

ABSTRACT
Hepatitis C virus (HCV) is a single-stranded positive-sense RNA hepatotropic virus. Despite cellular defenses, HCV is able to replicate in hepatocytes and to establish a chronic infection that could lead to severe complications and hepatocellular carcinoma. An important player in subverting the host response to HCV infection is the viral nonstructural protein NS5A, which, in addition to its role in replication and assembly, targets several pathways involved in the cellular response to viral infection. Several unbiased screens identified nucleosome assembly protein 1-like 1 (NAP1L1) as an interaction partner of HCV NS5A. Here we confirmed this interaction and mapped it to the C terminus of NS5A of both genotype 1 and 2. NS5A sequesters NAP1L1 in the cytoplasm, blocking its nuclear translocation. However, only NS5A from genotype 2 HCV, and not that from genotype 1, targets NAP1L1 for proteosome-mediated degradation. NAP1L1 is a nuclear chaperone involved in chromatin remodeling, and we demonstrated the NAP1L1-dependent regulation of specific pathways involved in cellular responses to viral infection and cell survival. Among those, we showed that lack of NAP1L1 leads to a decrease of RELA protein levels and a strong defect of IRF3 TBK1/IKKε-mediated phosphorylation, leading to inefficient RIG-I and Toll-like receptor 3 (TLR3) responses. Hence, HCV is able to modulate the host cell environment by targeting NAP1L1 through NS5A.
IMPORTANCE Viruses have evolved to replicate and to overcome antiviral countermeasures of the infected cell. Hepatitis C virus is capable of establishing a lifelong chronic infection in the liver, which could develop into cirrhosis and cancer. Chronic viruses are particularly able to interfere with the cellular antiviral pathways by several different mechanisms. In this study, we identified a novel cellular target of the viral nonstructural protein NS5A and demonstrated its role in antiviral signaling. This factor, called nucleosome assembly protein 1-like 1 (NAP1L1), is a nuclear chaperone involved in the remodeling of chromatin during transcription. When it is depleted, specific signaling pathways leading to antiviral effectors are affected. Therefore, we provide evidence for both a novel strategy of virus evasion from cellular immunity and a novel role for a cellular protein, which has not been described to date.
KEYWORDS: HCV, NAP1L1, innate immunity, IRF3, NF-κB, RIG-I, TLR3, NF-κB, hepatitis C virus
INTRODUCTION
Hepatitis C virus (HCV) is a member of the Flaviviridae family, genus Hepacivirus, with a single-stranded positive-polarity RNA genome of approximately 9.6 kb (1). Seven viral genotypes (1 to 7) with important differences in geographical distribution, pathogenesis, and response to treatment have been identified (2). The HCV life cycle is entirely cytoplasmic and involves entry, uncoating, and translation of the viral RNA to a polyprotein that is processed by proteolysis into 3 structural (core, E1, and E2) and 7 nonstructural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins. Viral RNA translation and replication and subsequent steps of particle assembly and release takes place associated with remodeled intracellular membranes and lipid droplets (LD) (3–5). Nonstructural proteins are required for RNA replication, with NS3-NS4A being the helicase/proteinase and NS5B being the viral RNA-dependent RNA polymerase. In addition, HCV nonstructural proteins have also been implicated in perturbing cell signaling and mediating immune evasion. Among them, NS5A has been implicated in subverting several cellular pathways and as a candidate viral oncogene.
NS5A is a 447-amino-acid (aa) (genotype 2a [gt2a]) phosphoprotein that is found associated with endoplasmic reticulum (ER) membranes through an N-terminal amphipathic helix (aa 1 to 27). The rest of the polypeptide is hydrophilic and consists of the large amino-terminal domain I followed by two smaller, more variable domains, II and III. Domains I and II are essential for viral genome replication, while domain III is dispensable for genome replication but is required for viral particle assembly via interaction with core (6–8). A cluster of phosphorylated serine residues at positions 452, 454, and 457 (strain JFH1, gt2a) at the very C-terminal end of NS5A is responsible for this interaction. In addition to a direct role in HCV genome replication and assembly, NS5A makes an important contribution to modulating the host cell environment. NS5A is a promiscuous protein and interacts with several host factors, thus affecting different signaling pathways that control the cell cycle, apoptosis, and the interferon (IFN) response to viral infection (reviewed in references 9 and 10).
Chronic infection with HCV is a major risk factor for the development of hepatocellular carcinoma (HCC) (11). The leading hypothesis is that malignant transformation of hepatocytes occurs through increased liver cell turnover induced by chronic liver injury and regeneration, in a context of inflammation and oxidative DNA damage. However, increasing experimental evidence suggests that HCV might also contribute to malignant transformation of hepatocytes through the direct action of viral proteins on cellular transformation pathways (12). Liver-specific expression in transgenic mice of the full viral polyprotein (core to NS5B) at low levels, comparable to those found in patients, has been shown to induce HCC without inflammation (13). Mice transgenic for NS5A alone may also develop liver cancer, depending on the genetic background of the mice (14, 15). Furthermore, expression of NS5A in NIH 3T3 fibroblasts promoted anchorage-independent growth and tumor formation in nude mice (16, 17). These data support a direct role of HCV proteins, and NS5A in particular, in the development of HCC.
In a recent attempt to identify host factors that associate with a number of innate immune-modulating viral proteins, Pichlmair et al. screened novel proteins that interact with HCV NS5A (18). Careful inspection of the data led to the unexpected observation that NS5A interacted with the human nucleosome assembly protein 1-like 1 (NAP1L1 or hNAP1). The interaction of NS5A with NAP1L1, or with the highly homologous NAP1L4, was independently identified in at least another three independent reports (19–21).
NAP1L1 was originally identified in HeLa cells as the human homolog of the yeast nucleosome assembly protein 1 (NAP-1) (22). NAP1L1 is a 391-aa polypeptide characterized by nuclear import/export sequences and histone and protein binding domains (23). NAP1L1 is a chaperone and nucleo-cytoplasmic shuttling factor that facilitates the delivery and incorporation of two histone H2A-H2B dimers to complete the nucleosome (reviewed in references 24, 25, and 26). NAP1L1 is involved in the regulation of transcription, cell cycle progression, incorporation and exchange of histone variants, and promotion of nucleosome sliding. In addition, NAP1L1 has been shown to interact with several host and viral factors, including the coactivator p300 and E2 of papillomaviruses (27–29). NAP1L1 interacts with the human immunodeficiency virus type 1 (HIV-1) Tat transactivator and enhances HIV-1 trans-activation (30, 31). NAP1L1 family proteins are localized in the cytoplasm, but inhibition of nuclear export results in their accumulation in the nuclei, indicating a shuttling activity that has also been implicated in the delivery of histones to the nucleus as part of their chaperoning activity. NAP1L1 is involved in the process of nucleosome depletion during embryonic stem cell differentiation (32). Knockdown of NAP1L1 enhances the differentiation of induced pluripotent stem cells (iPSC) into functional cardiomyocytes (33). NAP1L1 expression has also been shown to be elevated in several cancers (34–36).
In this work, we confirmed the interaction of NS5A with NAP1L1 and its colocalization in the cytoplasm of cells replicating HCV. The interaction could be mapped to the carboxy terminus of domain III of NS5A, which is shared among all HCV genotypes. However, only NS5A from genotype 2, and not that from genotype 1, targets NAP1L1 for proteosome-mediated degradation. Transcriptome RNA sequencing (RNA-seq) of NAP1L1-depleted cells shows dysregulation of a number of genes from pathways of innate immunity and cell survival. Among those, we show that depletion of NAP1L1 leads to the downmodulation of NF-κB and to a strong downregulation of IRF3 phosphorylation mediated by the kinase TBK1/IKKε. Both the RIG-I and Toll-like receptor 3 (TLR3) pathways were affected by NAP1L1 depletion. We conclude that HCV is able to modulate the host cell environment by targeting NAP1L1 through NS5A. We believe that this may represent a novel strategy deployed by HCV to evade cellular antiviral responses and possibly to maintain a chronic infection, thus contributing to the development of HCC.
RESULTS
HCV NS5A binds NAP1L1.
In independent reports, the nonstructural protein 5A (NS5A) from human hepatitis C virus (HCV) has repeatedly been found to interact with the nucleosome assembly protein NAP1L1 (18–21). However, such studies did not map the interaction, and additional functional studies were not conducted. To confirm the interaction, we generated a full-length FLAG-tagged NS5A (f-NS5A) (N terminus) derived from JFH1 genotype 2a (gt2a). Cotransfection of f-NS5A with hemagglutinin (HA)-NAP1L1 in HEK 293T cells followed by anti-FLAG affinity chromatography resulted in the detection of HA-NAP1L1 (Fig. 1A). As a positive control for the interaction, we used the human immunodeficiency virus Tat protein, which has been previously described to bind NAP1L1 (30, 31). To study the interaction of NS5A with NAP1L1 at the endogenous levels, we took advantage of the JFH1 subgenomic replicon (SGR-JFH1/Luc), which efficiently replicates in hepatocytes. As shown in Fig. 1B, the interaction was preserved, albeit with a very low efficiency of immunoprecipitation (IP), which could be explained by an effect of NS5A on the stability of the protein (see below). Furthermore, extensive colocalization of the two proteins was observed in the cytoplasm (Fig. 1C). Interestingly, the extent of the colocalization increased from approximately 10% of NS5A-expressing cells at 48 h postelectroporation (hpe) to more than 70% at 72 hpe (not shown). We repeated the experiment with a subgenomic replicon that also expresses HCV core (Luc-JFH1ΔE1/E2) (37). In this case, NS5A drives the localization of NAP1L1 to subcellular locations reminiscent of lipid droplets (Fig. 1C, lower panels). However, the interaction between NS5A and core was not required for the NS5A to bind NAP1L1, as demonstrated in the experiments conducted in the absence of core (Fig. 1A, B, and C [upper panels]).
FIG 1.

HCV NS5A binds NAP1L1. (A) Coimmunoprecipitation of FLAG-tagged NS5A and HA-NAP1L1 in HEK 293T cells. Transfected cells were lysed, coimmunoprecipitated with anti-FLAG–agarose beads, and blotted against anti-FLAG or anti-HA antibodies as indicated. A plasmid encoding HIV-1 FLAG-tagged Tat was used as positive control. IgL, immunoglobulin light chain; IgH, immunoglobulin heavy chain. (B) NS5A interacts with endogenous NAP1L1 during HCV replication. Huh7-Lunet cells were electroporated with subgenomic SGR-JFH1/Luc or mock transfected. At 72 hpe, cell lysates were incubated with anti-NAP1L1 antibodies or with matching irrelevant IgGs. Input and co-IP samples were then immunoblotted with anti-NAP1L1 and anti-NS5A antibodies as indicated. (C) NS5A and NAP1L1 colocalize during HCV replication. Huh7-Lunet cells were either mock electroporated or treated with the subgenomic HCV replicon SGR-JFH1/Luc or with SGR-FK-Luc-JFH1/ΔE1-E2 and fixed at 72 hpe. Indirect immunofluorescence analysis was performed with anti-NAP1L1 (green) and anti-NS5A (red) antibodies and corresponding fluorescent secondary antibodies (scale bars, 10 μm). Colocalization is shown in the merge channel (Pearson's correlation coefficients of 0.658 for SGR-JFH1/Luc and 0.731 for SGR-FK-Luc-JFH1/ΔE1-E2). The inset shows a high-magnification image. Diffused, cytoplasmic localization of NAP1L1 in mock-treated cells is also shown for comparison (top panels).
HCV NS5A from gt2 mediates NAP1L1 proteosomal degradation.
In order to investigate whether the interaction with NAP1L1 was shared by NS5A from other HCV genotypes, we performed an IP with genotype 1 (gt1) Con1- and H77-derived proteins. Surprisingly, NS5A from gt1 isolates was able to immunoprecipitate higher levels of NAP1L1 than JFH1 NS5A (Fig. 2A). Furthermore, cotransfection of HA-NAP1L1 with increasing amounts of JFH1 NS5A (gt2) resulted in a decrease of NAP1L1 levels, while Con1 NS5A (gt1) did not (Fig. 2B). Hence, NS5A from JFH1 (gt2), but not those from Con1 or H77 (gt1), resulted in the degradation of NAP1L1. Treatment with the inhibitor MG132 partially rescued the JFH1 NS5A degradation of NAP1L1, indicating proteasome involvement (Fig. 2C). Finally, cycloheximide treatment of SGR-JFH1/Luc-transfected cells showed a progressive decrease of endogenous NAP1L1, although with less efficiency than with transfection of NS5A alone (Fig. 2B), possibly related to the different levels of NS5A expression under the two experimental conditions. Degradation of NS5A could be rescued by MG132, further highlighting the targeting of NAP1L1 for proteosome-mediated degradation also at physiological levels during HCV replication (Fig. 2D). The major difference between JFH1-derived NS5A and those of the other HCV genotypes resides in a 20-aa insertion at the carboxy-terminal end of the protein (Fig. 2E), which also accounts for the differences in molecular weight observed (Fig. 2A).
FIG 2.

HCV JFH1 NS5A targets NAP1L1 for proteasome-mediated degradation. (A) HCV NS5A proteins from Con1 and H77 interact with NAP1L1. HEK 293T cells were transfected with FLAG-tagged NS5A from the indicated HCV genotypes. Transfected cells were lysed, coimmunoprecipitated with anti-FLAG–agarose beads, and blotted against anti-FLAG or anti-HA antibodies. (B) HCV NS5A from JFH1 degrades NAP1L1. 293T cells were transfected with an equal amount of HA-NAP1L1 (5 μg) and increasing amounts of FLAG-tagged NS5A from JFH1 or from Con1 (0, 2.5, 5, and 10 μg). After 24 h, cell lysates were immunoblotted with anti-FLAG, anti-HA, and antiactin antibodies. (C) HCV NS5A-mediated degradation is proteasome dependent. HEK 293T cells were cotransfected with HA-NAP1L1 and FLAG-NS5A, treated with the MG132 (5 μM) for 14 h, and lysed. Samples were run and immunoblotted with anti-HA, anti-FLAG, and antiactin antibodies. (D) HCV replication induces NAP1L1 degradation. Huh7-Lunet cells were electroporated with SGR-JFH1/Luc RNA or mock transfected. Cells were treated with 200 μM cycloheximide (CHX) for 1 and 3 h and lysed. Samples were run and immunoblotted with anti-HA, anti-FLAG, and antiactin antibodies. (E) CLUSTAL O (1.2.4) multiple-sequence alignment of NS5A proteins from different HCV genotypes. The asterisks indicates all the sequences that share the same amino acid. Gaps are indicated by dashes. The cluster of serines implicated in NS5A binding is boxed.
We conclude that NAP1L1 binding is shared among NS5A derived from g1 and gt2 genotypes but that gt2 NS5A has the additional feature of being able to target NAP1L1 for proteosome-mediated degradation.
The carboxy terminus of NS5A is required for NAP1L1 binding.
In order to map the interaction, NS5A from JFH1 was divided in three domains, domain I (D1) (corresponding to nucleotides [nt] 2003 to 2189), domain II (D2) (nt 2226 to 2314), and domain III (D3) (nt. 2328 to 2442), all linked in various combinations to the N-terminal amphipathic helix for membrane tethering and FLAG tagged at the N terminus (Fig. 3A). As shown in Fig. 3B, only constructs maintaining D3 could interact with NAP1L1. Therefore, NS5A was further truncated from the C terminus to generate proteins with progressive deletions. With this approach, we found that deletions starting from the acidic motif 458-EEDD-461 (AH-D3.5 in Fig. 3C) completely abolished the interaction with NAP1L1. Since the acidic motif is a perfect consensus for casein kinase 2-mediated serine phosphorylation (S-D/E-X-E/D), we reasoned that NAP1L1 interacts with the C-terminal cluster of serines already implicated in the interaction with HCV core (6–8). Therefore, we tested both the delB deletion mutant with deletion of aa 2419 to 2433 (residues are numbered according to the positions within the original JFH1 polyprotein corresponding to aa 443 to 457 of NS5A) (7) and the triple mutant of cluster 3B (CL3B/SA or m2) (corresponding to S2428/2430/2433A, aa S452/454/457A of NS5A) (6). Co-IP analysis showed loss of interaction for both mutants (Fig. 3D), further suggesting that these residues are crucial for NAP1L1 interaction. To further demonstrate that the colocalization requires the interaction of NS5A with NAP1L1 through the C-terminal serine-rich region, we exploited two subgenomic constructs, SGR-delB, and SGR-m2 (6, 7). NS5A from both constructs localized in clusters in the cytoplasm, like the wild-type replicon, but did not colocalize with NAP1L1, which remained diffused in the cytoplasm (Fig. 3E). We conclude that NAP1L1 binds to the same cluster of serine residues at the carboxy terminus of NS5A as the HCV core protein.
FIG 3.

NAP1L1 interacts with the extreme carboxy terminus of NS5A. (A) Diagram of NS5A and mutants produced in this work. The full length of JFH1 NS5A is shown with the FLAG tag at the N terminus, the amphipathic helix (AH) for membrane tethering, and domains I, II, and III with the low-complexity sequences (LCS) I and II. AH-fused domain III (AH-D3-FL) is deleted from the C terminus in 6 fragments (AH-D3.1 to AH-D3.6). The amino acid sequence of the C-terminal region of NS5A encompassing S452/454/457A (serines are in bold) is also shown, together with the sequences of the deletion mutant NS5A-delB and the triple S→A mutant NS5A-m2. (B) NAP1L1 binds domain III of NS5A. HEK 293T cells were transfected with the indicated domains fused to AH together with HA-NAP1L1. Transfected cells were lysed, coimmunoprecipitated with anti-FLAG–agarose beads, and blotted against anti-FLAG or anti-HA antibodies. Asterisks indicate the positions of the NS5A mutants that are distinguishable from the heavy (IgH) and light (IgL) immunoglobulin chains. (C) NAP1L1 binds the extreme C terminus of NS5A. HEK 293T cells were transfected with expression plasmids encoding NS5A domain III fused to AH carrying progressive deletions from the C terminus as indicated in panel A. Co-IP was conducted as for panel B. (D) NS5A mutants delB and m2 do not bind NAP1L1. HEK 293T cells were transfected with the expression plasmids for NS5A mutants delB and m2 described in panel A. Co-IP was conducted as for panel B. (E) NS5A mutants delB and m2 lose colocalization with NS5A. Huh7-Lunet cells were electroporated with SGR-JFH1/Luc and with mutants SGR-JFH1/Luc_m2 and SGR-JFH1/Luc_delB and fixed at 72 hpe. Indirect immunofluorescence analysis was performed with anti-NAP1L1 (green) and anti-NS5A (red) antibodies and corresponding fluorescent secondary antibodies (scale bars, 10 μm). Colocalization is shown in the merge channel.
NAP1L1 is not required for HCV replication and infectivity in Huh7 cells.
In order to assess the potential role of NAP1L1 in HCV replication, we overexpressed the enhanced yellow fluorescent protein (EYFP)-tagged version of NAP1L1 in Huh7-Lunet cells by lentiviral vector (LV) transduction. NAP1L1-EYFP had the expected cytoplasmic localization and colocalized with NS5A when cells were transfected with SGR-JFH1 RNA (Fig. 4A). However, there was no difference between cells overexpressing NAP1L1-EYFP or EYFP alone in the levels of luciferase from SGR-JFH/Luc, which is a measure of HCV genome replication (Fig. 4B). Next, we efficiently depleted NAP1L1 by using lentiviral vectors delivering a specific short hairpin RNA (shRNA) (Fig. 4C). Again, Huh7-Lunet cells replicated SGR-JFH/Luc under conditions of NAP1L1 depletion as well as under mock conditions (Fig. 4D). To further confirm these results, we introduced full-length genomic HCV JFH1 RNA into Huh7-Lunet cells which had been treated with shRNA as described above. Cell culture supernatants at the indicated time points were used to infect naive Huh7.5 cells to measure infectivity. As shown in Fig. 4E, HCV infectivity also was not affected by NAP1L1 depletion in Huh7-Lunet cells. Finally, we investigated the replication of HCV SGR-JFH/Luc carrying the m2 mutation of NS5A. As shown in Fig. 4F, wild-type and m2 replicons replicated equally well in Huh7-Lunet cells compared to the nonreplicative control mutated in the polymerase NS5B GND.
FIG 4.

HCV does not require NAP1L1 for replication and infectivity. (A) NS5A and NAP1L1-EYFP colocalize during HCV replication. Huh7-Lunet cells were transduced with a lentiviral vector (LV) expressing NAP1L1-EYFP and then either mock electroporated or treated with the subgenomic HCV replicon SGR-JFH1/Luc and fixed at 72 hpe. Indirect immunofluorescence analysis was performed with anti-NS5A antibody (red) (scale bars, 10 μm). (B) NAP1L1 overexpression does not affect HCV genome replication. Huh7-Lunet cells were transduced with a lentiviral vector (LV) expressing EYFP or NAP1L1-EYFP at an efficiency of >85% on average as measured by cytofluorimetric analysis. Cells were then electroporated with the HCV SGR JFH1/Luc RNA, and luciferase was monitored at the indicated time points. Values are normalized to the luciferase signal at 4 h postelectroporation. Averages for 3 independent replicates are shown with standard deviations. (C) Depletion of NAP1L1 by shRNA. Huh7-Lunet cells were transduced with an LV expressing shRNA targeting NAP1L1 (shNAP1L1) or a nontargeting control (shCTRL). After selection with puromycin, cells were electroporated with HCV SGR JFH1/Luc RNA, and protein levels were detected by WB. (D) Depletion of NAP1L1 does not affect HCV genome replication. Huh7-Lunet cells were treated as for panel C, and the luciferase signal was measured as for panel B. (E) Depletion of NAP1L1 does not affect HCV infectivity. Huh7-Lunet cells treated with shRNAs as for panel C were electroporated with full-length HCV JFH1 RNA. At the indicated time points, the supernatant was collected and used to infect naive Huh7.5 cells at various dilutions. The 50% tissue culture infective dose (TCID50) was then calculated by counting cells stained with the NS5A antiserum. (F) Mutagenesis of the NAP1L1 binding site of NS5A does not affect HCV genome replication. Huh7-Lunet cells were electroporated with HCV SGR JFH1/Luc RNA from the wild type (wt), the m2 mutant of NS5A, or the GND mutant of NS5B, which is replication defective. Luciferase was measured as for panel B.
We took the inverse approach to investigate the effect of NS5A on NAP1L1 activity. Under physiological conditions in cell culture, NAP1L1 is found predominantly in the cytoplasm (Fig. 1C). However, incubation of cells with the nuclear export inhibitor leptomycin B (LMB) results in the accumulation of NAP1L1 in the nucleus (Fig. 5A) (31). Therefore, we transfected cells with SGR-JFH1 and its mutant m2 and monitored the localization of endogenous NAP1L1 in the nucleus. As shown in Fig. 5A and B, SGR-driven NS5A, but not the m2 mutant, significantly inhibited nuclear localization of NAP1L1. To further analyze this phenotype in the context of HCV gt1, we also engineered the corresponding m2 mutations in Con1-derived NS5A (see Fig. 2E for an alignment of the carboxy terminus of NS5A from the different genotypes). As shown in Fig. 5C, Con1-derived NS5A m2 lost the ability to interact with NAP1L1, as expected. Interestingly, both JFH1- and Con1-derived NS5A equally inhibited nuclear translocation of NAP1L1 (Fig. 5D and E).
FIG 5.

The interaction with HCV NS5A inhibits NAP1L1 nuclear translocation. (A) SGR-JFH1 NS5A inhibits NAP1L1 nuclear localization. Huh7-Lunet cells were electroporated with HCV SGR-JFH1/Luc RNA, the mutant replicon SGR-JFH1/m2, or control SGR-JFH1/GND as indicated. At 63 hpe, cells were treated with 150 nM LMB for 9 h. Cells were then fixed and stained for NS5A and NAP1L1. Scale bars 10 μm. (B) Quantification of SGR-JFH1 NS5A inhibition of NAP1L1 nuclear localization. Three hundred cells treated as for panel A were visually scored for NAP1L1 nuclear localization in the presence of NS5A. The investigator was blinded to the group allocation during visual counting. Averages for 3 independent replicates are shown with standard deviations. (C) Con1 NS5A mutant m2 does not bind NAP1L1. HEK 293T cells were transfected with expression plasmids for Con1 NS5A and m2. Co-IP was conducted as for panel B. (D) Both JFH1- and Con1-derived NS5A proteins inhibit nuclear translocation of NAP1L1. Huh7-Lunet cells were transfected with expression plasmids for NS5A from JFH1 or Con1 and their respective m2 mutants. Cells were treated with LMB as described above, fixed, and stained for ectopic FLAG-tagged NS5A and endogenous NAP1L1. Scale bars, 10 μm. (E) Quantification of NS5A inhibition of NAP1L1 nuclear localization. Cells treated as for panel C were visually scored as described for panel B.
We conclude that NAP1L1 is not directly involved in HCV replication and infectivity but that NS5A from both genotypes affects NAP1L1 nuclear localization, possibly by sequestering NAP1L1 in the cytoplasm. Hence, we set to investigate the nuclear activity of NAP1L1.
Transcriptome analysis of NAP1L1-depleted cells.
NAP1L1 is a nucleosome chaperone involved in several nuclear processes, including transcription. Genome-wide analysis of yNAP1-deleted Saccharomyces cerevisiae showed that about 10% of all yeast open reading frames changed the transcription levels more than 2-fold (38). To investigate the transcriptome of hepatocytes, we depleted NAP1L1 with shNAP1L1 (Fig. 4C). Differential analysis of the shNAP1L1 transcriptome versus cells transduced with the control shRNA (shCTRL) showed significant upregulation of 144 genes (fold change of >2) and downregulation of 358 genes (fold change of <−2), with a false-discovery rate of less than 0.05 (data are derived from the most stringent DESEQ2 statistical analysis in Table S1, which also shows the EDGR statistical analysis of the same data for comparison). These numbers correspond to approximately 1% of the total reads in the analysis (46,623 reads), suggesting a good degree of specificity for the genes regulated by NAP1L1. To validate the sequencing data, we retested a number of modulated genes by reverse transcription-PCR (RT-PCR) (Fig. 6A). Ingenuity pathway analysis of downregulated genes indicated that top canonical pathways involved in cancer and signaling were mostly affected (Table S1). Interestingly, we also noticed downmodulation of interferon-stimulated genes (ISGs) such as IFITM3, GBP2, and UBD, as well as genes involved in interferon (IFN) transcriptional activation such as RELA (p65 subunit of NF-κB), c-Jun, and GEF2 (39, 40). To investigate whether the expression of the NAP1L1 target genes is reduced in HCV-infected cells we conducted a meta-analysis of published data obtained in a similar setting (41) (see Table S2 in the supplemental material). We found an overlap of 40 genes between the two analysis, with 5 upregulated genes, 27 downregulated genes, and 8 genes showing opposed regulation. Interestingly, among the overlapping downregulated genes we found again genes implicated in innate immunity regulation, such as RELB, c-Jun, and GEF2 (see Table S2). We also compared Huh7 cells stably replicating SGR JFH-1 with the homologous cured cells for the expression of some NAP1L1-regulated genes (not shown). We found that two significantly downregulated genes in the context of NAP1L1 depletion, GEF2 and IFTM1, were also downregulated under conditions that favor HCV replication. However, other genes were not affected (CFL2 and HEPACAM2) or showed opposite regulation (UBD).
FIG 6.
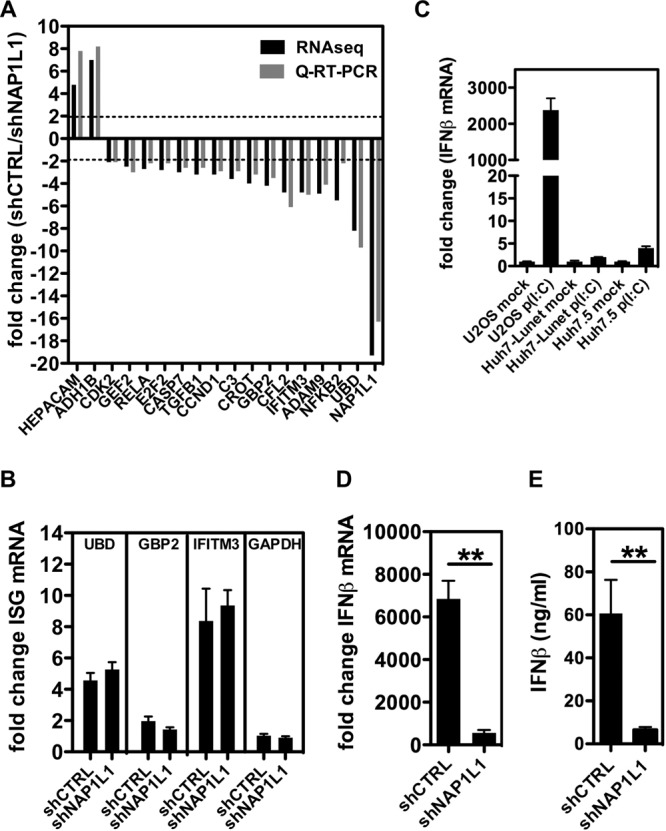
NAP1L1 is involved in the innate immune response. (A) Whole-genome transcriptome analysis in NAP1L1-depleted cells. Huh7-Lunet cells were treated with shNAP1L1/shCTRL, followed by RNA-seq analysis. Black bars show the levels of 15 downregulated genes (fold change, <−2) and 2 upregulated genes (fold change, >2), which were further validated by qRT-PCR (gray bars) normalized for β-actin. (B) Induction of ISGs is not NAP1L1 dependent. Huh7-Lunet cells were treated with 1000 U/ml of IFN-α for 8 h. UBD, GBP2, IFITM3, and GAPDH mRNAs were measured by qRT-PCR, normalized for β-actin in triplicate independent experiments. Shown are fold changes over basal, noninduced levels ± SD. (C) IFN induction following poly(I·C) transfection in different cell lines. U2OS, Huh7-Lunet, and Huh7.5 cells were transfected with 1 μg poly(I·C) for 8 h. IFN-β mRNA levels were measured by qRT-PCR, normalized for β-actin, and plotted against those for mock-treated cells (Lipofectamine). Average for 3 independent replicates are shown with standard deviations. (D) NAP1L1 depletion affects the induction of IFN-β mRNA by poly(I·C). U2OS cells treated with shNAP1L1/shCTRL for 3 days were transfected with 1 μg poly(I·C) for 8 h. IFN-β mRNA levels were measured as described above. (E) NAP1L1 depletion affects the induction of IFN-β by poly(I·C). U2OS cells were treated as for panel C. Secreted IFN-β protein was measured by a commercial enzyme-linked immunosorbent assay (ELISA) in triplicate and quantified against a standard curve, and results were plotted.
In order to explore the impact of NAP1L1 depletion on the interferon response pathway, we explored IFN-α-mediated induction of IFITM3, GBP2, and UBD (as well as IFIT1, IFIT3, OASL, interleukin-8 [IL-8], and CXCL11 [not shown]). We found that these genes were not impaired in their IFN-dependent induction by NAP1L1 depletion, ruling out a specific role for NAP1L1 in ISG transcriptional activation (Fig. 6B). Finally, in order to assess the effect of NAP1L1 depletion on the induction of IFN-β, we needed a different cell line than Huh7-derived cells. To this end, we used U2OS cells, which maintain an intact IFN signaling pathway following poly(I·C) transfection, compared to cells more permissive to HCV such as Huh7-Lunet or Huh7.5 (Fig. 6C) (42, 43). However, as shown in Fig. 6D and E, the IFN-β response to poly(I·C) transfection in U2OS cells was completely obliterated in the absence of NAP1L1, at both the mRNA and protein levels. Similar results were also obtained in the context of vesicular stomatitis virus infection (not shown). These data indicate that NAP1L1 is involved in regulating pathways that lead to interferon induction.
Involvement of NAP1L1 in the induction of IFN.
Pattern recognition receptors (PRRs) such as RIG-I/MDA5 and TLR3 respond to viral RNA agonists as well as to poly(I·C) by activating transcription factors such as NF-κB and IRF3. We observed previously that the NF-κB subunit RELA mRNA levels were reduced in the context of NAP1L1 depletion (Fig. 6A), and we confirmed this also at the protein level (Fig. 7A and B). Following activation, RELA translocates into the nucleus as a phosphorylated protein. As shown in Fig. 7A and C, phosphorylation of RELA occurs 1 to 4 h after poly(I·C) induction. Under conditions of NAP1L1 depletion, albeit under conditions of reduced RELA protein content, phosphorylation occurs normally up to 4 h posttreatment, when a slight decrease was consistently observed (Fig. 7A and C). Nuclear translocation of RELA showed a significant decrease in the context of NAP1L1 depletion following poly(I·C) induction (Fig. 7D and E). At variance with RELA, IRF3 protein levels were not changed by NAP1L1 depletion (Fig. 7A). However, IRF3 phosphorylation was profoundly affected (Fig. 7A and C), as was IRF3 nuclear translocation (Fig. 7F and G). Finally we wished to recapitulate the phenotype of NAP1L1 depletion using HCV NS5A. Wild-type NS5A inhibits TBK1-mediated activation of the IFN response (Fig. 7H), but the activity was rescued by the NS5A mutant that cannot bind NAP1L1. These results suggest that NAP1L1 depletion regulates the innate immune response by downmodulating RELA protein levels and by inhibiting IRF3 phosphorylation.
FIG 7.

NAP1L1 controls RELA levels and IRF3 activation. (A) NAP1L1 depletion affects RELA levels and IRF3 phosphorylation. U2OS cells were transduced with LV for shNAP1L1 or shCTRL and subsequently transfected with 1 μg poly(I·C) using Lipofectamine (lipo). Protein levels as indicated were monitored by WB at 1, 2, and 4 h posttransfection of poly(I·C). (B) NAP1L1 depletion decreases RELA protein levels. Blots as in panel A were quantified to measure RELA and IRF3 protein levels using ImageJ. Shown is the shNAP1L1/shCTRL ratio in cells not transfected with poly(I·C). Averages for 3 independent replicates are shown with standard deviations. (C) NAP1L1 depletion affects IRF3 phosphorylation. Blots as in panel A were quantified to measure RELA and IRF3 phosphorylation levels using ImageJ. Shown is the ratio of phosphorylated protein to total protein in cells transfected with poly(I·C). Averages for 3 independent replicates are shown with standard deviations. (D) NAP1L1 depletion reduces RELA nuclear translocation. U2OS cells were transduced with LV for shNAP1L1 or shCTRL and subsequently transfected with 1 μg poly(I·C) for 8 h. Cells were then fixed and stained for RELA. (E) NAP1L1 depletion reduces RELA nuclear translocation. Around 500 cells from the experiment for panel D were counted for each condition to calculate the percentage of RELA nuclear translocation. Averages for 3 independent replicates are shown with standard deviations. (F) NAP1L1 depletion reduces IRF3 nuclear translocation. U2OS cells were transduced with LV for shNAP1L1 or shCTRL and subsequently transfected with 1 μg poly(I·C) for 8 h. Cells were then fixed and stained for IRF3. (G) NAP1L1 depletion reduces IRF3 nuclear translocation. Around 500 cells from the experiment for panel F were counted for each condition to calculate the percentage of IRF3 nuclear translocation. Averages for 3 independent replicates are shown with standard deviations. (H) HCV NS5A inhibits TBK1-mediated activation of IFN-β. HEK 293T cells were transfected with expression vectors for FLAG-tagged TBK1, NS5A, or the NS5A-m2 mutant together with a reporter plasmid carrying the firefly luciferase (Fluc) gene under the control of the IFN-β promoter (pIFNβ-Luc) and the control pCMV-Renilla. Relative light units (RLUs) of luciferase activity were measured in quintuplicate independent experiments, normalized for Renilla, and represented as fold change over mock treatment ± SD.
Molecular basis of NAP1L1 depletion on IRF3 phosphorylation.
IRF3 phosphorylation is the consequence of a complex series of molecular events (Fig. 8A). PRRs such as RIG-I recognize the RNA agonist in the cytoplasm and bind the adaptor protein IPS-1/MAVS to trigger the downstream kinases TBK1/IKKε, which then phosphorylate IRF3. TLR3 instead recognizes the RNA agonist within endosomes and signals through the adaptor TIR-domain containing adaptor-inducing IFN-β (TRIF) to induce IRF3 phosphorylation. Since NAP1L1 depletion could affect each of these steps, we proceeded to dissect the whole signaling pathway. First, we took advantage of the constitutively active phosphomimetic IRF3-5D (44). Consistent with our interpretation, induction of IFN-β by IRF3-5D was not affected by NAP1L1 depletion (Fig. 8B and C). Conversely, depletion of NAP1L1 resulted in the inhibition of each step of the RIG-I pathway, from MAVS down to TBK1/IKKε kinases. With a similar approach, we could demonstrate that the TLR3 adaptor protein TRIF activity is inhibited by NAP1L1 depletion (Fig. 9A). To further investigate this pathway, we reconstituted the TLR3 pathway in Huh7-Lunet cells depleted of NAP1L1 (Fig. 9B and C). Stimulation with exogenous poly(I·C) induced high levels of the ISG IFIT1 (interferon-induced protein with tetratricopeptide repeats 1), which was severely affected by NAP1L1 depletion (Fig. 9D). We conclude that NAP1L1 controls both arms of innate sensing at the IRF3 crossroad.
FIG 8.
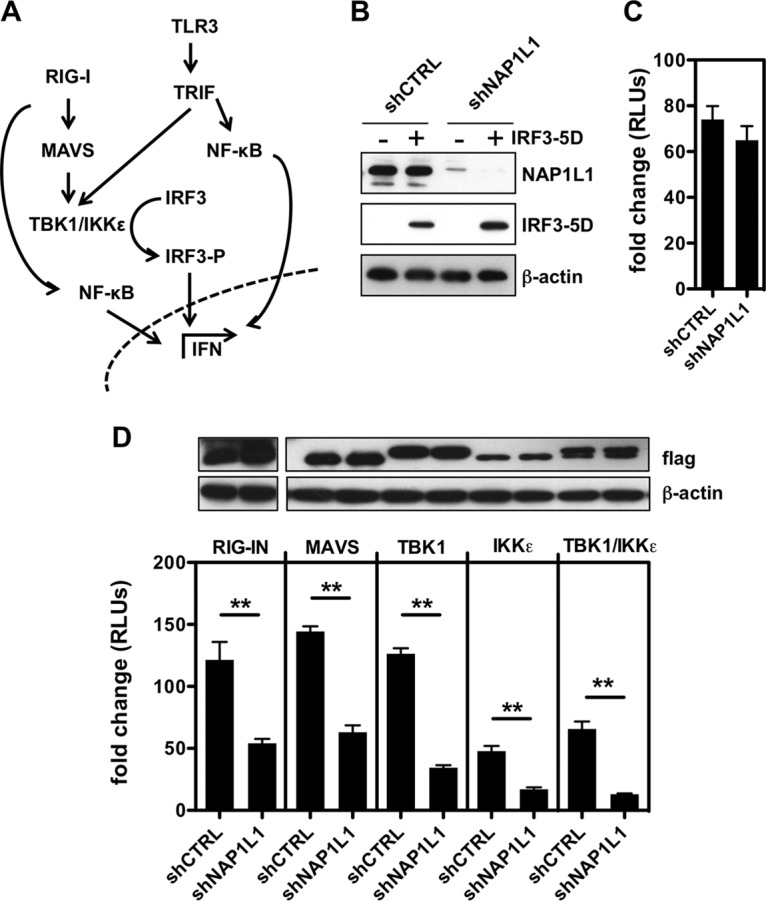
NAP1L1 controls IRF3 phosphorylation at the TBK1/IKKε level. (A) Schematic representation of the RIG-I and TLR3 pathways. Both lead to activation of NF-κB and phosphorylation of IRF3 through MAVS/TBK1/IKKϵ or TRIF, respectively. NF-κB and pIRF3 translocate to the nucleus and activate IFN-β and other ISGs. (B) NAP1L1 does not affect constitutive IRF3-5D activity. Huh7-Lunet cells were transduced with LV for shNAP1L1 or shCTRL and subsequently transfected with an expression vector for IRF3-5D, the reporter IFN-β-Luc, and the Renilla control. Cell lysates were blotted as indicated. (C) NAP1L1 does not affect constitutive IRF3-5D activity. Luciferase activity of cells from the experiment for Fig. 7H was measured in triplicate independent experiments, normalized for Renilla. Average values are shown with standard deviations. (D) Depletion of NAP1L1 affects TBK1/IKKε-mediated activation of IFN-β. HEK 293T cells were transduced with LV for shNAP1L1 or shCTRL and subsequently transfected with expression vectors for FLAG-tagged RIG-I, IPS-1/MAVS, TBK1, and IKKε together with the reporter IFN-β-Luc and the Renilla control. Cell lysates were blotted with anti-FLAG antibody as indicated. Luciferase activity was measured in triplicate independent experiments, normalized for Renilla, and represented as fold change over mock treatment ± SD.
FIG 9.

NAP1L1 controls the TLR3 pathway. (A) Depletion of NAP1L1 affects TRIF-mediated activation of IFN-β. Huh7-Lunet cells were transduced with LV for shNAP1L1 or shCTRL and subsequently transfected with expression vectors for TRIF together with the reporter IFN-β-Luc and the Renilla control. Luciferase activity was measured in triplicate independent experiments, normalized for Renilla, and represented as fold change over mock treatment ± SD. (B) Depletion of NAP1L1 by LV shRNA treatment. Huh7-Lunet cells were transduced with LV for shNAP1L1 or shCTRL and then with LV expressing TLR3 or EGFP as a control. Fifty micrograms of poly(I·C) was added to the medium and left for 24 h. IFN-β mRNA levels for NAP1L1 were measured by qRT-PCR, normalized for β-actin, and plotted against those for mock treatment. Averages for 3 independent replicates are shown with standard deviations. (C) Reconstitution of the TLR3 pathway. Cells were processed and TLR3 mRNA quantified as for panel B. (D) Depletion of NAP1L1 affects TLR3 signaling. Cells were processed and IFIT1 mRNA quantified as for panel B.
DISCUSSION
The coevolution of viruses with their host results in a number of defense strategies and countermeasures. Particularly for chronic infections, where the virus persists for long periods of time, a delicate equilibrium is established to permit limited virus replication in the context of a permissive cellular environment. HCV is highly successful at establishing a chronic infection, with about 80% of patients becoming chronically infected. In this work, we describe several lines of evidence that identify NAP1L1 as a key cellular effector of innate sensing and propose a novel mechanism that the virus deploys to subvert innate immunity in infected hepatocytes.
First, we confirmed that NAP1L1 is a bona fide interactor of NS5A. These data are in support of a series of independent observations from other groups that indicated NAP1L1 and/or NAP1L4 as a binding partner for NS5A but failed to identify a functional role (18–21). We mapped the interaction at the extreme carboxy terminus of NS5A, in a conserved motif encompassing three serine residues that have been implicated in the interaction of NS5A with core, which is essential for virus assembly but dispensable for HCV genome replication (Fig. 4F) (6–8). Interestingly, NAP1L1 has also been identified as a binding partner of core in the same proteomic screenings that identified it as a binding partner of NS5A (18, 21). Indeed, in the presence of core, we could visualize NAP1L1 on the surface of lipid droplets together with NS5A and core. However, interaction with core appears not to be essential for NAP1L1 and NS5A interaction, since experiments conducted in the absence of core showed efficient interaction and colocalization. Therefore, core and NAP1L1 bind independently to the same region of NS5A. Unfortunately, mutagenesis of the binding motif in NS5A results in the disruption of both core and NAP1L1 interactions and in a defect in assembly for viruses generated with these mutations, thus precluding their use unless these two interactions are uncoupled, if at all possible.
Next we questioned the functional role of the interaction. We discovered that NS5A from genotype 2 was able to bind and degrade NAP1L1 through a proteosome-dependent mechanism. NS5A from genotype 1 was also able to bind efficiently but was unable to degrade NAP1L1. Furthermore, wild-type NS5A from both genotypes, but not the mutated version defective for NAP1L1 binding, inhibited the nuclear relocalization of NAP1L1. It is well established that acutely infected patients respond well to IFN therapy, while in chronically infected ones the response to IFN is variable and depends on the viral genotype (39, 45). Patients infected with HCV genotypes 2 and 3 show a better response than those infected with genotypes 1 and 4, which correlates with higher levels hepatic ISG expression in HCV genotype 1- and 4-infected patient liver before therapy (46–49). The ability of different genotypes to subvert the innate response has been attributed to the genetic variability of NS3 and NS5A, which could affect their known activities in targeting innate immunity effectors such as MAVS (NS3) or protein kinase R (PKR) and possibly NAP1L1 (NS5A). For example, the levels of MAVS cleavage in vivo showed a positive correlation with the decrease of the interferon response (50). In that report, HCV genotypes 2 and 3 were more efficient than genotypes 1 and 4 in MAVS cleavage and blockage of the endogenous IFN system, which determines the response to treatment with pegylated IFN and ribavirin. Therefore, we could speculate that also the differential ability of NS5A from genotypes 1 (binding of NAP1L1) and 2 (binding and degradation of NAP1L1) contributes to the observed responses following IFN treatment. However, the interaction of NS5A and NAP1L1 appears to be functionally dominant over JFH1-dependent degradation. In fact, NS5A proteins from the two genotypes were observed to be equally efficient in blocking NAP1L1 translocation into the nucleus (Fig. 5D and E).
Depletion of NAP1L1 by shRNA, which recapitulates NS5A-mediated inhibition, resulted in the modulation of several genes at the transcription level. In particular, we noticed downmodulation of interferon-stimulated genes (ISGs), such as GBP2, IFITM3, and UBD, and of genes involved in the transcriptional activation of IFN-β, such as RELA, the p65 subunit of NF-κB. Indeed, depletion of NAP1L1 strongly affected poly(I·C)-mediated induction of IFN-β, while no effect was observed for IFN-β induction of ISGs. These findings restrict the functional role of NAP1L1 for HCV to the modulation of the innate sensing of the virus in infected cells. It is worthwhile to note that the latter experiments were conducted in U2OS cells, which are competent for the interferon response (43). Huh7-derived cell lines adapted for HCV growth are instead defective for the sensing of HCV replication (Fig. 6C) (42, 43). Hence, NS5A control of NAP1L1 results in the inhibition of the cellular innate response pathway leading to IFN-β transcription, which is appreciable only in cells that maintain this pathway active. This observation clearly explains why we failed to observe any effect of NAP1L1 overexpression or depletion on HCV replication and infectivity in Huh7-derived cells.
HCV infection triggers a number of innate immune pathways (39). The 5′-ppp and the poly(U/UC) sequence of viral RNA are potent activators of RIG-I signaling through MAVS/IPS-1, leading to the activation of the transcription factors IRF3 and NF-κB, which in turn drive transcription of IFN-β. HCV infection is also monitored in the host by the Toll-like receptors (TLRs). Viral RNA activates TLR3, and signals are transduced through the TIR-domain containing adaptor-inducing IFN-β (TRIF), leading to activation of the transcription factors IRF3 and NF-κB for the induction of innate immunity (51, 52). Another recently described sensor protein for HCV is the antiviral protein kinase R (PKR). Kinase-independent PKR signaling activates specific ISGs and IFN-β early during HCV infection (53). This signaling induces protein-protein interactions between PKR and MAVS, which has been previously described as a signaling adaptor protein also for PKR (54–56). Interestingly, all these pathways converge on the activation of transcription factors NF-κB and/or IRF3. We found that depletion of NAP1L1 not only results in a significant reduction of the mRNA and protein levels of NF-κB but also severely impairs IRF3 phosphorylation. Furthermore, nuclear translocation of both NF-κB and IRF3 following poly(I·C) stimulation of RIG-I is reduced when NAP1L1 is depleted. Therefore, NAP1L1 affects a step leading to IRF3 phosphorylation, a conclusion that is further substantiated by an experiment where IFN-β expression induced by a constitutively active phosphorylated form of IRF3 (IRF3-5D) remained unaffected by NAP1L1 depletion, which rules out inhibitory effects downstream of IRF3 phosphorylation. In order to understand at which step NAP1L1 depletion was inhibiting the pathway upstream of IRF3, we proceeded to dissect the major RIG-I-dependent axis leading to IFN-β expression. We could consistently observe that depletion of NAP1L1 reduces IFN-β expression induced by activated RIG-I, MAVS, TBK1, and IKKε. Hence, we can hypothesize that NAP1L1 depletion affects the RIG-I pathway at the level of TBK1/IKKε phosphorylation of IRF3. As mentioned above, at this step all three pathways of IFN-β activation by HCV, i.e., RIG-I, TLR3, and PKR, converge. Indeed, the TLR3 pathway was also inhibited by NAP1L1 depletion. Finally, to demonstrate that NS5A targeting of NAP1L1 is sufficient to inhibit this phosphorylation step, we showed that wild-type NS5A, but not NS5A mutants defective for NAP1L1 binding, are able to inhibit IFN-β induction by TBK1.
The master viral regulator of the HCV immune evasion program is the HCV NS3/4A protease. To regulate innate immune signaling, NS3/4A utilizes its protease domain to cleave key innate immune signaling adaptor proteins such as MAVS (57–60) and TRIF (61, 62). However, hepatitis A virus, a hepatotropic virus which does not usually become chronic, encodes a protease that also cleaves MAVS (63). Thus, MAVS/TRIF cleavage is probably necessary but not sufficient for viral chronicity. HCV also regulates PKR activity during viral infection. HCV has several PKR inactivation strategies that probably contribute to viral persistence in addition to NS3-NS4A cleavage of MAVS, which depend on the activity of NS5A and E2 (64–66). In this work we add another mechanism that could be deployed by HCV to subvert the host response to infection. We could not fully recapitulate the functional role of NAP1L1 in the infectious HCV life cycle due to a number of limitations of our experimental tools. First, mutations in NS5A that abolish NAP1L1 binding are not compatible with a fully infectious virus (6–8). Second, Huh7-derived cells that support HCV replication are impaired in the interferon response (Fig. 6C). Third, infection with full-length HCV would lead to several additional mechanisms of inhibition of the interferon response in addition to the NS5A/NAP1L1 axis, such as NS3/4A targeting MAVS and TRIF or NS2 inhibiting TBK/IKKε (39, 67). However, notwithstanding the limitations outlined above, we could clearly identify NAP1L1 as a target for HCV NS5A and define its novel role in the innate response. Notably, a very recent report confirmed the interaction of HCV NS5A with NAP1L1 and showed some effect of NAP1L1 depletion on viral replication in the context of cells stably harboring an SGR HCV (68). It is possible that chronically replicating HCV is somehow more sensitive to NAP1L1 depletion.
NAP1L1 is a cytoplasmic protein unless it is stimulated to translocate into the nucleus. Therefore, we initially hypothesized a direct participation of NAP1L1 in the TBK1/IKKε kinase complex that phosphorylates IRF3. However, we failed to immunoprecipitate NAP1L1 together with TBK1 and IKKε (not shown). Most probably, the activity of NAP1L1 is at the transcriptional level instead, as we observed for the downmodulation of NF-κB. NAP1L1 depletion and/or sequestration in the cytoplasm would result in the decrease of an as-yet-unknown cellular factor that promotes TBK1/IKKε phosphorylation of IRF3. Targeting general transcription factors to subvert innate sensing is not unusual. Several examples of viral proteins that target host cell transcription have been described. NSs from La Crosse encephalitis virus act downstream of IRF3 by specifically inhibiting RNA polymerase II (RNAPII)-mediated transcription by proteasomal degradation of the RBP1 subunit (69). Other bunyaviruses interfere with RNAPII CTD Ser2 phosphorylation or target TFIIH (70–72). The NS1 of the influenza A virus H3N2 subtype mimics a histone tail and suppresses hPAF1C-mediated transcriptional elongation of a subset of inducible genes involved in the antiviral response (73). Therefore, targeting transcription appears to be a generalized strategy to fine-tune transcriptional programs triggered by infection, which puts the cell in the optimal state to overcome the invaders' attack. HCV makes no exception and we demonstrate here that by targeting NAP1L1, it is able to control a subset of host genes, including key components of the antiviral innate sensing. It will be important to investigate at the transcriptional level the mechanism of action of NAP1L1 and to identify the factor(s) involved in TBK1/IKKε IRF3 phosphorylation that is downmodulated when NAP1L1 is depleted.
MATERIALS AND METHODS
Cells and viruses.
The human hepatocarcinoma Huh7 cell line, its derivative Huh7-Lunet (kindly provided by Ralf Bartenschlager, University of Heidelberg, Germany) (74), the HEK 293T cell line, and the osteosarcoma cell line U2OS were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics. Cell cultures were maintained at 37°C under 5% CO2. Cells were routinely tested for mycoplasma contamination.
In vitro-transcribed JFH1 RNA was introduced into Huh7-Lunet cells by electroporation as described previously (75). The supernatants from cells electroporated with JFH1 RNA were removed at the required time points and used to infect monolayers of naive Huh7.5 cells. Infected cells were detected at 3 days postinoculation by indirect immunofluorescence using a polyclonal NS5A antiserum. The 50% tissue culture infective dose (TCID50) was determined by limiting dilution assay (76).
IFN-α was obtained from the Biotechnology Development Group of the ICGEB.
Plasmids.
Plasmid pJFH1 was provided by T. Wakita (77). Plasmids encoding SGR-JFH1/Luc and the nonreplicating control SGR-JFH1/Luc-GND were described previously (78). Plasmid pFKLuc-JFH/ΔE1-E2 was provided by R. Bartenschlager (37), plasmid SGR-JFH/m2 was provided by T. Masaki (6), and plasmid SGR-JFH1/delB was provided by T. Tellinghuisen (7). The pHA-NAP1L1 and pNAP1L1-EYFP expression vectors were described previously (30, 31). pFLAG CMV-2 (Sigma-Aldrich) was used as the backbone for all NS5A constructs. FLAG-tagged NS5A from HCV JFH1, Con1, and H77 were cloned by PCR of the corresponding HCV genotypes. Mutagenesis of NS5A (JFH1 and Con1) was performed by PCR (detailed information is available on request). The reporter plasmid carrying the firefly luciferase (Fluc) gene under the control of the IFN-β promoter (pIFNβ-Luc) was provided by J. Jung (44). T. Fujita kindly provided FLAG-tagged expression vectors for RIG-I, RIG-I-N, TBK1, IKKε, IPS-1/MAVS, and HA-IRF3-5D. The control pCMV-Renilla was from Promega.
LV production and shRNA delivery.
Overexpression of NAP1-EYFP and control EYFP was obtained by lentiviral vector (LV) transduction on a pWPI backbone with blasticidin resistance (BLR), kindly provided by D. Trono. Lentiviral silencing vectors were derived from pLKO.1 TRC (Addgene). The control short hairpin RNA (shRNA) was the pLKO.1 scramble from Addgene, while for NAP1L1 targeting, a specific targeting sequence was designed and cloned into pLKO.1 TRC (shNAP1L1) using the oligonucleotides 5′-CCGGCCTATTCTGAAGCACTTGAAACTGCAGTTTCAAGTGCTTCAGAATAGGTTTTTG-3′ and 5′-GGATAAGACTTCGTGAACTTTGACGTCAAAGTTCACGAAGTCTTATCCAAAAACTTAA-3′.
An LV for TLR3 reconstitution together with its green fluorescent protein (GFP) control was obtained from Sam Wilson (MRC-University of Glasgow Centre for Virus Research). Packaging in HEK 293T was performed according to standard procedures using the packaging plasmid psPAX2 and pMD2. G (Addgene). Cell supernatants were filtered and kept at −80°C in small aliquots until use.
In vitro transcription and electroporation of HCV SGR RNA.
The HCV SGR constructs were linearized with XbaI and treated with mung bean nuclease as described previously (79). RNA was transcribed in vitro from linearized constructs using the MEGAscript T7 kit (Ambion). Synthesized RNA was treated with DNase I and transfected into cells by electroporation.
Polyinosinic·polycytidylic acid sodium salt [poly(I·C)] (InvivoGen) was also transfected into cells by lipofection (Lipofectamine Plus; Life Technologies) according to the manufacturer's instructions.
WB, IP, and IF.
Indirect immunofluorescence analysis (IF) and Western blotting (WB) were performed essentially as previously described (80). Immunoprecipitation (IP) was performed by lysis of cells in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 1% SDS, 1 M phenylmethylsulfonyl fluoride [PMSF], 1 mM EDTA, and proteinase inhibitors [cOmplete Mini; Roche]). Lysates were cleared by centrifugation and incubated with anti-FLAG M2 agarose beads (Sigma-Aldrich) or with anti-NAP1L1 antibody/IgG control and protein A/G agarose beads, washed several times in RIPA buffer, and eluted in SDS-PAGE sample buffer. The following antibodies were used in this study: a sheep polyclonal antibody against NS5A, kindly provided by M. Harris (1:200 for IF and 1:2,000 for WB) (81); a rabbit polyclonal antibody against human NAP1L1 (Ab33076; Abcam) (1:200 for IF; 1:1,000 for WB; and 1:100 for IP); a rabbit polyclonal antibody against human IRF3, kindly provided by T. Fujita (1:100 for IF); a rabbit monoclonal antibody against human IRF3 (4302; Cell Signaling) (1:500 for WB); a rabbit monoclonal antibody against phosphorylated human IRF3 (4947; Cell Signaling) (1:500 for WB); a rabbit monoclonal antibody against human NF-κB p65/RELA (8242; Cell Signaling) (1:100 for IF and 1:1,000 for WB); a rabbit monoclonal antibody against phosphorylated human NF-κB p65/RELA (3033; Cell Signaling) (1:1,000 for WB); a mouse monoclonal antibody against human β-actin conjugated with peroxidase (A3854; Sigma-Aldrich) (1:10,000 for WB); a mouse monoclonal antibody against the FLAG tag (F1804; Sigma-Aldrich) (1:1,000 for WB); and a mouse monoclonal antibody against the HA tag conjugated with peroxidase (H6533; Sigma-Aldrich) (1:10,000 for WB). Secondary antibodies conjugated with Alexa Fluor 488/594 were from Life Technologies (1:500 for IF), and peroxidase conjugates were from Dako (1:5,000 WB).
Luciferase assay and real-time qPCR.
Luciferase assays were conducted essentially as described previously (79, 80). For real-time quantitative reverse transcription-PCR (qPCR), total cellular RNA was extracted with the isol-RNA reagent (5 Prime) and treated with DNase I (Life Technologies), and 500 ng was then reverse transcribed with random primers and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Life Technologies). Quantification of mRNA was done by real-time PCR using the Kapa Sybr Fast qPCR kit on a CFX96 Bio-Rad thermocycler. Primers for amplification are available upon request.
Transcriptome analysis by RNA-seq.
Huh7-Lunet cells were transduced with shNAP1L1 or shCTRL in triplicate and incubated with puromycin for 3 days. Total RNA (isol-RNA lysis reagent; 5 Prime, Hamburg, Germany) was extracted. The quality of extracted RNA was checked by gel electrophoresis (ribosomal 18S and 28S), spectrophotometric analysis (260/280 > 1.8), and Agilent Bioanalyzer (RNA integrity number [RIN] ≥ 8). A cDNA library of poly(A)-containing mRNA molecules was prepared (TruSeq; Illumina) and sequenced on the Illumina platform (Hiseq2000 4-plex run, 50-bp reads, about 30 million reads/sample) at IGA Technology Services (Udine, Italy). Raw data were subjected to quality control (FastQC) and mapped against the human genome RNA reference from NCBI using CLCbio software.
The Bioconductor packages DESeq2 version 1.4.5 (82) and EdgeR (83) version 3.6.2 in the framework of R software version 3.1.0 were used to perform the differential gene expression analysis of mRNA-seq data. Both the packages are based on the negative binomial distribution (NB) to model the gene reads counts and shrinkage estimator to estimate the per-gene NB dispersion parameters. Specifically, we used rounded gene counts as input, and we estimated the per-gene NB dispersion parameter using the function DESeq for DESEQ2, while, for EdgeR we used the function calcNormFactors with the default parameters. To detect outlier data after normalization, we used the R package arrayQualityMetrix (84), and before testing differential gene expression, we dropped all genes with normalized counts below 14 to improve testing power while maintaining type I error rates. Estimated P values for each gene were adjusted using the Benjamini-Hochberg method (85). Genes with adjusted P values of <0.05 and absolute logarithmic base 2-fold changes of >1 were selected. Data were finally analyzed with the Ingenuity Pathway Analysis software. The significance values for the canonical pathway across the data set shown in the Table S1 in the supplemental material were calculated by Fisher's right-tailed exact test. The significance indicates the probability of association of molecules from our data set with the canonical pathway by random chance alone.
Statistics.
Three independent experiments in triplicate repeats were conducted for each condition examined, unless otherwise indicated in the figure legends. Mean values are shown with standard deviations (SD) and P values, measured with a paired two-tailed t test. Only significant P values are indicated by the asterisks above the graphs (**, P < 0.01 [highly significant]; *, P < 0.05 [significant]). Where asterisks are missing, the differences were calculated as nonsignificant.
Supplementary Material
ACKNOWLEDGMENTS
Work on flaviviruses in A.M.'s laboratory is supported by the Beneficentia Stiftung.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00880-17.
REFERENCES
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 4.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2013. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boulant S, Targett-Adams P, McLauchlan J. 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol 88:2204–2213. doi: 10.1099/vir.0.82898-0. [DOI] [PubMed] [Google Scholar]
- 6.Masaki T, Suzuki R, Murakami K, Aizaki H, Ishii K, Murayama A, Date T, Matsuura Y, Miyamura T, Wakita T, Suzuki T. 2008. Interaction of hepatitis C virus nonstructural protein 5A with core protein is critical for the production of infectious virus particles. J Virol 82:7964–7976. doi: 10.1128/JVI.00826-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tellinghuisen TL, Foss KL, Treadaway J. 2008. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035. doi: 10.1371/journal.ppat.1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macdonald A, Harris M. 2004. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol 85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 10.Ross-Thriepland D, Harris M. 2015. Hepatitis C virus NS5A: enigmatic but still promiscuous 10 years on! J Gen Virol 96:727–738. [DOI] [PubMed] [Google Scholar]
- 11.IARC. 2012. Hepatitis C virus. IARC Monogr Eval Carcinog Risks Hum 100(B):135–168. [Google Scholar]
- 12.McGivern DR, Lemon SM. 2011. Virus-specific mechanisms of carcinogenesis in hepatitis C virus associated liver cancer. Oncogene 30:1969–1983. doi: 10.1038/onc.2010.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA, Lemon SM. 2002. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 122:352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 14.Wang AG, Lee DS, Moon HB, Kim JM, Cho KH, Choi SH, Ha HL, Han YH, Kim DG, Hwang SB, Yu DY. 2009. Non-structural 5A protein of hepatitis C virus induces a range of liver pathology in transgenic mice. J Pathol 219:253–262. doi: 10.1002/path.2592. [DOI] [PubMed] [Google Scholar]
- 15.Majumder M, Steele R, Ghosh AK, Zhou XY, Thornburg L, Ray R, Phillips NJ, Ray RB. 2003. Expression of hepatitis C virus non-structural 5A protein in the liver of transgenic mice. FEBS Lett 555:528–532. doi: 10.1016/S0014-5793(03)01337-1. [DOI] [PubMed] [Google Scholar]
- 16.Gale M Jr, Kwieciszewski B, Dossett M, Nakao H, Katze MG. 1999. Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J Virol 73:6506–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, Steele R, Meyer K, Ray R, Ray RB. 1999. Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J Gen Virol 80:1179–1183. doi: 10.1099/0022-1317-80-5-1179. [DOI] [PubMed] [Google Scholar]
- 18.Pichlmair A, Kandasamy K, Alvisi G, Mulhern O, Sacco R, Habjan M, Binder M, Stefanovic A, Eberle CA, Goncalves A, Burckstummer T, Muller AC, Fauster A, Holze C, Lindsten K, Goodbourn S, Kochs G, Weber F, Bartenschlager R, Bowie AG, Bennett KL, Colinge J, Superti-Furga G. 2012. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 487:486–490. doi: 10.1038/nature11289. [DOI] [PubMed] [Google Scholar]
- 19.de Chassey B, Navratil V, Tafforeau L, Hiet MS, Aublin-Gex A, Agaugue S, Meiffren G, Pradezynski F, Faria BF, Chantier T, Le Breton M, Pellet J, Davoust N, Mangeot PE, Chaboud A, Penin F, Jacob Y, Vidalain PO, Vidal M, Andre P, Rabourdin-Combe C, Lotteau V. 2008. Hepatitis C virus infection protein network. Mol Syst Biol 4:230. doi: 10.1038/msb.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung HY, Gu M, Buehler E, MacDonald MR, Rice CM. 2014. Seed sequence-matched controls reveal limitations of small interfering RNA knockdown in functional and structural studies of hepatitis C virus NS5A-MOBKL1B interaction. J Virol 88:11022–11033. doi: 10.1128/JVI.01582-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramage HR, Kumar GR, Verschueren E, Johnson JR, Von Dollen J, Johnson T, Newton B, Shah P, Horner J, Krogan NJ, Ott M. 2015. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell 57:329–340. doi: 10.1016/j.molcel.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishimi Y, Kikuchi A. 1991. Identification and molecular cloning of yeast homolog of nucleosome assembly protein I which facilitates nucleosome assembly in vitro. J Biol Chem 266:7025–7029. [PubMed] [Google Scholar]
- 23.Park YJ, Luger K. 2006. The structure of nucleosome assembly protein 1. Proc Natl Acad Sci U S A 103:1248–1253. doi: 10.1073/pnas.0508002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park YJ, Luger K. 2006. Structure and function of nucleosome assembly proteins. Biochem Cell Biol 84:549–558. doi: 10.1139/o06-088. [DOI] [PubMed] [Google Scholar]
- 25.Loyola A, Almouzni G. 2004. Histone chaperones, a supporting role in the limelight. Biochim Biophys Acta 1677:3–11. doi: 10.1016/j.bbaexp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 26.Tyler JK. 2002. Chromatin assembly. Cooperation between histone chaperones and ATP-dependent nucleosome remodeling machines. Eur J Biochem 269:2268–2274. [DOI] [PubMed] [Google Scholar]
- 27.Asahara H, Tartare-Deckert S, Nakagawa T, Ikehara T, Hirose F, Hunter T, Ito T, Montminy M. 2002. Dual roles of p300 in chromatin assembly and transcriptional activation in cooperation with nucleosome assembly protein 1 in vitro. Mol Cell Biol 22:2974–2983. doi: 10.1128/MCB.22.9.2974-2983.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rehtanz M, Schmidt HM, Warthorst U, Steger G. 2004. Direct interaction between nucleosome assembly protein 1 and the papillomavirus E2 proteins involved in activation of transcription. Mol Cell Biol 24:2153–2168. doi: 10.1128/MCB.24.5.2153-2168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma N, Nyborg JK. 2008. The coactivators CBP/p300 and the histone chaperone NAP1 promote transcription-independent nucleosome eviction at the HTLV-1 promoter. Proc Natl Acad Sci U S A 105:7959–7963. doi: 10.1073/pnas.0800534105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vardabasso C, Manganaro L, Lusic M, Marcello A, Giacca M. 2008. The histone chaperone protein nucleosome assembly protein-1 (hNAP-1) binds HIV-1 Tat and promotes viral transcription. Retrovirology 5:8. doi: 10.1186/1742-4690-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Marco A, Dans PD, Knezevich A, Maiuri P, Pantano S, Marcello A. 2010. Subcellular localization of the interaction between the human immunodeficiency virus transactivator Tat and the nucleosome assembly protein 1. Amino Acids 38:1583–1593. doi: 10.1007/s00726-009-0378-9. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Gadue P, Chen K, Jiao Y, Tuteja G, Schug J, Li W, Kaestner KH. 2012. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 151:1608–1616. doi: 10.1016/j.cell.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong H, Yan Y, Fang B, Xue Y, Yin P, Li L, Zhang G, Sun X, Chen Z, Ma H, Yang C, Ding Y, Yong Y, Zhu Y, Yang H, Komuro I, Ge J, Zou Y. 2014. Knockdown of nucleosome assembly protein 1-like 1 induces mesoderm formation and cardiomyogenesis via notch signaling in murine-induced pluripotent stem cells. Stem Cells 32:1759–1773. doi: 10.1002/stem.1702. [DOI] [PubMed] [Google Scholar]
- 34.Nagata T, Takahashi Y, Ishii Y, Asai S, Nishida Y, Murata A, Koshinaga T, Fukuzawa M, Hamazaki M, Asami K, Ito E, Ikeda H, Takamatsu H, Koike K, Kikuta A, Kuroiwa M, Watanabe A, Kosaka Y, Fujita H, Miyake M, Mugishima H. 2003. Transcriptional profiling in hepatoblastomas using high-density oligonucleotide DNA array. Cancer Genet Cytogenet 145:152–160. doi: 10.1016/S0165-4608(03)00065-7. [DOI] [PubMed] [Google Scholar]
- 35.Kidd M, Modlin IM, Mane SM, Camp RL, Eick G, Latich I. 2006. The role of genetic markers–NAP1L1, MAGE-D2, and MTA1 in defining small-intestinal carcinoid neoplasia. Ann Surg Oncol 13:253–262. doi: 10.1245/ASO.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Schimmack S, Taylor A, Lawrence B, Alaimo D, Schmitz-Winnenthal H, Buchler MW, Modlin IM, Kidd M. 2014. A mechanistic role for the chromatin modulator, NAP1L1, in pancreatic neuroendocrine neoplasm proliferation and metastases. Epigenetics Chromatin 7:15. doi: 10.1186/1756-8935-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohkuni K, Shirahige K, Kikuchi A. 2003. Genome-wide expression analysis of NAP1 in Saccharomyces cerevisiae. Biochem Biophys Res Commun 306:5–9. doi: 10.1016/S0006-291X(03)00907-0. [DOI] [PubMed] [Google Scholar]
- 39.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiang HS, Zhao Y, Song JH, Liu S, Wang N, Terhorst C, Sharpe AH, Basavappa M, Jeffrey KL, Reinecker HC. 2014. GEF-H1 controls microtubule-dependent sensing of nucleic acids for antiviral host defenses. Nat Immunol 15:63–71. doi: 10.1038/ni.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luna JM, Scheel TK, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, Jacobson IM, Rice CM, Darnell RB. 2015. Hepatitis C virus RNA functionally sequesters miR-122. Cell 160:1099–1110. doi: 10.1016/j.cell.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol 79:2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miorin L, Albornoz A, Baba MM, D'Agaro P, Marcello A. 2012. Formation of membrane-defined compartments by tick-borne encephalitis virus contributes to the early delay in interferon signaling. Virus Res 163:660–666. doi: 10.1016/j.virusres.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 44.Lin R, Genin P, Mamane Y, Hiscott J. 2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol 20:6342–6353. doi: 10.1128/MCB.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaeckel E, Cornberg M, Wedemeyer H, Santantonio T, Mayer J, Zankel M, Pastore G, Dietrich M, Trautwein C, Manns MP, German Acute Hepatitis CTG. 2001. Treatment of acute hepatitis C with interferon alfa-2b. N Engl J Med 345:1452–1457. doi: 10.1056/NEJMoa011232. [DOI] [PubMed] [Google Scholar]
- 46.Sarasin-Filipowicz M, Oakeley EJ, Duong FH, Christen V, Terracciano L, Filipowicz W, Heim MH. 2008. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A 105:7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, Heathcote J, Edwards AM, McGilvray ID. 2005. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology 128:1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 48.Asselah T, Bieche I, Narguet S, Sabbagh A, Laurendeau I, Ripault MP, Boyer N, Martinot-Peignoux M, Valla D, Vidaud M, Marcellin P. 2008. Liver gene expression signature to predict response to pegylated interferon plus ribavirin combination therapy in patients with chronic hepatitis C. Gut 57:516–524. doi: 10.1136/gut.2007.128611. [DOI] [PubMed] [Google Scholar]
- 49.Dill MT, Duong FH, Vogt JE, Bibert S, Bochud PY, Terracciano L, Papassotiropoulos A, Roth V, Heim MH. 2011. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology 140:1021–1031. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 50.Bellecave P, Sarasin-Filipowicz M, Donze O, Kennel A, Gouttenoire J, Meylan E, Terracciano L, Tschopp J, Sarrazin C, Berg T, Moradpour D, Heim MH. 2010. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 51.Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. 2012. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55:666–675. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K. 2009. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol 83:9824–9834. doi: 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arnaud N, Dabo S, Akazawa D, Fukasawa M, Shinkai-Ouchi F, Hugon J, Wakita T, Meurs EF. 2011. Hepatitis C virus reveals a novel early control in acute immune response. PLoS Pathog 7:e1002289. doi: 10.1371/journal.ppat.1002289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanazawa N, Kurosaki M, Sakamoto N, Enomoto N, Itsui Y, Yamashiro T, Tanabe Y, Maekawa S, Nakagawa M, Chen CH, Kakinuma S, Oshima S, Nakamura T, Kato T, Wakita T, Watanabe M. 2004. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J Virol 78:9713–9720. doi: 10.1128/JVI.78.18.9713-9720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, Haque J, Reis L, Weissmann C, Williams BR. 1997. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. EMBO J 16:406–416. doi: 10.1093/emboj/16.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McAllister CS, Samuel CE. 2009. The RNA-activated protein kinase enhances the induction of interferon-beta and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem 284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foy E, Li K, Sumpter R Jr, Loo YM, Johnson CL, Wang C, Fish PM, Yoneyama M, Fujita T, Lemon SM, Gale M Jr. 2005. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A 102:2986–2991. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Foy E, Li K, Wang C, Sumpter R Jr, Ikeda M, Lemon SM, Gale M Jr. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 59.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 60.Baril M, Racine ME, Penin F, Lamarre D. 2009. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol 83:1299–1311. doi: 10.1128/JVI.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin R, Lacoste J, Nakhaei P, Sun Q, Yang L, Paz S, Wilkinson P, Julkunen I, Vitour D, Meurs E, Hiscott J. 2006. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol 80:6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. 2007. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A 104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gale MJ Jr, Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG. 1997. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 65.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. 1999. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science 285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 66.Noguchi T, Satoh S, Noshi T, Hatada E, Fukuda R, Kawai A, Ikeda S, Hijikata M, Shimotohno K. 2001. Effects of mutation in hepatitis C virus nonstructural protein 5A on interferon resistance mediated by inhibition of PKR kinase activity in mammalian cells. Microbiol Immunol 45:829–840. doi: 10.1111/j.1348-0421.2001.tb01322.x. [DOI] [PubMed] [Google Scholar]
- 67.Kaukinen P, Sillanpaa M, Nousiainen L, Melen K, Julkunen I. 2013. Hepatitis C virus NS2 protease inhibits host cell antiviral response by inhibiting IKKepsilon and TBK1 functions. J Med Virol 85:71–82. doi: 10.1002/jmv.23442. [DOI] [PubMed] [Google Scholar]
- 68.Goonawardane N, Gebhardt A, Bartlett C, Pichlmair A, Harris M. 14 June 2017. Phosphorylation of serine 225 in hepatitis C virus NS5A regulates protein-protein interactions. J Virol doi: 10.1128/JVI.00805-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verbruggen P, Ruf M, Blakqori G, Overby AK, Heidemann M, Eick D, Weber F. 2011. Interferon antagonist NSs of La Crosse virus triggers a DNA damage response-like degradation of transcribing RNA polymerase II. J Biol Chem 286:3681–3692. doi: 10.1074/jbc.M110.154799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le May N, Dubaele S, Proietti De Santis L, Billecocq A, Bouloy M, Egly JM. 2004. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell 116:541–550. doi: 10.1016/S0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- 71.Le May N, Mansuroglu Z, Leger P, Josse T, Blot G, Billecocq A, Flick R, Jacob Y, Bonnefoy E, Bouloy M. 2008. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog 4:e13. doi: 10.1371/journal.ppat.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas D, Blakqori G, Wagner V, Banholzer M, Kessler N, Elliott RM, Haller O, Weber F. 2004. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J Biol Chem 279:31471–31477. doi: 10.1074/jbc.M400938200. [DOI] [PubMed] [Google Scholar]
- 73.Marazzi I, Ho JS, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, Lee K, Garcia-Sastre A, Roeder RG, Tarakhovsky A. 2012. Suppression of the antiviral response by an influenza histone mimic. Nature 483:428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hope RG, McLauchlan J. 2000. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J Gen Virol 81:1913–1925. doi: 10.1099/0022-1317-81-8-1913. [DOI] [PubMed] [Google Scholar]
- 76.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 77.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Targett-Adams P, McLauchlan J. 2005. Development and characterization of a transient-replication assay for the genotype 2a hepatitis C virus subgenomic replicon. J Gen Virol 86:3075–3080. doi: 10.1099/vir.0.81334-0. [DOI] [PubMed] [Google Scholar]
- 79.Bartolomei G, Cevik RE, Marcello A. 2011. Modulation of hepatitis C virus replication by iron and hepcidin in Huh7 hepatocytes. J Gen Virol 92:2072–2081. doi: 10.1099/vir.0.032706-0. [DOI] [PubMed] [Google Scholar]
- 80.Albornoz A, Carletti T, Corazza G, Marcello A. 2014. The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation. J Virol 88:6611–6622. doi: 10.1128/JVI.03736-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Macdonald A, Crowder K, Street A, McCormick C, Saksela K, Harris M. 2003. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras-ERK pathway signaling. J Biol Chem 278:17775–17784. doi: 10.1074/jbc.M210900200. [DOI] [PubMed] [Google Scholar]
- 82.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCarthy DJ, Smyth GK. 2009. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25:765–771. doi: 10.1093/bioinformatics/btp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kauffmann A, Gentleman R, Huber W. 2009. arrayQualityMetrics—a bioconductor package for quality assessment of microarray data. Bioinformatics 25:415–416. doi: 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.