

Abstract
Molecular profiling of glioblastomas has revealed the presence of key signaling hubs that contribute to tumor progression and acquisition of resistance. One of these main signaling mechanisms is the NF-κB pathway, which integrates multiple extracellular signals into transcriptional programs for tumor growth, invasion, and maintenance of the tumor-initiating population. We show here that an extracellular protein released by glioblastoma cells, fibulin-3, drives oncogenic NF-κB in the tumor and increases NF-κB activation in peritumoral astrocytes. Fibulin-3 expression correlates with a NF-κB-regulated “invasive signature” linked to poorer survival, being a possible tissue marker for regions of active tumor progression. Accordingly, fibulin-3 promotes glioblastoma invasion in a manner that requires NF-κB activation both in the tumor cells and their microenvironment. Mechanistically, we found that fibulin-3 activates the metalloprotease ADAM17 by competing with its endogenous inhibitor, TIMP3. This results in sustained release of soluble TNFα by ADAM17, which in turn activates TNF receptors and canonical NF-κB signaling. Taken together, our results underscore fibulin-3 as a novel extracellular signal with strong activating effect on NF-κB in malignant gliomas. Because fibulin-3 is produced de novo in these tumors and is absent from normal brain we propose that targeting the fibulin-3/NF-κB axis may provide a novel avenue to disrupt oncogenic NF-κB signaling in combination therapies for malignant brain tumors.
Keywords: tumor microenvironment, glioma invasion, Tumor Necrosis Factor alpha, Extracellular matrix, CYLD, TACE, ADAM17, TIMP3
Glioblastomas (GBMs) are the most common primary tumors in the CNS, originating from transformed, glial-committed, progenitor cells1. Their invasive behavior and resistance to chemotherapy make them one of the solid tumors with worst prognosis2, 3, resulting in a median survival time of approximately 15 months4. GBMs can be classified into subtypes with well-defined transcriptional signatures and differences in invasive behavior, angiogenesis, and apoptotic resistance, all of which may help tailor targeted therapies more accurately5–7.
GBMs are driven by a relatively small number of major signaling hubs that regulate their phenotype and contribute to resistance and development of malignant features5, 8. One of these hubs is the Nuclear Factor-kappa B (NF-κB) pathway, which responds to multiple extracellular signals by activating transcriptional programs that mediate inflammatory and immune responses9. This pathway plays a key role in GBM growth and invasion via enhancement of cell proliferation and apoptotic resistance, release of pro-inflammatory cytokines, and upregulation of metalloproteases in the tumor microenvironment10, 11. Recent work has also demonstrated that NF-κB regulates the conversion of the proneural subtype of GBMs into the more aggressive mesenchymal subtype, as well as the radio-resistance of these aggressive GBMs12.
Expression of NF-κB-dependent genes is driven by the active forms of NF-κB transcription factors (RelA/p65 and NF-κB1/p105), which are upregulated in GBM specimens and glioma cells compared to normal astrocytes13, 14. NF-κB upregulation typically follows the activation of Tumor Necrosis Factor (TNF) and IL-1 receptors in response to cytokines, but this pathway can also be activated by cell-intrinsic mechanisms in GBM cells, mediated by EGFR/Akt signaling that can bypass inhibition of TNF receptors11, 15. However, there is little knowledge about the regulatory mechanisms that keep sustained NF-κB activity in mesenchymal GBMs and their microenvironment.
A majority of GBMs secrete the glycoprotein fibulin-3, which is a component of the fibrillar extracellular matrix (ECM) in connective tissues but is absent from normal brain16. Fibulin-3 expressed in high-grade gliomas gains a novel function as autocrine/paracrine enhancer of Notch signaling in tumor cells and adjacent endothelial cells, leading to increased tumor growth, invasion and angiogenesis17, 18. We and others have shown that fibulin-3 also promotes resistance to temozolomide treatment and is particularly upregulated in mesenchymal GBMs17, 19. Because fibulin-3 appears strongly associated with the malignant progression of gliomas we investigated if this protein could regulate NF-κB signaling and the downstream pro-tumoral effects of this pathway in GBM.
We report here that fibulin-3 has a novel and remarkable driving effect on canonical NF-κB signaling, acting not only in the tumor but also in peritumoral glia. Fibulin-3 expression correlates with an increased pro-invasive NF-κB signature in experimental models and clinical datasets; accordingly, this ECM protein promotes GBM invasion by ADAM17-dependent activation of NF-κB. These results suggest that fibulin-3 is a novel oncogeni factor regulating the major canonical NF-κB axis in GBM.
Results
Fibulin-3 regulates NF-κB signaling in GBM cells
Overexpression of fibulin-3 cDNA in a conventional GBM cell line (U251 cells16) caused remarkable activation of canonical NF-κB signaling, as observed by increased phosphorylation of the TNFR adapter protein RIP2, IKK kinases, and the transcription factor p65/RelA, as well as degradation of the NF-κB inhibitor IκBα (Fig. 1A,C). In addition, overexpression of fibulin-3 not only increased the levels of phospho-p65 but also the translocation of this transcription factor to the nucleus, mimicking the stimulation of NF-κB by TNFα (Fig. 1D,E). In contrast, when fibulin-3 expression was transiently downregulated in the same cells the phosphorylation of RIP2, IKK, and p65 was markedly decreased, indicating inhibition of NF-κB signaling (Fig. 1B,C). Strikingly, fibulin-3 knockdown also rendered this pathway insensitive to exogenous TNFα, reducing or preventing activation of RIP2, IKK and p65 even when TNFα was added to the cultures. The dominant-negative effect of fibulin-3 knockdown on TNFα-activated NF-κB signaling was also observed in mesenchymal GBM stem cells (Fig. S2A–C). We further confirmed that fibulin-3 specifically activated canonical (i.e., IκBα-dependent) NF-κB signaling because its effect was prevented by transfection of a dominant-negative “super-repressor” form of IκBα that prevents p65 phosphorylation (Fig. S2D). Moreover, fibulin-3 transfection did not increase the phosphorylation of RelB, a mediator of non-canonical NF-κB signaling (Fig. S2E).
Figure 1. Fibulin-3 activates canonical NF-κB signaling.
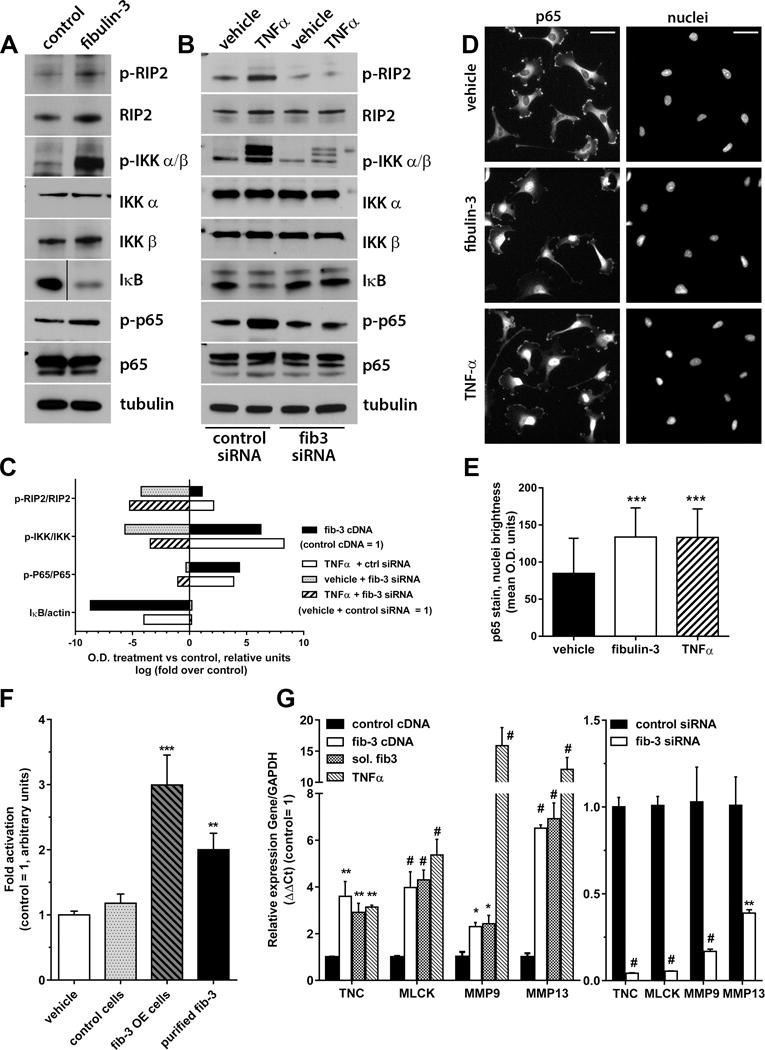
A) Fibulin-3 overexpression in U251 cells increased phosphorylation (p-) of NF-κB components (RIP2, IKK αβ, and p65) and degradation of IκBα. B) Conversely, fibulin-3 downregulation with siRNA prevented activation of NF-κB signaling even in presence of TNFα (10 ng/ml × 1 h). C) Quantification of Western blotting results from (A) and (B). The optical density (O.D.) for each phospho-protein band was normalized to its own total protein (or actin for IκB); bars represent fold-change (in log scale) in O.D. for each condition compared against their control (control transfections were given arbitrary O.D. value of 1). D) Treatment of U251 cells with fibulin-3 (2 h) or TNFα (30 min) increased the translocation of p65 to the nucleus. E) Quantification of nuclear p65 in cells from (D), co-localized with nuclear DAPI stain (*** p<0.001, one-way ANOVA). F) CNS1 cells carrying a NF-κB-dependent reporter were cultured with no treatments (vehicle), transfected with control or fibulin-3 cDNAs (control cells and fib-3 OE cells), or treated with purified fibulin-3 (300 ng/ml × 4h). Stimulation with transfected or exogenous fibulin-3 resulted in reporter activation (** p<0.01; ***p<0.001, one-way ANOVA). Similar results were observed in U251 cells (Fig. S2F). G) U251 cells were treated with TNFα, soluble purified fibulin-3 (sol. fib-3), or transfected with fibulin-3 cDNA or siRNA (or their controls), and processed for qRT-PCR. Fibulin-3 levels regulated the expression of the NF-κB-dependent genes TNC, MLCK, MMP9 and MMP13. Results were compared by one-way ANOVA for each gene (* p<0.05; ** p<0.01; # p<0.001).
Activation of NF-κB signaling by fibulin-3 resulted in increased NF-κB transcriptional activity, measured with a NF-κB luciferase reporter in U251 cells (Fig. S2F) and in a second GBM cell line, CNS1 (Fig. 1F). These results were observed after fibulin-3 overexpression as well as by adding soluble, purified fibulin-3 in the CNS1 cultures (Fig 1F). We also replaced the luciferase reporter gene with secreted alkaline phosphatase (SEAP), which allowed us to collect conditioned medium from transfected cells and monitor NF-κB transcriptional activity over time. Stimulation with fibulin-3 or TNFα increased SEAP release with similar fast kinetics and did not show additive effects, suggesting that they shared a common mechanism (Fig. S3A). More importantly, fibulin-3 knockdown reduced NF-κB-dependent SEAP release and significantly diminished the effects of TNFα (Fig. S3B), in agreement with our dominant negative effect observed in Fig. 1B.
We next verified whether fibulin-3 had specific impact on transcriptional targets of NF-κB associated with cell motility and adhesion because those processes are enhanced by fibulin-3 in GBM16. U251 cells overexpressing fibulin-3 or stimulated with soluble fibulin-3 showed increased expression of the pro-invasive genes tenascin-C (TNC), myosin light-chain kinase (MLCK) and the matrix metalloproteases MMP9 and MMP13, all of which were also upregulated by TNFα (Fig. 1G). Conversely, fibulin-3 knockdown caused significant downregulation of those genes. Together, these collective results demonstrated that fibulin-3 is a dominant regulator of NF-κB activity in GBM cells.
Fibulin-3 correlates with NF-κB upregulation in GBM and its microenvironment
To validate our results in vivo, we implanted invasive, fibulin-3-overexpressing, CNS1 cells intracranially in athymic mice. We have previously demonstrated that fibulin-3 overexpression leads to larger, more invasive, and more vascularized CNS1 tumors16, 18. Here, we focused on expression of p65 as marker of NF-κB activation in the tumor core and surrounding peritumoral tissue.
Analysis of tissue sections from similar-sized tumors revealed a significant increase of p65 in the core of fibulin-3-overexpressing tumors (Fig. 2A,B,G). Total expression of p65 was also increased in the border of these tumors but did not reach statistical significance (Fig. 2G). However, when we focused specifically on GFAP-positive astrocytes in the tumor border (Fig. 2C–F) we confirmed that those cells had significant increase of p65 in fibulin-3-overexpressing tumors (Fig. 2H). Analysis of higher magnification images revealed that the increased p65 significantly co-localized with nuclei both in the core and border of fibulin-3-overexpressing tumors, indicating increased activation and nuclear localization of this NF-κB transcription factor both in GBM and astroglial cells (Fig. 2J).
Figure 2. Fibulin-3 overexpression activates NF-κB in the tumor and its microenvironment.
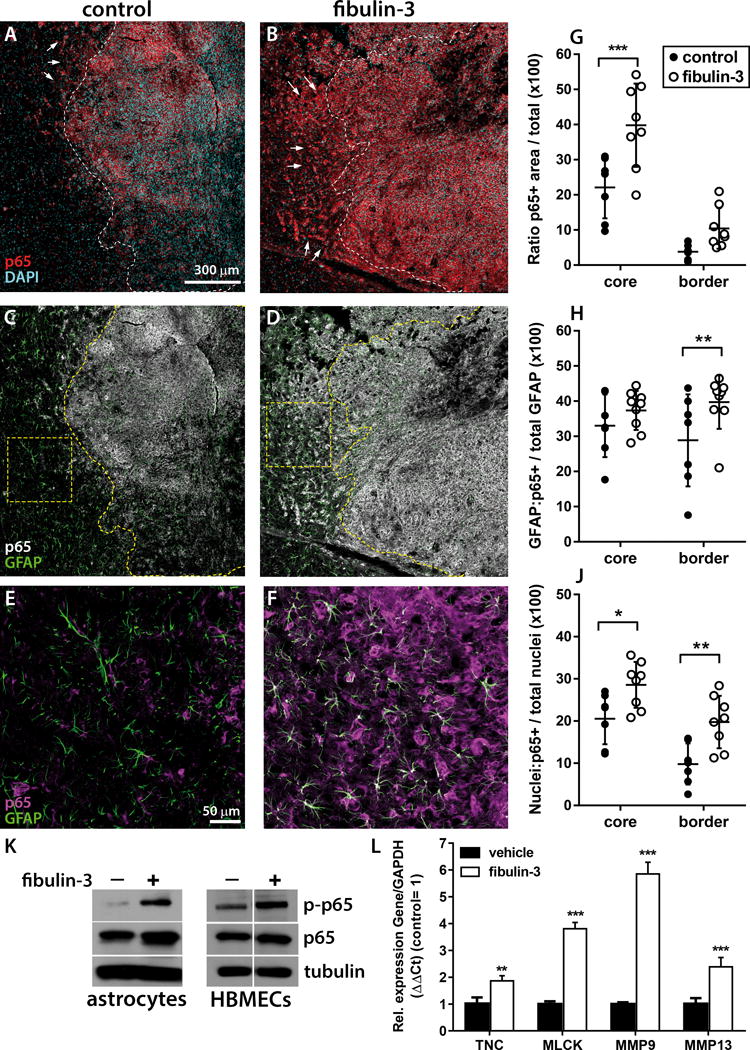
A–B) Representative low-magnification images of CNS1-derived intracranial tumors stained for total p65 (red) and cell nuclei (DAPI, light blue). The dashed line represents the boundary between the tumor core (defined by density of nuclei and GFP-expressing tumor cells, channel not shown) and border. Notice the considerable p65 staining beyond the boundary of fibulin-3-overexpressing tumors (arrows). C-D) Representative images as in (A–B) revealing intra- and peri-tumoral GFAP staining (green); p65 staining is shown in grayscale to avoid obscuring GFAP within the tumor core. E–F) Higher magnification images of the squares highlighted in panels (C) and (D). G–H) Quantification of p65-positive and GFAP-positive areas within (core) and around (border) the tumor mass. P65 staining was normalized to total tumor area and GFAP/P65-positive areas were normalized to total GFAP area. Results showed an increase in p65 expression in the tumor mass and in surrounding astroglia of fibulin-3-overexpressing tumors. J) Quantification of p65-positive nuclei in the tumor core and border, relative to total nuclei per area unit. Results show increased nuclear localization of p65 in fibulin-3-overexpressing tumors. Results in (G)–(J) were compared by repeated measures 2-way ANOVA with Sidak’s correction for multiple comparisons; * p<0.05; ** p<0.01; *** p<0.001. K) Soluble fibulin-3 (300 ng/ml × 1h) increased p65 phosphorylation in cultures of astrocytes and human brain microvascular endothelial cells (HBMECs). L) Soluble fibulin-3 also increased the expression of NF-κB-regulated genes in cultured astrocytes (*** p<0.01 by Student’s t-test for each gene).
To confirm that fibulin-3 could activate NF-κB signaling in tumor-associated neural cells, we added purified fibulin-3 to primary cultures of brain astrocytes and brain microvascular endothelial cells, none of which express endogenous fibulin-3. As expected, fibulin-3 increased phospho-p65 in both cell types (Fig. 2K). Moreover, fibulin-3 also increased the expression of NF-κB-dependent pro-invasive genes in astrocyte cultures (Fig. 2L) matching our previous result with GBM cells.
Fibulin-3 correlates with a pro-invasive NF-κB signature in GBM
Next, we studied the association of fibulin-3 (gene EFEMP1) with NF-κB transcription factors and NF-κB-dependent genes using transcriptomics data from the REMBRANDT and TCGA repositories. Fibulin-3 and its close homolog fibulin-4 (EFEMP2) showed significant positive correlation with canonical NF-κB transcription factors and the pro-invasive genes that we had assayed in Fig. 1G, except MMP13, which was expressed at very low levels in GBMs (Fig. S4A–B). Cluster analysis of those genes using TCGA microarray data revealed two well-defined patient populations with high and low gene co-expression (Fig. 3A) and significantly different survival curves (Fig. 3B).
Figure 3. Fibulin-3 correlates with a NF-κB-regulated invasive signature.
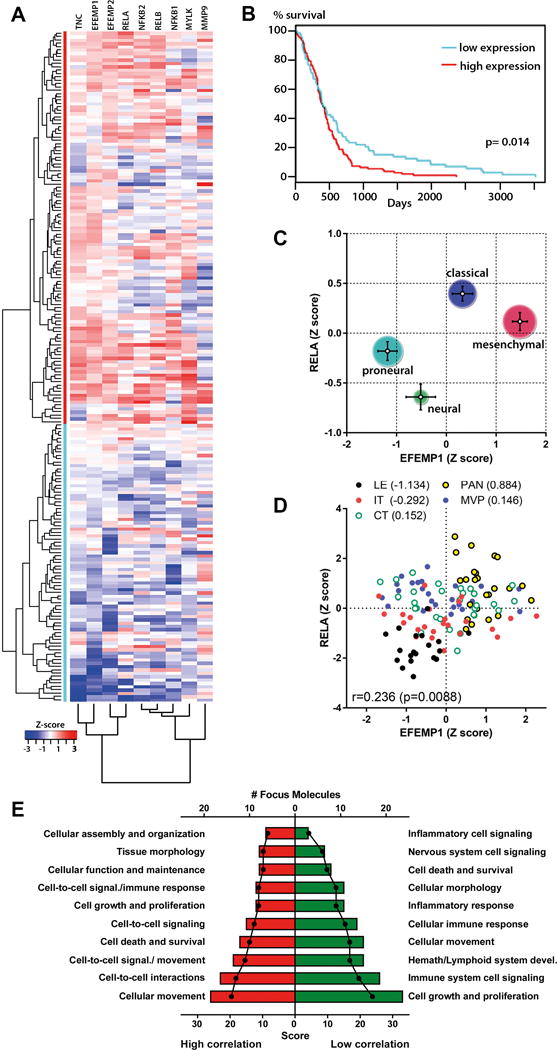
A) Gene expression clustering in GBMs using normalized TCGA data for EFEMP1, EFEMP2, NF-κB transcription factors, and fibulin-3-correlated pro-invasive genes tested in Fig. 1. Unsupervised cluster analysis revealed two major patient populations (red and blue lines) characterized by high and low co-expression of these genes. B) Kaplan-Meier analysis of those two populations showed significantly poorer survival in patients with higher co-expression of fibulin-3 and NF-κB-associated genes (p=0.014 by log-rank test). C) Co-expression of fibulin-3 (EFEMP1) and p65 (RELA) was higher in the mesenchymal and classical subtypes of GBMs compared to neural and proneural subtypes. Bars represent the standard error in Z-score for each subtype and the size of the circles represent the number of cases for subtype. D) Intratumoral expression of EFEMP1 and RELA from a set of 10 specimens with 5 histologically-defined regions: leading edge (LE); infiltrating tumor (IT); core tumor (CT); pseudopalisading cells around necrosis (PAN); and microvascular proliferation region (MVP). Numbers in parentheses are averaged Z scores for EFEMP1 and RELA expression. Results suggested preferential co-expression of both genes in perinecrotic areas of the tumor. E) NF-κB-regulated genes that were highly or poorly correlated with fibulin-3 (Table S-III) were aggregated into functional networks (Table S-IV). Networks were scored (red/green bars) based on the number of NF-κB-regulated genes found in each network (# Focus Molecules). NF-κB-regulated genes highly correlated with fibulin-3 were predominantly associated with cellular movement and cell-cell interactions, while genes not correlated with fibulin-3 were involved in cell growth and proliferation and immune responses.
Further analysis of fibulin-3 and NF-κB p65 (gene RELA) expression showed that the average co-expression of both genes was highest in the mesenchymal subtype of GBMs (Fig. 3C), which were the predominant subtype in the patient population with poorer survival. Moreover, analysis of intra-tumoral expression from an independent dataset (IVY Glioblastoma Atlas Project) confirmed the significant correlation of both genes (Fig. 3D) and their preferential co-expression in peri-necrotic pseudopalisade areas, which typically contain a “escaping” population of migratory glioma cells20.
Because the NF-κB pathway regulates the expression of hundreds of genes, we next asked if fibulin-3 expression had preferential correlation with specific transcriptional programs or acted as an indiscriminate NF-κB activator. To answer this we used publically available surveys to compile a list of 348 genes directly regulated by NF-κB (Suppl. Table S3) and correlated their expression against fibulin-3 using TCGA microarray data (Suppl. Table S3). We then chose two groups of 100 genes with the highest and lowest correlation against fibulin-3 and used Ingenuity Pathway Analysis to identify relevant pathways and cellular functions represented in each group (Suppl. Table S4). The analysis suggested that the top NF-κB-dependent cellular functions highly correlated with fibulin-3 are those involved in cell motility or invasion, matching the known function of fibulin-3 in GBM. On the other hand, NF-κB-regulated functions poorly correlated with fibulin-3 included cell proliferation and signaling in the nervous and immune systems (Fig. 3E). We further confirmed that genes identified as poorly correlated with fibulin-3 (e.g., cytokines IL-2, IL-12B, and IL-17) effectively did not change expression after fibulin-3 overexpression or knockdown in U251 cells (Fig. S4C).
The pro-invasive effects of fibulin-3 require NF-κB signaling
Fibulin-3 predominantly regulates cell adhesion and invasion in GBM16 and maintains the viability of the GBM stem cell population, even though it does not increase GBM cell proliferation17. Therefore, we next investigated if these phenotypic effects was mediated NF-κB as predicted from our in silico data.
We first measured the effect of fibulin-3 on cell adhesion and observed that fibulin-3-overexpressing U251 cells had increased adhesion to fibronectin-coated substrates, as expected (Fig. 4A). However, this effect was completely abolished when the cells were co-transfected with p65 siRNA or with the NF-κB “super-repressor” form of IκB. Next, we measured cell invasion using highly invasive CNS1 cells seeded on cultured brain slices, which provide an accurate ex vivo model for GBM dispersion in neural tissue21 (U251 cells are poorly invasive in vivo and do not disperse in cultured brain tissue). Fibulin-3-overexpressing CNS1 cells dispersed much further than control cells (Fig. 4B) but this effect was abolished by co-transfection of p65 siRNA (Fig. 4C) or by treating the cultures with the NF-κB inhibitor CAPE22 (Fig. 4D). A more specific NF-kB inhibitor, BAY-11-7082, was also assayed but caused unavoidable toxicity in the cells and cultured brain tissue (Fig. S5A). Together, results from Figs. 4A–D confirmed that fibulin-3 drives a NF-κB-dependent, pro-invasive mechanism in GBM cells. In contrast, although fibulin-3 knockdown reduced GBM stem cell viability as previously described17, this effect was independently rescued by TNFα (Fig. S5B), suggesting that fibulin-3 effects on GBM viability were probably not dependent on, or correlated with, NF-κB signaling, as predicted from Fig. 3E.
Figure 4. Fibulin-3 requires NF-κB for cell adhesion and invasion.
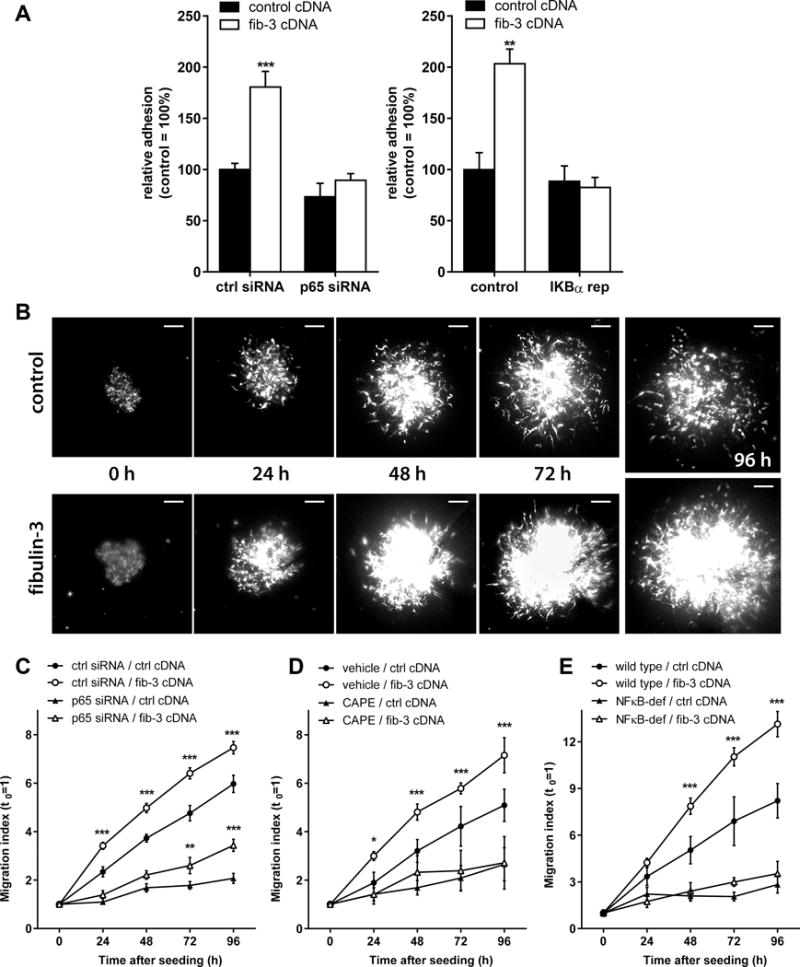
A) U251 cells were tested for adhesion to fibronectin-coated wells. Fibulin-3 cDNA expression increased cell adhesion but this effect was abolished by co-expression of p65 siRNA or IκBα super-repressor (IKBα rep) cDNA, both of which inhibited NF-κB activation (adhesion different from control at **p<0.01, ***p<0.001; one-way ANOVA). B) Representative images of control (ctrl) and fibulin-3-overexpressing (fib-3) CNS1 tumorspheres seeded and dispersing on mouse brain slices. C-D) The pro-invasive effect of fibulin-3 was highly attenuated or abolished when cells were transfected with p65 siRNA (C) or when NF-κB was inhibited with CAPE (20 μM) (D). E) CNS1 tumorspheres were also seeded on brain slices from NF-κB deficient mice (NFκB-def; TNFR1−/− RelA/p65−/−) or their wild-type littermates. NF-κB deficiency in brain tissue abolished cell invasion and the pro-invasive effect of fibulin-3. Results in (C) to (E) were analyzed by two-way ANOVA for repeated measures; asterisks indicate differences between fibulin-3-expressing cells and their respective control (** p<0.01; *** p<0.001).
Finally, we evaluated if the pro-invasive effect of fibulin-3 was tumor cell-intrinsic or could also involve NF-κB regulation in peritumoral brain tissue as suggested from our results in vivo (Fig. 2). We repeated our brain slice invasion assays using neonatal brain tissue from NF-κB-deficient mice lacking both TNFR1 and p65 expression23. Remarkably, CNS1 cells were unable to invade through NF-κB-deficient brain slices and the pro-invasive effect of fibulin-3 was completely suppressed (Fig. 4F). Taken together, our results suggest that the leading function of fibulin-3 as pro-invasive factor in GBMs is mediated by its ability to drive NF-κB-dependent “pro-invasion programs” both in tumor cells and their microenvironment.
NF-κB regulation by fibulin-3 involves control of CYLD expression and TACE activity
We finally investigated the mechanism of NF-κB activation by fibulin-3, focusing on possible mechanisms triggered by this ECM protein at the cell membrane level.
Fibulin-3 can compete with Notch inhibitors to directly activate Notch signaling in GBM, increasing the expression of Notch-regulated genes such as the transcription factors Hes-117, 18. Work in T-cell leukemia has shown that Hes-1 represses the expression of the de-ubiquitinase CYLD24, which is a NF-κB inhibitor that acts by preventing degradation of IκB25. Therefore, we reasoned that fibulin-3 stimulation of Notch could downregulate CYLD and indirectly activate NF-κB.
To test this hypothesis, we first confirmed that CYLD knockdown was sufficient to increase phosphorylation of p65 in U251 cells (Fig. 5A). Then, as predicted, we observed that fibulin-3 overexpression downregulated CYLD mRNA whereas fibulin-3 knockdown caused the opposite effect (Fig. 5B). Surprisingly, blockade of Notch signaling with the gamma-secretase inhibitor DAPT17 did not prevent the downregulation of CYLD (Fig. 5C) and the activation of NF-κB p65 (Fig. 5D) by fibulin-3. These results suggested that fibulin-3 may regulate Notch and NF-κB mechanisms independently in GBM. Even if CYLD-downregulation underlies, in part, the activation of NF-κB by fibulin-3, this mechanism does not depend on Notch signaling as postulated in other cancers. This prompted to investigate which other “nodes” could be regulated by fibulin-3 to result in joint but independent regulation of Notch and NF-κB pathways.
Figure 5. Fibulin-3 regulates the NFκB inhibitor CYLD in a Notch-independent manner.
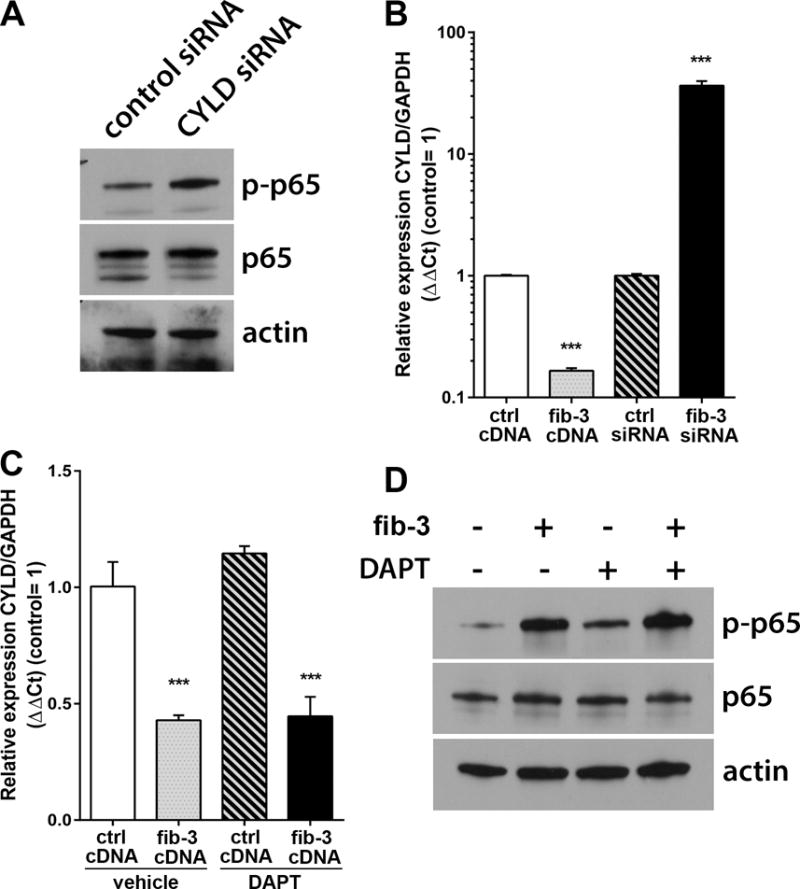
A) Knockdown of the enzyme CYLD (CYLD siRNA) in U251MG cells increased the phosphorylation of p65. B) Overexpression or knockdown of fibulin-3 in U251MG cells resulted in the opposite down- or up-regulation of CYLD mRNA. C) Overexpression of fibulin-3 continued to repress CYLD expression even when the cells were treated with the Notch inhibitor DAPT (25 μM). D) Increased phosphorylation of p65 by fibulin-3 was also resistant to Notch inhibition, suggesting that fibulin-3 regulates NF-κB signaling by a mechanism that does not require Notch activation. Results in (B) and (C): *** p<0.001 by one-way ANOVA.
An indirect mechanism by which fibulin-3 can activate Notch in GBM involves increased activity of the metalloproteases ADAM10 and ADAM1718, which cleave and activates Notch receptors26. These enzymes are also known as TNFα convertases (TACE) because one of their major functions is the cleavage of membrane-bound pro-TNFα to release soluble TNFα, which in turn activates TNF receptors and NF-κB signaling27. Therefore, we investigated if regulation of ADAM10/17 could be a mechanism by which fibulin-3 regulated NF-κB signaling in parallel and independent manner from Notch signaling.
Overexpression of fibulin-3 in U251 cells significantly increased the expression of ADAM17 mRNA, but not ADAM10 (Fig. 6A), as well as TACE enzymatic activity (Fig. 6B). TACE activity was also increased when purified fibulin-3 was added to lysates of GBM cells (Fig. 6B), suggesting that fibulin-3 directly regulated enzymatic activity of ADAM17 in addition to its expression. Accordingly, fibulin-3 stimulation of GBM cells increased the cleavage of membrane-bound pro-TNFα and the release of soluble TNFα to the culture medium (Fig. 6C). Moreover, inhibition of TACE activity with the small-molecule inhibitor TAPI-2 was sufficient to abolish the enhancing effect of fibulin-3 on p65 phosphorylation (Fig. 6D), suggesting that this was a key mechanism by which fibulin-3 regulated the NF-κB pathway.
Figure 6. Fibulin-3 stimulates ADAM17 to produce TNFα and activate NF-κB.
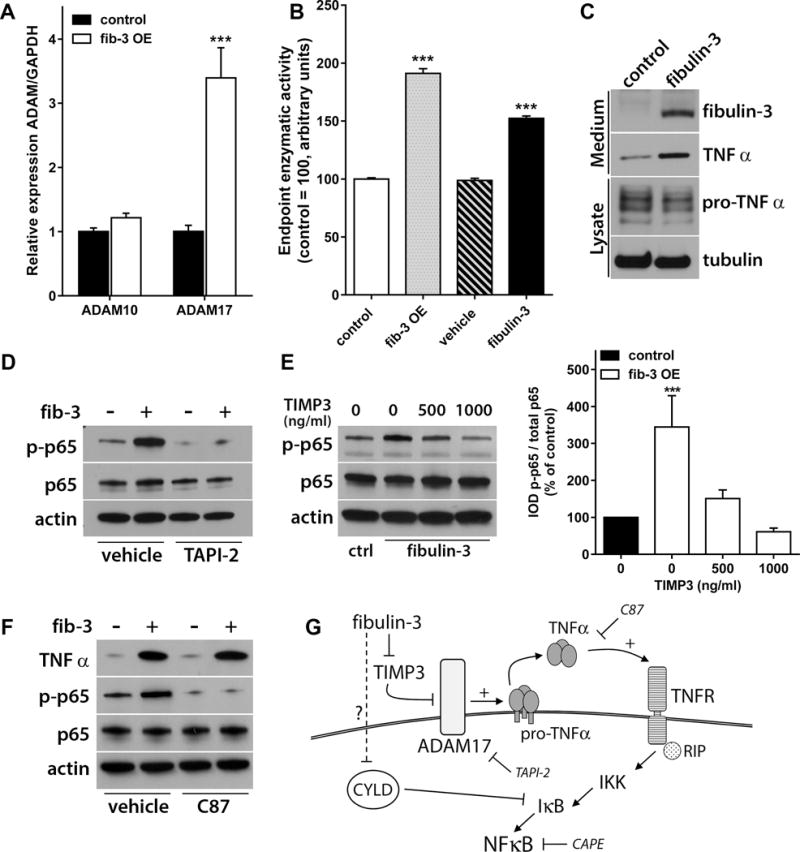
A) Fibulin-3 overexpression (OE) increased ADAM17 mRNA (*** p<0.001, Student’s t-test) but not ADAM10 mRNA in U251 cells. B) Lysates from fibulin-3-overexpressing cells or co-incubated with purified fibulin-3 for 6h also showed increased ADAM17/TACE enzymatic activity (*** p<0.001, one-way ANOVA). C) Fibulin-3 overexpression increased the cleavage of membrane-bound pro-TNFα and release of soluble TNFα, which can activate NF-κB. D) The enhancing effect of fibulin-3 on p65 phosphorylation (in U251 cells) was blocked by the ADAM17 inhibitor TAPI-2 (1 μM). E) Activation of NF-κB by fibulin-3 was also inhibited in a concentration-dependent manner by the ADAM17 endogenous inhibitor, TIMP3. The bar graph shows quantitative results from triplicate experiments (significant differences from control cells at *** p<0.001; one-way ANOVA). F) The small-molecule inhibitor C87 did not prevent activation of ADAM17 and release of soluble TNFα by fibulin-3, but blocked TNFα and downstream activation of NF-κB. G) Proposed mechanism of action of fibulin-3 to activate NF-κB via sustained activation of ADAM17 and accumulation of soluble TNFα.
Because ADAM17 seemed to be the TACE enzyme responsive to fibulin-3, we next investigated if these proteins interacted directly to activate NF-κB. We could not detect a significant association between fibulin-3 and ADAM17 by co-immunoprecipitation (Fig. S6), suggesting that fibulin-3 probably regulated other mediator protein(s) that interact with ADAM17. A well-known inhibitor of ADAM17 is the Tissue Inhibitor of Metalloproteases TIMP328, which also binds to fibulin-329 and can be “sequestered” by fibulin-3 in GBM18. Accordingly, the effect of fibulin-3 on NF-κB was inhibited by TIMP3 in a concentration-dependent manner (Fig. 6E), suggesting that fibulin-3 competes with TIMP3 to activate ADAM17. Finally, we used the TNFα small-molecule inhibitor C87, which does not prevent cleavage of pro-TNFα but inhibits the binding of released TNFα to TNF receptors and reduces NF-κB activation. Treatment of fibulin-3-overexpressing cells with C87 did not prevent the increase of soluble TNFα but strongly inhibited NF-κB activation (Fig. 6F). Together, these results strongly suggest a model in which fibulin-3 prevents TIMP3 inhibition of ADAM17, resulting in sustained release of TNFα that activates TNF receptors and therefore canonical NF-κB signaling (Fig. 6G).
Discussion
Dysregulated, constitutive activation of NF-κB signaling is a central mediator of inflammatory and anti-immune processes that correlate with tumor progression30, 31. In GBMs, sustained NF-κB activity is necessary for the maintenance of the tumor-initiating cell population32, 33 and for tumor progression towards a more aggressive phenotype12 that involves enhanced invasion34 and neo-angiogenesis35. NF-κB activation also occurs in tumor-associated astrocytes36 and microglia37–39, which contribute to tumor progression. Because NF-κB components are rarely mutated in adult GBMs40 this suggests that the pathway becomes constitutively activated by alterations in key regulatory mechanisms.
Our results suggest that fibulin-3 is a novel soluble signal that drives NF-κB signaling in GBMs and their microenvironment. Fibulin-3 is one of seven members of the fibulin family that are found in the basal lamina of blood vessels and the ECM of connective tissues41. These glycoproteins have been largely regarded as structural components necessary for the mechanical integrity of tissue42, 43 but their overexpression in cancer has revealed novel roles as soluble signals that facilitate cell adhesion, growth, and motility44.
The upregulation of fibulin-3 in cancer was first described in high-grade gliomas16 and pancreatic carcinomas45, after which this protein was identified as a pro-tumoral factor in several types of metastatic solid tumors46–51. Mechanistically, fibulin-3 has been shown to activate the EGFR/Akt axis in pancreatic cancer cells52 but this mechanism was not observed in GBM cells17, 53. Instead, we and others demonstrated that fibulin-3 directly activates the Notch pathway in GBM cells17, 19 and tumor-associated endothelial cells18, resulting in increased tumor invasion, angiogenesis, and chemoresistance. The current knowledge of fibulin-3 in GBMs underscores this ECM protein not only as a marker of tumor aggressiveness but more importantly as a promoter of malignant progression17, 19.
Here, we have demonstrated that the effects of fibulin-3 in GBM correlate with, and depend on, activation of canonical NF-κB. Fibulin-3 expression matches NF-κB upregulation in experimental models and clinical datasets, with preferential co-expression in mesenchymal GBMs and in tumor regions populated by invasive cells, all of which makes fibulin-3 a possible marker for regions of active tumor progression. Moreover, our in silico and in vitro data indicate that fibulin-3 promotes invasion in GBM by concerted NF-κB activation both in tumor cells and astroglia (Figs. 2 and 4), driving a NF-κB-dependent pro-invasive signature that includes genes for cell adhesion (tenascin-C), motility (MLCK) and ECM remodeling (metalloproteases). Overall, these results suggest that fibulin-3 may be among the key signals that stimulate and sustain NF-κB to favor the malignant progression of GBMs.
The molecular interactions of fibulin-3 with NF-κB regulatory proteins remain to be fully elucidated but our results already indicate that fibulin-3 likely activates NF-κB by preventing TIMP3 inhibition of ADAM17, therefore increasing the activity of this enzyme. This, in turn, increases the availability of soluble TNFα to bind TNF receptors and activate canonical NF-κB. This finding is of particular significance because ADAM17 is a “master protease” on the cell surface that regulates multiple signaling pathways54, 55 for tumor growth, invasion, angiogenesis, and immune escape56–59. Regulation of ADAM17 activity by fibulin-3 explains the ability of this ECM protein to activate both Notch (by cleavage of Notch-1 receptor) and NF-κB (by shedding of TNFα) in parallel and independent fashion, and suggests that there may be more pathways modulated by fibulin-3 in a similar manner. We acknowledge that some molecular effects of fibulin-3 may not be explained by ADAM17 regulation, such as the dominant inhibition of NF-κB signaling after fibulin-3 knockdown (Fig. 1). It is possible that fibulin-3 downregulation increases CYLD that in turn inhibits NF-κB signaling, but the mechanisms by which fibulin-3 regulates CYLD at the transcriptional level is yet unknown. Further work is needed to identify cell-surface mechanisms tonically regulated by fibulin-3 and affected by the loss of this protein.
NF-κB remains a challenging target for molecular therapies because of the multitude of cellular mechanisms controlled by this pathway, which raises concern for the adverse effects that may result from total NF-κB inhibition31, 32. New research has focused on inhibiting key NF-κB activating signals, thus disrupting NF-κB transcriptional programs with high-specificity and fewer off-target effects. The oncogenic effect of fibulin-3 on NF-κB is likely a GBM-specific function because fibulin-3 is neither soluble in normal tissues nor expressed in the brain16, 60. Therefore, specific targeting of fibulin-3/NF-κB interactions could inhibit tumor progression with potentially limited side effects. We suggest that targeting a fibulin-3/NF-κB axis may provide a novel avenue to improve combination therapies for GBM.
Materials and Methods
Cell cultures
GBM cell lines, GBM stem cells, primary mouse astrocytes, and human brain microvascular endothelial cells were cultured following standard methods, as described16–18. Human U251 cells were chosen for manipulation with fibulin-3 cDNA or siRNA because they have moderate endogenous levels of fibulin-3 among glioma lines16. Rat CNS1 cells were used to study invasion mechanisms because U251 cells are poorly invasive through brain tissue. All cells were authenticated using the “Cell Check” service (IDEXX-Research Animal Diagnostic Laboratory, Columbia MO). All supplements were removed from the cultures during in vitro tests requiring addition of fibulin-3 or TNFα.
DNA and RNA constructs
Fibulin-3 cDNA was cloned in the plasmid pcDNA3.1(+) (Life Technologies, Carlsbad CA) for transient transfections and subcloned into the lentiviral vector pCDH-EF1-copGFP (System Biosciences, Mountain View CA) for constitutive expression in GBM cells16. siRNA oligonucleotides against fibulin-3, p65, and CYLD (Qiagen, Valencia CA) were transfected at 100 pmol/5×105 cells and validated at the mRNA and protein levels (17 and Fig. S1). Cells were used in assays or processed 48 h after transfections. All transfections were performed with Lipofectamine-2000 (Life Technologies) following the manufacturer’s recommendations.
The NF-κB reporter plasmid pGL4.32/luc2P/NF-κB-RE was from Promega (Madison WI). The SEAP reporter plasmid was generated by removing the luciferase sequence in plasmid pGL4.32 and inserting the sequence of secreted alkaline phosphatase from plasmid pSEAP2-Control (Clontech, Mountain View CA). A plasmid carrying the cDNA for the degradation-resistant, super-repressor form of IκBα61 (pCMX-IκBα-M) was purchased from Addgene (Cambridge, MA).
Biochemical assays
Transfected cells were lysed and processed for semiquantitative RT-PCR or Western blotting using standard protocols16 (antibodies and primers are listed in Suppl. Tables S-I and S-II). Cultures were treated overnight with the gamma-secretase inhibitor DAPT (25 μM, Tocris Bioscience, Bristol UK) or the TACE inhibitor TAPI-2 (1 μM, Cayman Chemical, Ann Arbor MI). Cells were also treated with TNFα (10 ng/ml, Peprotech, Rocky Hill NJ), fibulin-3 (300 ng/ml, Origene, Rockville MD), or the TNFα inhibitor C87 (10 μM, Tocris) for 1 to 24 h before collection. For reporter assays a plasmid carrying Renilla luciferase was co-transfected as loading control.
To measure TACE activity, cells were lysed in 50 mM Tricine buffer (pH 7.5) containing 100 mM NaCl, 10 mM CaCl2, 1 mM ZnCl2 and 0.1% Triton X-100. Clarified lysates (50 μg total protein) were incubated with a fluorogenic ADAM10/17 substrate peptide (TACE substrate III) as described18.
Cell-based assays
To quantify nuclear translocation of p65, cells were treated with fibulin-3 or TNFα and processed for immunocytochemistry using an antibody against total p65. Nuclear translocation was quantified by co-localization of p65 with nuclear DAPI staining. For cell adhesion assays, U251 cells were dissociated and seeded (25,000 cells/well) on fibronectin-coated wells (5 μg/ml) for 1h. Adhered cells were fixed, stained with crystal violet and quantified as described16. For cell invasion assays, CNS1 cells were transfected with combinations of cDNAs and siRNAs and allowed to form tumorspheres for 48 before seeding on brain slices; cell dispersion was quantified by imaging methods as described16. The NF-κB inhibitors caffeic acid methyl ester (CAPE, 20 μM, Tocris) or BAY-11-7082 (0.5-5 μM Calbiochem, Billerica MA) were added to the slice culture medium for the duration of the experiments. To assess the role of microenvironmental NF-κB, spheroids were seeded on brain slices from neonatal NF-κB-deficient mice (TNFR1−/− p65−/−, selected from a heterozygous colony)23 provided by Dr. Denis Guttridge (The Ohio State University). To quantify GBM stem cell viability, dissociated cells were transfected with control or fibulin-3 siRNAs and plated in 96-well plates at a density of 2,000 cells/well. Cultures were spiked with TNFα (10 ng/ml) every other day after plating. Cell viability was assessed by ATP production using the CellTiter-Glo kit (Promega)17.
Animal procedures
All animal experiments were approved by the Animal Care and Use Committees at the Brigham and Women’s Hospital and SUNY Upstate Medical University. GFP-expressing CNS1 cells (5 × 104 cells in 2 μL) were implanted in the striatum of athymic mice as described16, 17. Brains were collected 20 days after tumor implantation and processed for cryo-sectioning at 20 μm. Sections were fixed in 4% paraformaldehyde and processed for immunohistochemistry to detect p65 and GFAP. Tumor boundaries were identified by GFP expression and high cellular density (nuclear DAPI staining). Sections were imaged using a confocal microscope Zeiss LSM710.
Clinical datasets and pathway analysis
GBM microarray data was obtained from REMBRANDT (Affymetrix HG-U133 v2.0+, N=214) and TCGA (Affymetrix Human Exon 1.0 ST, N=202, Affymetrix, HG-U133A, N=198 and Agilent G4502A, N=198) databases. Criteria from Ceccarelli et al.62 were used to exclude GBMs harboring 1p/19q co-deletion. Standard scores (Z scores) were calculated for fibulin-3 (EFEMP1), fibulin-4 (EFEMP2), NF-κB transcription factors (RELA, RELB, NFKB1, NFKB2) and pro-invasive genes (TNC, MLCK, MMP9, MMP13); expression of fibulin-3 was then correlated with each gene (Pearson’s correlation) across the different microarray platforms. Optimal heatmap clustering was calculated using the R’s package NBClust. Molecular subtypes of GBM were identified in the TCGA dataset using profiles provided by the GBM Bio-Discovery Portal (http://gbm-biodp.nci.nih.gov)63. RNAseq data (Z scores) for intratumoral expression of EFEMP1 and RELA in histologically-defined, microdissected regions from 10 GBM specimens (total N=122 samples) was obtained from the IVY Glioblastoma Atlas Project repository (http://glioblastoma.alleninstitute.org)
To perform correlation analysis between fibulin-3 and NF-κB-regulated genes we first reviewed publically available surveys of predicted and tested NF-κB targets (http://bioinfo.lifl.fr/NF-KB/ and http://www.bu.edu/nf-kb/gene-resources/target-genes)64, 65 to compile a list of 348 genes directly regulated by NF-κB. We next used TCGA GBM microarray data to correlate EFEMP1 with each NF-κB-regulated gene, followed by Bonferroni correction of p values for multiple comparisons. Correlations were filtered for significance (adjusted p<0.05) and sorted by Pearson’s Rho value to select 100 genes with the highest correlation with fibulin-3 (highest absolute Rho values) and 100 genes with the lowest correlation (Rho closest to zero) (Suppl. Table S-III). Each group of genes was separately analyzed using Ingenuity Pathway Analysis (Qiagen) to identify the most represented networks of signaling pathways and cellular functions. Those networks were scored based on how many genes from the original 100-gene list (“focus molecules”) were found in each network (Suppl. Table S-IV). This yielded two lists of ranked cellular functions containing NF-κB-regulated genes; one list for genes with highest correlation with fibulin-3 and another for genes with lowest correlation with fibulin-3.
Statistics
Experiments in vitro were repeated in triplicate with 3–5 independent replicates (8 replicates for invasion assays). Animals studies were performed in duplicate with N=5 per experimental condition (to detect differences at least twice as large as the S.D. of the groups, at power 80% and p<0.05). Blinding and randomization for animal studies followed the ARRIVE guidelines for animal research66. Grubb’s test was used to detect outlier results; all differences were deemed significant at p<0.05.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01CA152065) and the National Brain Tumor Society to MSV, and the Joel Gingras Jr. Research Fellowship from the American Brain Tumor Association to BH.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest
Bibliography
- 1.Zong H, Verhaak RG, Canoll P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev Mol Diagn. 2012;12:383–394. doi: 10.1586/erm.12.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watkins S, Sontheimer H. Unique biology of gliomas: challenges and opportunities. Trends Neurosci. 2012;35:546–556. doi: 10.1016/j.tins.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 4.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(Suppl 4):iv1–63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149:36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 10.Korkolopoulou P, Levidou G, Saetta AA, El-Habr E, Eftichiadis C, Demenagas P, et al. Expression of nuclear factor-kappaB in human astrocytomas: relation to pI kappa Ba, vascular endothelial growth factor, Cox-2, microvascular characteristics, and survival. Hum Pathol. 2008;39:1143–1152. doi: 10.1016/j.humpath.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 11.Puliyappadamba VT, Hatanpaa KJ, Chakraborty S, Habib AA. The role of NF-kappaB in the pathogenesis of glioma. Mol Cell Oncol. 2014;1:e963478. doi: 10.4161/23723548.2014.963478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhat KP, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell. 2013;24:331–346. doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–951. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- 14.Robe PA, Bentires-Alj M, Bonif M, Rogister B, Deprez M, Haddada H, et al. In vitro and in vivo activity of the nuclear factor-kappaB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res. 2004;10:5595–5603. doi: 10.1158/1078-0432.CCR-03-0392. [DOI] [PubMed] [Google Scholar]
- 15.Kusne Y, Carrera-Silva EA, Perry AS, Rushing EJ, Mandell EK, Dietrich JD, et al. Targeting aPKC disables oncogenic signaling by both the EGFR and the proinflammatory cytokine TNFalpha in glioblastoma. Science signaling. 2014;7:ra75. doi: 10.1126/scisignal.2005196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu B, Thirtamara-Rajamani KK, Sim H, Viapiano MS. Fibulin-3 Is Uniquely Upregulated in Malignant Gliomas and Promotes Tumor Cell Motility and Invasion. Mol Cancer Res. 2009;7:1756–1770. doi: 10.1158/1541-7786.MCR-09-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu B, Nandhu MS, Sim H, Agudelo-Garcia PA, Saldivar JC, Dolan CE, et al. Fibulin-3 promotes glioma growth and resistance through a novel paracrine regulation of Notch signaling. Cancer Res. 2012;72:3873–3885. doi: 10.1158/0008-5472.CAN-12-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nandhu MS, Hu B, Cole SE, Erdreich-Epstein A, Rodriguez-Gil DJ, Viapiano MS. Novel paracrine modulation of Notch-DLL4 signaling by fibulin-3 promotes angiogenesis in high-grade gliomas. Cancer Res. 2014;74:5435–5448. doi: 10.1158/0008-5472.CAN-14-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiddingh L, Tannous BA, Teng J, Tops B, Jeuken J, Hulleman E, et al. EFEMP1 induces gamma-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget. 2014;5:363–374. doi: 10.18632/oncotarget.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–927. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- 21.Ohnishi T, Matsumura H, Izumoto S, Hiraga S, Hayakawa T. A novel model of glioma cell invasion using organotypic brain slice culture. Cancer Res. 1998;58:2935–2940. [PubMed] [Google Scholar]
- 22.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alcamo E, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, et al. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-kappa B in leukocyte recruitment. J Immunol. 2001;167:1592–1600. doi: 10.4049/jimmunol.167.3.1592. [DOI] [PubMed] [Google Scholar]
- 24.Espinosa L, Cathelin S, D’Altri T, Trimarchi T, Statnikov A, Guiu J, et al. The Notch/Hes1 pathway sustains NF-kappaB activation through CYLD repression in T cell leukemia. Cancer Cell. 2010;18:268–281. doi: 10.1016/j.ccr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mathis BJ, Lai Y, Qu C, Janicki JS, Cui T. CYLD-mediated signaling and diseases. Curr Drug Targets. 2015;16:284–294. doi: 10.2174/1389450115666141024152421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29:5679–5695. doi: 10.1128/MCB.00406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rose-John S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol Res. 2013;71:19–22. doi: 10.1016/j.phrs.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- 29.Klenotic PA, Munier FL, Marmorstein LY, nand-Apte B. Tissue inhibitor of metalloproteinases-3 (TIMP-3) is a binding partner of epithelial growth factor-containing fibulin-like extracellular matrix protein 1 (EFEMP1). Implications for macular degenerations. JBiolChem. 2004;279:30469–30473. doi: 10.1074/jbc.M403026200. [DOI] [PubMed] [Google Scholar]
- 30.Baltimore D. NF-kappaB is 25. Nat Immunol. 2011;12:683–685. doi: 10.1038/ni.2072. [DOI] [PubMed] [Google Scholar]
- 31.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 32.Gray GK, McFarland BC, Nozell SE, Benveniste EN. NF-kappaB and STAT3 in glioblastoma: therapeutic targets coming of age. Expert Rev Neurother. 2014;14:1293–1306. doi: 10.1586/14737175.2014.964211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nogueira L, Ruiz-Ontanon P, Vazquez-Barquero A, Moris F, Fernandez-Luna JL. The NFkappaB pathway: a therapeutic target in glioblastoma. Oncotarget. 2011;2:646–653. doi: 10.18632/oncotarget.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westhoff MA, Zhou S, Nonnenmacher L, Karpel-Massler G, Jennewein C, Schneider M, et al. Inhibition of NF-kappaB signaling ablates the invasive phenotype of glioblastoma. Mol Cancer Res. 2013;11:1611–1623. doi: 10.1158/1541-7786.MCR-13-0435-T. [DOI] [PubMed] [Google Scholar]
- 35.Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger P, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene. 2012;31:4054–4066. doi: 10.1038/onc.2011.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JK, Jin X, Sohn YW, Jin X, Jeon HY, Kim EJ, et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014;353:194–200. doi: 10.1016/j.canlet.2014.07.034. [DOI] [PubMed] [Google Scholar]
- 37.Ellert-Miklaszewska A, Dabrowski M, Lipko M, Sliwa M, Maleszewska M, Kaminska B. Molecular definition of the pro-tumorigenic phenotype of glioma-activated microglia. Glia. 2013;61:1178–1190. doi: 10.1002/glia.22510. [DOI] [PubMed] [Google Scholar]
- 38.Kim YJ, Hwang SY, Han IO. Insoluble matrix components of glioma cells suppress LPS-mediated iNOS/NO induction in microglia. Biochem Biophys Res Commun. 2006;347:731–738. doi: 10.1016/j.bbrc.2006.06.149. [DOI] [PubMed] [Google Scholar]
- 39.Conti A, Guli C, La Torre D, Tomasello C, Angileri FF, Aguennouz M. Role of inflammation and oxidative stress mediators in gliomas. Cancers (Basel) 2010;2:693–712. doi: 10.3390/cancers2020693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bredel M, Scholtens DM, Yadav AK, Alvarez AA, Renfrow JJ, Chandler JP, et al. NFKBIA deletion in glioblastomas. N Engl J Med. 2011;364:627–637. doi: 10.1056/NEJMoa1006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobayashi N, Kostka G, Garbe JH, Keene DR, Bachinger HP, Hanisch FG, et al. A comparative analysis of the fibulin protein family. Biochemical characterization, binding interactions, and tissue localization. J Biol Chem. 2007;282:11805–11816. doi: 10.1074/jbc.M611029200. [DOI] [PubMed] [Google Scholar]
- 42.Argraves WS, Greene LM, Cooley MA, Gallagher WM. Fibulins: physiological and disease perspectives. EMBO Rep. 2003;4:1127–1131. doi: 10.1038/sj.embor.7400033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Vega S, Iwamoto T, Yamada Y. Fibulins: multiple roles in matrix structures and tissue functions. Cell Mol Life Sci. 2009;66:1890–1902. doi: 10.1007/s00018-009-8632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Obaya AJ, Rua S, Moncada-Pazos A, Cal S. The dual role of fibulins in tumorigenesis. Cancer Lett. 2012;325:132–138. doi: 10.1016/j.canlet.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 45.Seeliger H, Camaj P, Ischenko I, Kleespies A, De Toni EN, Thieme SE, et al. EFEMP1 expression promotes in vivo tumor growth in human pancreatic adenocarcinoma. Mol Cancer Res. 2009;7:189–198. doi: 10.1158/1541-7786.MCR-08-0132. [DOI] [PubMed] [Google Scholar]
- 46.En-lin S, Sheng-guo C, Hua-qiao W. The expression of EFEMP1 in cervical carcinoma and its relationship with prognosis. Gynecol Oncol. 2010;117:417–422. doi: 10.1016/j.ygyno.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 47.Song EL, Hou YP, Yu SP, Chen SG, Huang JT, Luo T, et al. EFEMP1 expression promotes angiogenesis and accelerates the growth of cervical cancer in vivo. Gynecol Oncol. 2011;121:174–180. doi: 10.1016/j.ygyno.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 48.Hwang CF, Chien CY, Huang SC, Yin YF, Huang CC, Fang FM, et al. Fibulin-3 is associated with tumour progression and a poor prognosis in nasopharyngeal carcinomas and inhibits cell migration and invasion via suppressed AKT activity. J Pathol. 2010;222:367–379. doi: 10.1002/path.2776. [DOI] [PubMed] [Google Scholar]
- 49.Pass HI, Levin SM, Harbut MR, Melamed J, Chiriboga L, Donington J, et al. Fibulin-3 as a blood and effusion biomarker for pleural mesothelioma. N Engl J Med. 2012;367:1417–1427. doi: 10.1056/NEJMoa1115050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Creaney J, Dick IM, Meniawy TM, Leong SL, Leon JS, Demelker Y, et al. Comparison of fibulin-3 and mesothelin as markers in malignant mesothelioma. Thorax. 2014;69:895–902. doi: 10.1136/thoraxjnl-2014-205205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Wei D, Zhao Y, Liu X, Zhang J. Overexpression of EFEMP1 correlates with tumor progression and poor prognosis in human ovarian carcinoma. PLoS One. 2013;8:e78783. doi: 10.1371/journal.pone.0078783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camaj P, Seeliger H, Ischenko I, Krebs S, Blum H, De Toni EN, et al. EFEMP1 binds the EGF receptor and activates MAPK and Akt pathways in pancreatic carcinoma cells. Biol Chem. 2009;390:1293–1302. doi: 10.1515/BC.2009.140. [DOI] [PubMed] [Google Scholar]
- 53.Hu Y, Gao H, Vo C, Ke C, Pan F, Yu L, et al. Anti-EGFR function of EFEMP1 in glioma cells and patient prognosis. Oncoscience. 2014;1:205–215. doi: 10.18632/oncoscience.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45:146–169. doi: 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newton RC, Solomon KA, Covington MB, Decicco CP, Haley PJ, Friedman SM, et al. Biology of TACE inhibition. Ann Rheum Dis. 2001;60(Suppl 3):iii25–32. doi: 10.1136/ard.60.90003.iii25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Chen L, Chen J, Hu W, Gao H, Xie B, et al. ADAM17 promotes U87 glioblastoma stem cell migration and invasion. Brain Res. 2013;1538:151–158. doi: 10.1016/j.brainres.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 57.Chen X, Chen L, Zhang R, Yi Y, Ma Y, Yan K, et al. ADAM17 regulates self-renewal and differentiation of U87 glioblastoma stem cells. Neurosci Lett. 2013;537:44–49. doi: 10.1016/j.neulet.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 58.Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G. A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro Oncol. 2014;16:382–391. doi: 10.1093/neuonc/not232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng X, Jiang F, Katakowski M, Lu Y, Chopp M. ADAM17 promotes glioma cell malignant phenotype. Mol Carcinog. 2012;51:150–164. doi: 10.1002/mc.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giltay R, Timpl R, Kostka G. Sequence, recombinant expression and tissue localization of two novel extracellular matrix proteins, fibulin-3 and fibulin-4. Matrix Biol. 1999;18:469–480. doi: 10.1016/s0945-053x(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 61.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 62.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Celiku O, Johnson S, Zhao S, Camphausen K, Shankavaram U. Visualizing molecular profiles of glioblastoma with GBM-BioDP. PLoS One. 2014;9:e101239. doi: 10.1371/journal.pone.0101239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 65.Shelest E, Kel AE, Goessling E, Wingender E. Prediction of potential C/EBP/NF-kappaB composite elements using matrix-based search methods. In Silico Biol. 2003;3:71–79. [PubMed] [Google Scholar]
- 66.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.