

ABSTRACT
Enterovirus 71 (EV71) can cause hand-foot-and-mouth disease (HFMD) in young children. Severe infection with EV71 can lead to neurological complications and even death. However, the molecular basis of viral pathogenesis remains poorly understood. Here, we report that EV71 induces degradation of gasdermin D (GSDMD), an essential component of pyroptosis. Remarkably, the viral protease 3C directly targets GSDMD and induces its cleavage, which is dependent on the protease activity. Further analyses show that the Q193-G194 pair within GSDMD is the cleavage site of 3C. This cleavage produces a shorter N-terminal fragment spanning amino acids 1 to 193 (GSDMD1–193). However, unlike the N-terminal fragment produced by caspase-1 cleavage, this fragment fails to trigger cell death or inhibit EV71 replication. Importantly, a T239D or F240D substitution abrogates the activity of GSDMD consisting of amino acids 1 to 275 (GSDMD1–275). This is correlated with the lack of pyroptosis or inhibition of viral replication. These results reveal a previously unrecognized strategy for EV71 to evade the antiviral response.
IMPORTANCE Recently, it has been reported that GSDMD plays a critical role in regulating lipopolysaccharide and NLRP3-mediated interleukin-1β (IL-1β) secretion. In this process, the N-terminal domain of p30 released from GSDMD acts as an effector in cell pyroptosis. We show that EV71 infection downregulates GSDMD. EV71 3C cleaves GSDMD at the Q193-G194 pair, resulting in a truncated N-terminal fragment disrupted for inducing cell pyroptosis. Notably, GSDMD1–275 (p30) inhibits EV71 replication whereas GSDMD1–193 does not. These results reveal a new strategy for EV71 to evade the antiviral response.
KEYWORDS: EV71, HFMD, 3C, GSDMD
INTRODUCTION
Enterovirus 71 (EV71) is a major etiological agent of hand-foot-and-mouth disease (HFMD) which affects infants and children younger than 5 years of age. Most of these infections are self-limited with mild symptoms, including fever and vesicular eruptions mainly on the skin of hands and feet and in the mouth. However, some patients show severe neurological diseases, including aseptic meningitis, acute flaccid paralysis, encephalitis, and pulmonary edema. Since it was first isolated in California in 1974 (1–3), EV71 infection has caused many large-scale epidemics in the world, especially in the Asia-Pacific region (1, 4–6). To date, there are no effective therapeutic drugs for EV71 infection.
EV71 belongs to the Enterovirus genus of the family Picornaviridae. The viral genome is approximately 7,500 nucleotides in length, with a single open reading frame that encodes a large precursor protein that is subsequently processed into three structural (VP0, VP1, and VP3) and seven nonstructural (2A, 2B, 2C, 3A, 3B, 3C, and 3D) proteins by the virus-encoded protease 2A or 3C (7). The three structural proteins and the RNA form provirions, in which VP0 is cleaved into VP2 and VP4 by RNA or 3CD, but the molecular mechanism is unclear (8). These proteases are also involved in a number of biological processes. For example, 2A inhibits interferon (IFN) responses by cleaving interferon receptor (IFNAR) or mitochondrial antiviral signaling (MAVS) protein (9, 10). 3C induces apoptosis through caspase activation (11) to facilitate viral spread or pathogenesis. In addition, the 3C protease induces apoptosis by inducing cleavage of PinX1, an intrinsic telomerase inhibitor, to facilitate EV71 release (12). Other important roles of 3C are to dampen immune responses by impeding the RIG-I-MAVS interaction or by cleaving a series of host factors, including TIR domain-containing adaptor inducing IFN-β (TRIF), interferon regulatory factor 7/9, and TAK1/TAB1/TAB2/TAB3 complex (13–17).
Our previous results showed that NLRP3 inflammasome activation plays a protective role against EV71 infection in vivo. Conversely, EV71 has developed mechanisms to antagonize inflammasome activation through cleavage of NLRP3 by the 2A and 3C proteases (18). The NLRP3 inflammasome is a caspase-1-activating complex, including NLRP3, ASC, and procaspase-1, which is triggered by most endogenous danger- and pathogen-associated molecules (19). Activated caspase-1 induces the maturation of interleukin-1β (IL-1β) and cell pyroptosis. Pyroptosis is a form of programmed necrosis that features pore formation in the plasma membrane, cell swelling, and lysis and releases cytoplasmic content to the extracellular environment (20–22). Its initiation depends on the activation of inflammatory caspases, such caspase-1, caspase-4, and caspase-5 in humans (23, 24), leading to the release of cytoplasmic contents. Recently, it has been reported that gasdermin D (GSDMD) mediates pyroptosis, which is cleaved and activated by caspase-1 and caspase-11 (24–27). The cleaved N-terminal fragment of GSDMD directly forms pores in the cytoplasmic membrane (28–33). It is believed that pyroptosis can inhibit and clear intracellular pathogens (34). However, whether viral replication or viral proteins inhibit pyroptosis is unclear.
Here, we report that EV71 infection decreases the expression of GSDMD. In this process, 3C directly induces the cleavage of GSDMD at the pair of Q193-G194, producing a nonfunctional GSDMD fragment consisting of amino acids (aa) 1 to 193 (GSDMD1–193). Importantly, we found that the GSDMD fragment consisting of aa 1 to 275 (GSDMD1–275) cleaved by caspase-1 inhibited EV71 replication, while GSDMD1–193 cleaved by 3C did not. These results suggest a previously unrecognized mechanism of EV71 to evade the antiviral response.
RESULTS
EV71 infection induces the degradation of GSDMD.
Recently, GSDMD has been identified as a key determinant of pyroptosis (24–27). To investigate the impact of EV71 infection, we analyzed the expression of GSDMD. THP-1, 293T, and rhabdomyosarcoma (RD) cells were mock infected or infected with EV71. At different time points postinfection, cell lysates were processed for Western blot analysis. As shown in Fig. 1, GSDMD was expressed in mock-infected THP-1 (Fig. 1A), 293T (Fig. 1B), and RD (Fig. 1C) cells. EV71 infection induced degradation of GSDMD in these cells. These results show that EV71 infection downregulates GSDMD.
FIG 1.

GSDMD is degraded in EV71-infected cells. THP-1, 293T, and RD cells were mock infected or infected with EV71 at an MOI of 10, 2, and 1, respectively. At the indicated time points, cell lysates were analyzed by Western blotting with antibodies for GSDMD, EV71, and β-actin.
EV71 3C associates with and cleaves GSDMD in mammalian cells.
We along with others have reported that 3C can mediate cleavage of host factors and affect their functions (12, 35, 36). To investigate whether EV71 3C targets GSDMD, we conducted cleavage experiments. 293T cells were transfected with increasing amounts of a plasmid expressing green fluorescent protein (GFP)-tagged 3C (GFP-3C), along with a plasmid expressing GSDMD, with a Flag or Myc tag fused at its N or C terminus, respectively. At 24 h after transfection, cell lysates were processed to detect 3C-mediated cleavage of GSDMD by Western blotting. We found that smaller protein bands (about 20 kDa of an N-terminal fragment detected by anti-Flag antibody and 32 kDa of a C-terminal fragment detected by anti-Myc antibody) appeared when GSDMD was cotransfected with GFP-3C but not with GFP alone (Fig. 2A). These small bands represent the N-terminal fragment (about 20 kDa) and the C-terminal fragment (about 32 kDa). Under this experimental condition, EV71 2A did not cleave GSDMD (Fig. 2B).
FIG 2.

The 3C but not 2A protease of EV71 cleaves GSDMD. (A) 293T cells were transfected with plasmids encoding Flag-GSDMD-Myc along with GFP (lane 1) or increasing amounts of GFP-3C (lanes 2 to 5). At 24 h after transfection, the cells were then processed for Western blotting with antibodies, as indicated, using a Li-Cor Odyssey dual-color system (Li-Cor, Lincoln, NE). Antibodies recognizing Flag-GSDMD-Myc (Flag, N terminus of GSDMD, 800 nm, green; Myc, C terminus of GSDMD, 680 nm, red) were used. The merged images of the two channels are shown below (yellow). 3C or GFP was detected by using GFP antibody. β-Actin was included as a loading control. (B) 293T cells were transfected with plasmids encoding Flag-GSDMD-Myc along with a control plasmid or pcDNA3.1-IRES-2A plasmid. At 24 h after transfection, cells were lysed and analyzed by Western blotting. (C) The 3C protease interacts with GSDMD. 293T cells were transfected with plasmids encoding Flag-GSDMD (lanes 3 and 4), GFP (lanes 1 and 3), or GFP-3C (lanes 2 and 4). The total amount of DNA was kept constant with an empty vector. At 24 h after transfection, cell lysates were immunoprecipitated with anti-Flag antibody. Immunoprecipitates and aliquots of cell lysates were then analyzed by Western blotting. WCL, whole-cell lysates; IB, immunoblotting.
To explore whether the 3C protease interacts with GSDMD, we performed an immunoprecipitation assay. 293T cells were transiently transfected with GFP-3C along with Flag-GSDMD. Cell extracts were immunoprecipitated with anti-Flag antibody, followed by Western blotting with anti-GFP antibody. As indicated in Fig. 2C, GSDMD associated with GFP-3C (Fig. 2C, lane 4) but not with GFP (Fig. 2C, lane 3). Taken together, these results show that 3C protease associates with GSDMD and induces its cleavage.
The protease activity of 3C is crucial for cleavage of GSDMD.
To explore whether the protease activity of EV71 3C is required for the cleavage of GSDMD, we evaluated the impact of rupintrivir, which is an inhibitor of 3C protease with a broad spectrum of activity against picornaviruses (37–39). As shown in Fig. 3A, expression of GFP-3C resulted in a cleaved GSDMD fragment in the absence of rupintrivir (lane 2). However, when cells were treated with rupintrivir, the cleavage of GSDMD was blocked (lane 4). The caspase inhibitor Z-VAD (benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) had no effects on 3C-mediated GSDMD cleavage (Fig. 3B), suggesting that the cleavage is induced by 3C but not by caspases. In order to further verify this issue, we carried out mutational analysis. His40, Glu71, and Cys147 are key amino acids which form the catalytic triad of EV71 3C (39). H40D and C147S substitutions in the active site of EV71 3C disrupt the protease activity. As shown in Fig. 3C, wild-type 3C, but not the GFP control, cleaved GSDMD (lanes 1 and 2). However, neither the H40D nor C147S mutant cleaved GSDMD, as measured by Western blotting (Fig. 3C, lanes 3 and 4). R84Q or V154S still cleaved GSDMD (Fig. 3C, lanes 5 and 6). These two mutants are abolished for the RNA binding activity but not the protease activity of 3C (15). All of these mutants were expressed at levels comparable to the level wild-type 3C, as determined by Western blotting (Fig. 3C). Hence, the protease activity of 3C is essential for GSDMD cleavage.
FIG 3.

The protease activity of 3C is needed for cleavage of GSDMD. (A and B) 293T cells were transfected with plasmids encoding Flag-GSDMD-Myc along with GFP or GFP-3C. At 4 h after transfection, cells were incubated with the protease inhibitor rupintrivir or caspase inhibitor Z-VAD, as indicated, for 24 h. Cell lysates were then processed for Western blotting. (C) 293T cells were transfected with plasmids encoding Flag-GSDMD-Myc along with GFP or GFP-3C variants, as indicated. Cell lysates were subjected to Western blot analysis with antibodies.
The Q193-G194 pair is a cleavage site of 3C within GSDMD.
In order to identify the potential cleavage site within GSDMD, we carried out mutational analysis. There are several glutamines which resemble the Q-G/Q-S sequence of proteolytic sites for the EV71 3C protease within GSDMD. Based on the sizes of the cleaved bands, the Q193-G194 and Q335-G336 pairs were tested as the potential cleaved sites for 3C protease (Fig. 4A). As such, we constructed the two mutants in which Q was replaced with an A residue. As shown in Fig. 4B, wild-type GSDMD was cleaved by GFP-3C, resulting in a 20-kDa band representing the N terminus and a 32-kDa band representing the C terminus of GSDMD (lane 2). However, the Q193A mutant was resistant to cleavage (Fig. 4B, lane 4); the two major cleaved fragments disappeared when it was coexpressed with 3C. The Q335A mutant was also cleaved by 3C (lane 6). These results show that the Q193-G194 pair is a major cleavage site of 3C within GSDMD.
FIG 4.
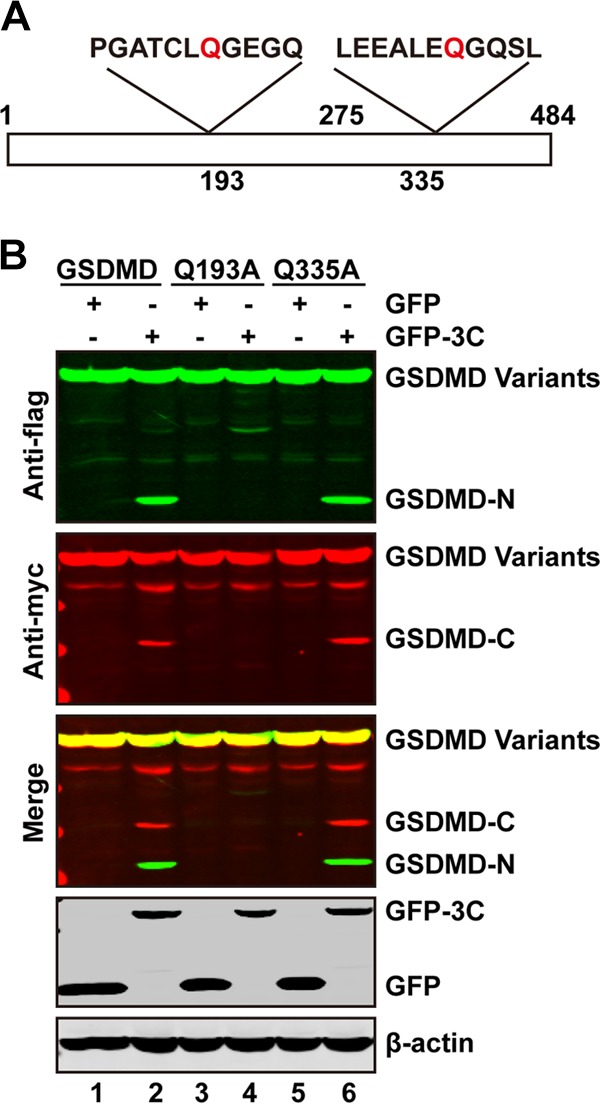
The Q193-G194 pair is the site of GSDMD cleavage. (A) Primary sequences of amino acids 187 to 197 or 329 to 339 with GSDMD. In these regions, glutamine was replaced with alanine. (B) 293T cells were transfected with plasmids encoding wild-type GSDMD or GSDMD variants along with GFP (lanes 1, 3, and 5) or GFP-3C (lanes 2, 4, and 6) as indicated. At 24 h after transfection, cell lysates were subjected to Western blotting with antibodies against Flag, Myc, GFP, and β-actin.
EV71 promotes the cleavage of GSDMD in infected cells.
The above data showed that EV71 induced the degradation of GSDMD in THP-1, 293T, and RD cells (Fig. 1A to C), but the cleaved fragments were not detectable in our system due to the low expression level of endogenous GSDMD. To address this issue, a plasmid encoding Flag-GSDMD-Myc was transfected into 293T cells. The cells were then infected with increasing doses of EV71. As shown in Fig. 5A, the cleaved fragments of the N terminus and C terminus were also seen in EV71-infected 293T cells. However, when the Q193A mutant was ectopically expressed in 293T cells, these cleaved fragments disappeared (Fig. 5B). Since NLRP3 mediates GSDMD cleavage at D275-G276 and GSDMD activation (25–27), we further evaluated the effect of EV71 3C. As shown in Fig. 5C, in 293T cells transfected with the inflammasome components, caspase-1 cleaved GSDMD at D275-G276, resulting in GSDMD1–275 and GSDMD276–484. However, addition of GFP-3C resulted in the shorter N-terminal fragment GSDMD1–193 and the C-terminal fragment GSDMD194–484. Such a shift in the GSDMD cleavage pattern indicates that EV71 3C precluded GSDMD activation by the inflammasome. Together, these results suggest that EV71 infection induces cleavage of GSDMD in a 3C-dependent manner.
FIG 5.
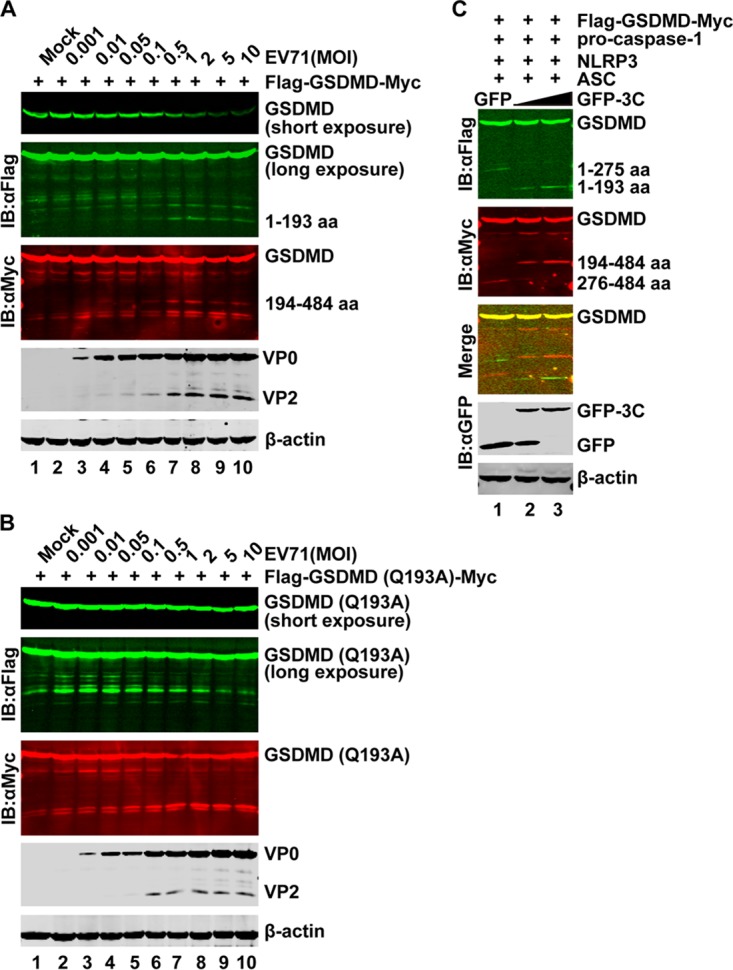
GSDMD is cleaved in EV71-infected cells. (A and B) 293T cells were transfected with plasmid encoding GSDMD (A) or GSDMD-Q193A (B) for 24 h. Cells were then mock infected or infected with an increasing dose of EV71. At 24 h after infection, cell lysates were analyzed by Western blotting using antibodies against Flag, Myc, EV71, and β-actin. (C) 293T cells were transfected with plasmids encoding GSDMD, NLRP3, ASC, and procaspase-1 and a plasmid expressing enhanced GFP (PEGFP) or PEGFP-3C. At 24 h after transfection, cell lysates were subjected to Western blotting.
The cleavage fragment of GSDMD by 3C is unable to induce the pyroptosis.
GSDMD1–275 cleavage by caspase-1 induces pyroptosis by forming a large plasma membrane pore (28–33). To evaluate the effect of EV71 3C on GSDMD, we assessed the cell viability and cell death of different cleavage fragments of GSDMD. First, we constructed the plasmids which express GSDMD1–275 and GSDMD276–484, two GSDMD fragments that mimic cleavage products by caspase-1. In parallel, we constructed GSDMD1–193 and GSDMD194–484, two GSDMD fragments that mimic cleavage by EV71 3C (Fig. 6A). 293T cells were transfected with these constructs individually. Figure 6B shows that GSDMD1–275 but not wild-type GSDMD induced cell death, with typical pyroptosis morphological features. However, GSDMD1–193 did not have such an effect. The same results were observed in cell viability and cell death analyses. GSDMD1–275 triggered pyroptosis (Fig. 6C and D), while other fragments did not. The protein expression levels were verified by Western blotting (Fig. 6E). However, the expression level of GSDMD1–275 was not detectable, in agreement with the hypothesis that the protein might have a toxic effect on the host cell.
FIG 6.

The cleavage fragments of GSDMD by 3C cannot induce the pyroptosis. (A) Schematic diagrams of GSDMD deletion mutant cleavage by caspase-1 or 3C protease. (B) Pyroptosis induced by GSDMD mutants. 293T cells were transfected with Flag-tagged wild-type GSDMD or a cleaved fragment of GSDMD. FL, full-length. (C and D) Assays for the wild-type or cleaved fragments of GSDMD in pyroptosis of 293T cells. (E) Expression of GSDMD mutants. ***, P < 0.001.
Amino acids T239 and F240 are key sites for pyroptosis induced by GSDMD1–275.
Based on the above results, we speculated that GSDMD mediates pyroptosis through an active motif located between amino acids 193 and 275. To test this, we constructed the a series of GSDMD deletion mutants (aa 1 to 203, 1 to 213, 1 to 223, 1 to 233, 1 to 243, 1 to 253, and 1 to 263) (Fig. 7A). These mutants were expressed in 293T cells for 24 h. As shown in Fig. 7B, the mutants consisting of aa 1 to 243, 1 to 253, and 1 to 263 could also induce cell death in 293T cells, in contrast to GSDMD1–275. However, the mutant constructs consisting of aa 1 to 193, 1 to 203, 1 to 213, 1 to 223, and 1 to 233 could not induce cell death of 293T cells, indicating that the active site(s) of GSDMD1–275 is located between amino acids 234 and 243. Similar results were obtained in cell viability and cell death assays (Fig. 7C and D). Lactate dehydrogenase (LDH) release was detectable in cells expressing fragments consisting of aa 1 to 243, 1 to 253, 1 to 263, and 1 to 275. Western blot analysis detected the expression of inactive but not activated mutants (Fig. 7E).
FIG 7.

The domain spanning amino acids 234 to 243 is a determinant for GSDMD-mediated pyroptosis. (A) Schematic diagrams of deletion mutants of GSDMD. (B) Pyroptosis induced by deletion mutants of the N terminus of GSDMD. (C and D) Assays for wild-type GSDMD and its variants in pyroptosis of 293T cells. (E) Expression of GSDMD and the N terminus of GSDMD and its deletion mutants. **, P < 0.01; ***, P < 0.001.
To define the putative activity site(s) of GSDMD1–275, we constructed additional point mutants, as indicated in Fig. 8A. As shown in Fig. 8B, all of the mutants, except T239D and F240D, can induce cell death (Fig. 8B), indicating that T239 and F240 are the critical sites for pyroptosis induced by GSDMD1–275. The same results were noted in cell viability and cell death assays (Fig. 8C and D). The expression of only these two mutants can be detected by Western blotting (Fig. 8E). Collectively, these data indicate that the T239 and F240 residues in GSDMD are critical for cell pyroptosis mediated by the N terminus of GSDMD.
FIG 8.

The T239 and F240 amino acids of the N terminus of GSDMD are necessary for its induced pyroptosis. (A) Primary sequences of amino acids 234 to 243 within the N terminus of GSDMD. In this region, each amino acid was replaced with aspartic acid. (B) Pyroptosis induced by point mutants of the N terminus of GSDMD. (C and D) Assays of the wild-type GSDMD, GSDMD1–275, and GSDMD1–275 point mutants in pyroptosis of 293T cells. (E) Expression of GSDMD, GSDMD1–275, and the GSDMD1–275 point mutants. ***, P < 0.001.
Amino acids T239 and F240 within GSDMD determine EV71 replication.
Finally, we asked whether GSDMD cleavage by 3C or caspase-1 is linked to EV71 replication. Specifically, we transfected plasmids expressing GSDMD variants, including the fragments consisting of aa 1 to 275, 276 to 484, 1 to 193, and 194 to 484, into the RD cells. The cells were then infected with EV71 for 24 h, and RNA was extracted for real-time reverse transcription-PCR (real-time RT-PCR) analysis. As shown in Fig. 9A, EV71 replication decreased in cells transfected with a vector expressing GSDMD1–275 compared to that in cells with a control vector. However, wild-type GSDMD displayed little effect on viral replication. Other cleavage fragments of GSDMD had no inhibitory effect on viral replication. We further evaluated the effects of these fragments on viral production by plaque assay. As shown in Fig. 9B and C, the results of the plaque assay are consistent with the data from real-time RT-PCR, with only GSDMD1–275 significantly inhibiting EV71 production. Next, we investigated whether pyroptosis induced by GSDMD1–275 is important for inhibiting EV71 production. As the T239 and F240 residues are critical amino acids for cell pyroptosis induced by GSDMD1–275, we examined the effects of the two mutants T239D and F240D on viral replication, which could not induce pyroptosis. As illustrated in Fig. 9D, unlike the wild-type GSDMD1–275, neither T239D nor F240D inhibited viral RNA replication. Similar results were obtained by plaque assay (Fig. 9E). These results demonstrate that overexpression of GSDMD1–275 inhibits EV71 replication, which is dependent on its pyroptosis activity. However, GSDMD1–193 cleaved by 3C cannot inhibit viral production.
FIG 9.

The effect of GSDMD mutations on EV71 replication. (A) RD cells transfected with plasmids encoding GSDMD and its variants as indicated. An empty vector was used as a control. At 24 h after transfection, cells were infected with EV71. After 24 h, total RNA was extracted, and the viral RNA levels of EV71 were evaluated by quantitative real-time PCR using SYBR green. Data are expressed as fold change of the EV71 mRNA level relative to that of the control vector. (B and C) RD cells were treated as described in panel A. Then plaque assays of viruses were performed on RD cells at 37°C. Monolayers were stained with crystal violet at 72 h postinfection. Data shown are representative of three independent experiments. (D) The effects of GSDMD1–275 point mutants on EV71 replication. RD cells were transfected with plasmids as indicated. At 24 h after transfection, cells were infected with EV71. After 24 h, the viral RNA levels were detected by real-time RT-PCR as described in for panel A. (E) Plaque assay of viral growth. **, P < 0.01; ***, P < 0.001.
DISCUSSION
Several studies have suggested that EV71 suppresses antiviral immunity (9, 10, 13–17). In addition to inhibition of type I IFN induction, EV71 has evolved strategies to counter inflammasome activation through cleavage of NLRP3 by viral proteases 2A and 3C (18). Although incompletely deciphered, these observations highlight the importance of inflammatory responses in the control of EV71 infection. In the present studies, we found that the 3C protein of EV71 targets GSDMD, which is a key effector of pyroptosis. Notably, EV71 3C mediates specific proteolytic cleavage within GSDMD, resulting in its inactivation. Our work suggests that the interaction of EV71 3C and GSDMD is an interface that may determine EV71 replication or pathogenesis.
EV71 3C is a protease that primarily processes the EV71 precursor protein for replication. Our experimental data suggest that it also cleaves GSDMD. Therefore, GSDMD represents a novel cellular target of EV71 3C. In support of this notion, EV71 3C associated with and cleaved GSDMD when it was expressed in mammalian cells. Consistently, EV71 induced proteolytic cleavage of GSDMD in infected cells. Indeed, protease-dead mutants of 3C were unable to mediate GSDMD cleavage whereas mutants defective in RNA binding functioned effectively. Furthermore, rupintrivir but not Z-VAD inhibited GSDMD cleavage by EV71 3C. On the other hand, expression of GSDMD1–275 induced pyroptosis. This occurred in parallel with reduced viral replication. These results suggest a biological connection of EV71 and pyroptosis.
As a key component in pyroptosis, GSDMD usually stays in an inactive form. Upon activation by caspases, GSDMD undergoes proteolytic cleavage and releases its N-terminal domain, GSDMD1–275 (28–32). As a result, this induces pyroptosis by causing extensive pore formation on the cell plasma membrane (28, 29, 31–33, 40, 41). Mutational analyses suggest that T239D and F240D substitutions had no effect whereas T239D and F240D substitutions abrogated GSDMD activity, indicating that T239 and F240 within GSDMD are essential for inducing pyroptosis. Although the underlying mechanism is unknown, we suspect that the region with T239 and F240 likely serves as a functional motif. An attractive possibility is that it may be involved in oligomerization of GSDMD. Alternatively, this motif may be required for membrane insertion of GSDMD. Additional work is needed to test these possibilities.
It is noteworthy that EV71 3C mediates GSDMD cleavage at the Q193-G194 pair. We found that a mutation in an additional potential site, as represented by the Q335A substitution, had no visible effect on GSDMD cleavage. Therefore, EV71 3C appears to have a single cleavage site within GSDMD. Interestingly, when expressed in mammalian cells, EV71 3C induced a 20-kDa protein fragment, GSDMD1–193. However, unlike the GSDMD1–275 that mimics the cleavage product of caspases (aa 1 to 275), this fragment failed to induce pyroptosis or reduce viral replication. This is attributable to the elimination of the functional motif from GSDMD by EV71 3C. Collectively, these results support the argument that cleavage at the Q193-G194 pair by EV71 3C is a viral mechanism to perturb pyroptosis mediated by GSDMD. Future work will investigate its role in viral pathogenesis.
MATERIALS AND METHODS
Cells and viruses.
Human rhabdomyosarcoma (RD) cells and embryonic kidney HEK-293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco), supplemented with 10% heated-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Human monocytic leukemia THP-1 cells were cultured in RPMI 1640 medium contained 10% heated-inactivated fetal bovine serum (FBS; HyClone, Logan, UT). For EV71 infection of THP-1 cells, phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 macrophages were used. To make macrophages, THP-1 cells were differentiated by exposure to 200 ng/ml PMA (Sigma-Aldrich, St. Louis, MO, USA) for 12 h. The medium was replaced, and the cells were allowed to recover for 24 h. Then the cells were used for infection. All of these cells were purchased from the ATCC and cultured at 37°C in a 5% CO2 humidified atmosphere. Enterovirus 71 infection was carried out as described previously (15–17).
Plasmids and antibodies.
The plasmid expressing GSDMD was purchased from OriGene. The variants were constructed by site-directed mutagenesis using PFU DNA polymerase (Stratagene, La Jolla, CA). First, three plasmids encoding full-length GSDMD were constructed with different tags added, including the Flag tag at the N terminus, the Myc tag at the C terminus, or a Flag tag and Myc tag located at the N terminus and C terminus of GSDMD, respectively. Then, the deletion mutants (including fragments of aa 1 to 275, 276 to 484, 1 to 193, and 194 to 484) were constructed based on the full-length GSDMD plasmid. The deletion mutants (including fragments of aa 1 to 203, 1 to 213, 1 to 223, 1 to 233, 1 to 243, 1 to 253, and 1 to 263) and point mutants (including D234K, K235D, K236D, Q237D, R238D, T239D, F240D, Q241D, P242D, and P243D) of the fragment of aa 1 to 275 were constructed based on the plasmid containing the fragment of aa 1 to 275. All of these mutants were verified by nucleotide sequencing analysis.
Antibodies against Flag, Myc, GFP, and β-actin were purchased from Sigma (St. Louis, MO). Mouse anti-enterovirus 71 was purchase from Chemicon (Billerica, MA). Rabbit anti-GSDMD (G7422) was purchased from Sigma.
Cell cytotoxicity and viability assay.
Cell death and cell viability were analyzed by using a CytoTox 96 nonradioactive cytotoxicity assay kit (G1780) and a CellTiter-Glo luminescent cell viability assay (G7571) according to the manufacturer's instructions.
Real-time reverse transcription-PCR.
As for RD cells, at 24 h after transfection, cells were mock infected or infected with EV71 at a multiplicity of infection (MOI) of 1 PFU per cell. After 24 h, the medium was discarded, and total RNA was extracted from cells by using TRIzol reagent (Invitrogen, Carlsbad, CA). The concentration of samples was measured by a NanoDrop instrument (ND-1000). The samples were treated with DNase I (Pierce, Rockford, IL), and reverse transcription was carried out using a Superscript cDNA synthesis kit (Invitrogen) according to the manufacturer's instructions. cDNA samples were subjected to real-time PCR by using SYBR green. Results are reported as fold change using the ΔΔCT (where CT is threshold cycle) method.
Western blot analysis.
Cells were collected by centrifuging at 5,000 rpm for 5 min at 4°C and lysed in radioimmunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 25 mM Tris (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, and 1 mM EDTA with protease inhibitor cocktail (Roche, Indianapolis, IN). The lysates were centrifuged at 16,000 × g for 10 min at 4°C. Cell lysates were electrophoresed on SDS-PAGE gels and transferred to a nitrocellulose membrane (Pall, Port Washington, NY). The membranes were blocked with 5% nonfat dry milk and then probed with the primary antibodies indicated in the figures at 4°C overnight. The membranes were incubated with the corresponding IRDye Fluor 800-labeled IgG or IRDye Fluor 680-labeled IgG secondary antibody (Li-Cor, Inc., Lincoln, NE) and scanned by using an Odyssey infrared imaging system at a wavelength of 700 to 800 nm and analyzed with Odyssey software.
Immunoprecipitation.
Transfected cells were lysed with RIPA buffer, and then lysates were incubated with anti-Flag antibody (Sigma, St. Louis, MO) in 500 μl of RIPA buffer at 4°C overnight on a rotator in the presence of protein A/G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). The next day, immunocomplexes captured on the beads were washed and analyzed by Western blotting.
Plaque assay.
RD cells (2 × 105 per well) were seeded in 24-well plates with growth medium (DMEM–10% FBS). The next day, growth medium was removed, and 200 μl of serial dilutions of EV71 stocks was added to wells (virus stock was serially diluted by 1:10). Then the 24-well plates were incubated at 37°C for 2 h. One milliliter per well of 1.2% Avicel (R-591; FMC) (one volume of 2.4% Avicel with the same volume of 2× DMEM) overlay medium was added to the plate (42). After 72 h, cells were fixed and stained with crystal violet.
Statistics.
A two-tailed Student's t test was used for two-group comparisons. P values of <0.05, <0.01, and <0.001 were considered significant.
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (81672032, 81225014, and 31270200), the Program for Changjiang Scholars and Innovative Research Team in Universities (IRT13007), the Changjiang Scholars Program (J.W.), CAMS Innovation Fund for Medical Sciences (2016-I2M-1-014), and the National Institute of Allergy and Infectious Diseases of the United States (AI112755).
X.L., B.H., and J.W. designed the research; X.L., Z.Z., X.X., and J.Q. performed the experiments; X.L., Z.Z., B.H., and J.W. analyzed data; X.L., B.H., and J.W. wrote the manuscript. All authors reviewed the manuscript.
REFERENCES
- 1.Xing W, Liao Q, Viboud C, Zhang J, Sun J, Wu JT, Chang Z, Liu F, Fang VJ, Zheng Y, Cowling BJ, Varma JK, Farrar JJ, Leung GM, Yu H. 2014. Hand, foot, and mouth disease in China, 2008–12: an epidemiological study. Lancet Infect Dis 14:308–318. doi: 10.1016/S1473-3099(13)70342-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang CC, Liu CC, Chang YC, Chen CY, Wang ST, Yeh TF. 1999. Neurologic complications in children with enterovirus 71 infection. N Engl J Med 341:936–942. doi: 10.1056/NEJM199909233411302. [DOI] [PubMed] [Google Scholar]
- 3.Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH. 2010. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10:778–790. doi: 10.1016/S1473-3099(10)70194-8. [DOI] [PubMed] [Google Scholar]
- 4.Chan LG, Parashar UD, Lye MS, Ong FG, Zaki SR, Alexander JP, Ho KK, Han LL, Pallansch MA, Suleiman AB, Jegathesan M, Anderson LJ. 2000. Deaths of children during an outbreak of hand, foot, and mouth disease in Sarawak, Malaysia: clinical and pathological characteristics of the disease. For the Outbreak Study Group. Clin Infect Dis 31:678–683. doi: 10.1086/314032. [DOI] [PubMed] [Google Scholar]
- 5.Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shih SR. 1999. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med 341:929–935. [DOI] [PubMed] [Google Scholar]
- 6.McMinn P, Stratov I, Nagarajan L, Davis S. 2001. Neurological manifestations of enterovirus 71 infection in children during an outbreak of hand, foot, and mouth disease in Western Australia. Clin Infect Dis 32:236–242. doi: 10.1086/318454. [DOI] [PubMed] [Google Scholar]
- 7.McMinn PC. 2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev 26:91–107. doi: 10.1111/j.1574-6976.2002.tb00601.x. [DOI] [PubMed] [Google Scholar]
- 8.Kiener TK, Jia Q, Lim XF, He F, Meng T, Chow VT, Kwang J. 2012. Characterization and specificity of the linear epitope of the enterovirus 71 VP2 protein. Virol J 9:55. doi: 10.1186/1743-422X-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He ML. 2012. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J Virol 86:3767–3776. doi: 10.1128/JVI.06687-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z. 2013. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9:e1003231. doi: 10.1371/journal.ppat.1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li ML, Hsu TA, Chen TC, Chang SC, Lee JC, Chen CC, Stollar V, Shih SR. 2002. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 293:386–395. doi: 10.1006/viro.2001.1310. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Yao Y, Chen Y, Xu X, Lin Y, Yang Z, Qiao W, Tan J. 2017. Enterovirus 71 3C promotes apoptosis through cleavage of PinX1, a telomere binding protein. J Virol 91:e02016-16. doi: 10.1128/JVI.02016-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP, Hsu JT. 2011. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis 203:1784–1790. doi: 10.1093/infdis/jir174. [DOI] [PubMed] [Google Scholar]
- 14.Lei X, Han N, Xiao X, Jin Q, He B, Wang J. 2014. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J Virol 88:9830–9841. doi: 10.1128/JVI.01425-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lei X, Liu X, Ma Y, Sun Z, Yang Y, Jin Q, He B, Wang J. 2010. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84:8051–8061. doi: 10.1128/JVI.02491-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. 2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol 85:8811–8818. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J. 2013. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol 87:1690–1698. doi: 10.1128/JVI.01855-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Lei X, Xiao X, Yang C, Lu W, Huang Z, Leng Q, Jin Q, He B, Meng G, Wang J. 2015. Reciprocal regulation between enterovirus 71 and the NLRP3 inflammasome. Cell Rep 12:42–48. doi: 10.1016/j.celrep.2015.05.047. [DOI] [PubMed] [Google Scholar]
- 19.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH. 2014. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fink SL, Cookson BT. 2007. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol 9:2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x. [DOI] [PubMed] [Google Scholar]
- 21.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–192. doi: 10.1038/nature13820. [DOI] [PubMed] [Google Scholar]
- 23.Jorgensen I, Rayamajhi M, Miao EA. 2017. Programmed cell death as a defence against infection. Nat Rev Immunol 17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi J, Gao W, Shao F. 2017. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42:245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 26.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 27.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. 2015. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Muller DJ, Broz P, Hiller S. 2016. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC. 2016. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, Han J. 2016. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26:1007–1020. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen I, Zhang Y, Krantz BA, Miao EA. 2016. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med 213:2113–2128. doi: 10.1084/jem.20151613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. 2010. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei X, Xiao X, Wang J. 2016. Innate immunity evasion by enteroviruses: insights into virus-host interaction. Viruses 8:E22. doi: 10.3390/v8010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weng KF, Li ML, Hung CT, Shih SR. 2009. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog 5:e1000593. doi: 10.1371/journal.ppat.1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Song Z, Qin B, Zhang X, Chen L, Hu Y, Yuan Z. 2013. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antiviral Res 97:264–269. doi: 10.1016/j.antiviral.2012.12.029. [DOI] [PubMed] [Google Scholar]
- 38.Zhang XN, Song ZG, Jiang T, Shi BS, Hu YW, Yuan ZH. 2010. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection. World J Gastroenterol 16:201–209. doi: 10.3748/wjg.v16.i2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, Wang J, Wang M, Jin Q, Cui S. 2011. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol 85:10021–10030. doi: 10.1128/JVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaidt MM, Hornung V. 2016. Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J 35:2167–2169. doi: 10.15252/embj.201695415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aglietti RA, Dueber EC. 2017. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol 38:261–271. doi: 10.1016/j.it.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Yin Y, Xu Y, Ou Z, Su L, Xia H. 2015. A simple and highly repeatable viral plaque assay for enterovirus 71. J Basic Microbiol 55:538–541. doi: 10.1002/jobm.201400330. [DOI] [PubMed] [Google Scholar]