/journal.pone.0015420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. Journal of hepatology. 2003;39:978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 33.Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kincaid B, Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front Aging Neurosci. 2013;5:48. doi: 10.3389/fnagi.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim M, Lee JS, Oh JE, Nan J, Lee H, Jung HS, Chung SS, et al. SIRT3 Overexpression Attenuates Palmitate-Induced Pancreatic beta-Cell Dysfunction. PLoS One. 2015;10:e0124744. doi: 10.1371/journal.pone.0124744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med. 2010;49:1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dou X, Wang Z, Yao T, Song Z. Cysteine aggravates palmitate-induced cell death in hepatocytes. Life Sci. 2011;89:878–885. doi: 10.1016/j.lfs.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buler M, Aatsinki SM, Izzi V, Hakkola J. Metformin reduces hepatic expression of SIRT3, the mitochondrial deacetylase controlling energy metabolism. PLoS One. 2012;7:e49863. doi: 10.1371/journal.pone.0049863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi T, Fan GQ, Xiao SD. SIRT3 reduces lipid accumulation via AMPK activation in human hepatic cells. J Dig Dis. 2010;11:55–62. doi: 10.1111/j.1751-2980.2009.00416.x. [DOI] [PubMed] [Google Scholar]
- 40.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 41.Liangpunsakul S, Sozio MS, Shin E, Zhao Z, Xu Y, Ross RA, Zeng Y, Crabb DW. Inhibitory effect of ethanol on AMPK phosphorylation is mediated in part through elevated ceramide levels. Am J Physiol Gastrointest Liver Physiol. 2010;298:G1004–12. doi: 10.1152/ajpgi.00482.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–88. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 43.Sun Y1, Ren M, Gao GQ, Gong B, Xin W, Guo H, Zhang XJ, Gao L, Zhao JJ. Chronic palmitate exposure inhibits AMPKalpha and decreases glucose-stimulated insulin secretion from beta-cells: modulation by fenofibrate. Acta Pharmacol Sin. 2008;29:443–50. doi: 10.1111/j.1745-7254.2008.00717.x. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez-Marcos PJ, Jeninga EH, Canto C, Harach T, de Boer VC, Andreux P, Moullan N, et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci Rep. 2012;2:425. doi: 10.1038/srep00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu Z, Bourdi M, Li JH, Aponte AM, Chen Y, Lombard DB, Gucek M, et al. SIRT3-dependent deacetylation exacerbates acetaminophen hepatotoxicity. EMBO Rep. 2011;12:840–846. doi: 10.1038/embor.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.