


Abstract
Triplex-forming oligodeoxynucleotide 15mers, designed to bind in the antiparallel triple-helical binding motif, containing single substitutions (Z) of the four isomeric αN7-, βN7-, αN9- and βN9-2-aminopurine (ap)-deoxyribonucleosides were prepared. Their association with double-stranded DNA targets containing all four natural base pairs (X-Y) opposite the aminopurine residues was determined by quantitative DNase I footprint titration in the absence of monovalent metal cations. The corresponding association constants were found to be in a rather narrow range between 1.0 × 106 and 1.3 × 108 M–1. The following relative order in Z × X-Y base-triple stabilities was found: Z = αN7ap: T-A > A-T> C-G ∼ G-C; Z = βN7ap: A-T > C-G > G-C > T-A; Z = αN9ap: A-T = G-C > T-A > C-G; and Z = βN9ap: G-C > A-T > C-G > T-A.
INTRODUCTION
Strong and sequence-specific triple-helix formation of oligonucleotides with genomic DNA can selectively interfere with gene expression at the level of transcription and is therefore of interest in medicinal chemistry and biotechnology (1–2). However, the molecular recognition of a DNA duplex via a triplex forming oligonucleotide (TFO) in either the parallel (3–4) or the antiparallel (5–6) binding motif is limited to homopurine–homopyrimidine DNA sequence tracts. Despite considerable efforts over the past decade to overcome this sequence restriction by TFO design, a general solution to the problem still remains elusive (7).
A number of third-strand design options to overcome this sequence restriction have been investigated in the past. In order to recognize DNA duplexes containing purine–pyrimidine block sequence intervals, TFOs binding via a combination of the purine and the pyrimidine motif (8–9), as well as TFOs with 3′–3′- or base–base junctions were studied (10–15). Furthermore, TFOs containing extended nucleobases spanning a complete Watson–Crick base pair in the major groove (16,17), and base-modified TFOs selectively recognizing pyrimidine bases (18–22), were investigated. In all of these cases, either the affinity of the corresponding TFOs to their target, or the selectivity in base pair recognition, or both did not fulfill the required criteria for broad application in DNA-based therapy and diagnosis.
A particularly intriguing conceptual approach is the design of TFOs with a structural switch, enabling the bases to cross the major groove and target consecutive purine bases on either strand of the target duplex. One such switch model consists of the consecutive use of α- and β-configured nucleotides (α,β-switch model). The α,β-switch model had been proposed earlier (23), and was recently investigated experimentally in the context of alternate strand G recognition by α-, and β-4-guanidinopyrimidine-containing oligonucleotides (24,25), without success, however. In this communication we report on an alternative design and experimental evaluation of such switch TFOs.
MATERIALS AND METHODS
Oligonucleotide synthesis
Oligonucleotide synthesis was performed on a Pharmacia Gene Assembler Special connected to a Compaq ProLinea 3/25-zs PC. All syntheses were performed using the 1.3 µmol cycle with coupling times of 2 min for the 2-aminopurine building blocks. The phosphoramidite building blocks of the four isomeric 2-aminopurine (ap) nucleoside analogs αN7ap, βN7ap, αN9ap and βN9ap (Fig. 1) were prepared as described by us previously (26). Phosphoramidite solutions (0.1 M in MeCN) were equal in concentration to those used for the synthesis of natural oligodeoxynucleotides. The activator 1H-tetrazole was replaced in all syntheses by 5-(benzylthio)-1H-tetrazole (0.25 M in MeCN) (27). Average coupling yields, monitored by the on-line trityl assay, were >98% for all four 2-aminopurine phosphoramidites. Removal of the protecting groups and detachment from the solid support was effected in concentrated NH3 solution (1–2 ml) at 55°C for 18–20 h. The crude oligomers were purified by MonoQ ion-exchange HPLC, desalted over Sep-Pak C-18 cartridges (Waters) and their purity and composition confirmed by Electrospray-MS analysis. Table 1 contains synthetic and analytical data of the oligonucleotides described here. All natural DNA sequences used in this study were prepared according to standard phosphoramidite chemistry and purified by HPLC.
Figure 1.

Chemical structures of the isomeric 2-aminopurine nucleosides αN7ap, βN7ap, αN9ap and βN9ap used in this study.
Table 1. Synthesis and analytical data of oligonucleotides 1–8.
| Sequence | Isolated yield OD (260 nm) ([%]) | Electrospray-MS [M-H]– | |
| |
|
m/z (calc.) |
m/z (found) |
| 1 d(GGTGG-αN7ap-GGTTGTGGT) | 52 (29) | 4734.7 | 4734.9 |
| 2 d(GGTGG-βN7ap-GGTTGTGGT) | 80 (45) | 4734.7 | 4734.6 |
| 3 d(GGTGG-αN9ap-GGTTGTGGT) | 28 (16) | 4734.7 | 4732.1 |
| 4 d(GGTGG-βN9ap-GGTTGTGGT) | 69 (38) | 4734.7 | 4734.5 |
| 5 d(GGTGGTGGTTGTGGT) | 71 (40) | 4727.9a | 4727.7a |
aMS measured in ESI+ mode; m/z given is for [M+H]+.
Tm measurements
UV-melting experiments were performed on a Varian-Cary-3E UV/Vis spectrometer equipped with a temperature controller unit and connected to a PC running the Varian WinUV software; temperature gradient 0.5°C/min; data-point collection in intervals of ∼1°C; below 20°C, the cell compartment was flushed with N2 to avoid condensation of H2O on the UV cells; the transition temperature Tm was determined as the maximum of the first derivative of the melting curve using the software package Origin™ v5.0.
Preparation of plasmid pSPP4C1
All DNA and cell manipulations followed standard protocols (28). All aqueous solutions for DNA manipulations were prepared with Milli-Q water (Millipore). Escherichia coli XL-1 Blue competent cells were from Stratagene. Sonicated calf thymus DNA, T4 polynucleotide kinase and Sequenase v.2.0 were from AP Biotech. The restriction enzymes were from Roche Diagnostics. T4 DNA ligase was from Sigma. [α-32P]dATP (3000 Ci/mmol, 250 µCi) was from Hartmann Analytik GmbH. Buffers (if not provided with the enzymes) were made from Fluka (Microselect grade) or Sigma chemicals. Synthetic oligonucleotides were prepared as described before at the 0.2 µmol scale using standard β-cyanoethyl phosphoramidites and solid supports (AP Biotech). Modified coupling times of 2 min were used. Oligonucleotides were purified by denaturing polyacrylamide gel electrophoresis.
The DNA duplex containing the four triplex target cassettes was prepared starting with the following set of overlapping oligonucleotides: 5′-CGACGACCCGGGACGCTCGACAGGCAGGAGAAGGAGGAGGGTATCAAGACTTCG-3′; 5′-CAGGAGAAGGCGGAGGCAACTCACGTTCGGTAGGAGAAGGTGGAGGTCTTGAGCTGGCA-3′; 5′-ACAGGAGAAGGGGGAGGTACACTGGCCATGACCAAGCTTGCGCTCTGCTGAAGAAGCCAG-3′; 3′-GCTGCTGGGCCCTGCGAGCTGTCC-5′; 3′-GTCCTCTTCCTCCTCCCATAGTTCTGAAGCGTCCTCTTCCGCCTCCGTTG-5′; 3′-AGTGCAAGCCATCCTCTTCCACCTCCAGAACTCGACCGTTGTCCTCTTCCCCCTCCATGTG-5′; 3′-ACCGGTACTGGTTCGAACGCGAGACGACTTCTTCGGTC-5′.
With the exception of the two 5′-fragments, all oligonucleotides were phosphorylated using ATP and T4 polynucleotide kinase. An equimolar mixture of all oligonucleotides was then equilibrated and ligated using T4 ligase. The resulting duplex was digested with HindIII and AvaI and ligated into the large HindIII–AvaI restriction fragment of pUC18 (→pSPP4C1). Competent E.coli XL-1 Blue cells were transformed with the ligated plasmid, and plasmid DNA from ampicillin-resistant white colonies was isolated. The presence of the desired insert of the selected clone was determined by restriction analysis and forward and reversed Sanger dideoxy sequencing. Two mutations (CG→TA at position 55 and CG→GC at position 112 of the original duplex) were detected. The mutations, however, were outside of the target cassettes. The plasmid was isolated on a preparative scale using a Maxi Prep Kit (Qiagen) affording 380 µg of pSPP4C1.
3′-End-labeling of DNA fragments
In a typical end-labeling experiment, 6 µg of plasmid pSPP4C1 were digested with restriction endonucleases EcoRI and PvuII and the products were separated by agarose gel electrophoresis. The desired band was excised and extracted with the QiaexII Extraction Kit (Qiagen). The restriction fragment was then ethanol-precipitated and redissolved in H2O. 3′-End-labeling was performed with [α-32P]dATP using Sequenase (v2.0). Excess unlabeled nucleotide triphosphates (dTTP, dATP) were added after labeling to ensure complete fill in. Non-incorporated nucleotide was removed by gel filtration over NICK columns (AP Biotech). Finally, the labeled fragment was ethanol-precipitated. From ∼6 µg of plasmid, a total Cerenkov radioactivity of 4 000 000–15 000 000 c.p.m. was obtained.
DNase I footprint titrations
These experiments were performed according to published protocols (29,30). Typically, TFOs were dissolved in H2O to a concentration of 25 µM and this solution was serially diluted to a total of 10 concentrations spanning the desired range for the experiment (final concentrations of 300 pM to 10 µM). H2O was added to adjust the volume to 18 µl. To each tube, 27 µl of a stock solution containing the labeled restriction fragment of pSPP4C1 (20 000 c.p.m.) together with 9 µl 5× association buffer (50 mM bis-Tris–HCl, 50 mM MgCl2, 5 mM spermine tetrahydrochloride, pH 7.2) and H2O were added (final solution conditions: 10 mM bis-Tris–HCl, 10 mM MgCl2, 1 mM spermine). After 7 days equilibration at 20°C, 2.5 µg sonicated calf thymus DNA was added. The digestion was started by the addition, at room temperature of 5 µl of a solution containing 24 U DNase I (from a 10 U/µl stock solution) in 120 µl digestion buffer [50 mM CaCl2, 50 mM MgCl2, 10 mM bis-Tris–HCl pH 7, 5% glycerine, 10 µM non-specific sequence 5′d(TATAATTTAA)3′]. After 1.5 min, the digestion was quenched by the addition of 8.3 µl of a solution containing 1.4 M NaCl, 0.14 M EDTA pH 8, 0.35 µg/ml glycogene. 20 µl 3 M NaOAc pH 5.2, and 150 µl cold ethanol were added. After 3 h at –20°C, the tubes were centrifuged (30 min, 16 000 g, 4°C). The pellets were washed with 80 µl cold 70% ethanol, centrifuged (15 min), solved in 20 µl H2O, lyophilized and dissolved in 7 µl of 80% formamide loading buffer. After denaturation (10 min, 85°C), the samples were loaded on an 8% denaturing polyacrylamide gel and separated by electrophoresis in 1× TBE buffer at 1900 V for 90 min. After drying on a slab drier (80°C, 1 h) the gel was exposed to a storage PhosphorImager screen (Molecular Dynamics) overnight at ambient temperature.
Quantification and data analysis
Quantification of the footprint titrations was performed as described (30), using ImageQuant v3.3 software (Molecular Dynamics). For the determination of the association constants of the studied triple helices, the following equilibrium and the corresponding equilibrium constant Ka were considered:
D + SS ↔ T
Ka = [T]/([SS][D])
where D, SS and T represent the duplex, single-strand and triplex, respectively.
The apparent fractional occupancy θapp can be calculated as:
θapp = 1 – [(Dn,XY/Dn,out)/(DR,XY/DR,out)]
with Dn,XY being the optical density of target XY (XY = AT, CG, GC or TA) in lane n, Dn,out being the optical density of a reference region of lane n (the optical density of the reference region should be more or less constant for all lanes of the gel). DR,XY is the optical density of target XY in the reference lane (no third strand) and DR,out is the optical density of the reference region in the reference lane. The obtained θapp can then be fitted to a sigmoidal curve using the following formula:
Ifit = θmin,XY + (θmax,XY – θmin,XY) Ka [c/(1 + Ka × c)]
θmin,XY, θmax,XY and Ka being three variable parameters and c the total third strand concentration. The association constant is Ka.
RESULTS AND DISCUSSION
Design of switch TFOs
In order to target the base adenine on either strand of a DNA duplex, we set out to study the triplex-forming properties of TFOs containing the four isomeric forms of the 2-aminopurine deoxyribonucleotides αN7ap, βN7ap, αN9ap and βN9ap, depicted in Figure 1. By molecular modeling we have identified two switch models, in which the position of the C(1′) atoms between two consecutive third strand residues are located in the center of the major groove and within an acceptable distance for linkage via a phosphodiester unit. One consists of the consecutive arrangement of αN7ap and βN7ap nucleotides (α,β-switch) and the other of the consecutive arrangement of βN7- and βN9 -configured purine nucleotides (N7,N9-switch) (Fig. 2). Both switch models are expected to be compatible with the antiparallel (purine) triplex binding motif, in which the location of the C(1′) atoms of the third strand deoxyribose units are also located towards the center of the major groove (23).
Figure 2.
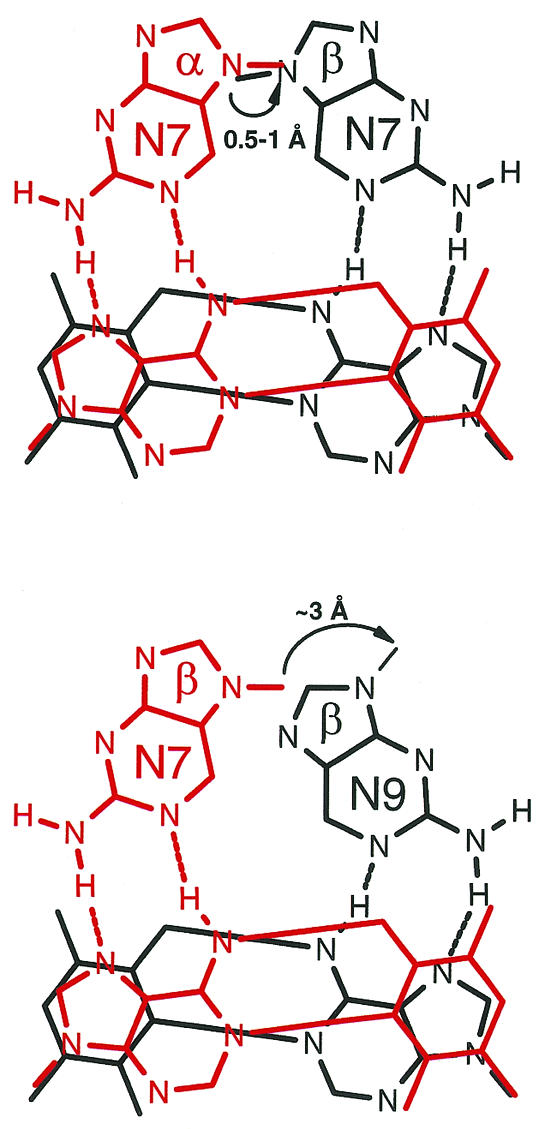
Schematic view of consecutive, alternate strand recognition of adenine bases on a DNA target duplex by aminopurine bases of a TFO, using the α/β-switch model (top) or the N7/N9-switch model (bottom). For clarity, only the heteroatoms of the purine bases are drawn.
The DNA target sequence we chose is based on the human epidermal growth hormone receptor (HER2) promoter sequence. This promoter region contains a 15 nt polypurine region which is suitable for triple helix formation. Antiparallel triplex formation with G/T-containing TFOs at this target site was investigated by Matteucci and co-workers (31). They showed by DMS footprint analysis that full protection of this site can be achieved in the absence of KCl at 1 µM concentration of TFO.
UV-melting experiments
In order to investigate triplex formation of ap-containing TFOs to the intact HER2 target sequence, we prepared the following TFOs and the DNA target duplex shown in Figure 3. In initial experiments we screened the triple helix formation potential of TFOs 1, 2, 3 and 5 (as reference) to the corresponding target duplex D1 by UV-melting curve analysis. As can be seen from Figure 4, both the βN7ap- and αN9ap-containing TFOs 2 and 3 bind to their target, while no triplex formation is observed in the case of the αN7ap-containing TFO 1. Although there is no clear sigmoidal transition for melting of the third strands 2, 3 and 5 from the target duplex, we conclude that triplexes were formed in these cases from the fact that there is a clear increase in UV absorption before the melting of the double helix in duplex–TFO mixtures, and from the equal or even higher hyperchromicity relative to the duplex melting in the absence of a TFO. From these experiments it can be qualitatively inferred that both βN7ap and αN9ap can replace T in T × A-T base-triple formation in the antiparallel binding motif while αN7ap cannot. This is in accord with model considerations.
Figure 3.

Sequences of the duplex target used in the UV-melting curve analyses as well as TFOs 1–5 containing the different isomeric 2-aminopurine nucleosides.
Figure 4.

UV-melting curves (280 nm) of duplex D1 with TFOs 1, 2, 3 and 5 in 90 mM Tris–borate pH 7.5, 10 mM MgCl2 (strand concentration = 1.5 µM). Superposition of consecutive heating–cooling–heating cycles in the presence [circles, crosses, inverted triangles and open squares in (A), (B), (C) and (D), respectively] and absence (closed squares) of TFO.
DNase I footprint titration experiments
In order to assess the potential of the aminopurine-containing oligonucleotides 1–4 to recognize a T-A inversion site, and in order to evaluate their binding selectivity, a DNase I footprint titration assay (29,32) was developed. A plasmid (pSPP4C1) containing a DNA insert with the four triplex target cassettes derived from the HER2 promoter sequence (as outlined in Fig. 5, top) was prepared and used as a source of target DNA. The 247 bp EcoRI––PvuII restriction fragment of this plasmid was 3′-end-labeled (pyrimidine-rich strand) and incubated with increasing concentrations of oligonucleotides 1–5 (300 pM to 10 µM), in 10 mM bis-Tris–HCl pH 7.2, 10 mM MgCl2, 1 mM spermine at 20°C for 7 days, and then digested with DNase I. The generated reaction products were separated by denaturing gel electrophoresis and visualized by storage phosphor autoradiography. Figure 5 (bottom) shows the autoradiograms of the duplex incubated with oligonucleotide 1 (left part of the gel) and oligonucleotide 4 (right part of the gel), as an example.
Figure 5.

UV-melting curves (280 nm) of duplex D1 with TFOs 1, 2, 3 and 5 in 90 mM Tris–borate pH 7.5, 10 mM MgCl2 (strand concentration = 1.5 µM). Superposition of consecutive heating–cooling–heating cycles in the presence [circles, crosses, inverted triangles and open squares in (A), (B), (C) and (D), respectively] and absence (closed squares) of TFO.
The association constants (Ka) obtained for the oligonucleotides 1–5 are shown in Table 2 and the corresponding standard free energies of triplex formation (ΔG°) are listed in Table 3. Possible spatial arrangements of all 16 base triples are given in Figure 6. These arrangements are based on the standard model for an antiparallel triple helix, considering anti-conformation around all nucleosidic bonds, and neglecting rare tautomeric or protonated forms of the aminopurine base.
Table 2. Association constants Ka (in M–1) reported as the mean (± standard error) of at least three measurements for the triplexes formed by the oligonucleotides 1–5 with the four target cassettes on duplex SPP4C1, where X is on the purine strand and Y is on the pyrimidine strand.
| X·Y | |||||
| A·T | C·G | G·C | T·A |
| T | 1.7 (±0.8)·108 | 1.4 (±0.7)·107 | 2.3 (±1.0)·107 | 5.5 (±4.7)·106 | |
| Z | βN7ap | 5.2 (±0.6)·107 | 2.2 (±1.0)·107 | 7.5 (±6.1)·106 | 5.8 (±5.6)·106 |
| αN7ap | 1.1 (±0.5)·107 | 6.9 (±3.1)·106 | 7.3 (±2.5)·106 | 4.2 (±2.6)·107 | |
| βN9ap | 2.0 (±0.9)·107 | 2.3 (±1.1)·106 | 1.2 (±0.8)·108 | 1.0 (±0.5)·106 | |
| αN9ap | 1.3 (±0.4)·108 | 2.7 (±1.2)·107 | 1.3 (±0.6)·108 | 5.5 (±2.5)·107 |
For experimental conditions see Figure 5.
Table 3. Standard free energies of triplex formation (ΔG°) at 20°C [kcal·mol–1] derived from the association constants listed in Table 1.
| X·Y | |||||
| A·T | C·G | G·C | T·A |
| T | –11.1 (±0.2) | –9.6 (±0.3) | –9.9 (±0.1) | –9.1 (±0.4) | |
| Z | βN7ap | –10.4 (±0.1) | –9.9 (±0.2) | –9.3 (±0.3) | –9.1 (±0.4) |
| αN7ap | –9.5 (±0.2) | –9.2 (±0.2) | –9.3 (±0.1) | –10.3 (±0.3) | |
| βN9ap | –9.9 (±0.2) | –8.6 (±0.2) | –10.9 (±0.3) | –8.1 (±0.2) | |
| αN9ap | –10.9 (±0.2) | –10.0 (±0.2) | –10.9 (±0.3) | –10.4 (±0.3) |
Figure 6.


Spatial arrangement of all 16 base-triples investigated. For details see text.
By taking a closer look at the values in Table 2, it clearly appears that within the chosen sequence context, there is no strong discrimination between the different arrangements Z × X-Y. In almost all cases, binding to each target cassette could be observed. The Ka values all lie within two orders of magnitude. This seems not to be unusual for the purine motif (32) but is significantly different compared to the pyrimidine motif, where mismatched base arrangements are subject to a stronger energetic discrimination (33). As expected, the reference TFO 5 forms particularly stable triplexes with its parent adenine target. Furthermore, triple helical complexes containing the T × G-C and T × C-G have intermediate stability. This latter case was also reported by Greenberg and Dervan (32) and was attributed to specific, single hydrogen bonding contacts. In the present work under the given buffer conditions (10 mM bis-Tris–HCl pH 7.2, 10 mM MgCl2, 1 mM spermine) the T × G-C base-triple is even slightly more stable than the T × C-G base-triple, which can eventually be rationalized by a bifurcated hydrogen bond between the N7 and O6 atoms of the guanine base.
The fact that the αN9ap-containing TFO 3 gives the highest stability with the target sequence X-Y = A-T is probably due to the fact that this aminopurine nucleotide unit has its C(1′) atom very close to the position adopted by those of T and G within a normal, antiparallel (G/T) motif, thus giving rise to minimal distortion of the backbone. This can be inferred from molecular modeling experiments on a αN9ap × A-T triple helix (34). Based on this reasoning, the lower affinities of the TFO 2, containing βN7ap, for the A-T base pair, and 4, containing βN9ap, for the T-A base pair, can also be understood. The resulting large distortions of the backbone induced by the 5′Gp(βN7ap)3′ and 5′(βN7ap)pG3′ steps seem to have a detrimental effect on stability.
A very rewarding result of the DNase I footprint experiments was the recognition of a T-A inversion site by TFO 1, containing an αN7ap unit, as the major binding site. The Ka shows a clear, although not very pronounced, preference for the T-A over the A-T target. As in the case of the A-T recognition by TFO 3, the position of the C(1′) atom of αN7ap is near to the position adopted by the C(1′) atom of G and T in the third strand, thus constituting nearly isomorphic base arrangements. We note, however, that the stability of TFO 1, binding to the T-A target, is less by a factor of ∼4 relative to the canonical triplex formed by 5 with the A-T target. Among other factors this could be due to a loss of intrastrand stacking interactions in the G-αN7ap-G step of the TFO, and thus points towards a general problem in the switch models (Fig. 2) concerning affinity of third strands to their target (see Conclusions).
Another interesting result is the high affinity exhibited by both the αN9- and βN9ap-containing TFOs 3 and 4 for the G-C target. Both sequences bind with similar Ka (1.2–1.3 × 108 M–1). In contrast to the low specificity of the αN9ap-containing TFO 3, the βN9ap-containing TFO 4 is specific for the G-C target site with an energetic advantage of 1.0–2.8 kcal·mol–1 over the other three target sites. Besides the possible base arrangement given in Figure 6, the relatively high stability of the βN9ap × G-C base triple may alternatively be rationalized by invoking protonation of the N1 postition of the 2-aminopurine base, thus forming two hydrogen bonds with guanine (Fig. 7A). In fact, it is known from studies by Sowers et al. (35) on the βN9ap-C mispair, which is at the origin of transition mutations in vivo, that a protonated 2-aminopurine base is engaged in this base pair (35). This could also explain the high stability of the αN9ap × G-C base triple. In this case, the hydrogen bonds are formed between N7(G) and H-N2(ap) and between O6(G) and H-N1(ap) (Fig. 7B).
Figure 7.

Hypothetical βN9apH+d× G-C (A) and αN9apH+ × G-C (B) base-triples.
CONCLUSIONS
From the data obtained we draw the following conclusions on the utility of 2-aminopurine nucleosides for antiparallel triple-helix formation. (i) βN7ap acts as a replacement for T (or A) in recognizing an A-T base pair. (ii) Due to the poor discrimination between the different target base-pairs X-Y, αN9ap could eventually be further developed into a ‘universal base’ for antiparallel triple-helix formation. Together with the intrinsic fluorescence properties of this nucleoside (which the N7 series is lacking) this could lead to useful diagnostic probes. (iii) The preferential recognition of a G-C base pair by βN9ap could in principal open the way to guanine replacement in TFOs. This is of considerable interest given the fact that one of the severe limitations of the antiparallel triple-helix approach lies in the formation of G-tetrad structures of the G-rich TFOs, thus evading duplex target recognition.
The structural basis for the αN7ap unit recognizing a T-A inversion, as set forth in the switch concept, still needs approval by high resolution structural analysis of a corresponding triplex. The question on how many inversion sites could potentially be recognized by αN7ap-containing TFOs within a duplex target sequence also still remains open at this point. We note again that the stability of TFO 1, binding to the T-A target, is less by a factor of ∼4, compared to the canonical triplex formed by 5 with the A-T target. Thus, it seems that attaining high affinity is the most important challenge in future TFO design. In this context it will be of considerable interest to combine newly discovered bases for purine–pyrimidine base-inversion recognition, as the one presented here, with overall triplex stability enhancing features, e.g. the 2-aminoethoxy sugar modification (36,37), the grafting of positive charges onto the bases (20), or both (38), in order to successfully remove the target sequence constraints in the triple-helix approach.
Acknowledgments
ACKNOWLEDGEMENT
We would like to thank the Swiss National Science Foundation for financial support of this project.
References
- 1.Fox K.R. (2000) Targeting DNA with Triplexes. Curr. Med. Chem., 7, 17–37. [DOI] [PubMed] [Google Scholar]
- 2.Neidle S. (1997) Recent developments in triple-helix regulation of gene expression. Anticancer Drug Des., 12, 433–442. [PubMed] [Google Scholar]
- 3.Moser H.E. and Dervan,P.B. (1987) Sequence specific cleavage of double helical DNA by triple helix formation. Science, 238, 645–650. [DOI] [PubMed] [Google Scholar]
- 4.François J.-C., Saison-Behmoaras,T. and Hélène,C. (1988) Sequence-specific recognition of the major groove of DNA by oligodeoxynucleotides via triple helix formation. Footprinting studies. Nucleic Acids Res., 16, 11431–11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beal P.A. and Dervan,P.B. (1991) Second structural motif for recognition of DNA by oligonucleotide directed triple-helix formation. Science, 251, 1360–1363. [DOI] [PubMed] [Google Scholar]
- 6.Durland R.H., Kessler,D.J., Gunnell,S., Duvic,M., Pettitt,B.M. and Hogan,M.E. (1991) Binding of triple helix forming oligonucleotides to sites in gene promoters. Biochemistry, 30, 9246–9255. [DOI] [PubMed] [Google Scholar]
- 7.Gowers D.M. and Fox,K.R. (1999) Towards mixed sequence recognition by triple helix formation. Nucleic Acids Res., 27, 1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun J.S., De Bizemont,T., Duval-Valentin,G., Montenay-Garestier,T. and Hélène,C. (1991) Extension of the range of recognition sequences for triple helix formation by oligonucleotides containing guanines and thymines. C.R. Acad. Sci. III, 313, 585–590. [PubMed] [Google Scholar]
- 9.Beal P.A. and Dervan,P.B. (1992) Recognition of double helical DNA by alternate strand triple helix formation. J. Am. Chem. Soc., 114, 4976–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horne D.A. and Dervan,P.B. (1990) Recognition of mixed-sequence duplex DNA by alternate-strand triple-helix formation. J. Am. Chem. Soc., 112, 2435–2437. [Google Scholar]
- 11.Ono A., Chen,C.-N. and Kan,L.-S. (1991) DNA triplex formation of oligonucleotide analogues consisting of linker groups and octamer segments that have opposite sugar-phosphate backbone polarities. Biochemistry, 30, 9914–9921. [DOI] [PubMed] [Google Scholar]
- 12.De Bizemont T., Duval-Valentin,G., Sun,J.-S., Bisagni,E., Garestier,T. and Hélène,C. (1996) Alternate strand recognition of double-helical DNA by (T,G)-containing oligonucleotides in the presence of a triple helix-specific ligand. Nucleic Acids Res., 24, 1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou B.-W., Marchand,C., Asseline,U., Thuong,N.T., Sun,J.-S., Garestier,T. and Hélène,C. (1995) Recognition of alternating oligopurine/oligopyrimidine tracts of DNA by oligonucleotides with base-to-base linkages. Bioconjugate Chem., 6, 516–523. [DOI] [PubMed] [Google Scholar]
- 14.Asseline U. and Thuong,N.T. (1994) 5′-5′ tethered oligonucleotides via nucleic bases: a potential new set of compounds for alternate strand triple-helix formation. Tetrahedron Lett., 35, 5221–5224. [Google Scholar]
- 15.Asseline U. and Thuong,N.T. (1993) Oligonucleotides tethered via nucleic bases. A potential new set of compounds for alternate strand triple-helix formation. Tetrahedron Lett., 34, 4173–4176. [Google Scholar]
- 16.Lehmann T.E., Greenberg,W.A., Liberles,D.A., Wada,C.K. and Dervan,P.B. (1997) Triple-helix formation by pyrimidine oligonucleotides containing nonnatural nucleosides with extended aromatic nucleobases: Intercalation from the major groove as a method for recognizing C.G and T.A base-pairs. Helv. Chim. Acta, 80, 2002–2022. [Google Scholar]
- 17.Huang C.-Y., Cushman,C.D. and Miller,P.S. (1993) Triplex formation by an oligonucleotide containing N4-(3-amidopropyl)cytosine. J. Org. Chem., 58, 5048–5049. [Google Scholar]
- 18.Stilz H.-U. and Dervan,P.B. (1993) Specific recognition of CG base pairs by 2-deoxynebularine within the purine·purine·pyrimidine triple-helix motif. Biochemistry, 32, 2177–2185. [DOI] [PubMed] [Google Scholar]
- 19.Amosova O. and Fresco,J.R. (1999) A search for base analogs to enhance third-strand binding to ‘inverted’ target base-pairs of triplexes in the pyrimidine/parallel motif. Nucleic Acids Res., 27, 4632–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gowers D.M., Bijapur,J., Brown,T. and Fox,K.R. (2000) DNA triple helix formation at target sites containing several pyrimidine interrupts: stabilization by protonated cytosine or 5-(1-propargylamino)dU. Biochemistry, 38, 13747–13758. [DOI] [PubMed] [Google Scholar]
- 21.Prévot-Halter I. and Leumann,C. (1999) Selective recognition of a C-G base-pair in the parallel DNA triple-helical binding motif. Bioorg. Med. Chem. Lett., 9, 2657–2660. [DOI] [PubMed] [Google Scholar]
- 22.Marfurt J. and Leumann,C. (1998) Evidence for CH…O hydrogen bond assisted recognition of a pyrimidine base in the parallel DNA triple helical motif. Angew. Chem. Inl. Ed Engl., 37, 175–177. [Google Scholar]
- 23.Thuong N.T. and Hélène,C. (1993) Sequence specific recognition and modification of double helical DNA by oligonucleotides. Angew. Chem. Intl. Ed. Engl., 32, 666–690. [Google Scholar]
- 24.Doronina S.O., Blanalt-Feidt,S. and Behr,J.-P. (1999) DNA recognition by non-natural oligonucleotides. Nucl. Nucl., 18, 1617–1618. [Google Scholar]
- 25.Doronina S.O. and Behr,J.-P. (1998) Synthesis of 4-guanidinopyrimidine nucleosides for triple helix-mediated guanine and cytosine recognition. Tetrahedron Lett., 39, 547–550. [Google Scholar]
- 26.Parel S.P. and Leumann,C.J. (2000) Synthesis and pairing properties of oligodeoxynucleotides containing N7-(purine-2-amine deoxynucleosides). Helv. Chim. Acta, 83, 2514–2526. [Google Scholar]
- 27.Wu X. and Pitsch,S. (1998) Synthesis and pairing properties of oligoribonucleotide analogues containing a metal-binding site attached to β-d-allofuranosyl cytosine. Nucleic Acids Res., 26, 4315–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning:A Laboratory Manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY.
- 29.Brenowitz M., Senear,D.F., Shea,M.A. and Ackers,G.K. (1986) Quantitative DNase footprint titration: a method for studying protein–DNA interactions. Methods Enzymol., 130, 132–181. [DOI] [PubMed] [Google Scholar]
- 30.Priestley E.S. and Dervan,P.B. (1995) Sequence composition effects on the energetics of triple helix formation by oligonucleotides containing a designed mimic of protonated cytosine. J. Am. Chem. Soc., 117, 4761–4765. [Google Scholar]
- 31.Milligan J.F., Krawczyk,S.H., Wadwani,S. and Matteucci,M.D. (1993) An antiparallel triple helix motif with oligodeoxynucleotides containing 2′-deoxyguanosine and 7-deaza-2′-deoxyxanthosine. Nucleic Acids Res., 21, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenberg W.A. and Dervan,P.B. (1995) Energetics of sixteen triple helical complexes which vary at a single position within a purine motif. J. Am. Chem. Soc., 117, 5016–5022. [Google Scholar]
- 33.Best G.C. and Dervan,P.B. (1995) Energetics of formation of sixteen triple helical complexes which vary at a single position within a pyrimidine motif. J. Am. Chem. Soc., 117, 1187–1193. [Google Scholar]
- 34.Parel S.P. (2000) 2-Aminopurine nucleotides in DNA-triplex formation: selective recognition of a T-A base-pair in the antiparallel recognition motif. PhD Thesis, University of Berne, Switzerland.
- 35.Sowers L.C., Boulard,Y. and Fazakerley,G.V. (2000) Multiple structures for the 2-aminopurine–cytosine mispair. Biochemistry, 39, 7613–7620. [DOI] [PubMed] [Google Scholar]
- 36.Blommers M.J.J., Natt,F., Jahnke,W. and Cuenoud,B. (1998) Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides: the solution structure of an intramolecular triplex obtained by NMR spectroscopy. Biochemistry, 37, 17714–17725. [DOI] [PubMed] [Google Scholar]
- 37.Cuenoud B., Casset,F., Hüsken,D., Natt,F., Wolf,R.M., Altmann,K.-H., Martin,P. and Moser,H.E. (1998) Dual recognition of double-stranded DNA by 2′-aminoethoxy-modified oligonucleotides. Angew. Chem. Intl. Ed. Engl., 37, 1288–1291. [DOI] [PubMed] [Google Scholar]
- 38.Sollogoub M., Dominguez,B., Fox,K.R. and Brown,T. (2000) Synthesis of a novel bis-amino-modified thymidine monomer for use in DNA triplex stabilisation. Chem. Commun., 2315–2316. [Google Scholar]