

Abstract
Objective:
To identify rare coding variants segregating with late-onset Alzheimer disease (LOAD) in Caribbean Hispanic families.
Methods:
Whole-exome sequencing (WES) was completed in 110 individuals from 31 Caribbean Hispanic families without APOE ε4 homozygous carriers. Rare coding mutations segregating in families were subsequently genotyped in additional families and in an independent cohort of Caribbean Hispanic patients and controls. SRCAP messenger RNA (mRNA) expression was assessed in whole blood from mutation carriers with LOAD, noncarriers with LOAD, and healthy elderly controls, and also from autopsied brains in 2 clinical neuropathologic cohort studies of aging and dementia.
Results:
Ten ultra-rare missense mutations in the Snf2-related CREBBP, activator protein (SRCAP), were found in 12 unrelated families. Compared with the frequency in Caribbean Hispanic controls and the Latino population in the Exome Aggregation Consortium, the frequency of SRCAP mutations among Caribbean Hispanic patients with LOAD was significantly enriched (p = 1.19e-16). mRNA expression of SRCAP in whole blood was significantly lower in mutation carriers with LOAD, while the expression in whole blood and in the brain was significantly higher in nonmutation carriers with LOAD. Brain expression also correlated with clinical and neuropathologic endophenotypes.
Conclusions:
WES in Caribbean Hispanic families with LOAD revealed ultra-rare missense mutations in SRCAP, a gene expressed in the brain and mutated in Floating-Harbor syndrome. SRCAP is a potent coactivator of the CREB-binding protein and a regulator of DNA damage response involving ATP-dependent chromatin remodeling. We hypothesize that increased expression in LOAD suggests a compensatory mechanism altered in mutation carriers.
Progress has been made in understanding the genetics of late-onset Alzheimer disease (LOAD),1,2 but gaps in its genetic influence still need investigation. Common variants play a role in disease risk, but functionally important rare or ultra-rare variants may help to explain the remaining heritability2,3 undetected by genome-wide association studies. Sequencing of large families multiply affected by LOAD increases the ability to detect novel variants conferring risk.
The frequency of LOAD among Caribbean Hispanic multiplex families from the Dominican Republic was 5 times higher than expected for similarly aged individuals in a non-Hispanic white population from the United States,4 and inbreeding was a significant predictor of LOAD in this population after adjusting for APOE-ε4 genotype, an established genetic risk factor.5
To identify novel variants associated with the risk of LOAD, we conducted whole-exome sequencing in 31 Caribbean Hispanic families (table e-1 at Neurology.org/ng) with 4 or more affected individuals, no mutations in known AD genes, specifically PSEN1, PSEN2, or APP, and no APOE ε4 homozygotes. For each family, we sequenced at least 2 affected and 1 unaffected member aged 65 years or older.
METHODS
Sample selection.
Families were recruited as a part of a 15-year family-based study with institutional review board (IRB) approval based in the Dominican Republic.6 Thirty-one families (98 affected and 12 unaffected individuals) were selected for sequencing (mean age at onset was 74.8 wa8.3 years, and 63.1% were women) (table e-1, a–c). All family members had standard neuropsychological tests and neurologic examinations to verify their clinical status and for diagnoses based on NINCDS-ADRDA criteria.7,8
Postmortem human brain samples.
Data were obtained from 2 clinical neuropathologic cohort studies: the ROS9 and the MAP.10 The IRB of Rush University Medical Center previously approved both studies. Clinical evaluations were used to determine NINCDS-ADRDA7,8 criteria for dementia annually.11–13 At death, a clinical diagnosis opinion was provided by a neurologist.14 Neuropathologic evaluations included neuritic plaques, diffuse plaques, and neurofibrillary tangles in 5 cortical regions, scaled and averaged to obtain a composite score.15 Participants who met intermediate or high likelihood were rendered pathologic diagnosis of LOAD.16,17
WHOLE-EXOME SEQUENCING
Sample preparation.
Qiagen's Gentra Puregene and FlexiGene kits were used to extract high-molecular-weight DNA from fresh or frozen (<−80°C) samples. DNA from saliva was isolated using prepIT.L2P (DNA Genoteck Inc., Ottawa, ON, Canada). Cell lines from lymphocytes (in 13 probands) were used when high-quality blood DNA was not available. The concentration of DNA was determined using a NanoDrop spectrophotometer.
Sequencing.
The Illumina TruSeq DNA preparation kit was used to prepare and index genomic DNA libraries. Custom oligonucleotide baits in the TruSeq Exome Enrichment kit were used to capture coding regions and splice sites and amplified according to the Illumina protocol. The DNA samples were multiplexed in batches of 12 samples with index “barcode” primers. These were sequenced using the Illumina Genome Analyzer IIx, HiSeq 2000, and MiSeq platforms (illumina.com) as paired-end reads over 82–307 cycles. Demultiplexing by barcode retrieved individual samples from sequencing pools. We obtained a high coverage across the samples at an average depth of >60× per sample.
Follow-up genotyping.
Putative variants were confirmed and population frequencies estimated by genotyping the discovery samples, additional family members, and unrelated controls of the same ancestry (table e-1). Allele frequencies of novel variants were estimated from 1,949 unrelated patients and 318 healthy elderly controls similar in age and ancestry.6 Genotypes were generated using the KASP genotyping technology, which uses allele-specific PCR for accurate calling of single nucleotide polymorphisms (SNPs) and Indels.18
Analytical methods.
Burrows-Wheeler Aligner19 was used to align sequence reads to the reference genome build 37. Sequencing data quality control (QC) was performed using the Genome Analysis Toolkit (GATK),20 followed by variant calling using the UnifiedGenotyper and VariantRecalibrator modules. Variants that passed QC were annotated by ANNOVAR21 that included functional prediction by SIFT22 and PolyPhen.23
STATISTICAL METHODS
Association tests.
Variants that were validated by follow-up genotyping were tested for association with LOAD using generalized estimating equations (GEEs), which accounts for the familial correlation. We adjusted for age and sex using data from the families and unrelated healthy controls. We used GEE to conduct single variant and burden tests.
For joint analyses of multiple variants, we summed the number of rare variant alleles found in all individuals and tested association using GEE, adjusting for familial correlation, age and sex, and APOE ε4. The p value threshold required to define statistical significance using a rare variant burden analysis for 20,000 genes would be 2.5 × 10−6. However, this would be difficult to achieve by assessing rare or ultra-rare variants in a data set of this size. Thus, to determine whether or not the variants discovered were enriched in families with AD in subsequent analyses, we used data from the Exome Aggregation Consortium (ExAC) (exac.broadinstitute.org) combined with the Caribbean Hispanic controls.
Exome Aggregation Consortium.
The ExAC database24 contains whole-exome data from 60,706 unrelated adults sequenced as part of various disease-specific (excluding AD) and population genetic studies from 6 ethnic groups. For disease association analyses, we compared the allele frequencies of suspected variants in the healthy controls from the Caribbean Hispanic cohort to the Latino, Caucasian, and African subpopulations of the ExAC database using a Fisher exact test to avoid differences based on ancestry. Subsequent disease-associated analyses were only conducted when there were no statistically significant differences in allele frequency between the Caribbean Hispanic controls and the ExAC subpopulations. To confirm associations of genetic variants with LOAD, we compared the variant allele frequencies by selecting 1 patient from each Caribbean Hispanic family with both unaffected unrelated controls and ExAC combined using a Fisher exact test.
Gene expression in the postmortem human brain.
RNA-Seq data came from the 541 ROS-MAP postmortem human brain samples (average age at death was 88.4 NA6.7 years; 63.0% were women and 97.4 self-identified Caucasian ancestry). RNA was extracted from the gray matter of frozen dorsolateral prefrontal cortex tissue using the Qiagen miRNeasy mini kit (Cat. no. 217004) and the RNase-free DNase Set (Cat. no. 79254) and quantified using NanoDrop. RNA-Seq library was prepared on the Broad Institute's Genomics Platform using the strand-specific dUTP method25 with poly-A selection.26 Sequencing was performed on the Illumina HiSeq with 101bp paired-end reads and minimal coverage of 50M reads. Reads were aligned to the reference genome (GRCh37/hg19) using Bowtie and gene expression, measured as fragments per kilobase per million fragments mapped (FPKM), and estimated using RSEM software. Quantile normalization was applied to the FPKM calls, with batch effect removed using Combat.27 Logarithm base 2 transformation was applied to the gene expression level prior to the analyses.
The Student t test compared brain expression levels between LOAD and controls. Regression models were used to examine the association of gene expression with LOAD and with level of cognition proximate to death. Regression models were used to examine the relation between gene expression and a postmortem diagnosis of LOAD or with LOAD pathology. All models were adjusted for age at death, sex, education, postmortem interval RNA integrity number, and APOE ε4 status. Analyses were performed using SAS software, version 9.3, of the SAS(R) system for Linux.
RESULTS
Whole-exome sequencing.
Variant calling, recalibration, and application of QC filters resulted in 290,623 single nucleotide variants (SNVs) and indels called across the 110 individuals from the 31 families (figure 1). The mean depth of sequence coverage was over 60-fold across individuals. We prioritized damaging SNVs by SIFT22 or by PolyPhen28 and indels in 78 genes segregating with LOAD in families (figure e-1, A and B), which were confirmed by genotyping in the discovery families, additional family members (265 affected and 61 unaffected individuals), and a set of 318 unrelated controls of similar age and ancestry. Using GEE adjusted for age, sex, and APOE genotype, we found 22 (28%) of the 78 selected variants (in 22 genes) significantly associated with LOAD (p = 6.4e-03 corrected for multiple testing) (table e-2). Only 4 of these 22 genes were known to be expressed in brain-ADCY6, CIT, SRCAP, and SVOPL. The variants in SRCAP, ADCY6, and SVOPL were absent in unaffected Caribbean Hispanic individuals, but only SRCAP met criteria for further analyses by having multiple, putatively damaging variants segregating in more than 1 of the 31 families.
Figure 1. Workflow of the experiment and yield at each step.
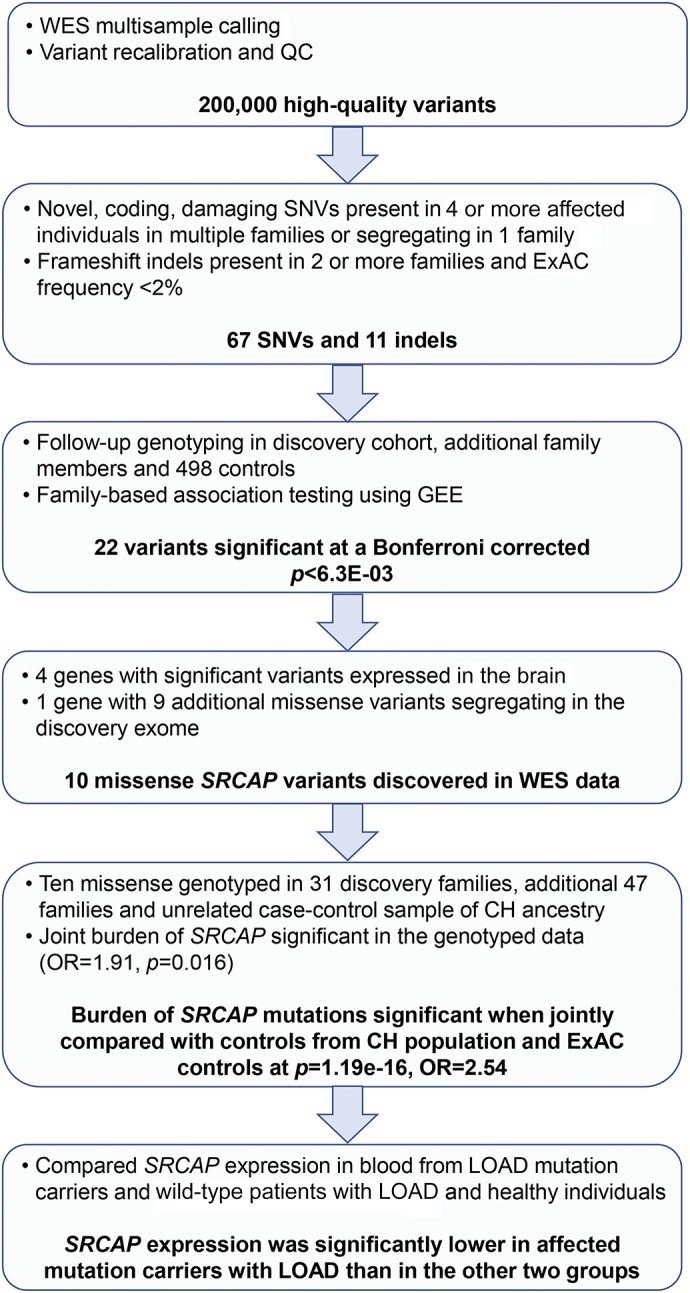
CH = Caribbean Hispanic; ExAC = Exome Aggregation Consortium; GEE = generalized estimating equation; OR = odds ratio; QC = quality control; SNV = single nucleotide variant; WES = whole-exome sequencing.
We observed 10 ultra-rare missense mutations, defined as having an allele frequency ≤0.5%, in SRCAP in 12 families (table 1). Seven of these mutations were predicted to be damaging by either PolyPhen or SIFT and had a Combined Annotation Dependent Depletion29 >10. Twenty-four (24.4%) of the 98 affected individuals sequenced within the 31 families were carriers of SRCAP mutations. This was significantly higher than the expected frequency based on data from ExAC database24 (1.79 expected mutations; p = 1.38e-05 using a binomial test).
Table 1.
Missense variants in SRCAP found in WES of Caribbean Hispanic families
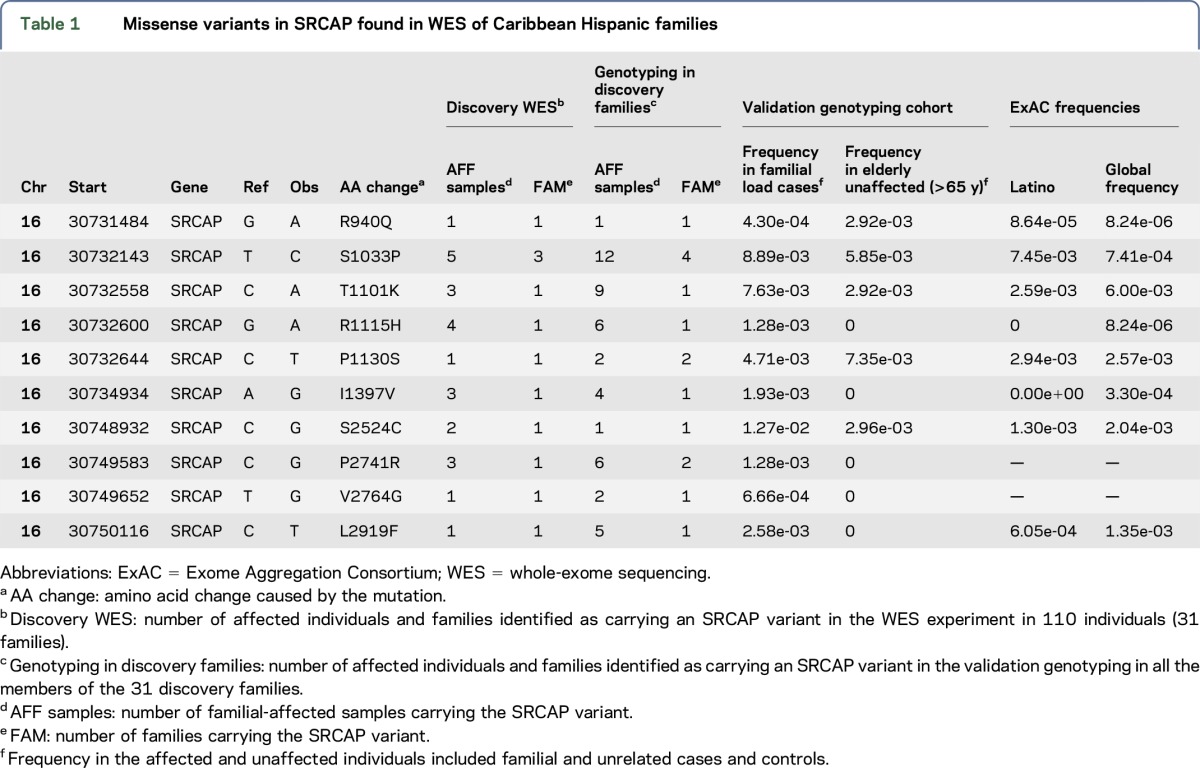
All 10 SRCAP mutations were genotyped in additional members of the 31 families and segregated with affection status but with incomplete penetrance (figures e-3, A–D and e-4). Of the 265 patients with LOAD in the 31 families, 47 patients (17.67%) carried at least 1 of the 10 missense mutations in SRCAP compared with 2 of the 61 (3.2%) unaffected individuals from these families. Variant p.R1115H segregated with LOAD in the largest sibship in an age-dependent manner (supplementary methods, figure e-2, tables e-3, a and b) and was absent in Caribbean Hispanic controls and ExAC individuals of European, Latino, and African ancestry. p.R1115H was also found within 0.35 MB of a linkage peak (logarithm (base 10) of odds = 1.8). A single patient was homozygous for an SRCAP p.S2524C mutation and had an earlier age at onset compared with the average age in the cohort (63 vs 73.8 years). In family 1755 (figure 2), there were 2 compound heterozygous carriers (p.L2919F and p.S1033P) with earlier ages at onset (55 and 59 years) compared with LOAD patients without SRCAP mutations (p = 0.023) or patients who had a single SRCAP mutation (p = 0.018). Of interest, no unaffected family member or unrelated control was a homozygous or compound heterozygous carrier.
Figure 2. Segregation pattern of missense SRCAP mutations in pedigree 1755.

Patients 99 and 9 are compound heterozygotes with ages at onset of 59 and 55 years, respectively. Four other affected siblings who were heterozygous (3 p.L2919F and 1 p.S1033P) for SRCAP mutation had ages at onset of 58, 58, 72, and 76 years, respectively. Import ID: Subject ID; AAO_Affected_Else_AgeLast_Seen: Age at onset of LOAD or age at last examination for healthy individuals. LOAD = late-onset Alzheimer disease.
A joint burden analysis within the 31 discovery families using GEE adjusted for familial correlation, age, sex, and APOE genotype found a 6-fold increase in the risk of LOAD (odds ratio [OR] = 5.94; 95% confidence interval [CI] 1.59–22.27, p = 8.2e-03). Adding the additional 47 families resulted in a 2.5-fold risk of LOAD in families (OR = 2.54; 95% CI 1.03–6.21, p = 0.04). We then tested the joint burden of the 10 SRCAP variants in all the genotyped individuals in both sets of families, 1,949 unrelated LOAD cases and 318 unrelated elderly controls (table e-1b). The risk of LOAD associated with SRCAP remained increased (OR = 1.92; 95% CI 1.13–3.28, p = 0.016) after including APOE ε4 in the model (OR = 1.82; 95% CI 1.04–3.19, p = 0.036).
To reach genome-wide significance (p = 2.5e-06) in a gene-based test, assuming an OR of 2.0 and 5% causal SNPs and β = 0.8, a gene of average size, such as SRCAP, requires an extremely large sample size (N = 42,000). Therefore, we compared the combined genotyped data mentioned above with data from the ExAC database.24 Using a Fisher exact test, we found no statistically significant differences in the allele frequencies of the SRCAP variants in healthy Caribbean Hispanic controls compared with the Latino, African American, and white, non-Hispanics in the ExAC database. We then compared the frequencies of SRCAP variants of LOAD patients with Caribbean Hispanic controls alone or with ExAC Latino controls, ExAC African American controls, or ExAC white, non-Hispanic controls. Using one mutation carrier per family, we compared the number of variant alleles in the LOAD patients with the number of alleles in the ExAC database. Two variants, p.P2741R and p.V2764G, were not found in the ExAC database (table 1). We used the total number of alleles genotyped or sequenced for the 10 mutations as the denominator for the Fisher exact test. The enrichment of SRCAP variants in the Caribbean Hispanic patients with LOAD was highly significant at p = 1.19e-16. Similarly, the enrichment was statistically significant for white, non-Hispanics, and the African American ethnic groups in the ExAC database (table e-4; p = 1.51e-21 for white, non-Hispanics; p = 3e-04 for African Americans). Five of the 10 SRCAP mutations were not found in the Caribbean Hispanic controls, and 4 of these were absent in the ExAC Latino data (table 1). Restricting analyses to these 5 SNPs only, the enrichment remained significant p = 3.55e-07.
SRCAP is highly intolerant toward loss of function and missense mutations.24,30 SRCAP's intolerance score (2.23) in the ExAC ranks in the 18th percentile and in the top 0.15 and 0.3 percentiles of genes ranked by the Residual Variation Intolerance Score (RVIS) using the ExAC and Exome Sequencing Project data sets, respectively.
Replication in the AD Neuroimaging Initiative data set.
Using data from the Alzheimer Disease Neuroimaging Initiative (ADNI) data set,31 we repeated the analysis of the exonic regions of SRCAP. The analyses included 213 patients with LOAD, 304 individuals with mild cognitive impairment (MCI), and 214 healthy controls of white, non-Hispanic ancestry. We found a higher frequency of nonsynonymous mutations in LOAD and MCI cases (10.4%) vs controls (7.4%) representing a 40% enrichment. Because of the sample size, we once again compared the frequency of nonsynonymous mutations in LOAD with the ExAC database. Of interest, there was a higher frequency in LOAD vs ExAC controls (OR = 1.78, p = 2.3e-04).
SRCAP gene expression.
Among the ROS-MAP cohorts, 218 of 541 (40.3%) individuals were diagnosed with LOAD at death, and compared with nondemented persons, SRCAP expression was significantly higher (OR = 4.9 [2.1–11.6], p = 0.0002) (figure 3A). Individuals with higher SRCAP expression also had lower cognitive scores prior to death (β = −0.96, p < 0.0001) (figure 3B). Similar associations were observed in episodic, semantic and working memory, and perceptual speed (table e-5). Using the clinical diagnosis, higher SRCAP expression was associated with greater likelihood of meeting the NIA-Reagan criteria for AD (OR = 4.0 [1.7–9.2], p = 0.0015) (figure 3C). Individuals with higher SRCAP expression had a greater burden of pathology (β = 0.3, p < 0.0001) (figure 3D). We also accessed whole-genome sequencing data from 63 participants with brain expression. Two of 69 individuals sequenced carried 1 of the 10 SRCAP missense mutations (S1033P and L2919F), and both were diagnosed with mild cognitive impairment.
Figure 3. Association of expression level of SRCAP in autopsy brain tissue with (A) AD clinical diagnoses, (B) cognitive performance, (C) AD pathologic diagnoses, and (D) global AD pathology.

AD = Alzheimer disease.
Mutation-specific gene expression.
Relative messenger RNA (mRNA) expression of SRCAP (supplementary methods) in whole blood was measured in 5 mutation carriers with LOAD, 12 individuals without mutations with LOAD, and 12 elderly controls without dementia, all unrelated. After normalization against GAPDH, SRCAP mRNA expression was significantly lower in the mutation carriers with LOAD than in the other 2 groups (figure 4) (LOAD carriers 0.74, LOAD noncarriers 1.128, and controls 0.99, p = 0.004). We compared the levels of exogenous mature wild-type SRCAP mRNA in HEK293 cells to the one carrying an ultra-rare mutation (p.P2741R; minor allele frequency = 0.00128 in Caribbean Hispanic cases and absent in ExAC). mRNA expression of mutant SRCAP (p.P2741R) was downregulated by 40% relative to wild-type SRCAP.
Figure 4. Comparison of SRCAP mRNA expression in affected mutation carriers, affected noncarriers, and controls.
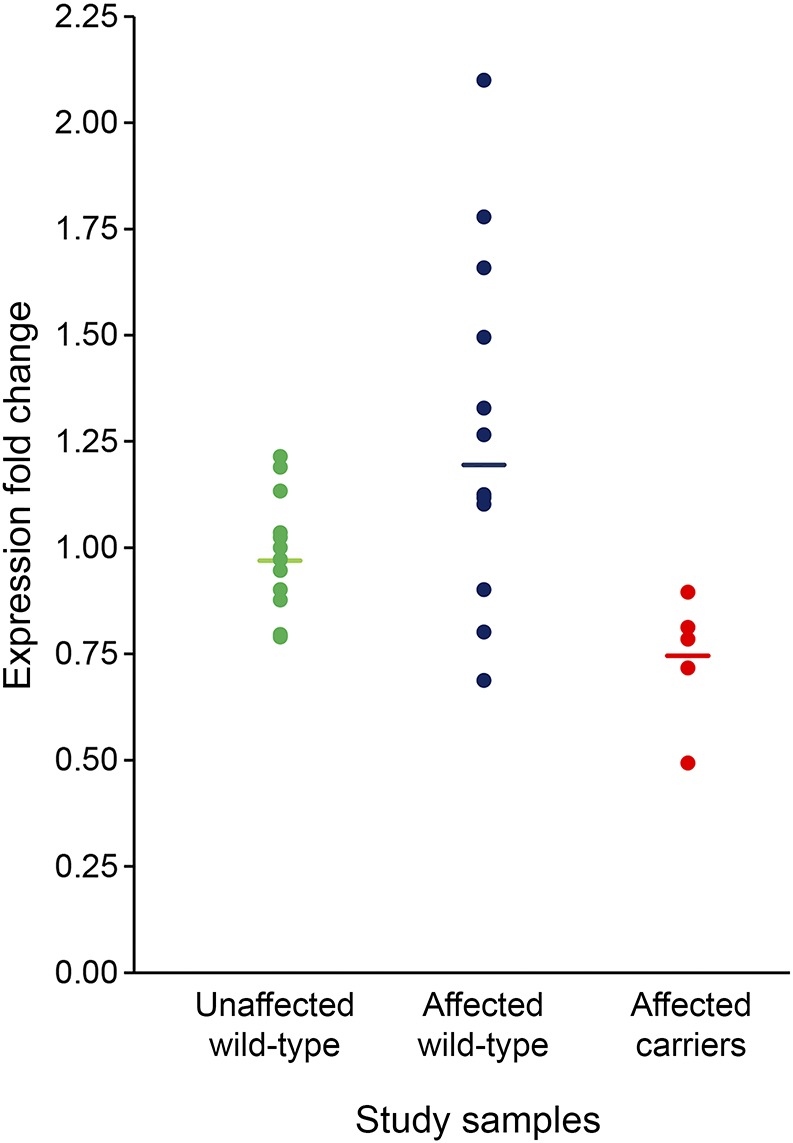
mRNA = messenger RNA.
DISCUSSION
A statistically significant enrichment of SRCAP ultra-rare mutations was found among patients within Caribbean Hispanic families multiply affected by LOAD. These mutations were observed to segregate imperfectly with disease consistent with the presence of phenocopies in large LOAD families. Reduced mRNA expression of SRCAP was found in whole blood of mutation carriers compared with noncarriers and controls. Higher SRCAP expression was expressed in noncarriers in both blood of Caribbean Hispanic patients with LOAD and brains of Caucasian patients with LOAD, irrespective of differences in ancestry. Pathologic evidence indicates that SRCAP expression is altered in LOAD with or without mutations, correlating with characteristic clinical manifestations. Taken together, the combined data implicate a putative role for SRCAP in LOAD.
Snf2-related cyclic AMP-responsive element–binding protein (SRCAP) binds to the CREB-binding protein (CBP), which influences the transcription of CREB. CREB and the related transcription factors are involved in memory retention32 and consolidation by hippocampal neurogenesis.33 In AD-transgenic mice and in humans with LOAD, CREB and CBP are decreased or disrupted by the accumulation of amyloid-β.34–36 Reduced levels have also been observed for pCREB, CBP, and related CREB co-activators p300 and cAMP-dependent protein kinase in the human AD brain and blood compared with elderly controls.37 We also observed reduced mRNA expression of SRCAP in mutation carriers compared with healthy elderly controls and unrelated, noncarriers with LOAD, and a reduction in SRCAP expression in cell lines transfected with SRCAP mutations. We reported an association between episodic memory and SNP rs2526690 in CBP as well as nominal significance for common variants in CREB1 and RBAP48 in the CREB pathway.38
SRCAP regulates the CREB pathway by catalyzing H2A.Z into chromatin, which is required for gene expression and interactions with co-activators of CREB such as CBP.39,40 In the absence of mutations, the increased mRNA expression of SRCAP in postmortem LOAD brain suggests a compensatory response to promote CREB activation in the presence of Aβ accumulation. Paradoxically, in persons with SRCAP mutations, disruption of the binding of SRCAP to CBP would decrease CREB-mediated transcription in brain over the lifetime. CREB may have neuroprotective qualities against Aβ toxicity, and persistent downregulation of CREB resulting from decreased SRCAP could promote neurodegeneration.41 This suggests that SRCAP has a regulatory role in the CREB pathway in LOAD, regardless of the presence or absence of SRCAP mutations. Ample evidence indicates disruption of CREB transcription by Aβ, but the effects of these ultra-rare mutations on protein function and the LOAD phenotype will need to be fully elucidated.
Truncating mutations in SRCAP cause Floating-Harbor syndrome,42 a childhood disorder characterized by short stature, delayed speech, and facial abnormalities. The majority of SRCAP mutations reduced expression and create a nonfunctioning protein. Mutations in CBP, the substrate of SRCAP activation, cause a developmental disorder similar to Floating-Harbor syndrome and Rubinstein-Taybi syndrome.
This study does have limitations. We found a modest increase in the risk of LOAD among carriers of these ultra-rare SRCAP mutations in Caribbean Hispanic families, which makes replication difficult. Validation and replication efforts in other ethnic backgrounds would be essential to generalize the findings.
Supplementary Material
GLOSSARY
- CBP
CREB-binding protein
- CI
confidence interval
- DLPFC
dorsolateral prefrontal cortex
- ExAC
Exome Aggregation Consortium
- FPKM
fragments per kilobase per million fragments mapped
- GEE
generalized estimating equation
- IRB
institutional review board
- LOAD
late-onset Alzheimer disease
- MCI
mild cognitive impairment
- mRNA
messenger RNA
- OR
odds ratio
- QC
quality control
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
Conception and design of the study: B.N.V., G.T., S.B., R. Lefort, D.A.B., P.L.D.J., E.R., P.S.G.-H., and R.M. Acquisition and analysis of data: B.N.V., R. Lefort, P.L.N., D.R.-D., J.H.L., R.C., L.Y., D.A.B., P.L.D.J., M.M., R. Lantigua, and R.M. Drafting the manuscript or figures: B.N.V., G.T., E.R., P.S.G.-H., and R.M.
STUDY FUNDING
Supported by grants from the National Institute on Aging and the NIH, RF1AG015473, R01AG037212, P50AG008702 (R.M.), P30AG10161, RF1AG1518, R01AG17917, R01AG36936, and U01AG46152 (D.A.B. and P.L.D.J.).
DISCLOSURE
B.N. Vardarajan has served on the scientific advisory board of the Immuneering Corporation. G. Tosto has received research support from NIH/NIA, Columbia University Alzheimer's Disease Research Center (ADRC), and the Department of Defense. R. Lefort has received research support from Columbia University Alzheimer's Disease Research Center (ADRC) and the Alzheimer's Association. L. Yu has received research support from NIH/NIA. D.A. Bennett has served on the scientific advisory boards of Vigorous Minds, Takeda Pharmaceuticals, and AbbVie; has served on the editorial boards of Neurology, Current Alzheimer Research, and Neuroepidemiology; and has received research support from NIH. P.L. De Jager has served on the scientific advisory boards of TEVA Neuroscience, Sanofi/Genzyme, and Celgene; has received speaker honoraria from Biogen Idec, Source Healthcare Analytics, Pfizer Inc., and TEVA; has served on the editorial boards of the Journal of Neuroimmunology, Neuroepigenetics, and Multiple Sclerosis; and has received research support from Biogen, Eisai, UCB, Pfizer, Sanofi/Genzyme, NIH, and the National MS Society. S. Barral and D. Reyes-Dumeyer report no disclosures. P.L. Nagy has been an employee of MNG Laboratories. J.H. Lee has received research support from NIH/NIA, NIH/NCATS, and BrightFocus Foundation. R. Cheng, M. Medrano, and R. Lantigue report no disclosures. E. Rogaeva has received research support from the Ontario Research Fund and the W. Garfield Weston Foundation. P. St George-Hyslop has received research support from the Canadian Institute of Health Research, Medical Research Council, and Wellcome Trust. R. Mayeux has received research support from NIH. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. . Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonsson T, Stefansson H, Steinberg S, et al. . Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guerreiro R, Wojtas A, Bras J, et al. . TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vardarajan BN, Faber KM, Bird TD, et al. . Age-specific incidence rates for dementia and Alzheimer disease in NIA-LOAD/NCRAD and EFIGA families: National Institute on Aging Genetics Initiative for Late-Onset Alzheimer Disease/National Cell Repository for Alzheimer Disease (NIA-LOAD/NCRAD) and Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA). JAMA Neurol 2014;71:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vardarajan BN, Schaid DJ, Reitz C, et al. . Inbreeding among Caribbean Hispanics from the Dominican Republic and its effects on risk of Alzheimer disease. Genet Med 2015;17:639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JH, Cheng R, Barral S, et al. . Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol 2011;68:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Aggarwal NT, et al. . Decision rules guiding the clinical diagnosis of Alzheimer's disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006;27:169–176. [DOI] [PubMed] [Google Scholar]
- 9.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the Religious orders study. Curr Alzheimer Res 2012;9:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the rush memory and aging project. Curr Alzheimer Res 2012;9:646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson RS, Beckett LA, Barnes LL, et al. . Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193. [PubMed] [Google Scholar]
- 12.Wilson RS, Boyle PA, Yu L, Segawa E, Sytsma J, Bennett DA. Conscientiousness, dementia related pathology, and trajectories of cognitive aging. Psychol Aging 2015;30:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson R, Barnes L, Bennett D. Assessment of lifetime participation in cognitively stimulating activities. J Clin Exp Neuropsychol 2003;25:634–642. [DOI] [PubMed] [Google Scholar]
- 14.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–2204. [DOI] [PubMed] [Google Scholar]
- 15.Bennett DA, Wilson RS, Schneider JA, et al. . Apolipoprotein E epsilon4 allele, AD pathology, and the clinical expression of Alzheimer's disease. Neurology 2003;60:246–252. [DOI] [PubMed] [Google Scholar]
- 16.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009;66:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997;56:1095–1097. [DOI] [PubMed] [Google Scholar]
- 18.Abrams KR, Gillies CL, Lambert PC. Meta-analysis of heterogeneously reported trials assessing change from baseline. Stat Med 2005;24:3823–3844. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKenna A, Hanna M, Banks E, et al. . The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 23.Adzhubei IA, Schmidt S, Peshkin L, et al. . A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lek M, Karczewski KJ, Minikel EV, et al. . Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin JZ, Yassour M, Adiconis X, et al. . Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 2010;7:709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adiconis X, Borges-Rivera D, Satija R, et al. . Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods 2013;10:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007;8:118–127. [DOI] [PubMed] [Google Scholar]
- 28.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013;Chap 7. Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saykin AJ, Shen L, Foroud TM, et al. . Alzheimer's Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimers Dement 2010;6:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lakhina V, Arey RN, Kaletsky R, et al. . Genome-wide functional analysis of CREB/long-term memory-dependent transcription reveals distinct basal and memory gene expression programs. Neuron 2015;85:330–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ortega-Martinez S. A new perspective on the role of the CREB family of transcription factors in memory consolidation via adult hippocampal neurogenesis. Front Mol Neurosci 2015;8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duclot F, Meffre J, Jacquet C, Gongora C, Maurice T. Mice knock out for the histone acetyltransferase p300/CREB binding protein-associated factor develop a resistance to amyloid toxicity. Neuroscience 2010;167:850–863. [DOI] [PubMed] [Google Scholar]
- 35.Pugazhenthi S, Wang M, Pham S, Sze CI, Eckman CB. Downregulation of CREB expression in Alzheimer's brain and in Abeta-treated rat hippocampal neurons. Mol Neurodegener 2011;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satoh J, Tabunoki H, Arima K. Molecular network analysis suggests aberrant CREB-mediated gene regulation in the Alzheimer disease hippocampus. Dis Markers 2009;27:239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartolotti N, Bennett DA, Lazarov O. Reduced pCREB in Alzheimer's disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol Psychiatry 2016;21:1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barral S, Reitz C, Small SA, Mayeux R. Genetic variants in a “cAMP element binding protein” (CREB)-dependent histone acetylation pathway influence memory performance in cognitively healthy elderly individuals. Neurobiol Aging 2014;35:2881.e7–2881.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong MM, Cox LK, Chrivia JC. The chromatin remodeling protein, SRCAP, is critical for deposition of the histone variant H2A.Z at promoters. J Biol Chem 2007;282:26132–26139. [DOI] [PubMed] [Google Scholar]
- 40.Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM, Sweatt JD. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 2014;515:582–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monroy MA, Ruhl DD, Xu X, Granner DK, Yaciuk P, Chrivia JC. Regulation of cAMP-responsive element-binding protein-mediated transcription by the SNF2/SWI-related protein, SRCAP. J Biol Chem 2001;276:40721–40726. [DOI] [PubMed] [Google Scholar]
- 42.Nikkel SM, Dauber A, de Munnik S, et al. . The phenotype of Floating-Harbor syndrome: clinical characterization of 52 individuals with mutations in exon 34 of SRCAP. Orphanet J Rare Dis 2013;8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.