

Abstract
Toxin-induced liver diseases lack effective therapies despite increased understanding of the role factors such as an overactive innate immune response play in the pathogenesis of this form of hepatic injury. Pentamidine is an effective antimicrobial agent against several human pathogens, but studies have also suggested that this drug inhibits inflammation. This potential anti-inflammatory mechanism of action, together with the development of a new oral form of pentamidine isethionate VLX103, led to investigations of the effectiveness of this drug in the prevention and treatment of hepatotoxic liver injury. Pretreatment with a single injection of VLX103 in the D-galactosamine (GalN) and lipopolysaccharide (LPS) model of acute, fulminant liver injury dramatically decreased serum alanine aminotransferase levels, histological injury, the number of terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL)-positive cells and mortality as compared to vehicle-injected controls. VLX103 decreased GalN/LPS induction of TNF but had no effect on other proinflammatory cytokines. VLX103 prevented the proinflammatory activation of cultured hepatic macrophages and partially blocked liver injury from GalN/TNF. In GalN/LPS-treated mice, VLX103 decreased activation of both the mitochondrial death pathway and downstream effector caspases 3 and 7 which resulted from reduced c-Jun N-terminal kinase activation and initiator caspase 8 cleavage. Delaying VLX103 treatment for up to 3 h after GalN/LPS administration was still remarkably effective in blocking liver injury in this model. Oral administration of VLX103 also decreased hepatotoxic injury in a second more chronic model of alcohol-induced liver injury, as demonstrated by decreased serum alanine and aspartate aminotransferase levels and numbers of TUNEL-positive cells.
Conclusion
VLX103 effectively decreases toxin-induced liver injury in mice and may be an effective therapy for this and other forms of human liver disease.
Keywords: alcoholic liver disease, galactosamine, pentamidine, tumor necrosis factor, VLX103
Toxin-induced hepatocyte injury is a common etiology of human liver disease. Despite an increased understanding of the pathophysiology of toxin-induced liver injury, therapies for the major forms of human hepatotoxicity, such as alcohol-induced liver disease, remain inadequate or non-existent. Central to the pathogenesis of toxin-induced hepatocyte injury and cell death is hepatocellular damage from proinflammatory factors generated by the innate immune response. Inflammatory mediators of toxic liver injury include cytokines such as TNF and reactive oxygen species that are injurious to toxin-damaged hepatocytes.(1,2) Hepatotoxins also cause intestinal permeabilization(3,4) which leads to increased delivery to the liver of gut bacterial products such as lipopolysaccharide (LPS) that stimulate hepatic macrophages to produce TNF and other factors that lead to liver injury.(5,6) Macrophage-produced TNF triggers hepatocyte apoptosis through TNF receptor 1. The cascade of events in this hepatocyte death pathway includes initiator caspase 8 activation, cleavage of the pro-apoptotic Bcl-2 family member Bid to tBid, and translocation of tBid to the mitochondrial membrane to trigger the release of apoptogenic factors such as cytochrome c that activate effector executioner caspases 3 and 7.(7) The hepatic inflammatory response is therefore a potential therapeutic target to prevent activation of this death pathway in toxin-induced liver disease.
Pentamidine is an antimicrobial agent with well-established effectiveness and safety in several human protozoan diseases.(8) Pentamidine may have anti-inflammatory properties that contribute to its efficacy against infectious pathogens. Pentamidine inhibits TNF production from LPS stimulation or Pneumocystis carinii infection in cultured alveolar macrophages, although the effect is partial.(9,10) The absence of effects on TNF mRNA or intracellular protein levels in these studies suggested that the mechanism could be inhibition of secretion.(9) Pentamidine also decreases serum TNF and IL-6 levels after high-dose LPS injection in rodents.(11,12) The effectiveness of pentamidine in other inflammatory responses has not been examined with the exception of the ability of the drug to mitigate dextran sodium sulphate-induced murine colitis.(13)
The importance of inflammation in the pathogenesis of hepatotoxic liver injury, together with the development of a new, oral form of pentamidine isethionate VLX103, suggested that this drug might be effective in toxin-induced liver disease. To examine this possibility, we determined the effects of this drug on both acute fulminant liver injury from galactosamine (GalN)/LPS and a more chronic and milder alcohol model of hepatic injury. LPS stimulation of cytotoxic TNF has been implicated in the pathogenesis of liver injury in these models.(14,15) VLX103 significantly decreased liver injury in both models. Mechanistic studies in the GalN/LPS model revealed that VLX103 moderately reduced TNF production and blocked caspase 8 and mitochondrial death pathway activation. VLX103 was protective even when given well after the initiation of GalN/LPS-induced hepatic injury. These findings indicate that VLX103 may be an effective therapy for toxin-induced human liver disease.
Materials and Methods
Animal Models
C57BL/6J male and female mice were maintained under 12 h light/dark cycles with unlimited access to food and water. Mice were feed a standard chow diet (LabDiet, St. Louis, MO; PicoLab® Rodent Diet 20 #5053) at all times except during alcohol diet feeding. Mice were housed in static cages with ¼ inch corncob bedding, individual water bottles and cotton nestlets for enrichment. During alcohol diet feeding cages also contained a paper igloo. Littermates were used as vehicle-treated controls for all experiments.
GalN/LPS liver injury was induced in 10-14 week old male mice by an intraperitoneal injection of 100 μg/kg of LPS (E. coli 0111:B4) and 700 mg/kg of D-galactosamine (Sigma-Aldrich, St. Louis, MO) dissolved in phosphate-buffered saline (PBS), as previously performed.(16) The GalN/LPS mortality studies were performed with an LPS dose of 10 μg/kg. For the pretreatment studies, an equal volume of PBS vehicle or pentamidine isethionate (VLX103, supplied by Verlyx Pharma Inc, Montreal, QC, Canada) at a dose of 25, 40 or 50 mg/kg was injected intraperitoneally 30 min prior to GalN/LPS administration. For the post-treatment experiments, PBS or VLX103 40 mg/kg was injected at various times after GalN/LPS.
Additional C57BL/6J male mice were injected with GalN plus TNF, as previously described.(2) Mice were pretreated with PBS or VLX103 40 mg/kg intraperitoneally. At 30 min after this pretreatment, the mice were injected with GalN intraperitoneally, and then another 30 min later injected intravenously with 10 ug/kg of mouse recombinant TNF (PeproTech, Rocky Hill, NJ).
Studies of alcohol-induced injury employed a modified NIAAA alcohol model.(17) Female 10-week old mice were administered 5 days of liquid control diet (Bio-Serv, Flemington, NJ; #F1259SP). Mice were then randomized to receive a 5% liquid ethanol diet (Bio-Serv; #F1258SP), or pair-fed an equal caloric amount of the control diet. At this time the mice were started on a daily gavage of normal saline vehicle or VLX103 75 mg/kg dissolved in normal saline. After ten days of control/ethanol diet feeding, a single gavage of ethanol 5 g/kg, or an equal volume and caloric intake of maltose dextrin, was administered to the mice. Mice were sacrificed 9 h after gavage. All animal studies were approved by the Animal Care and Use Committees of the Albert Einstein College of Medicine and Emory University School of Medicine and followed the National Institutes of Health guidelines for animal care.
Serum Assays
Serum alanine (ALT) and aspartate aminotransferases (AST) were measured by commercial kits (TECO Diagnostics, Anaheim, CA).
Histology
Hematoxylin and eosin-stained livers were evaluated for the amount of hepatic parenchyma with apoptosis/necrosis or inflammation by a single pathologist in a blinded fashion on a semi-quantitative scale of: 0, absent; 0.5, minimal; 1, mild; 1.5, mild to moderate; 2, moderate; 2.5, moderate to severe; 3, severe; and 4 markedly severe.
Terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) assay
TUNEL positive cells in liver sections were stained with the commercial kit DeadEnd Colorimetric System (Promega, Madison, WI). Under light microscopy, the numbers of TUNEL positive cells in 10 randomly selected high power fields (200× magnification) were counted per liver section.
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from livers with the commercial kit RNeasy Plus (QIAGEN, Valencia, CA). Reverse transcription was performed with 1 μg of RNA in an Eppendorf Mastercycler (Hamburg, Germany) using a high capacity cDNA reverse transcription kit (ABI, Foster City, CA). Annealing of primers was done at 25°C for 10 min, followed by elongation at 37°C for 2 h and inactivation of the enzyme at 85°C for 5 min. Negative controls (no added transcriptase) were performed in parallel. Real-time PCR was performed in duplicate in a 7500 Fast Real-Time PCR System (ABI). The primer sequences in Supplemental Table 1 were purchased from Integrated DNA Technologies (Coralville, IA). PCR was carried out using Power SYBR Green Master Mix (ABI). Taq polymerase was activated at 95°C for 10 min. The cycling parameters were denaturation at 95°C for 30 sec and extension at 60°C for 1 min (for 40 cycles). Data analysis was performed using the 2-ΔΔCT method for relative quantification. Samples were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Cytokine assay
Cytokine levels were measured in serum samples with a multiplex kit (MCYTOMAG-70K; EMD Millipore, Billerica, MA) and Luminex® xMAP® Technology.
Macrophage isolation and treatments
Bone marrow cells were isolated from untreated C57BL/6J mice and differentiated into bone marrow derived macrophages (BMDM), as previously described.(18) BMDM were pretreated with VLX103 at various concentrations for 1 h, administered LPS (10 ng/ml) and IFNγ (100 ng/ml) (R&D Systems, Minneapolis, MN), and harvested at 12 h for qRT-PCR.
Hepatic macrophages were isolated by Nycodenz gradient from untreated C57BL/6J mice and cultured, as previously described.(19) Following overnight culture, the cells were pretreated with 10 μM VLX103 and 1 h later treated with LPS and IFNγ for harvest at 6 h for qRT-PCR analysis.
Protein isolation and western blotting
Total protein and mitochondrial and cytosolic protein fractions were isolated from mouse liver and western blotting performed, as previously described.(20,21) Membranes were probed with the following antibodies: caspase 3 (Cell Signaling, Beverly, MA; #9665), caspase 7 (Cell Signaling; #9492), poly (ADP-ribose) polymerase (PARP; Cell Signaling; #9542), tubulin (Cell Signaling; #2148), cytochrome c (BD Biosciences, San Jose, CA; #556433), cytochrome oxidase (Abcam, Cambridge, MA; #14744), caspase 8 (R&D Systems; #AF1650), p50 NF-κB (Santa Cruz Biotechnology, Dallas, TX; #SC-114), p65 NF-κB (Santa Cruz; #SC-109), lamin A/C (Cell Signaling; #2032), cIAP1 (Enzo Life Sciences, Farmingdale, NY; #ALX-803-335-C100), cIAP2 (Santa Cruz; #SC-7944), superoxide dismutase 2 (SOD2) (Enzo Life Sciences; #ADI-SOD-111-D), Bcl-XL (Cell Signaling; #2764), phospho-c-Jun N-terminal kinase (JNK; Cell Signaling; #9252), phospho-JNK (Cell Signaling; #4671) and Bid (kind gift of Xiao-Ming Yin, University of Indiana).
Caspase 8 activity
Caspase 8 activity was detected with a fluorometric commercial kit (Biovision, Milpitis, CA). Reactions were carried out on 200 ug protein at 37°C for 2 h, and the samples were read in a fluorometer with 400-nm excitation and 505-nm emission filters.
Triglyceride content
Hepatic triglyceride content was determined using a kit according to the manufacturer's instructions (Sigma) and values normalized to wet liver weight.
Statistical analysis
Numerical results are reported as means ± S.E. and derived from at least three independent experiments unless otherwise indicated. The unpaired Student's t-test was used to assess significance between control and treated groups. Survival rates were compared by the Tukey multiple comparison test. Statistical significance was defined as P<0.05.
Results
VLX103 prevents liver injury from GalN/LPS
To examine the possible efficacy of VLX103 treatment in hepatotoxic liver injury, we determined whether pretreatment altered acute liver injury from GalN/LPS. C57BL/6J mice were injected with PBS vehicle or 25 or 40 mg/kg of VLX103 30 min prior to GalN/LPS. In C57BL/6J mice, GalN/LPS-induced liver injury is detectable at 4 h and becomes severe by 6 h at which time lethality ensues. VLX103 pretreatment remarkably improved gross liver appearance after GalN/LPS with an absence of the typical signs of hemorrhage (Fig. 1A). Liver injury as determined by serum ALT levels was markedly decreased by both VLX103 concentrations (Fig. 1B). Blinded, histological grading confirmed that VLX103 significantly reduced liver injury at 6 h. (Fig. 1C and 1D). Hepatocyte death was decreased as determined by significantly reduced numbers of TUNEL-positive cells in VLX103-treated livers (Fig. 2A and 2B). VLX103 pretreatment therefore dramatically reduced liver injury from GalN/LPS.
Fig. 1.
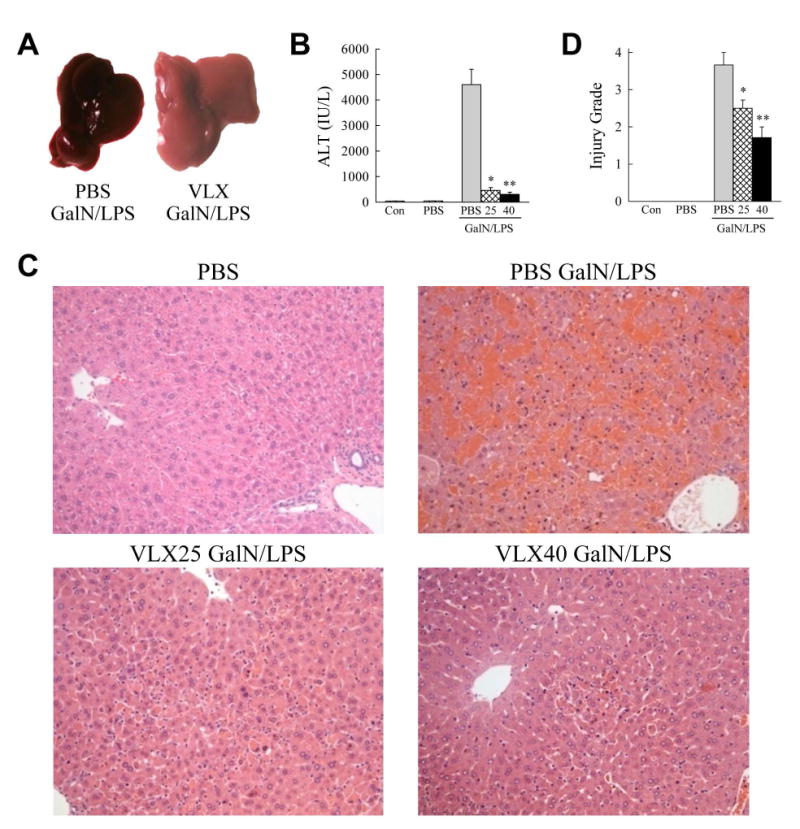
VLX103 prevents GalN/LPS-induced liver injury. (A) Images of PBS- and VLX103-treated livers 6 h after GalN/LPS. (B) Serum ALT levels in uninjected controls (Con), mice injected with PBS alone, and 6 h GalN/LPS-injected mice pretreated with PBS or 25 or 40 mg/kg of VLX103 (*P<0.0001, **P<0.0000001 as compared to PBS + GalN/LPS; n=4-15). (C) Representative hematoxylin and eosin stained livers sections from untreated and GalN/LPS-treated mice (200×). (D) Histological grades of liver injury in untreated and GalN/LPS-treated mice (*P<0.02, **P<0.001 as compared to PBS + GalN/LPS; n=3-7).
Fig. 2.
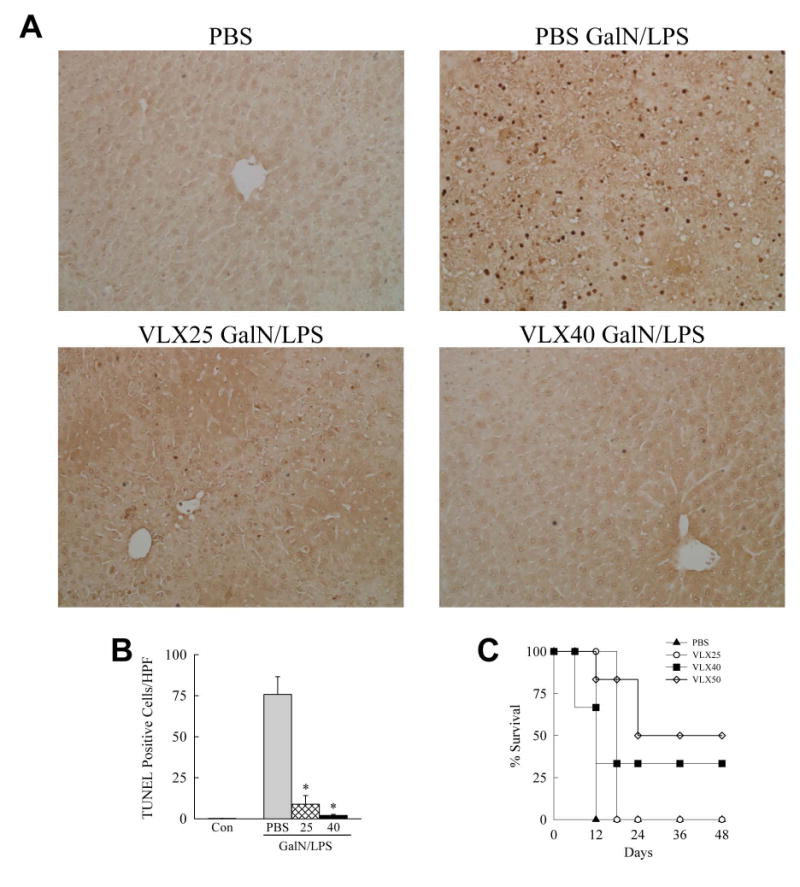
VLX103 decreases cell death and increases survival. (A) TUNEL-stained livers from mice with the indicated treatments (200×). (B) Quantification of TUNEL-positive cells per high power field (HPF) (*P<0.0002 as compared to PBS + GalN/LPS; n=6-7). (C) Percentage survival after pretreatment with PBS or the indicated VLX103 concentrations and GalN/LPS (P<0.004 for 40 mg/kg and P<0.001 for 50 mg/kg; n=6-12).
VLX103 decreases GalN/LPS-induced mortality
GalN/LPS triggers fulminant hepatic failure and death. To determine whether VLX103 merely delayed liver injury, or had a sustained protective effect that prevented mortality, survival was examined. PBS-pretreated mice all died within 12 h of GalN/LPS, whereas VLX103-treated mice had a dose-dependent improvement in survival (Fig. 2C). Mice pretreated with 25 mg/kg had a prolonged survival time but succumbed to death within 18 h. Mice receiving the 40 or 50 mg/kg doses had a significantly improved survival with 50% surviving with the highest dose (Fig. 2C). Subsequent GalN/LPS studies were largely performed with the 40 mg/kg dose which significantly blocked both liver injury and improved survival.
VLX103 reduces the induction of TNF
GalN/LPS liver injury is mediated by hepatocyte sensitization to cytotoxicity from LPS-induced macrophage cytokines particularly TNF.(14,22) To determine whether VLX103 was protective through TNF down regulation, hepatic cytokine mRNA induction was assayed by qRT-PCR. Tnf mRNA levels after GalN/LPS treatment were decreased significantly by 47% at 1 h, but unchanged at 2 and 4 h in VLX103-treated mice (Fig. 3A). VLX103 had no significant effect on Ifnγ, Il-1β or Il-6 mRNA levels (Fig. 3A). Consistent with the mRNA findings, serum TNF levels were decreased significantly by 54% at 1 h but unaffected at later times, and other cytokines were unchanged by VLX103 (Fig. 3B). Decreased liver injury from VLX103 was therefore associated with an early decrease in TNF but not other proinflammatory cytokines.
Fig. 3.
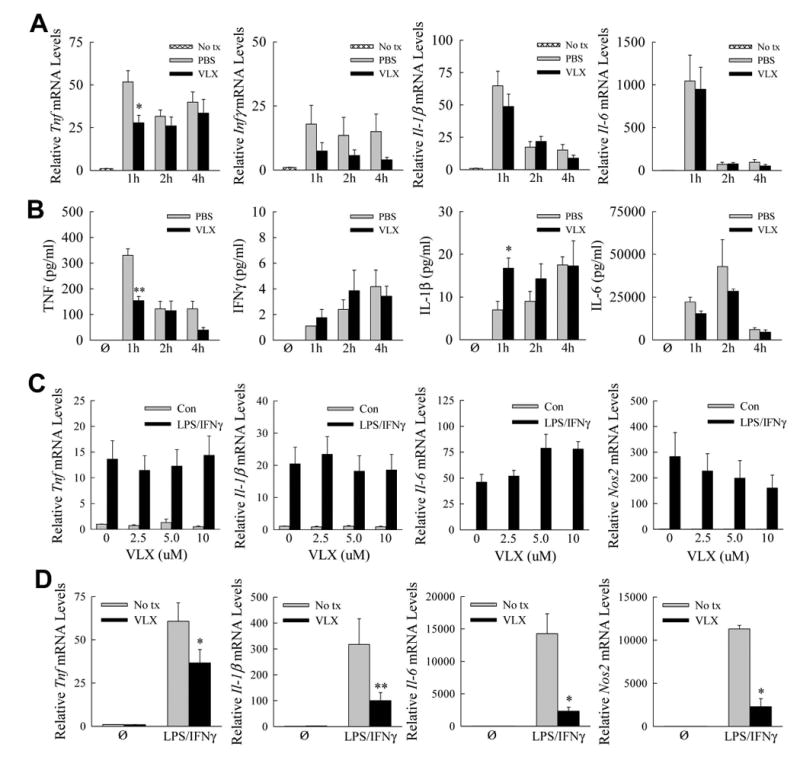
Effects of VLX103 on proinflammatory cytokines. (A) Relative hepatic mRNA levels for Tnf, Ifnγ, Il-1β, and Il-6 as determined by qRT-PCR in untreated mice (No tx) and PBS- and VLX103-pretreated mice at the indicated hours after GalN/LPS administration (*P<0.01 as compared to PBS-treated at the same time point; n=5-6). (B) Serum levels of TNF, IFNγ, IL-1β and IL-6 in untreated mice (Ø) and PBS- and VLX103-pretreated mice after GalN/LPS (*P<0.05, **P<0.0001 as compared to PBS-treated at the same time point; n=4-8). (C) BMDM were not pretreated (0) or pretreated with 2.5 -10.0 μM VLX103 and then left untreated (Con) or were treated with LPS/IFNγ for 12 h. Relative mRNA levels are shown for the genes indicated (n=3-9). (D) Relative mRNA levels for the indicated genes in cultured hepatic macrophages that were not pretreated (No tx) or pretreated with VLX103 and then not treated (Ø) or treated with LPS/IFNγ for 6 h (*P<0.04, **P<0.01 as compared to cells that did not receive VLX103; n=4-5).
VLX103 decreases hepatic macrophage activation and TNF-induced liver injury
The finding of decreased TNF production, together with reports that pentamidine is anti-inflammatory in alveolar macrophages,(9,10) suggested that VLX103 may directly suppress injurious proinflammatory hepatic macrophage activation. Cultured BMDM were pretreated with increasing VLX103 concentrations and stimulated with LPS/IFNγ for polarization into a proinflammatory M1 phenotype.(18) VLX103 failed to inhibit induction of the cytokine genes Tnfα, Il-1β, and Il-6 or M1-marker gene Nos2 (Fig. 3C). In hepatic macrophages, however, the drug did inhibit proinflammatory activation as indicated by significant reductions in these same genes (Fig. 3D).
To determine whether TNF suppression is a mechanism of VLX103 hepatoprotection, drug effect was examined in GalN-sensitized mice co-treated with TNF. VLX103 pretreatment partially but significantly reduced liver injury from GalN/TNF as determined by decreased ALT levels and TUNEL staining (Fig. 4A and 4B). There was a trend to improved histology which did not reach statistical significance (Fig. 4C). Although significant, the reduction in liver injury was markedly less than in the GalN/LPS model (Fig. 1 and 2). Together these in vitro and in vivo findings demonstrate that a partial mechanism of VLX103's hepatoprotective effect is through inhibition of macrophage generation of TNF.
Fig. 4.
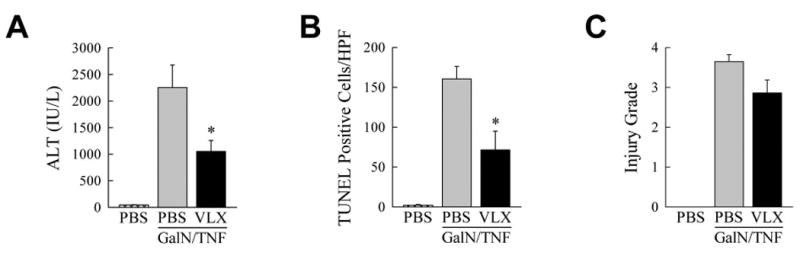
VLX103 decreases GalN/TNF liver injury. (A) ALT levels in PBS-injected controls, and mice pretreated with PBS or VLX103 and then GalN/TNF for 5 h (*P<0.02, compared to mice treated with PBS + GalN/TNF; n=4-7). (B) Numbers of TUNEL-positive cells in the same mice (*P<0.01, compared to mice treated with PBS + GalN/TNF; n=4-7). (C) Histological grades of liver injury (n=4-7).
VLX103 blocks activation of the mitochondrial death pathway
Hepatic toxicity from GalN/LPS occurs through the classical TNF death cascade of caspase 8-mediated Bid cleavage into tBid which translocates to mitochondria to trigger cytochrome c release, effector caspase cleavage and apoptosis.(7) Decreased TUNEL positivity suggested that VLX103 blocked TNF-induced apoptosis at some point along this pathway. Supportive of this idea was that VLX103 induced a dose-dependent decrease in the active, cleaved forms of effector caspases 3 and 7 at 6 h after GalN/LPS (Fig. 5A). Deceased caspase activity was confirmed by parallel findings of a dose-dependent reduction in cleavage of the caspase substrate PARP in VLX103-treated livers (Fig. 5A).
Fig. 5.
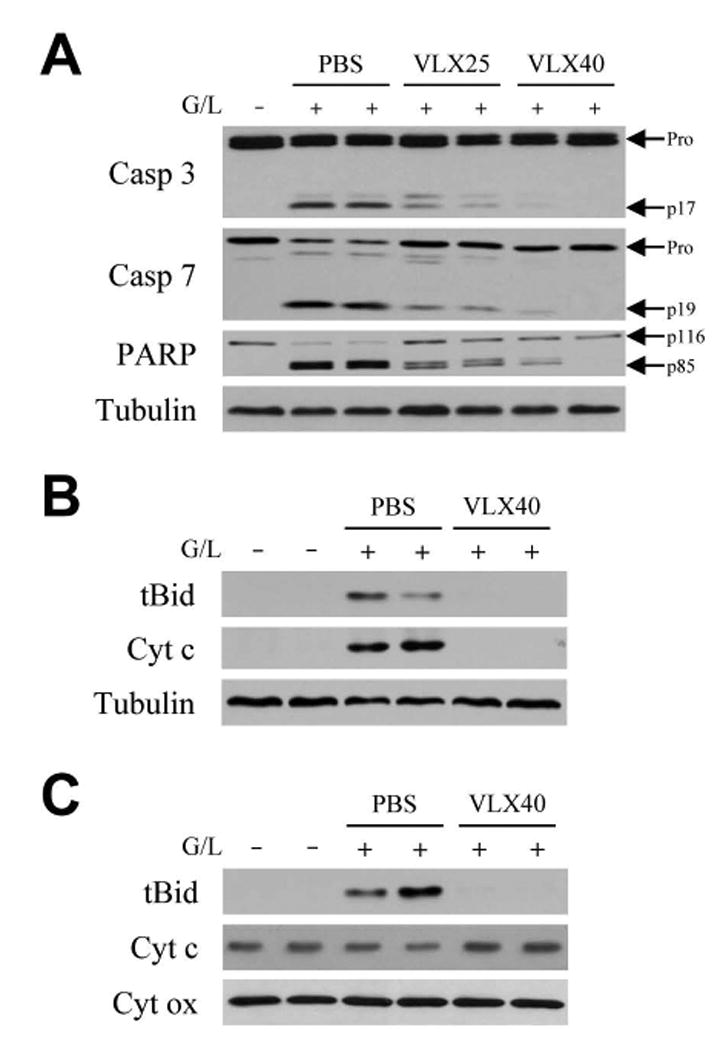
VLX103 blocks effector caspase cleavage and mitochondrial death pathway activation. (A) Immunoblots of total hepatic protein from untreated (G/L -) or 6 h GalN/LPS-treated (G/L +) mice that were pretreated with PBS, or VLX103 at 25 (VLX25) or 40 mg/kg (VLX40). Blots were probed for caspase 3 (Casp 3), caspase 7 (Casp 7), PARP, and tubulin as a loading control. Arrows indicate procaspases (Pro), cleaved caspase 3 (p17) and 7 (p19) forms, and intact (p116) and cleaved (p85) PARP. (B) Cytosolic fractions from the same mice pretreated with PBS or VLX103 40 mg/kg and immunoblotted for tBid, cytochrome c (Cyt c) and tubulin. (C) Immunoblots of mitochondrial protein isolates for tBid, cytochrome c and cytochrome oxidase (Cyt ox) as a loading control. Results are representative of 3 independent experiments.
Examination of the upstream mitochondrial pathway that triggers effector caspase cleavage revealed that VLX103 prevented its activation. Cytosolic tBid cleavage was blocked by VLX103 (Fig. 5B), and tBid was virtually absent in mitochondria from VLX103-treated livers in contrast to the high levels in PBS-treated mice (Fig. 5C). As a result, the decrease in mitochondrial levels of cytochrome c (Fig. 5C), and its release into the cytoplasm (Fig. 5B), were blocked in VLX103-treated mice. VLX103 therefore decreased apoptosis from GalN/LPS by mitigating mitochondrial death pathway and effector caspase activation.
VLX103 inhibits caspase 8 activation
The marked decrease in mitochondrial death pathway activation suggested that VLX103 blocked hepatocyte death from GalN/LPS through inhibition of upstream caspase 8. Increased hepatic caspase 8 activity was detectable in PBS-treated mice at 2-6 h after GalN/LPS treatment, but activity was decreased significantly at 4 and 6 h by VLX103 (Fig. 6A). Immunoblots confirmed that cleavage of caspase 8 to its active p18 form was decreased by VLX103 (Fig. 6B). VLX103 therefore blocked caspase 8 activation, preventing Bid cleavage and mitochondrial death pathway activation that induce liver injury from GalN/LPS.
Fig. 6.
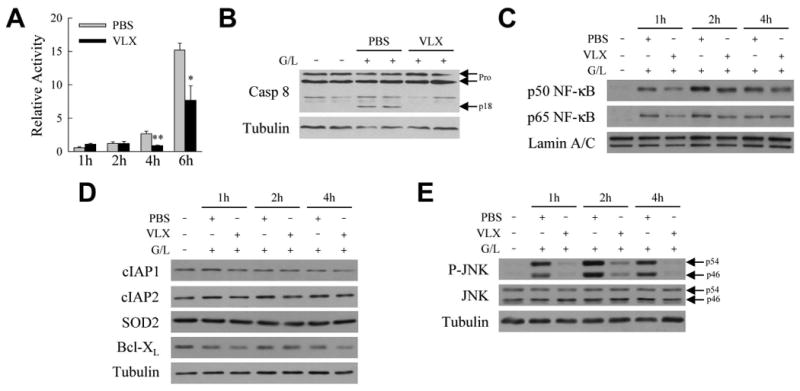
VLX103 blocks caspase 8 activation. (A) Hepatic caspase 8 activity relative to untreated mice in mice pretreated with PBS or VLX103 40 mg/kg and GalN/LPS for the hours indicated (*P<0.01, **P<0.002 as compared to PBS-treated at the same time point; n=4-6). (B) Immunoblots of total hepatic protein from untreated and 4 h GalN/LPS-treated mice pretreated with PBS or VLX103 40 mg/kg probed for caspase 8 (Casp 8) and tubulin. Arrows indicate the procaspases (Pro) and cleaved caspase 8 (p18). (C) Nuclear protein fractions from untreated and GalN/LPS-treated mice with PBS or VLX 40 mg/kg pretreatment immunoblotted for the phosphorylated p50 and p65 subunits of NF-κB and the loading control lamin A/C. (D) Immunoblots of total liver protein from the same mice for cIAP1, cIAP2, SOD2 and Bcl-XL. (E) Total liver protein immunoblots probed for phospho-JNK (P-JNK), total JNK and tubulin. The p54 and p46 isoforms of JNK are indicated by arrows. Results of B-E are representative of 3 independent experiments.
TNF induction of hepatocyte caspase 8 activation is down regulated by NF-κB which curtails JNK phosphorylation.(21,23) Examination of VLX103's effect on NF-κB signaling revealed that hepatic NF-κB activation occurred in both vehicle- and VLX103-treated mice as indicated by nuclear translocation of the phosphorylated p50 and p65 NF-κB subunits (Fig. 6C). Levels of proteins that mediate hepatocyte resistance to TNF-dependent apoptosis such as cIAP1, cIAP2, SOD2 and Bcl-XL(7) were unaffected by VLX103 (Fig. 6D). Despite the absence of an effect on NF-κB activation, VLX103 did markedly reduce levels of JNK activation as indicated by decreased levels of phosphorylated JNK (Fig. 6E).
VLX103 treatment is hepatoprotective post-GalN/LPS administration
For human liver disease a therapeutic agent must be effective when administered after the onset of injury. We therefore determined whether VLX103 was hepatoprotective when administered after the initiation of GalN/LPS-induced injury. Although the greatest reduction in hepatic injury was afforded by VLX103 pretreatment, administration of this agent even at 2 and 3 h after GalN/LPS significantly decreased liver injury. Serum ALT levels were reduced 75% by VLX103 given 2-3 h after GalN/LPS (Fig. 7A). TUNEL staining revealed a diminished but still significant decrease in hepatocyte death with VLX103 post-treatment (Fig. 7B). VLX103 treatment 3 h post-GalN/LPS was highly effective in reducing cytosolic (Fig. 7C) and mitochondrial levels of tBid, and mitochondrial cytochrome c loss (Fig. 7D) and release into the cytosol (Fig. 7C). As a result VLX103 post-treatment was highly effective in decreasing caspase 3 and 7 and PARP cleavage (Fig. 7E). These findings indicate that VLX103 is therapeutic when administered significantly after GalN/LPS, even immediately before the onset of liver injury.
Fig. 7.
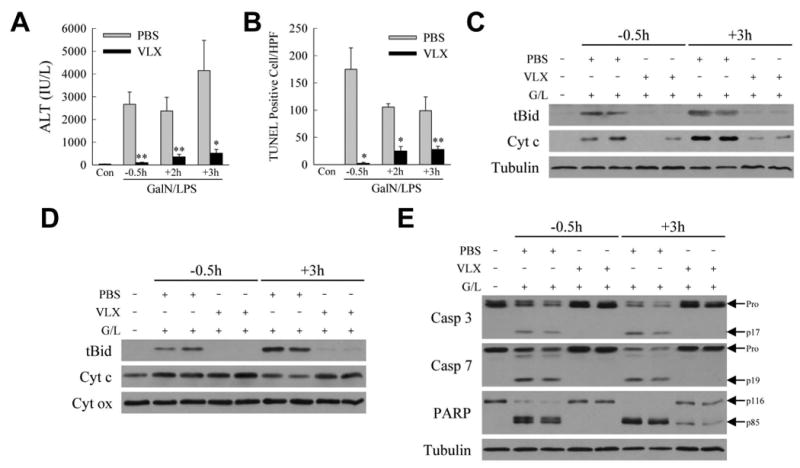
Post-GalN/LPS VLX103 treatment decreases liver injury. Serum ALT levels (A), and numbers of TUNEL positive cells (B), in mice treated with VLX103 40 mg/kg 0.5 h before (-0.5 h) or 2 or 3 h after (+2 h and +3 h) GalN/LPS (*P<0.05, **P<0.01 as compared to PBS-treated at the same time point; n=4-7). (C) Immunoblots of cytosolic protein fractions from the same mice probed for tBid, cytochrome c (Cyt c) and tubulin. (D) Immunoblots of mitochondrial protein fractions with cytochrome oxidase (Cyt ox) as the loading control. (E) Total liver protein from the mice immunoblotted for caspase 3 (Casp 3), caspase 7 (Casp 7), PARP, and tubulin. The pro- and cleavage-forms of caspase 3 and 7 and PARP are indicated. Immunoblots are representative of 3 independent experiments.
VLX103 protects against alcoholic liver injury
The therapeutic potential of oral VLX103 was examined in a more chronic and non-lethal modified NIAAA model of alcoholic liver disease.(17) Mice received a daily gavage of vehicle or VLX103 (75 mg/kg) during 10 days of ethanol feeding, and then a binge dose of alcohol by gavage for analysis 9 h later. VLX103 decreased hepatic triglyceride content by 34% (Fig. 8A). In vehicle-treated mice serum ALT and AST increased 3.3- and 2.5-fold respectively, but levels were significantly decreased 50% by VLX103 (Fig. 8B and 8C). Histologically there was a non-significant trend to decreased steatosis, but differences were not detected in injury or inflammation, as the levels of both histological features were low with alcohol feeding (Fig. 8D). A reduction in liver injury was confirmed by findings of a significant decrease in TUNEL-positive cells with VLX103 (Fig. 8E) in association with decreased proinflammatory cytokine gene expression (Fig. 8F). Oral VLX103 was therefore effective in decreasing hepatotoxic liver injury from alcohol.
Fig. 8.
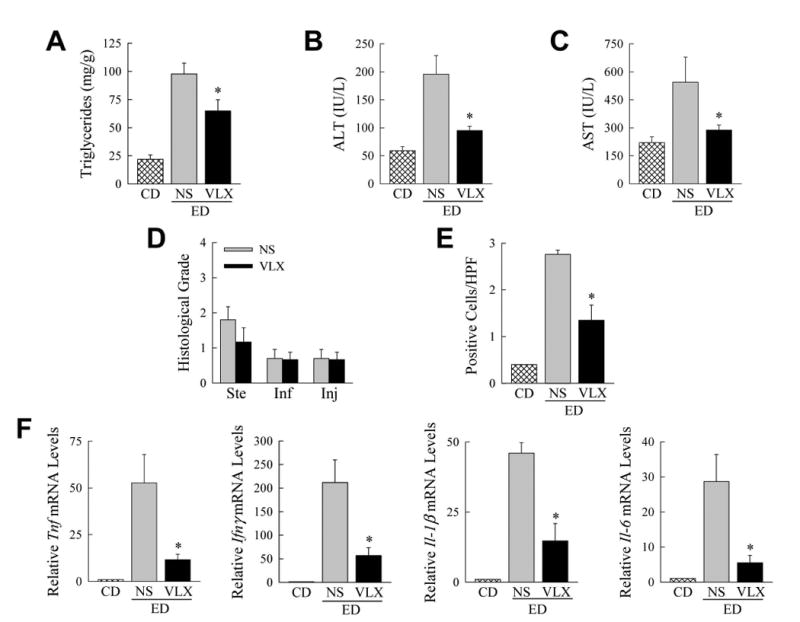
VLX103 blocks alcohol-induced liver injury. (A) Hepatic triglyceride content in mice that received control (CD) or ethanol diet (ED) with normal saline (NS) or VLX103 (75 mg/kg) by gavage (*P<0.01 as compared to ethanol diet-fed, normal saline-treated; n=7-9). Serum ALT (B) and AST (C) levels in the same mice (*P<0.005 as compared to ethanol diet-fed, normal saline-treated; n=7-9). (D) Histologic grades of steatosis (Ste), inflammation (Inf) and injury (Inj) (n=5-6). (E) Numbers of TUNEL-positive cells (*P<0.004 as compared to ethanol diet-fed, normal saline-treated; n=7-9). (F) Relative hepatic mRNA levels for Tnf, Ifnγ, Il-1β, and Il-6 (*P<0.02 as compared to ethanol diet-fed, normal saline-treated; n=3-4).
Discussion
Inflammatory mediators such as TNF promote hepatocellular injury in alcoholic hepatitis, ischemia/reperfusion, nonalcoholic steatohepatitis, viral hepatitis and fulminant hepatic failure.(7) Anti-inflammatory therapies might therefore be effective in liver diseases. The results of studies to date however, such as investigations blocking TNF, have been disappointing. Pentamidine is an antimicrobial agent used safely in humans for many years.(8) The mechanism of pentamidine's antimicrobial effect is unknown. In vitro macrophage and rodent studies have suggested that one mechanism of the drug's actions may be an anti-inflammatory effect.(9-13) In the present study we demonstrate for the first time that pentamidine significantly reduces hepatotoxic liver injury. Particularly significant findings from these studies are that VLX103 reduced GalN/LPS injury when administered after cell death initiation and induced lasting hepatoprotection that dramatically increased survival.
Consistent with prior studies in nonhepatic cells,(9-11) VLX103 decreased proinflammatory cytokine production. Hepatic TNF protein secretion was reduced in response to GalN/LPS, an effect that occurred in part at the transcriptional level, in contrast with previous in vitro studies indicating that pentamidine acts post-translationally.(9,24) TNF inhibition was partial with a 54% decrease at 1 h after GalN/LPS and no effect at later times. The reduction in TNF may reflect a direct anti-inflammatory effect on macrophages as VLX103 inhibited hepatic macrophage activation in vitro including TNF induction. Interestingly this finding was specific for hepatic macrophages, as VLX103 exerted no anti-inflammatory effect on BMDM. This discrepancy raises the interesting therapeutic possibility that VLX103 specifically targets tissue macrophages without systemic depression of the innate immune response, thereby potentially avoiding infectious complications that have doomed anti-TNF therapy in human alcoholic liver disease. The effects of VLX103 on cultured hepatic macrophages were different than with GalN/LPS as Il-1β and Il-6 were inhibited in vitro but not in the GalN/LPS mouse model. However, all of these cytokines were decreased by VLX103 in the in vivo alcohol model. Suppression of macrophage activation may have been less complete in the acute fulminant GalN/LPS model because of more extensive macrophage stimulants that partially overrode the anti-inflammatory effects of VLX103.
Alternatively, pentamidine binds LPS,(25) and VLX103 may inactivate LPS thereby removing the stimulus for immune cell activation through TLR4 signaling. The fact that induction of Il-1β and Il-6 was unaffected by VLX103 in GalN/LPS, however, indicates that the drug did not block the LPS initiation of inflammation. Although the reduction in TNF may partially explain VLX103's protective effects, this mechanism cannot be the sole mode of action as VLX103 treatment delayed to 3 h after GalN/LPS, a time long after LPS has triggered a proinflammatory cytokine response, was still highly effective. Consistent with the fact that an anti-TNF effect in part explains the efficacy of VLX103 is that delayed VLX103 administration was less effective than pretreatment. Findings of a significant but reduced protective effect of VLX103 in GalN/TNF-induced injury also support the conclusion that the hepatoprotective mechanisms of this drug are only partially due to TNF inhibition. VLX103 must therefore act through mechanisms other than direct anti-inflammatory effects on immune cells and/or LPS neutralization in this model of acute fulminant injury.
VLX103 also reduced liver injury from a chronic plus acute binge alcohol model. This study is notable for establishing drug efficacy in a second hepatotoxic injury model and for demonstrating the effectiveness of oral VLX103 in liver injury. In contrast to GalN/LPS, in the alcohol model VLX103 had a generalized anti-inflammatory effect as indicated by a significant reduction in multiple cytokines. These results are concordant with the generalized in vitro anti-inflammatory effect of VLX103 on hepatic macrophages. The different in vivo findings may be the result of a greater reduction in LPS due to the use of oral VLX103 in the alcohol model as opposed to intraperitoneal delivery for GalN/LPS. The reduction in triglycerides might be secondary to this anti-inflammatory effect as cytokines such as TNF promote excessive lipid accumulation.(26)
VLX103 also inhibited the hepatocyte extrinsic death pathway that is triggered by TNF. TNF-induced apoptosis results from type 1 TNF receptor binding which recruits a complex that triggers initiator caspase 8 activation by self-cleavage. Caspase 8 cleaves Bid into tBid which with other Bcl-2 family members initiates permeabilization of the outer mitochondrial membrane and release of proteins such as cytochrome c that activate effector caspases 3 and 7.(7) In the GalN/LPS model, VLX103 decreased activation of all elements of this pathway. That VLX103 affected initial caspase 8 cleavage, suggests that the drug acts at this level of the cascade. However, the reduction in caspase 8 cleavage was only partial, whereas inhibition of the mitochondrial death pathway as measured by cytochrome c release was virtually complete, suggesting that either partial caspase 8 inhibition was sufficient to achieve this effect or VLX103 acted upon several steps in the death cascade. Supportive of the effect being strictly at the level of caspase 8 inhibition is that caspase 8-dependent Bid cleavage was almost totally blocked.
The mechanism of caspase 8 activation from GalN/LPS is sustained JNK over activation.(21,23,27) VLX103 led to a reduction in activated, phosphorylated JNK in response to GalN/LPS indicating that the drug blocked caspase 8 activation through this mechanism. TNF-inducible NF-κB signaling down regulates JNK activation,(23) but NF-κB activation and levels of protective proteins were unaffected by VLX103. This finding suggests that VLX103 is a direct JNK inhibitor, but studies in cultured hepatocytes revealed no direct inhibitory effect of this compound on JNK phosphorylation (data not shown). Elucidation of the effects of VLX103 on JNK signaling will require additional study.
The findings demonstrate a novel therapeutic effect of pentamidine in toxin-induced liver injury that likely occurs through several mechanisms. The ability of VLX103 to have beneficial effects on both immune cell cytokine production and hepatocellular death pathways indicate a possible unique therapeutic strength of the drug, and suggest that VLX103 may have more widespread applications than toxin-induced liver injury. Supportive of this concept are that VLX103 reduced liver injury in a mouse model of nonalcoholic fatty liver disease.(28) In light of these findings, and the established human safety profile of pentamidine, VLX103 warrants further investigation as a possible therapy in human liver disease.
Supplementary Material
Acknowledgments
We thank Xiao-Ming Yin for the Bid antibody.
Supported by research grants from Verlyx Pharma Inc and NIH grants R01DK044234 and R01AA022601 (MJC).
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BMDM
bone marrow-derived macrophages
- GalN
D-galactosamine
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- SOD2
superoxide dismutase 2
- TUNEL
terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling
Footnotes
Conflict of Interest: This work was supported in part by research grants from Verlyx Pharma Inc, François Ravenelle is an employee of the company, and Mark J. Czaja is a Scientific Advisory Board member.
References
- 1.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol. 2007;22(Suppl 1):S45–48. doi: 10.1111/j.1440-1746.2006.04646.x. [DOI] [PubMed] [Google Scholar]
- 2.Amir M, Zhao E, Fontana L, Rosenberg H, Tanaka K, Gao G, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013;20:878–887. doi: 10.1038/cdd.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bjarnason I, Peters TJ, Wise RJ. The leaky gut of alcoholism: possible route of entry for toxic compounds. Lancet. 1984;1:179–182. doi: 10.1016/s0140-6736(84)92109-3. [DOI] [PubMed] [Google Scholar]
- 4.Li GZ, Wang ZH, Cui W, Fu JL, Wang YR, Liu P. Tumor necrosis factor alpha increases intestinal permeability in mice with fulminant hepatic failure. World J Gastroenterol. 2012;18:5042–5050. doi: 10.3748/wjg.v18.i36.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Czaja MJ, Xu J, Alt E. Prevention of carbon tetrachloride-induced rat liver injury by soluble tumor necrosis factor receptor. Gastroenterology. 1995;108:1849–1854. doi: 10.1016/0016-5085(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 6.Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mann A, Czaja MJ, Schattenberg JM. TNF signaling. In: Dufour JF, Clavien PA, editors. Signaling Pathways in Liver Diseases. 3rd. Hoboken: John Wiley & Sons; 2015. pp. 186–202. [Google Scholar]
- 8.Pearson RD, Hewlett EL. Pentamidine for the treatment of Pneumocystis carinii pneumonia and other protozoal diseases. Ann Intern Med. 1985;103:782–786. doi: 10.7326/0003-4819-103-5-782. [DOI] [PubMed] [Google Scholar]
- 9.Corsini E, Craig WA, Rosenthal GJ. Modulation of tumor necrosis factor release from alveolar macrophages treated with pentamidine isethionate. Int J Immunopharmacol. 1992;14:121–130. doi: 10.1016/0192-0561(92)90022-d. [DOI] [PubMed] [Google Scholar]
- 10.Corsini E, Dykstra C, Craig WA, Tidwell RR, Rosenthal GJ. Pneumocystis carinii induction of tumor necrosis factor-a by alveolar macrophages: modulation by pentamidine isethionate. Immunol Lett. 1992;34:303–308. doi: 10.1016/0165-2478(92)90228-g. [DOI] [PubMed] [Google Scholar]
- 11.Chen YL, Le Vraux V, Giroud JP, Chauvelot-Moachon L. Anti-tumor necrosis factor properties of non-peptide drugs in acute-phase responses. Eur J Pharmacol. 1994;271:319–327. doi: 10.1016/0014-2999(94)90789-7. [DOI] [PubMed] [Google Scholar]
- 12.Rosenthal GJ, Craig WA, Corsini E, Taylor M, Luster MI. Pentamidine blocks the pathophysiologic effects of endotoxemia through inhibition of cytokine release. Toxicol Appl Pharmacol. 1992;112:222–228. doi: 10.1016/0041-008x(92)90191-t. [DOI] [PubMed] [Google Scholar]
- 13.Esposito G, Capoccia E, Sarnelli G, Scuderi C, Cirillo C, Cuomo R, et al. The antiprotozoal drug pentamidine ameliorates experimentally induced acute colitis in mice. J Neuroinflammation. 2012;9:277. doi: 10.1186/1742-2094-9-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nowak M, Gaines GC, Rosenberg J, Minter R, Bahjat FR, Rectenwald J, et al. LPS-induced liver injury in D-galactosamine-sensitized mice requires secreted TNF-a and the TNF-p55 receptor. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1202–R1209. doi: 10.1152/ajpregu.2000.278.5.R1202. [DOI] [PubMed] [Google Scholar]
- 15.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, et al. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Singh R, Xiang Y, Greenbaum LE, Czaja MJ. Nuclear factor κB up-regulation of CCAAT/enhancer-binding protein β mediates hepatocyte resistance to tumor necrosis factor α toxicity. Hepatology. 2010;52:2118–2126. doi: 10.1002/hep.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilyas G, Zhao E, Liu K, Lin Y, Tesfa L, Tanaka KE, et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1β. J Hepatol. 2016;64:118–127. doi: 10.1016/j.jhep.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Schattenberg JM, Rigoli RM, Storz P, Czaja MJ. Hepatocyte resistance to oxidative stress is dependent on protein kinase C-mediated down-regulation of c-Jun/AP-1. J Biol Chem. 2004;279:31089–31097. doi: 10.1074/jbc.M404170200. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from JNK2-dependent activation of caspase-8 and the mitochondrial death pathway. J Biol Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Car BD, Eng VM, Schnyder B, Ozmen L, Huang S, Gallay P, et al. Interferon g receptor deficient mice are resistant to endotoxic shock. J Exp Med. 1994;179:1437–1444. doi: 10.1084/jem.179.5.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Lo CR, Czaja MJ. NF-βB inhibition sensitizes hepatocytes to TNF-induced apoptosis through a sustained activation of JNK and c-Jun. Hepatology. 2002;35:772–778. doi: 10.1053/jhep.2002.32534. [DOI] [PubMed] [Google Scholar]
- 24.Rosenthal GJ, Corsini E, Craig WA, Comment CE, Luster MI. Pentamidine: an inhibitor of interleukin-1 that acts via a post-translational event. Toxicol Appl Pharmacol. 1991;107:555–561. doi: 10.1016/0041-008x(91)90318-9. [DOI] [PubMed] [Google Scholar]
- 25.David SA, Bechtel B, Annaiah C, Mathan VI, Balaram P. Interaction of cationic amphiphilic drugs with lipid A: implications for development of endotoxin antagonists. Biochim Biophys Acta. 1994;1212:167–175. doi: 10.1016/0005-2760(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 26.Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298:G107–G116. doi: 10.1152/ajpgi.00391.2009. [DOI] [PubMed] [Google Scholar]
- 27.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFα- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 28.Zhao E, Ilyas G, Ravenelle F, Tanaka KE, Czaja MJ. Oral pentamidine (VLX103) is an effective treatment for high fat diet-induced NASH in mice. Hepatology. 2016;64(Suppl):798A. [Abstract] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.