

Abstract
Atrial fibrillation (AF) is the most common arrhythmia in clinical practice and is a major cause of morbidity and mortality. The upregulation of TRP channels is believed to mediate the progression of electrical remodelling and the arrhythmogenesis of the diseased heart. However, there is limited data about the contribution of the TRP channels to development of AF. The aim of this study was to investigate leukocyte TRP channels gene expressions in non-valvular atrial fibrillation (NVAF) patients. The study included 47 NVAF patients and 47 sex and age matched controls. mRNA was extracted from blood samples, and real-time polymerase chain reaction was performed for gene expressions by using a dynamic array system. Low levels of TRP channel expressions in the controls were markedly potentiated in NVAF group. We observed marked increases in MCOLN1 (TRPML1), MCOLN2 (TRPML2), MCOLN3 (TRPML3), TRPA1, TRPM1, TRPM2, TRPM3, TRPM4, TRPM5, TRPM6, TRPM7, TRPM8, TRPC1, TRPC2, TRPC3, TRPC4, TRPC5, TRPC6, TRPC7, TRPV1, TRPV2, TRPV3, TRPV4, TRPV5, TRPV6, and PKD2 (TRPP2) gene expressions in NVAF patients (P < 0.05). However, there was no change in PKD1 (TRPP1) gene expression. This is the first study to provide evidence that elevated gene expressions of TRP channels are associated with the pathogenesis of NVAF.
Introduction
Atrial fibrillation (AF) is the most common arrhythmic disorder associated with increased risk of stroke, heart failure, dementia and cardiovascular mortality1. The prevelance of AF is increasing with age, and it is a growing public health problem2. Pathophysiology of AF is a complex process including structural alterations in the atrium and electrophysiological abnormalities. Atrial fibrosis and inflammation makes the atrial tissue a substrate prone to AF3. Local ectopic firing and multiple wavelets propagating in atrial tissue can initate and maintain AF4, 5. The etiology of AF involved a complex interaction of environmental factors with genetic factors6, 7. Because the utility of conventional antiarrhythmic agents that target cardiac ion channels is limited by inefficacy and side effects, new treatment strategies are required8, 9. Altered Ca2+ handling is a cruical process in AF pathophysiology, and may be a target for antiarrythmic therapy10, 11.
Transient receptor potential (TRP) channels consist of a large number of nonselective cation channels with variable degree of Ca2+-permeability. The 28 mammalian TRP channel proteins can be grouped into six subfamilies based on protein sequence homology: TRPC (canonical), TRPM (melastatin), TRPV (vanilloid), TRPP (polycystin), TRPA (ankyrin), and TRPML (mucolipin)12, 13. The majority of these TRP channels are expressed in different cell types including both excitable and nonexcitable cells of the cardiovascular system. TRP channels are not voltage gated but are activated by a variety of stimuli including pressure, shear stress, mechanical stretch, oxidative stress, membrane-receptor stimulation, hypertrophic signals, inflammation products, and thermal or sensory stimuli12, 13. All functionally characterized TRP channels are permeable to calcium except monovalan cation selective TRPM4 and TRPM512, 13. TRP channels also contribute to endothelial cell apoptosis and cardiac fibrosis via fibroblast differentiation13, 14. Accumulating studies revealed that TRP subfamilies are involved in differentiation of cardiac fibroblasts in most cardiac diseases and atrial electrical remodeling in AF patients15–17. In cardiac myocytes or experimental studies, several TRP channels have been shown to be involved in arrhythmogenesis13. However, which type of TRP channels participates in AF is not exactly known in humans. In this study, we aimed to investigate whether peripheral leukocyte TRP channel gene expressions are associated with the devepment of nonvalvular atrial fibrillation (NVAF), as a reflection of inflammatory status.
Materials and Methods
Patients
A total of 47 NVAF patients followed up in Gaziantep 25 Aralik State Hospital were enrolled in this study. All of the patients had NVAF on surface electrocardiogram. Exclusion criterias were valvular heart disease, heart failure, coronary artery disease, peripheral artery disease, diabetes mellitus, thyroid disorder, kidney failure, autoimmune disorder, pregnancy and cancer. Patients who had any cardiac intervention or an ablation procedure for AF management were also excluded. A total of 47 sex and age matched controls were recruited to the study. The control group consisted of healthy individuals who had no history of AF or cardiac arrhythmias. Hypertension was defined as systolic blood pressure of >140 mm Hg and diastolic blood pressure of >90 mm Hg, in a sitting position, on ≥3 different occasions. Dyslipidemia was defined according to the third report of the National Cholesterol Education Program18. Subjects stopped taking medications for at least 12 h prior to venous blood sample collection. All blood samples were obtained between 9:00 and 10:00 AM. Medications used by the patients are given in Table 1. The study was approved by the Gaziantep University Clinical Research Ethics Committee (Decision no:2015/194), written informed consent prior to participation in the study was obtained from patients and healthy volunteers according to the Declaration of Helsinki.
Table 1.
Baseline demographic and clinical characteristics of patients with NVAF and controls.
| Controls (n = 47) | NVAF Patients (n = 47) | P value | |
|---|---|---|---|
| Age (years) | 59.21 ± 7.43 | 58.78 ± 7.92 | 0.7866 |
| Gender | |||
| Male (n, %) | 25 (53.2) | 24 (51.1) | 0.8364 |
| Female (n, %) | 22 (46.8) | 23 (48.9) | |
| Smoking status | |||
| Current (n, %) | 7 (14.9) | 12 (25.5) | 0.3018 |
| Never (n, %) | 33 (70.2) | 26 (55.3) | |
| Past (n, %) | 7 (14.9) | 9 (19.2) | |
| BMI (kg/m2) | 24.87 ± 4.59 | 26.75 ± 5.51 | 0.0756 |
| Systolic BP (mm Hg) | 119.87 ± 8.96 | 132.39 ± 12.76 | <0.0001 |
| Diastolic BP (mm Hg) | 78.69 ± 9.32 | 86.10 ± 14.72 | 0.0045 |
| Total cholesterol (mg/dl) | 145.62 ± 17.05 | 187.78 ± 36.87 | <0.0001 |
| Low density lipoprotein cholesterol (mg/dl) | 98.65 ± 13.62 | 133.86 ± 33.91 | <0.0001 |
| High density lipoprotein cholesterol (mg/dl) | 44.75 ± 8.90 | 41.34 ± 9.83 | 0.0812 |
| Triglyceride (mg/dl) | 124.06 ± 24.73 | 170.02 ± 55.92 | <0.0001 |
| Comorbidities | |||
| Hypertension (n, %) | — | 8 (17.0) | |
| Dyslipidemia (n, %) | — | 6 (12.8) | |
| Medications | |||
| Antiplatelets (n, %) | — | 25 (53.2) | |
| Anticoagulants (n, %) | — | 18 (38.3) | |
| β-blockers (n, %) | — | 5 (10.6) | |
| ACEIs/ARBs (n, %) | — | 5 (10.6) | |
| Calcium channel blockers (n, %) | — | 4 (8.5) | |
| Digoxin (n, %) | — | 2 (4.3) | |
Values are presented as mean ± SD or as percentage. NVAF, non-valvular atrial fibrillation; BMI, body mass index; BP, blood pressure; ACEIs, angiotensin-converting enzymes inhibitors; ARBs, angiotensin II receptor blockers.
Blood Samples
Peripheral venous blood samples (5 ml) were collected by venipuncture into sterile siliconized Vacutainer tubes with 2 mg/ml disodium ethylenediaminetetraacetic acid. All samples were stored at −20 °C until use.
cDNA Synthesis and Gene Expression
mRNA was isolated from leukocytes by using β-mercaptoethanol, and stored at −80 °C until use. cDNA was produced with the Qiagen miScript Reverse Transcription Kit according to manufacturer’s protocol. PCR was performed by BioMark HD system (Fluidigm, South San Francisco, CA, USA) with TRP channel primers, and β-actin (ACTB, housekeeping gene). We screened 26 TRP channel genes [TRPA1, TRPC1-7, TRPM1-8, TRPV1-6, MCOLN1-3 (TRPML1-3), and PKD2 (TRPP2)] and PKD1 (TRPP1) for this expression study. Data were analyzed using the 2−ΔΔCt method, according to the formula: ΔCt = CtTRP − CtACTB, where Ct = threshold cycle.
Statistical analyses
Results are expressed as the mean ± SD, SEM or percentage. For comparisons of the differences between mean values of two groups, the unpaired Student’s t test was used. Chi-square test was used for calculation of the significance of differences in categorical data. The gene expression analysis was performed by using online program, QIAGEN GeneGlobe (http://www.qiagen.com/geneglobe). Student’s t test was used to compare gene expression data. Statistical analysis was performed using GraphPad Instat version 3.05 (GraphPad Software Inc., San Diego, CA, USA). All probability values were based on two-tailed tests. P values less than 0.05 were considered to be statistically significant.
Results
Demographic and clinical characteristics of the study population are presented in Table 1. The prevalence of cardiovascular risk factors, including hypertension, lipid profiles, smoking, and body mass index for the control and NVAF groups are shown in Table 1. Compared with the controls, the average age, genders, percentages of smokers, and BMI in the NVAF group were similar. Blood pressure, total cholesterol, LDL cholesterol, and TG levels were all greater among NVAF subjects. There was no marked difference in HDL cholesterol levels between the groups (Table 1). Eight (17.0%) patients had hypertension, and 6 (12.8%) had dyslipidemia. While about two-third of the patients 61.7% (n = 29) were on a single medication, 34.0% (n = 16) of them were on two drugs.
All the TRP genes studied were upregulated in leukocytes of NVAF patients. Gene expression analysis showed that MCOLN1 (TRPML1), MCOLN2 (TRPML2), and MCOLN3 (TRPML3) mRNA contents in leukocytes were augmented in NVAF patients when compared to the control groups (P < 0.05, Fig. 1). There were also elevations in TRPM8, TRPC6, TRPV5, TRPV4, TRPA1, TRPC3, TRPV6, TRPC4, and TRPV2 gene expressions in NVAF patients (P < 0.05, Fig. 2). Higher TRPM3, PKD2 (TRPP2), TRPM7, TRPV1, TRPM6, TRPV3, TRPM5, TRPM4, TRPM1, TRPC7, TRPC2, TRPC1, TRPM2, and TRPC5 gene expressions were detected in NVAF patients (P < 0.05, Figs 3 and 4). However, PKD1 gene expression was not changed in patients with NVAF when compared to controls (Fig. 4).
Figure 1.
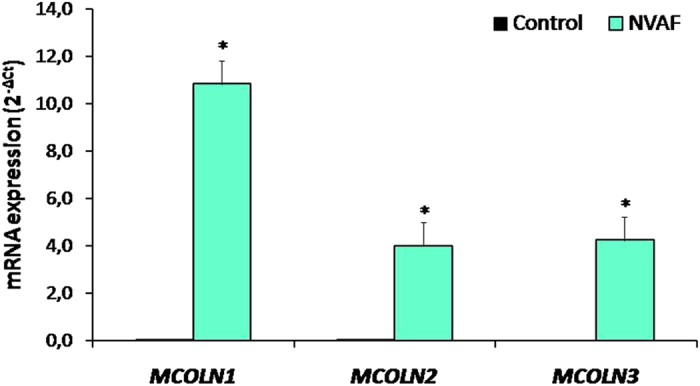
Comparison of the peripheral blood mRNA MCOLN1 (TRPML1), MCOLN2 (TRPML2), and MCOLN3 (TRPML3) expressions in healthy controls (n = 47, solid bars) and in patients with non-valvular atrial fibrillation (NVAF, n = 47, open bars). Values are given as mean ± SEM, *P = 0.0060, P = 0.0217, and P = 0.0001 values were obtained for MCOLN1 (TRPML1), MCOLN2 (TRPML2), and MCOLN3 (TRPML3), respectively.
Figure 2.
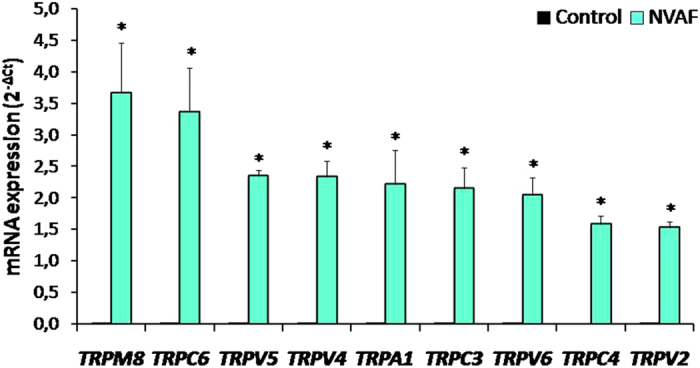
Comparison of the peripheral blood mRNA TRPM8, TRPC6, TRPV5, TRPV4, TRPA1, TRPC3, TRPV6, TRPC4, and TRPV2 expressions in healthy controls (n = 47, solid bars) and in patients with non-valvular atrial fibrillation (NVAF, n = 47, open bars). Values are given as mean ± SEM, *P = 0.0191, P < 0.0001, P < 0.0001, P < 0.0001, P = 0.0147, P < 0.0001, P < 0.0001, P < 0.0001, and P < 0.0001 values were obtained for TRPM8, TRPC6, TRPV5, TRPV4, TRPA1, TRPC3, TRPV6, TRPC4, and TRPV2, respectively.
Figure 3.
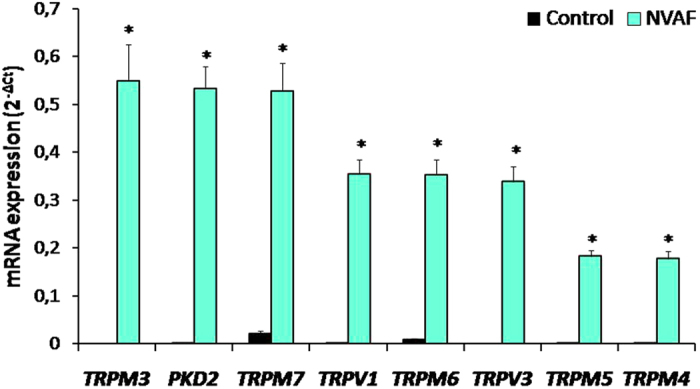
Comparison of the peripheral blood mRNA TRPM3, PKD2 (TRPP2), TRPM7, TRPV1, TRPM6, TRPV3, TRPM5, and TRPM4 expressions in healthy controls (n = 47, solid bars) and in patients with non-valvular atrial fibrillation (NVAF, n = 47, open bars). Values are given as mean ± SEM, *P = 0.0001, P < 0.0001, P < 0.0001, P < 0.0001, P < 0.0001, P < 0.0001, P < 0.0001, and P < 0.0001 values were obtained for TRPM3, PKD2 (TRPP2), TRPM7, TRPV1, TRPM6, TRPV3, TRPM5, and TRPM4, respectively.
Figure 4.
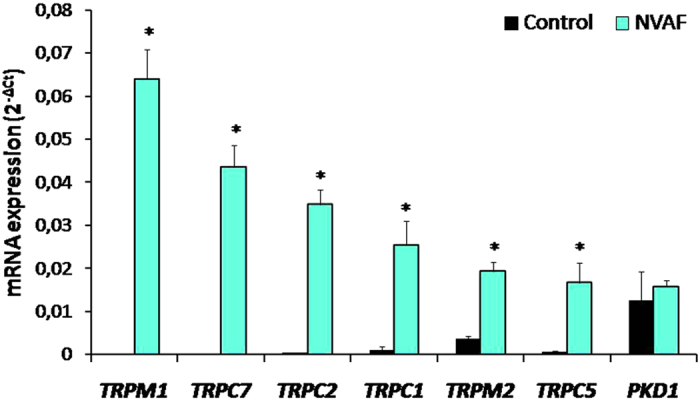
Comparison of the peripheral blood mRNA TRPM1, TRPC7, TRPC2, TRPC1, TRPM2, TRPC5, and PKD1 expressions in healthy controls (n = 47, solid bars) and in patients with non-valvular atrial fibrillation (NVAF, n = 47, open bars). Values are given as mean ± SEM, *P < 0.0001, P < 0.0001, P < 0.0001, P = 0.0040, P = 0.0001, P < 0.0001, and P = 0.3083 values were obtained for TRPM1, TRPC7, TRPC2, TRPC1, TRPM2, TRPC5, and PKD1 respectively.
Discussion
The present study evaluated 26 TRP channel gene expressions in patients with NVAF and compared to healthy controls in this study. Elevated [TRPA1, TRPC1-7, TRPM1-8, TRPV1-6, MCOLN1-3 (TRPML1-3), and PKD2 (TRPP2)] gene expressions in circulating leukocytes were observed in patients with NVAF. To the best of our knowledge, this is the first study to investigate the TRP channel gene expressions in relation to NVAF.
Several TRP channels are functionally expressed in the immune cells including lymphocytes, monocytes and macrophages19, 20. Several TRP channels are also expressed on leukocytes, but the functional roles of these channels are still unclear.
Expressions of TRPM4, TRPV5 and TRPV6 in normal human leukocytes were not found by Northern blot analysis21–23. Additionally, Northern-blot analysis showed that a faint signal of TRPM6 was detected in leukocytes24. However, we were able to detect the expressions of these channels in our real-time PCR assay. In the present study, TRPV5 showed the highest expression, whereas TRPV3 demonstrated the lowest expression patterns among the TRPV genes in leukocytes. Our findings are in agreement with data from Spinsanti et al.25, who showed that TRPV3 was the least expressed gene among the TRPV1-4 genes in human leukocytes.
TRPM4 is a monovalent nonselective cation channel permeable to Na+, and K+, but not to Ca2+ 26. Atrial myocytes from Trpm4−/− mice display a shorter action potential27. TRPM7 knockdown suppresses endogenous TRPM7 currents and Ca2+ influx in atrial fibroblasts and inhibits transforming growth factor-β1-induced fibroblast proliferation, differentiation, and collagen production28. Since fibrosis is one of the major detrimental factors for AF, TRPM7-mediated Ca2+ signals may play a pivotal role in fibroblast differentiation and fibrogenesis in human AF28. We have found that both TRPM4 and TRPM7 gene expressions were markedly upregulated in leukocytes of the NVAF patients.
TRPM6 is suggested to be responsible for systemic Mg2+ homeostasis in humans29. Also, the function of TRPM7 is modulated by TRPM6, and the TRPM6 kinase may be involved in tuning the phenotype of the TRPM7/M6 channel complex30. We have noted significant augmentations in other TRPM gene expressions in our study. Contributions of these channels to the genesis of AF are currently unknown in humans.
TRPC channels may play a key role in regulation of cardiac pacemaking, conduction, ventricular activity, and contractility during cardiogenesis31. The overexpression of TRPC3 enhances the store-operated calcium entry32, 33. The increased TRPC3 gene expression, which was observed in the present study, together with consecutive increase of calcium influx may account for the activation of leukocytes that has been described in patients with atrial fibrillation34. We have detected increases in all TRPC channel gene expressions in leukocytes obtained from NVAF patients, but significances of these upregulations are unknown.
TRPV1 is found to express on the endoplasmic reticulum/sarcoplasmic reticulum and the mitochondria35. Therefore, intracellular TRPV1 may control calcium level both inside the organelles and in the cytoplasm. TRPV1 is involved in systemic inflammatory response such as phagocytosis by macrophages, nitric oxide and reactive oxygen species (ROS) production, and cytokine production36. Regulation of the relative expression levels of TRPV5 and/or TRPV6 may affect the Ca2+ transport kinetics and Ca2+-dependent functions, such as proliferation and differentiation, in lymphocytes37. We have detected marked increases in all TRPV channel gene expressions in leukocytes of the NVAF patients.
Our study is the first to show that there were significant TRPML mRNA expressions in leukocytes of NVAF patients. Expression level of MCOLN1 (TRPML1) gene was found to be high. TRPML1, a Ca2+-permeable non-selective cation channel that localizes to late endosomes and lysosomes38, 39, is also activated by ROS in in vitro to regulate autophagy40. TRPML1 appears to be ubiquitously expressed, but it is not known to be specifically involved in AF.
Deletion of Pkd2 (Trpp2) in mice can cause abnormal heart development13. PKD2 related proteins form Ca2+-permeable channel with PKD1, an 11 transmembrane protein, which is also known as TRPP1, but it is not a TRP protein. PKD1 is thought to interact with TRPP2, which functions as a receptor for mechanical stimuli such as shear stress41. Moreover, PKD1 and TRPP2 can interact with and amplify Ca2+ release from inositol trisphosphate receptors in the endoplasmic reticulum42. Although we have observed an increase in PKD2 (TRPP2) gene expression, no change was noted with PKD1 (TRPP1) in this study. However, contribution of PKD2 (TRPP2) to the pathogenesis of AF has not been studied yet.
Current data suggest that inflammation is associated with the development of AF34, 43–45. Indeed, white blood cell count is significantly higher in patients with AF, and it is significantly and independently associated with AF46. Leukocyte activation has been regarded to have a critical role in the pathogenesis of AF34. There is evidence that atrial neutrophil infiltration is enhanced in atrial appendage sections of patients with persistent AF47. The physiological role of TRP genes in human peripheral blood cells has yet to be determined, but it has been hypothesized that, under pathological conditions, their upregulation may be an indicator of inflammation at a secondary site.
Accumulating evidence suggests oxidative stress may play an important role in the induction and maintenance of AF43. The type 2 ryanodine receptor oxidation resulting from mitochondrial-derived ROS in atrial myocytes leads to increased sarcoplasmic reticulum Ca2+ leak contributing to the pathogenesis of AF44. Furthermore, serum oxidative stress marker levels are elevated in patients with AF45. Modification of cysteine residues by ROS has been shown to alter the activity of TRP channels48, 49. There is evidence that TRPC5, TRPA1, TRPV1, TRPV3, and TRPV4 channels are modulated by covalent modification of cysteine residues by ROS and/or reactive nitrogen species (RNS)48–50. TRPA1, TRPV1 and TRPC5 channels are directly activated by oxidizing agents through cysteine modification; whereas, TRPM2 channel is indirectly activated by production of ADP-ribose49. TRPM7 overexpression can enhance levels of ROS and nitric oxide51. TRPM2, TRPM7, TRPC5, and TRPV1 are activated by ROS and RNS48. TRPM7 can contribute to hydrogen peroxide-induced cardiac fibrosis52. Wuensch et al.53 showed that oxidative stress increases the expression of both TRPC3 and TRPC6 mRNA in human monocytes. Collectively, these data may imply that TRP channels are involved in the pathogenesis of AF through activated leukocytes which promotes the inflammatory or immune cascade.
The gene expression profiles in this study were measured in isolated leukocytes, but the ideal tissue for study AF is the left atrium. This is considered main limitation of this study. However, it should be pointed out that this group patient with NVAF has no indication for cardiac operation. Additionally, Lin et al.54 examined the association of whole blood gene expression with AF in a large community-based cohort, and identified seven genes statistically significantly up-regulated with prevalent AF. Raman et al.55 also evaluated peripheral blood gene expression in patients with persistent AF that underwent electrical cardioversion. In a recent study, peripheral monocyte toll-like receptor (TLR) expression levels have been investigated and higher levels of TLR-2 and TLR-4 expressions were detected in patients with AF56. The increased leukocyte TRP gene expressions observed in this study should be studied at the cardiac level.
In conclusion, the results of the present study revealed that TRP channels gene expressions are upregulated in leukocytes of the NVAF patients. Our findings showed that TRPML genes are strongly expressed in NVAF patients. Our data may imply that TRP channels may be effective targets for prevention or prophylaxis of AF. The findings of this study may lead to development of more effective approaches for treatment of AF. Further investigations are needed for understanding the role of TRP channels in AF.
Acknowledgements
The authors wish to thank Ebru Temiz (Gaziantep University, Department of Medical Biology) for her contribution to gene expression analysis.
Author Contributions
I.V.D. designed and performed experiments. I.V.D., F.Y., E.V., E.S., F.P. collected and analysed the data. H.G., B.C. interpreted results of the study. S.D. did statistical analyses, prepared figures, and wrote the paper. All authors edited and revised manuscript and approved final submission of manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Benjamin EJ, et al. Prevention of atrial fibrillation: report from a national heart, lung, and blood institute workshop. Circulation. 2009;119:606–618. doi: 10.1161/CIRCULATIONAHA.108.825380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rich MW. Epidemiology of atrial fibrillation. J. Interv. Card. Electrophysiol. 2009;25:3–8. doi: 10.1007/s10840-008-9337-8. [DOI] [PubMed] [Google Scholar]
- 3.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol. Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 4.Haissaguerre M, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998;339:659–666. doi: 10.1056/NEJM199809033391003. [DOI] [PubMed] [Google Scholar]
- 5.Haissaguerre M, et al. Driver domains in persistent atrial fibrillation. Circulation. 2014;130:530–538. doi: 10.1161/CIRCULATIONAHA.113.005421. [DOI] [PubMed] [Google Scholar]
- 6.Magnani JW, et al. Atrial fibrillation: current knowledge and future directions in epidemiology and genomics. Circulation. 2011;124:1982–1993. doi: 10.1161/CIRCULATIONAHA.111.039677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tucker NR, Ellinor PT. Emerging directions in the genetics of atrial fibrillation. Circ. Res. 2014;114:1469–1482. doi: 10.1161/CIRCRESAHA.114.302225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 2011;121:2955–2968. doi: 10.1172/JCI46315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nattel S. From guidelines to bench: implications of unresolved clinical issues for basic investigations of atrial fibrillation mechanisms. Can. J. Cardiol. 2011;27:19–26. doi: 10.1016/j.cjca.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Harada M, et al. Atrial fibrillation activates AMP-dependent protein kinase and its regulation of cellular calcium handling: Potential Role in Metabolic Adaptation and Prevention of Progression. J. Am. Coll. Cardiol. 2015;66:47–58. doi: 10.1016/j.jacc.2015.04.056. [DOI] [PubMed] [Google Scholar]
- 11.Voigt N, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flockerzi V, Nilius B. TRPs: truly remarkable proteins. Handb. Exp. Pharmacol. 2014;222:1–12. doi: 10.1007/978-3-642-54215-2_1. [DOI] [PubMed] [Google Scholar]
- 13.Yue Z, et al. Role of TRP channels in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2015;308:H157–H182. doi: 10.1152/ajpheart.00457.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yue L, Xie J, Nattel S. Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2011;89:744–753. doi: 10.1093/cvr/cvq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harada M, et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation. 2012;126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thodeti CK, Paruchuri S, Meszaros JGA. TRP to cardiac fibroblast differentiation. Channels (Austin) 2013;7:211–214. doi: 10.4161/chan.24328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang YH, et al. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflugers Arch. 2013;465:1439–1449. doi: 10.1007/s00424-013-1291-3. [DOI] [PubMed] [Google Scholar]
- 18.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA285, 2486–2497 (2001). [DOI] [PubMed]
- 19.Billeter AT, Hellmann JL, Bhatnagar A, Polk HC., Jr. Transient receptor potential ion channels: powerful regulators of cell function. Ann. Surg. 2014;259:229–235. doi: 10.1097/SLA.0b013e3182a6359c. [DOI] [PubMed] [Google Scholar]
- 20.Bertin S, Raz E. Transient Receptor Potential (TRP) channels in T cells. Semin. Immunopathol. 2016;38:309–319. doi: 10.1007/s00281-015-0535-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wissenbach U, et al. Expression of CaT-like, a novel calcium-selective channel, correlates with the malignancy of prostate cancer. J. Biol. Chem. 2001;276:19461–19468. doi: 10.1074/jbc.M009895200. [DOI] [PubMed] [Google Scholar]
- 22.Xu XZ, Moebius F, Gill DL, Montell C. Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc. Natl. Acad. Sci. U S A. 2001;98:10692–10697. doi: 10.1073/pnas.191360198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nilius B, et al. Voltage dependence of the Ca2+-activated cation channel TRPM4. J. Biol. Chem. 2003;278:30813–30820. doi: 10.1074/jbc.M305127200. [DOI] [PubMed] [Google Scholar]
- 24.Walder RY, et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet. 2002;31:171–174. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 25.Spinsanti G, et al. Quantitative Real-Time PCR detection of TRPV1-4 gene expression in human leukocytes from healthy and hyposensitive subjects. Mol. Pain. 2008;4 doi: 10.1186/1744-8069-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Launay P, et al. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407. doi: 10.1016/S0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 27.Simard C, Hof T, Keddache Z, Launay P, Guinamard R. The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential. J. Mol. Cell.Cardiol. 2013;59:11–19. doi: 10.1016/j.yjmcc.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 28.Du J, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 2010;106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chubanov V, Gudermann T. TRPM6. Handb. Exp. Pharmacol. 2014;222:503–520. doi: 10.1007/978-3-642-54215-2_20. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, et al. The TRPM6 kinase domain determines the Mg·ATP sensitivity of TRPM7/M6 heteromeric ion channels. J. Biol. Chem. 2014;289:5217–5227. doi: 10.1074/jbc.M113.512285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sabourin J, Robin E, Raddatz E. A key role of TRPC channels in the regulation of electromechanical activity of the developing heart. Cardiovasc. Res. 2011;92:226–236. doi: 10.1093/cvr/cvr167. [DOI] [PubMed] [Google Scholar]
- 32.Shlykov SG, Yang M, Alcorn JL, Sanborn BM. Capacitative cation entry in human myometrial cells and augmentation by hTrpC3 overexpression. Biol. Reprod. 2003;69:647–655. doi: 10.1095/biolreprod.103.015396. [DOI] [PubMed] [Google Scholar]
- 33.Liu D, et al. Transient receptor potential channels in essential hypertension. J. Hypertens. 2006;24:1105–1114. doi: 10.1097/01.hjh.0000226201.73065.14. [DOI] [PubMed] [Google Scholar]
- 34.Patel P, Dokainish H, Tsai P, Lakkis N. Update on the association of inflammation and atrial fibrillation. J. Cardiovasc. Electrophysiol. 2010;21:1064–1070. doi: 10.1111/j.1540-8167.2010.01774.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhao R, Tsang SY. Versatile roles of intracellularly located TRPV1 channel. J. Cell. Physiol. 2017;232:1957–1965. doi: 10.1002/jcp.25704. [DOI] [PubMed] [Google Scholar]
- 36.Fernandes ES, et al. TRPV1 deletion enhances local inflammation and accelerates the onset of systemic inflammatory response syndrome. J. Immunol. 2012;188:5741–5751. doi: 10.4049/jimmunol.1102147. [DOI] [PubMed] [Google Scholar]
- 37.Vassilieva IO, et al. Expression of transient receptor potential vanilloid channels TRPV5 and TRPV6 in human blood lymphocytes and Jurkat leukemia T cells. J. Membr. Biol. 2013;246:131–140. doi: 10.1007/s00232-012-9511-x. [DOI] [PubMed] [Google Scholar]
- 38.Wang W, Zhang X, Gao Q, Xu H. TRPML1: an ion channel in the lysosome. Handb. Exp. Pharmacol. 2014;222:631–645. doi: 10.1007/978-3-642-54215-2_24. [DOI] [PubMed] [Google Scholar]
- 39.Dong XP, et al. PI3,5P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun. 2010;1 doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016;7 doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng J. Molecular mechanism of TRP channels. Compr. Physiol. 2013;3:221–242. doi: 10.1002/cphy.c120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mekahli D, et al. Polycystin-1 and polycystin-2 are both required to amplify inositol-trisphosphate-induced Ca2+ release. Cell. Calcium. 2012;51:452–458. doi: 10.1016/j.ceca.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Gutierrez A, Van Wagoner DR. Oxidant and inflammatory mechanisms and targeted therapy in atrial fibrillation: An update. J. Cardiovasc. Pharmacol. 2015;66:523–529. doi: 10.1097/FJC.0000000000000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie W, et al. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015;5 doi: 10.1038/srep11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimano M, et al. Reactive oxidative metabolites are associated with atrial conduction disturbance in patients with atrial fibrillation. Heart Rhythm. 2009;6:935–940. doi: 10.1016/j.hrthm.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 46.Tekin G, et al. Mean platelet volume in patients with nonvalvular atrial fibrillation. Blood Coagul. Fibrinolysis. 2013;24:537–539. doi: 10.1097/MBC.0b013e32835facb3. [DOI] [PubMed] [Google Scholar]
- 47.Friedrichs K, et al. Induction of atrial fibrillation by neutrophils critically depends on CD11b/CD18 integrins. PLoS One. 2014;9 doi: 10.1371/journal.pone.0089307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozai D, Ogawa N, Mori Y. Redox regulation of transient receptor potential channels. Antioxid. Redox Signal. 2014;21:971–986. doi: 10.1089/ars.2013.5616. [DOI] [PubMed] [Google Scholar]
- 49.Ogawa N, Kurokawa T, Mori Y. Sensing of redox status by TRP channels. Cell. Calcium. 2016;60:115–122. doi: 10.1016/j.ceca.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 50.Yoshida T, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 51.Su LT, et al. TRPM7 activates m-calpain by stress-dependent stimulation of p38 MAPK and c-Jun N-terminal kinase. J. Mol. Biol. 2010;396:858–869. doi: 10.1016/j.jmb.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo JL, et al. Transient receptor potential melastatin 7 (TRPM7) contributes to H2O2-induced cardiac fibrosis via mediating Ca2+ influx and extracellular signal-regulated kinase 1/2 (ERK1/2) activation in cardiac fibroblasts. J. Pharmacol. Sci. 2014;125:184–192. doi: 10.1254/jphs.13224FP. [DOI] [PubMed] [Google Scholar]
- 53.Wuensch T, et al. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes. 2010;59:844–849. doi: 10.2337/db09-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin H, et al. Whole blood gene expression and atrial fibrillation: the Framingham Heart Study. PLoS One. 2014;9 doi: 10.1371/journal.pone.0096794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raman K, et al. Whole blood gene expression differentiates between atrial fibrillation and sinus rhythm after cardioversion. PLoS One. 2016;11 doi: 10.1371/journal.pone.0157550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gurses KM, et al. Monocyte Toll-like receptor expression in patients with atrial fibrillation. Am. J. Cardiol. 2016;117:1463–1467. doi: 10.1016/j.amjcard.2016.02.014. [DOI] [PubMed] [Google Scholar]