

ABSTRACT
Hepatitis C virus (HCV) primarily infects hepatocytes, which are highly polarized cells. The relevance of cell polarity in the HCV life cycle has been addressed only in distantly related models and remains poorly understood. Although polarized epithelial cells have a rather simple morphology with a basolateral and an apical domain, hepatocytes exhibit complex polarization structures. However, it has been reported that some selected polarized HepG2 cell clones can exhibit a honeycomb pattern of distribution of the tight-junction proteins typical of columnar polarized epithelia, which can be used as a simple model to study the role of cell polarization in viral infection of hepatocytes. To obtain similar clones, HepG2 cells expressing CD81 (HepG2-CD81) were used, and clones were isolated by limiting dilutions. Two clones exhibiting a simple columnar polarization capacity when grown on a semipermeable support were isolated and characterized. To test the polarity of HCV entry and release, our polarized HepG2-CD81 clones were infected with cell culture-derived HCV. Our data indicate that HCV binds equally to both sides of the cells, but productive infection occurs mainly when the virus is added at the basolateral domain. Furthermore, we also observed that HCV virions are released from the basolateral domain of the cells. Finally, when polarized cells were treated with oleic acid and U0126, a MEK inhibitor, to promote lipoprotein secretion, a higher proportion of infectious viral particles of lower density were secreted. This cell culture system provides an excellent model to investigate the influence of cell polarization on the HCV life cycle.
IMPORTANCE Hepatitis C is a major health burden, with approximately 170 million persons infected worldwide. Hepatitis C virus (HCV) primarily infects hepatocytes, which are highly polarized cells with a complex organization. The relevance of cell polarity in the HCV life cycle has been addressed in distantly related models and remains unclear. Hepatocyte organization is complex, with multiple apical and basolateral surfaces. A simple culture model of HepG2 cells expressing CD81 that are able to polarize with unique apical and basolateral domains was developed to study HCV infection. With this model, we demonstrated that HCV enters and exits hepatocytes by the basolateral domain. Furthermore, lower-density viral particles were produced under conditions that promote lipoprotein secretion. This cell culture system provides a useful model to study the influence of cell polarization on HCV infection.
KEYWORDS: cell polarity, hepatitis C virus, virus egress, virus entry
INTRODUCTION
With approximately 170 million persons infected worldwide, hepatitis C is a major health burden that can lead to chronic liver disease, often resulting in cirrhosis and hepatocellular carcinoma. Hepatitis C virus (HCV) is primarily transmitted through percutaneous routes and by unsafe injections with HCV-contaminated needles, with vertical and sexual transmission contributing little to HCV spread. Once in the bloodstream, HCV has direct access to hepatocytes, its major cellular target.
Hepatocytes comprise 60% of the total liver cells and are the main contributor to liver functions, such as lipid and glucose metabolism regulation, glycogen storage, plasma protein and bile acid production, and detoxification (1). To perform these functions, hepatocytes are polarized with functionally and morphologically distinct membrane domains. The lateral domains are involved in cell-cell contact, and the basal domains are in contact with the underlying space of Disse. The apical domains of adjacent cells make up a narrow lumen in which the bile is secreted. Hepatocytes ensure the blood-biliary barrier and mediate the vectorial exchange of macromolecules between the two spaces. The apical and basolateral domains are separated by tight junctions and have specific protein and lipid compositions. Tight junctions, formed by multiprotein complexes, ensure the barrier functions to prevent the diffusion of components between the apical and basolateral domains of cells. They also regulate paracellular permeability across epithelia (2). Most epithelial cells have a columnar organization with single apical and basolateral domains. In contrast, hepatocyte organization is complex, with multiple basolateral and apical poles.
HCV is a small enveloped virus with a single-stranded positive RNA genome of 9.6 kb. The virus takes advantage of many aspects of lipid metabolism in different steps of its life cycle (3, 4). In infected patients, virions found in the very-low- and low-density fractions of plasma display the highest infectivity (5). The low density of viral particles is due to their association with very-low-density lipoproteins (VLDL) or low-density lipoproteins (LDL) to form the so-called lipoviroparticles (LVP). Indeed, in addition to the genome, capsid, and E1 and E2 envelope glycoproteins, lipoprotein components have also been found in LVP. These molecules include cholesterol esters, triglycerides, and several apolipoproteins (6–9). Nevertheless, the detailed mechanisms of LVP formation remain unclear.
HCV entry into hepatocytes is a complex, multistep, and slow process (10). Two tight-junction proteins, claudin-1 (CLDN1) and occludin (OCLN), are among the many players involved in HCV entry (11, 12). Initial attachment of the virus to the cell surface involves glycosaminoglycan and/or lipoprotein receptors (LDLR). After binding to the cell surface, the virus undergoes a series of interactions with specific molecules. Data gathered over the years suggest that HCV first encounters the scavenger receptor class B type I (SR-BI), leading to conformational and composition changes in the viral particle and exposure of the binding site to the tetraspanin CD81. Then, lateral diffusion of virus-CD81 complexes promotes their association with CLDN1, whereas the exact role of OCLN in HCV entry remains unclear. The virus is then internalized by a clathrin-dependent pathway, followed by fusion of the viral envelope with early endosome membranes to deliver the viral genome into the host cell (13, 14). Other proteins have also been reported to contribute to the regulation of HCV entry (reviewed in reference 10).
Because the HCV entry process is complex and involves two tight-junction proteins, polarization of hepatocytes likely adds additional constraints on early steps of the HCV life cycle. Furthermore, polarization also likely affects the release of virions in the appropriate compartment. A robust cell culture model with appropriate polarization properties to study HCV is still lacking. It has been known for a long time that HepG2 cells exhibit polarization properties, and clones of HepG2 cells have been isolated that can polarize with a simple columnar morphology, like epithelial cells, when grown on a semipermeable support (15, 16). Here, we developed a simple culture model of HepG2 cells expressing CD81 (HepG2-CD81) able to differentiate into columnar polarized cells with unique apical and basolateral domains. With this model, we demonstrated that HCV entry and release take place at the basolateral domain of hepatocytes. Furthermore, lower-density viral particles were produced in polarized HepG2-CD81 cells under conditions that promote lipoprotein secretion. This cell culture system provides a novel model to study the influence of cell polarization on the HCV life cycle.
RESULTS
Isolation and characterization of HepG2-CD81 clones.
To obtain cells that could differentiate into columnar polarized monolayers, HepG2-CD81 cells were used, and clones were isolated by limiting dilutions. A first screen of the cells was performed based on the staining of the tight-junction marker zonula occludens 1 (ZO-1) on confluent cells. We selected clones showing ZO-1 recruitment at cell-cell contact for further characterization of their columnar polarization capacity. The cells were then grown on semipermeable supports in William's medium supplemented with 1% dimethyl sulfoxide (DMSO).
Intracellular transport of proteins needs to be polarized to target a protein to the correct membrane surface or extracellular space. Since one main function of hepatocytes is the synthesis and secretion of serum components, including human serum albumin (HSA) or lipoproteins, we monitored the amounts of HSA and apolipoprotein E (apoE) secreted in the basolateral chamber over time to test the functionality of cell polarization. As shown in Fig. 1A, we identified two groups of clones. In the first group (top row), two clones, 15 and 1SC3, secreted high levels of HSA (>80%) for 12 days before the monolayer started to age and the basolateral secretion of HSA decreased. The total amount of HSA secreted by these clones increased until day 12 before reaching a plateau. Clone 1 also showed a high level of secretion of HSA in the basolateral chamber, but the cells were polarized for a shorter period. The other clones, falling in the second group (middle row), presented a weaker level of polarization, with secretion of HSA in the basolateral chamber below 70%. apoE secretion in the basolateral medium was analyzed at days 6 and 9, and the profile of secretion observed for each clone was consistent with the results obtained with HSA secretion (Fig. 1B). We next analyzed the localization of the tight-junction protein ZO-1 and of two proteins partitioning on the different membrane domains, CD26 (also called dipeptidylpeptidase IV), an apical marker, and the transferrin receptor (TfR), a basolateral marker (Fig. 2). To facilitate the discrimination of the transferrin receptor expressed at the cell surface from the intracellular pool, a transferrin receptor with a hemagglutinin (HA) tag (HA-TfR) in the extracellular domain was expressed, and its cell surface expression was analyzed. In Fig. 2A and B, three different z sections of the cells are presented for clone 1SC3. In Fig. 2C are depicted the z sections displayed in column 1, 2, and 3 of Fig. 2A and B. In polarized clone 1SC3, ZO-1 was recruited at cell-cell contacts, showing staining characteristic of ZO-1 association with tight junctions in well-differentiated polarized epithelial cells (Fig. 2A and B, z sections 2). CD26 was found on the uppermost cell surface (Fig. 2A and D, x-z projection) but was not detected on the cell surface below the tight junctions (Fig. 2A, z section 3). In contrast, the transferrin receptor was detected only on the basolateral membrane surface (Fig. 2B, z section 3, and D, x-z projection). These data confirm the formation of distinct membrane domains. Similar results were obtained with clone 15 (data not shown).
FIG 1.

Polarization of different HepG2-CD81 clones grown on transwell inserts was induced with 1% DMSO. Apical and basolateral media were collected every day over a 21-day period. Secreted HSA (A) and apoE (B) were quantified by ELISA. The percentages of basolateral secretion over the total secreted quantity (black bars), as well as the total quantities of HSA secreted in both chambers (white bars), are presented for each clone and are expressed as the means of the results of three independent experiments. The error bars represent standard errors of the mean (SEM).
FIG 2.

(A and B) Clone 1SC3 cells were polarized and transduced or not with lentiviral vector to express HA-TfR. The cells were fixed 6 days after inducing polarization and processed for membrane staining of CD26 (A) and HA (B), followed by permeabilization and staining of the ZO-1 tight-junction protein and nuclei with DAPI. z stacks of xy sections of the cells were acquired by confocal microscopy, and 3 different z sections are presented. (C) Schematic drawing of the positions of the chosen sections. (D) x-z projections of the cells stained for CD26 and ZO-1 (top) and TfR and ZO-1 (bottom). CD26 and TfR are shown in green, ZO-1 in red, and nuclei in blue.
In conclusion, polarized clones 15 and 1SC3 grown on semipermeable supports have the morphology of epithelial cells, with one apical domain and one basolateral domain separated by tight junctions. Based on the secretion of HSA, the polarization of the cells was optimal between days 5 and 10 after induction of polarization.
HepG2-CD81 clone susceptibility to HCV infection.
It has been shown that the amount of CD81 expressed at the cell surface is important for HCV entry (17). However, HepG2 cells only weakly support HCV replication because they poorly express the liver-specific micro-RNA 122 (miR122) (18). Therefore, we analyzed the susceptibility of our clones to HCV infection by measuring the viral titer after infection with a virus stock produced in Huh-7 cells. The assay was performed in nonpolarized cells. Titers varying between 3.2 ± 0.13 and 4.63 ± 0.13 log10 focus-forming units (FFU)/ml were obtained after infection of our clones, whereas a titer of 5.45 ± 0.19 log10 FFU/ml was measured in Huh-7 cells using the same stock of HCV grown in cell culture (HCVcc), indicating that our clones are all susceptible to HCV infection, albeit with varying levels of replication efficiency (Fig. 3A). It is worth noting that the clones that were not selected in our first screen because of their lack of ZO-1 recruitment at cell-cell contacst were not susceptible to HCV infection (data not shown). Although they showed a 1.5 log10 decrease in susceptibility to HCV infection compared to Huh-7 cells, clones 15 and 1SC3 were further investigated in this study due to their relevant polarization properties and their susceptibility to HCV. Expression of miR122 in these two clones was confirmed by quantitative real-time reverse transcription (RT)-PCR. In the parental cell line, HepG2-CD81, miR122 was detectable, but its relative expression level compared to Huh-7 cells was very low (0.3%). As expected, we could not detect any miR122 expression in HEK293T cells (not shown). Clone 1SC3 showed a level of miR122 expression similar to that in Huh-7 cells, whereas clone 15 showed only around 40% of the miR122 level observed in Huh-7 cells.
FIG 3.

(A) The same viral stock was titrated on different nonpolarized HepG2-CD81 clones by counting FFU. The results are expressed as log10 FFU/ml, and the error bars indicate SEM from three independent experiments. (B) The relative expression of miR122 was determined by RT-quantitative PCR (qPCR) in Huh-7, clone 15, and clone 1SC3 cells and the HepG2-CD81 parental cell line. The results are expressed as relative miR122 expression using Huh-7 cells as a calibrator.
Infection of polarized cell clones takes place at the basolateral domain.
HCV is thought to cross the fenestrated endothelium to enter the space of Disse, where virions can make contact with the basolateral domain of hepatocytes. However, it has never been proved experimentally that HCV infects cells at the basolateral domain. Polarization of cells on semipermeable supports offers the advantage of having independent access to the different poles of the cells. To determine which membrane domain is targeted for HCV entry, we infected polarized cells either from the apical or from the basolateral domain. In control nonpolarized cells (Huh-7 or nonpolarized clones 15 and 1SC3), HCV could infect cells equally from both domains (Fig. 4A). In contrast, in polarized cells, HCV predominantly infected cells when administered at the basolateral domain. To confirm that HCV genomes detected upon inoculation in polarized cells reflect HCV infection, the same experiment was performed in the presence of 1 nM daclatasvir to inhibit HCV replication. As shown in Fig. 4A, strong inhibition of infection of the polarized clones was observed after apical or basolateral infection. To determine if polarized infection of clones 15 and 1SC3 is specific to HCV or can be applied to other viruses, we analyzed infection of polarized cells by human coronavirus 229E (HCoV-229E). HCoV-229E relies on aminopeptidase N (APN), an apically located protein, to initiate entry into the host cells. As expected, infection occurred predominantly when the virus was applied at the apical domain (Fig. 4B). Differences in apical versus basolateral infection were statistically significant for clone 15; however, for clone 1SC3, the differences did not reach significance.
FIG 4.

(A) HCV infection of polarized cells. Polarized cells (clones 15 and 1SC3) and nonpolarized cells (clone 1SC3, clone 15, and Huh7 cells) grown on transwell inserts were infected either at the basolateral or at the apical domain. The polarized cells were incubated in the presence or absence of 1 nM daclatasvir after inoculation. Thirty hours later, the cells were lysed and total RNAs were extracted. HCV genomes were measured by quantitative RT-PCR. The results are expressed as RNA copies per nanogram of total RNA, and the error bars indicate the SEM of the results of three independent experiments. The data were analyzed by Student's t test (**, P < 0.01). (B) HCoV-229E infection of polarized cells. Polarized cells (clones 15 and 1SC3) were infected at the apical or basolateral domain with a recombinant HCoV-229E expressing Renilla luciferase. At 6 h postinfection, the cells were lysed and luciferase activities were quantified. The results are expressed as relative light units (RLU). The error bars indicate the SEM of the results of three independent experiments. The data were analyzed by Student's t test (**, P < 0.01).
The difference in infection efficiency between inoculation at the basolateral and at the apical domain could result either from a defect in binding at the apical pole or from restriction of the localization of at least one (co)receptor at the basolateral domain. To address this question, we first quantified virus attachment to each membrane domain. Purified virus was incubated at 4°C at either the apical or basolateral domain of polarized or nonpolarized cells, and bound viruses were measured by quantitative RT-PCR. We did not detect any difference in the initial attachment of the virus to the apical or basolateral surfac of the cells (Fig. 5A), supporting the idea that cell polarization has no effect on HCV attachment. The same experiment was performed in the presence of heparin to confirm that our binding assay was able to detect differences in binding efficiencies. As expected, heparin inhibited apical as well as basolateral binding of the virus (Fig. 5B).
FIG 5.

(A) Polarized cells (clones 15 and 1SC3) and nonpolarized cells (clone 1SC3, clone 15, and Huh-7 cells) were incubated with purified virions at 4°C for 2 h at the apical or basolateral domain. Then, the cells were lysed and total RNAs were extracted. HCV genomes were measured by quantitative RT-PCR. The results are expressed as RNA copies per nanogram of total RNA, and the error bars indicate the SEM of the results of three independent experiments. (B) Binding of purified virus in the presence or absence of 500 µg/ml heparin.
We next analyzed the cell surface distributions of the four major HCV coreceptors under polarized conditions. To facilitate their detection, polarized cells were transduced with lentiviral expression vectors for CD81, OCLN, or CLDN1 fused to a fluorescent protein (green fluorescent protein [GFP] or cerulean [Cer]), whereas SR-BI was detected by immunostaining of the endogenous protein. As shown in Fig. 6A and E, cell surface staining of SR-BI was observed at both domains of the cells (z sections 1, 2, and 3), whereas CD81 was present only at the basolateral pole (Fig. 6B, z sections 2 and 3, and E). CLDN1 mainly colocalized with ZO-1 at cell-cell contacts, where tight junctions are formed, but it was also weakly detected at the basolateral surface (Fig. 6C, z sections 2 and 3). Under our experimental conditions, OCLN was not detected outside the tight junctions (Fig. 6D). The localization of the four receptors is compatible with the mode of infection of HCV, and the absence of CD81 expression on the apical surface is likely responsible for the restriction of infection at the basolateral domain.
FIG 6.

(A to D) Polarized clone 15 cells were transduced with lentiviral expression vector for SR-BI (A), CD81-GFP (B), CLDN1-Cer (C), or OCLN-GFP (D). The cells were fixed and processed for cell surface staining of SR-BI. Then, the cells were fixed and stained for ZO-1 and nuclei. z stacks of xy sections of the cells were acquired by confocal microscopy, and 3 different z sections are presented. (E) x-z projections of the cells stained for SR-BI or expressing CD81-GFP, CLDN1-Cer, or OCLN-GFP. SR-BI, CD81-GFP, CLDN1-Cer, and OCLN-GFP are shown in green, ZO-1 in red, and nuclei in blue.
Newly synthesized progeny virus is preferentially released from the basolateral domain.
Bile duct damage is observed in chronic hepatitis C. In some infected patients, it has been shown that HCV RNA can be detected in the bile and that bile duct epithelial cells can be infected by HCV (19, 20). Bile viral loads can result from HCV secretion either from infected hepatocytes or from infected bile duct epithelial cells. We therefore investigated HCV secretion in polarized cells to determine if the virus undergoes a vectorial secretion or if HCV can be equally secreted into the bile and the blood. Therefore, 48 h after infection at the basolateral domain of polarized cells, apical and basolateral supernatants were collected and viral production was quantified. As seen in Fig. 7A, more than 90% of HCV particles were secreted from the basolateral pole. Indeed, 3.09 ± 0.08 and 3.6 ± 0.24 log10 FFU/ml were released from the basolateral pole for clone 15 and clone 1SC3, respectively, whereas only 2.3 ± 0.22 and 2.1 ± 0.31 log10 FFU/ml were produced in the apical chamber. Since we cannot exclude the possibility that noninfectious viral particles might also be secreted from the apical pole, we also quantified HCV genome secreted from both poles of infected polarized cells. However, as shown in Fig. 7B, the release of HCV genome paralleled that of infectious particles. Altogether, these data indicate that HCV secretion is polarized and that hepatocytes release HCV particles from their basolateral domains.
FIG 7.
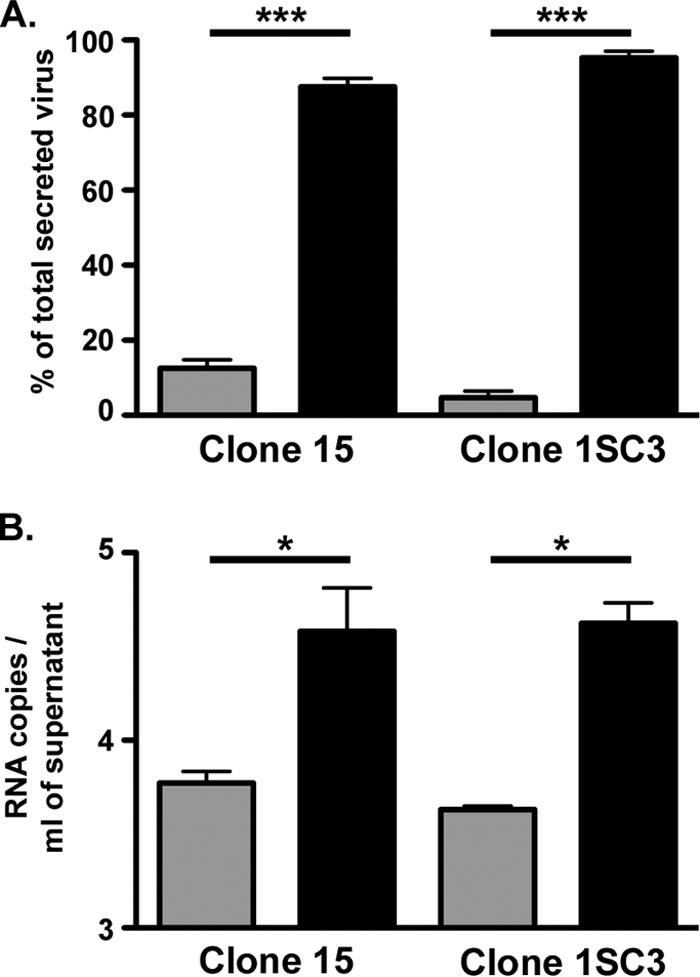
(A) Polarized clone 15 and 1SC3 cells were infected at the basolateral membrane; 48 h later, apical (gray bars) and basolateral (black bars) supernatants were collected and infectious titers were determined. The results are expressed as percentages of the total secreted virus. (B) HCV genomes were measured by quantitative RT-PCR. The results are expressed as RNA copies per milliliter of supernatant. The error bars indicate the SEM of the results of three independent experiments. The data were analyzed by Student's t test (*, P < 0.05; ***, P < 0,001).
Low-density viral particles are secreted from polarized cells.
Due to their association with lipoproteins, HCV particles exhibit a rather low density. However, the buoyant density of virions produced in vivo is lower than that of viral particles generated in hepatocytic cell lines (21), indicating that cell culture systems do not completely recapitulate the production of fully mature viral particles. A suitable cellular model to study the detailed mechanisms of VLDL and LVP formation is still lacking. It has recently been shown that HepG2 treatment with oleic acid (OA) combined with the MEK/ERK inhibitor U0126 can stimulate the production of VLDL, but without affecting the density of secreted HCV particles (22). Here, we analyzed the density of HCV virions produced in polarized cells treated with OA and U0126. In untreated polarized cells, the density profile of secreted virions was the same as that of HCV particles released from nonpolarized Huh-7 cells (Fig. 8A and B, gray lines). As previously shown (22), treatment of Huh-7 cells with OA and U0126 did not modify the density profile of HCV virions (Fig. 8B, black line). In contrast, we observed a shift toward lower densities for HCV particles released from clone 15 cells treated with OA and U0126 (Fig. 8A, black line). Indeed 21% of the total virus secreted by treated polarized cells had a density of 1.07 g/ml, whereas in control polarized cells, the virus peaked at a density ranging from 1.10 g/ml to 1.13 g/ml. These results indicate that cell polarization combined with OA and U0126 treatment promotes the formation of LVP of lower density.
FIG 8.

Polarized clone 15 cells and nonpolarized Huh-7 cells were infected with HCV. Twenty-four hours later, the medium was replaced and the cells were incubated for another 24 h in the presence (black lines) or absence (gray lines) of 10% OA-BSA and 1 μM U0126. Basolateral supernatants were collected and separated on an iodixanol buoyant-density gradient. Twelve fractions were collected from top (fraction 1) to bottom (fraction 12) and analyzed for their density and infectivity by titration. The error bars indicate the SEM of the results of three independent experiments.
DISCUSSION
In the absence of appropriate models, the role of cell polarity in the HCV life cycle remains poorly defined. This is due to the specific polarization structures of hepatocytes, which exhibit a complex pattern of distribution of the tight-junction proteins and therefore do not allow the physical separation of the apical and basolateral domains under cell culture conditions. Here, we selected HepG2-CD81 cell clones exhibiting a simple columnar polarization capacity when grown on a semipermeable support. We showed that these HepG2-CD81 cell clones are able to polarize with unique apical and basolateral domains. With the help of this cell culture system, we also showed that HCV entry and release take place at the basolateral domain of hepatocytes. In addition, when polarized cells were treated with OA and U0126, a MEK inhibitor, lower-density viral particles were secreted. Therefore, this cell culture system provides a unique model with which to decipher the influence of cell polarization on the HCV life cycle.
Most studies concerning HCV have been performed in hepatoma cells derived from Huh-7 cell lacking polarization properties. However, hepatocyte polarity likely affects the HCV life cycle. First, at the entry step, cell polarity can affect the cell surface distribution of HCV coreceptors, as shown in this work. Polarization can also affect the route of virus internalization. Indeed, for influenza virus, the use of clathrin-mediated endocytosis is more predominant in polarized cells than in nonpolarized cells, in which the virus can also enter by clathrin-independent endocytosis (23). The release of newly synthesized virions can also be influenced by polarization of the intracellular trafficking. Cell polarization can therefore also affect virus propagation in the liver.
To address the role of hepatocyte polarity, culture models have been developed to stimulate cellular polarization of Huh-7 cells. First, when they are grown in rotating-wall vessels that stimulate microgravity, Huh-7 cells are able to form multilayers and exhibit relocalization of polarity markers, but without forming bile canaliculus-like structures (24). In contrast, the formation of canaliculus-like structures has been observed in spheroids that develop when Huh-7 cells are grown in Matrigel (25). In both three-dimensional (3D) models, Huh-7 cells were susceptible to HCV to the same extent as cells grown in 2D models. HepG2 cells are also able to form bile canaliculus-like structures that are closed by tight junctions, and therefore, HepG2-CD81 cells have been used to study HCV despite lower levels of replication in these cells (26). All these models have provided valuable information regarding cell polarity, but because they have a hepatocytic organization with multiple apical and basolateral membrane domains, the specific function of each pole regarding HCV entry or release is difficult to address. An attempt was made to investigate these specific functions by using the Caco-2 human colon carcinoma cell line, which is able to polarize in cell culture (27). Unfortunately, levels of HCV infection in these cells are low, and surprisingly, HCV infects Caco-2 cells at their apical domains.
In this work, we isolated two HepG2-CD81 clones with polarization properties similar to those of epithelial cells, allowing independent access to the basolateral or apical pole. It has been reported that HepG2 cells lack expression of the liver-specific microRNA miR122, necessary for HCV replication. However, a MIR122 gene is present in HepG2 cells, suggesting that under some conditions it may be expressed (28). Our two clones, selected for their polarization properties, could be infected by HCV, but infectivities were lower than for Huh-7 cells. Indeed, infectivities of 4.06 ± 0.19 and 4.19 ± 0.13 log10 FFU/ml were observed in clone 15 and clone 1SC3, respectively, compared to 5.45 ± 0.19 in Huh-7 cells. Accordingly, HCV RNA loads in infected polarized cells were also approximately 1.5 to 2 log10 units below viral genome loads in Huh-7 cells. When we analyzed miR122 expression, we detected very low but detectable levels of miR122 expression in HepG2-CD81 cells. However, we observed higher levels of expression of miR122 in clones 15 and 1SC3, suggesting that by isolating clones by limiting dilution, cells that have a higher miR122 expression level than the general cell population were selected. Therefore, the ectopic expression of miR122 was not needed in these cell clones to achieve a good level of HCV replication. However, despite a level of miR122 similar to that in Huh-7 cells, clone 1SC3 presented lower infectivity, suggesting that other factors, such as HCV (co)receptor expression, may account for the differences observed.
Cellular polarization does not restrict HCV infection. Indeed, when polarized HepG2-CD81 cells were infected at the basolateral domain, the level of HCV infection was similar to infection levels obtained in nonpolarized cells (Fig. 4). Similar results have been obtained previously in Huh-7 or Caco-2 cells (24, 27). However, under our experimental conditions, productive infections were observed only when the virus was added at the basolateral domain of the cells. In contrast, virus attachment was not restricted to this membrane domain. Our results differ from those obtained in Caco-2 cells, in which HCV preferentially infects cells from the apical domain (24, 27). This difference likely results from the use of cell lines with different origins; Caco-2 cells have an intestinal epithelial origin. Inefficiency of HCV infection at the apical domain of polarized HepG2-CD81 cells can be explained by the specific localization of CD81 at the basolateral domain of the cells. It has also been reported previously that in polarized epithelial cells, CD81 is localized at the basolateral membrane of the cells (29); however, in hepatocyte or Caco-2 cells, CD81 is detected on both poles (27). The differences observed in protein partitioning between the two membrane domains in different cell lines could result from the extent of polarization achieved by the cell lines.
Upon infection, HCV circulates in the bloodstream and crosses the sinusoidal endothelium to reach the space of Disse, where the virus encounters the basolateral surface of the hepatocyte. Therefore, it is not surprising that the virus is able to take advantage of this membrane domain to invade cells. In polarized HepG2-CD81 cells, CLDN1 and OCLN were mainly localized at the tight junctions, but we cannot exclude extrajunctional pools of these proteins that were not detected. Tight junctions form a physical barrier that regulates the transport of ions, solutes, and cells across the paracellular space, but they are also involved in accumulation and anchoring of signaling proteins and play a key role in connecting the cytoskeleton and intercellular junctions. Tight junctions are the target of several viruses and bacteria. For instance, coxsackievirus B virus infects enterocytes via internalization at the tight junctions (30). So far, the role of tight junctions in HCV entry remains poorly understood, but our polarized HepG2-CD81 cell model will be a unique tool to address this question.
In polarized HepG2-CD81 cells, HCV was predominantly secreted from the basolateral domain. Over the years, valuable progress has been made regarding HCV assembly and secretion; however, the detailed mechanisms are not yet elucidated. Th virus is secreted as an LVP, interacting with lipoproteins and apolipoproteins. Therefore, HCV likely usurps the VLDL production machinery, but it remains unclear how this interplay takes place. For the correct delivery of serum proteins and bile acids in the appropriate compartment, the intracellular trafficking is also polarized. It is known that VLDL are secreted at the basolateral domain of hepatocytes and delivered into the blood. Furthermore, in our experimental setting, we found that apoE undergoes vectorial secretion, with more than 80% of the protein released in the basolateral chamber. Accordingly, secretion of HCV also followed the same pathway, since more than 90% of released infectious virions were secreted from the basolateral domain of the cells.
HepG2 cells have a defect in VLDL biosynthesis that can be partially corrected by inhibition of the MEK/ERK pathway (22). A study by Jammart et al. demonstrated that when treated with a combination of OA and U0126, a MEK/ERK inhibitor, HepG2 cells secrete lipoproteins with lower density; however, these conditions did not affect LVP density (31). In contrast, our polarized HepG2-CD81 cells released HCV particles with lower density, suggesting that polarization of cells modifies their intracellular trafficking, which promotes lipid sorting and favors virus-lipid interactions.
In conclusion, our study sheds light on the role of hepatocyte polarization in HCV entry and egress, and our HepG2-CD81 cell culture system provides a unique model to decipher the interplay between HCV infection and cell polarization. Finally, this simple polarized cell culture model also provides the opportunity to produce and characterize viral particles that exhibit biophysical properties similar to those of patient virus.
MATERIALS AND METHODS
Cell culture.
HEK293T/17 cells (ATCC), Huh-7 cells (32), and HepG2-CD81 cells (kindly provided by F. L. Cosset, ENS, Lyon, France) were cultured in Dulbecco's modified Eagle medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum and 2 mM GlutaMax (Life Technologies).
Antibodies.
The following antibodies were used in this work: anti-ZO-1 (Zymed; Life Technologies), anti-CD26 (M-A261; AbD; Serotec), anti-SR-BI (BD Transduction Laboratories), and anti-HA (3F10; Roche).
Virus.
The virus used in this study was based on the JFH1 strain engineered to reconstitute the A4 epitope in E1 (33) with titer-enhancing mutations (34). Plasmids carrying the full-length genome were digested with XbaI and treated with mung bean nuclease (New England BioLabs). In vitro transcriptions were performed using a Megascript kit (Ambion) according to the manufacturer's protocol. Viruses were rescued by electroporation of Huh-7 cells with 10 μg of in vitro-transcribed RNA, as described in reference 35. Viruses were further amplified to increase viral titers. HCV infectivity in the viral stocks was determined by counting FFU, as previously reported (35). For virus purification, highly infectious viral supernatants were collected and cleared by centrifugation. Viruses were concentrated by overnight precipitation with 8% polyethylene glycol (Fluka Chemie AG) and centrifugation at 13,000 × g for 20 min. The pellets were resuspended in 1 ml of cold phosphate-buffered saline (PBS) and layered on top of a 10 to 50% continuous iodixanol gradient (Optiprep; Proteogenix). The gradients were ultracentrifuged for 16 h at 160,000 × g at 4°C in an SW41 rotor. Twelve 1-ml fractions were collected. The density, titer, and HCV RNA content of each fraction were determined. The two most infectious fractions were used for binding experiments. Recombinant HCoV-229E expressing Renilla luciferase (HCoV-229E-Ren), kindly provided by V. Thiel (Bern University), has been described previously (36).
Cell polarization.
To induce the development of cell polarity, HepG2-CD81 cells were seeded on transwell permeable supports (polyester membranes with 3-μm pores; Corning). After 48 h, when the cells were confluent, the medium was replaced with William's medium (Life Technologies) supplemented with 10% fetal bovine serum, 2 mM GlutaMax, 10 ng/ml gentamicin, and 1% DMSO (polarization medium) to induce polarization (day 0). Maximum cell polarity was achieved between days 5 and 10.
Human serum albumin and apoE secretion.
Cells were grown on transwell permeable supports for 21 days. The culture media of the apical and basolateral chambers were collected every day, and fresh medium was added. Supernatants were centrifuged for 1 min at 2,000 rpm to remove cell debris. HSA secreted in the apical and basolateral media was quantified with an enzyme-linked immunosorbent assay (ELISA), as previously described (16). apoE secretion was quantified at days 6 and 9 after inducing polarization by using a human apoE ELISA kit (MabTech), as recommended by the manufacturer.
Lentivirus transduction.
pTRIP-CD81-GFP, pTRIP-SRBI, and pTRIP-cerulean-CD81 were kindly provided by C. M. Rice (Rockefeller University) (12), and pTRIP-OCLN-GFP was described previously (36).
pTRIP-Ha-Tfr was constructed by excision of the mCherry-CD81 coding sequence in pTRIP-mcherry-CD81 between the NheI and XhoI sites. The coding sequence of the transferrin receptor with an HA tag was PCR amplified using pCDNA3-tfR-Ha (37) as a template with forward primer 5′-GTCTCGAGTCAAGCGTAATCTGGAACATCGTATGGGTA-3′ and reverse primer 5′-TGGCTAGCACCATGAAGTGCCTTTTGTACTTAGCCTTT-3′ and cloned in the digested vector. To produce the lentivirus stocks, HEK293T/17 cells were plated in 6-well plates. The next day, the cells were transfected with 500 ng of pTRIP vector, 400 ng gag-pol expressing vector, and 100 ng of vesicular stomatitis virus (VSV) G protein expression vector with Turbofect (Thermofischer) according to the manufacturer's instructions. Virus stocks were collected 48 h after transfection and filtered.
At day 5 after polarization induction, the cells were transduced by adding lentivirus to the apical chamber for 2 h. Then, the cells were washed, and fresh polarization medium was added to each chamber. The cells were fixed 36 h later with 3% paraformaldehyde in PBS and processed for indirect immunofluorescence.
Immunofluorescence.
Polarized cells were fixed and permeabilized with 0.5% Triton X-100 diluted in PBS. Then, the cells were blocked for 10 min in PBS containing 10% goat serum and incubated with anti-ZO-1 (Zymed, Life Technologies) with or without anti-CD26 diluted in blocking solution for 30 min. Then, the cells were washed 3 times in PBS for 5 min each time and were incubated with fluorescent secondary antibodies (cyanine 3-conjugated goat anti-rabbit and Alexa 488-conjugated goat anti-mouse; Jackson ImmunoResearch) and DAPI (4′,6-diamidino-2-phenylindole) diluted in blocking solution. The cells were washed, and the permeable membranes were mounted between a coverslip and a slide with 4-88 Mowiol-based mounting medium.
For membrane staining of HA-TfR and SR-BI, fixed cells were blocked in 10% goat serum in PBS and incubated with anti-HA or anti-SR-BI diluted in blocking buffer for 30 min prior to permeabilization. Then, the cells were permeabilized and further processed for staining of ZO-1 as described above.
Confocal microscopy was carried out with an LSM780 confocal microscope (Zeiss) using a 63× oil immersion objective with a 1.4 numerical aperture. Signals were sequentially collected using single fluorescence excitation and acquisition settings to avoid crossover. Eleven to 21 confocal planes separated by 0.4 μm were acquired. The image stacks were processed using ImageJ, and final figures were assembled using Adobe Photoshop Elements version 6.0.
Relative miR122 expression.
Total RNA was extracted from confluent cells using a mirVana miRNA isolation kit according to the manufacturer's recommendations (Life Technologies). Reverse transcription was performed by using the TaqMan Advanced miRNA cDNA synthesis kit according to the manufacturer's instructions. Then, miR122 and miR423, an endogenous control, expression levels were quantified by real-time quantitative PCR using the TaqMan Advanced miRNA assay according to the manufacturer's recommendations. We used the ΔΔCT method with miR423 as the endogenous control and Huh-7 cells as the calibrator.
HCV infection of polarized cells.
At day 5 after polarization induction, cells were infected by incubating the virus either at the basolateral domain or at the apical domain for 1 h at 37°C at a multiplicity of infection (MOI) of 0.25. Then, the cells were washed and fresh polarization medium with or without 1 nM daclatasvir was added in both chambers. At 30 h postinfection, total cellular RNA was extracted by using the Nucleospin RNA kit (Macherey-Nagel) as recommended by the manufacturer, and HCV genomes were measured by quantitative RT-PCR as previously described (38). Control, nonpolarized cells were seeded on a transwell permeable support and infected in parallel with polarized cells at 48 h after seeding, when the cells were confluent.
HCoV-229E infection of polarized cells.
At day 5 after induction of polarization, cells were infected by incubating the virus either at the basolateral domain or at the apical domain for 1 h at 37°C. At 6 h postinfection, the cells were lysed and luciferase activities were quantified by using the Renilla luciferase assay system (Promega) according to the manufacturer's instructions.
Virus attachment.
Purified virus was diluted in cold DMEM-HEPES (DMEM without bicarbonate containing 25 mM HEPES buffer). The diluted virus was bound to the apical or basolateral domain of polarized cells for 1 h at 4°C. Then, the cells were washed with cold PBS on ice, and total cell RNAs were extracted by using a Nucleospin RNA kit (Macherey-Nagel) as recommended by the manufacturer. HCV genomes were measured by quantitative RT-PCR as previously described (38).
Secretion.
At day 5 after inducing cell polarization, the cells were infected at the basolateral domain at 37°C for 1 h. Then, the cells were washed, and fresh polarization medium was added to both chambers. Forty-eight hours later, the supernatants from both chambers were collected. HCV infectivity in each supernatant (apical and basolateral) was quantified by counting the FFU, as previously reported (35). RNAs were extracted by using a QIAamp viral RNA kit (Qiagen) to measure HCV genomes in the supernatant by real-time quantitative RT-PCR, as previously described (38).
Density profiles of virions secreted in the presence of MEK inhibitors.
Clone 15 polarized cells grown on transwell plates were infected by incubating the virus at the basolateral domain for 1 h at 37°C. Then, the cells were washed, and fresh polarization medium was added to both chambers. Twenty-four hours later, the medium was replaced with polarization medium containing 1 μM U0126 and 10% (vol/vol) OA-bovine serum albumin (BSA). The basolateral media of 3 transwells for each condition were collected 24 h later and pooled before loading on a 10 to 50% iodixanol gradient. The gradients were ultracentrifuged for 16 h at 160,000 × g at 4°C in a SW41 rotor. Twelve 1-ml fractions were collected, and the titer and density of each fraction were determined. For nonpolarized cells, clone 15 cells or Huh-7 cells were infected, and 24 h later, the cells were incubated in culture medium supplemented with 1% DMSO, 1 μM U0126, and 10% (vol/vol) OA-BSA.
Graphs and statistics.
Prism v5.0c software (GraphPad Software Inc., La Jolla, CA) was used to prepare graphs and to determine the statistical significance of differences between data sets.
ACKNOWLEDGMENTS
We thank F. L. Cosset, C. M. Rice, V. Thiel, and T. Wakita for providing essential reagents. The immunofluorescence analyses were performed with the help of the imaging core facility of the BioImaging Center, Lille, Nord-de-France.
This work was supported by a Marie Curie International Reintegration Grant (PIRG-GA-2009-256300) and by the French National Agency for Research on AIDS and Viral Hepatitis (ANRS) and the ANR through the ERA-NET Infect-ERA program (ANR-13-IFEC-0002-01).
S.B., K.S., Y.R., and J.D. conceived and designed the experiments; S.B., A.D., and L.F. performed the experiments; S.B., K.S., Y.R., and J.D. analyzed the data; and S.B. and J.D. wrote the paper.
REFERENCES
- 1.Treyer A, Müsch A. 2013. Hepatocyte polarity. Compr Physiol 3:243–287. doi: 10.1002/cphy.c120009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steed E, Balda MS, Matter K. 2010. Dynamics and functions of tight junctions. Trends Cell Biol 20:142–149. doi: 10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Grassi G. 2016. Hepatitis C virus relies on lipoproteins for its life cycle. World J Gastroenterol 22:1953. doi: 10.3748/wjg.v22.i6.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuhara T, Ono C, Puig-Basagoiti F, Matsuura Y. 2015. Roles of lipoproteins and apolipoproteins in particle formation of hepatitis C virus. Trends Microbiol 23:618–629. doi: 10.1016/j.tim.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 5.André P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Bréchot C, Paranhos-Baccalà G, Lotteau V. 2002. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol 76:6919–6928. doi: 10.1128/JVI.76.14.6919-6928.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merz A, Long G, Hiet M-S, Brügger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piver E, Boyer A, Gaillard J, Bull A, Beaumont E, Roingeard P, Meunier J-C. 2017. Ultrastructural organisation of HCV from the bloodstream of infected patients revealed by electron microscopy after specific immunocapture. Gut 66:1487–1495. doi: 10.1136/gutjnl-2016-311726. [DOI] [PubMed] [Google Scholar]
- 8.Meunier J-C, Russell RS, Engle RE, Faulk KN, Purcell RH, Emerson SU. 2008. Apolipoprotein c1 association with hepatitis C virus. J Virol 82:9647–9656. doi: 10.1128/JVI.00914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mancone C, Steindler C, Santangelo L, Simonte G, Vlassi C, Longo MA, D'Offizi G, Di Giacomo C, Pucillo LP, Amicone L, Tripodi M, Alonzi T. 2011. Hepatitis C virus production requires apolipoprotein A-I and affects its association with nascent low-density lipoproteins. Gut 60:378–386. doi: 10.1136/gut.2010.211292. [DOI] [PubMed] [Google Scholar]
- 10.Douam F, Lavillette D, Cosset F-L. 2015. The mechanism of HCV entry into host cells. Prog Mol Biol Transl Sci 129:63–107. doi: 10.1016/bs.pmbts.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 12.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouillé Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meertens L, Bertaux C, Dragic T. 2006. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol 80:11571–11578. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decaens C, Durand M, Grosse B, Cassio D. 2008. Which in vitro models could be best used to study hepatocyte polarity? Biol Cell 100:387–398. doi: 10.1042/BC20070127. [DOI] [PubMed] [Google Scholar]
- 16.Snooks MJ, Bhat P, Mackenzie J, Counihan NA, Vaughan N, Anderson DA. 2008. Vectorial entry and release of hepatitis A virus in polarized human hepatocytes. J Virol 82:8733–8742. doi: 10.1128/JVI.00219-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koutsoudakis G, Herrmann E, Kallis S, Bartenschlager R, Pietschmann T. 2007. The level of CD81 cell surface expression is a key determinant for productive entry of hepatitis C virus into host cells. J Virol 81:588–598. doi: 10.1128/JVI.01534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narbus CM, Israelow B, Sourisseau M, Michta ML, Hopcraft SE, Zeiner GM, Evans MJ. 2011. HepG2 cells expressing microRNA miR-122 support the entire hepatitis C virus life cycle. J Virol 85:12087–12092. doi: 10.1128/JVI.05843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yanaga K, Yoshizumi T, Uchiyama H, Okano S, Takenaka K, Sugimachi K. 1997. Detection of hepatitis C virus RNA in bile. Am J Gastroenterol 92:1927–1928. [PubMed] [Google Scholar]
- 20.Haruna Y. 2001. Detection of hepatitis C virus in the bile and bile duct epithelial cells of hepatitis C virus-infected patients. Hepatology 33:977–980. doi: 10.1053/jhep.2001.23435. [DOI] [PubMed] [Google Scholar]
- 21.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, Rice CM. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci U S A 103:3805–3809. doi: 10.1073/pnas.0511218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai J, Qiu W, Kohen-Avramoglu R, Adeli K. 2007. MEK-ERK inhibition corrects the defect in VLDL assembly in HepG2 cells: potential role of ERK in VLDL-ApoB100 particle assembly. Arterioscler Thromb Vasc Biol 27:211–218. doi: 10.1161/01.ATV.0000249861.80471.96. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Whittaker GR. 2014. Influenza entry pathways in polarized MDCK cells. Biochem Biophys Res Commun 450:234–239. doi: 10.1016/j.bbrc.2014.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sainz B, Tencate V, Uprichard SL. 2009. Three-dimensional Huh7 cell culture system for the study of hepatitis C virus infection. Virol J 6:103. doi: 10.1186/1743-422X-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molina-Jiménez F, Benedicto I, Dao Thi VL, Gondar V, Lavillette D, Marin JJ, Briz O, Moreno-Otero R, Aldabe R, Baumert TF, Cosset F-L, López-Cabrera M, Majano PL. 2012. Matrigel-embedded 3D culture of Huh-7 cells as a hepatocyte-like polarized system to study hepatitis C virus cycle. Virology 425:31–39. doi: 10.1016/j.virol.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 26.Mee CJ, Harris HJ, Farquhar MJ, Wilson G, Reynolds G, Davis C, van IJzendoorn SCD, Balfe P, McKeating JA. 2009. Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J Virol 83:6211–6221. doi: 10.1128/JVI.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mee CJ, Grove J, Harris HJ, Hu K, Balfe P, McKeating JA. 2008. Effect of cell polarization on hepatitis C virus entry. J Virol 82:461–470. doi: 10.1128/JVI.01894-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamad IAY, Fei Y, Kalea AZ, Yin D, Smith AJP, Palmen J, Humphries SE, Talmud PJ, Walker AP. 2015. Demonstration of the presence of the “deleted” MIR122 gene in HepG2 cells. PLoS One 10:e0122471. doi: 10.1371/journal.pone.0122471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yáñez-Mó M, Tejedor R, Rousselle P, Sánchez-Madrid F. 2001. Tetraspanins in intercellular adhesion of polarized epithelial cells: spatial and functional relationship to integrins and cadherins. J Cell Sci 114:577–587. [DOI] [PubMed] [Google Scholar]
- 30.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 31.Jammart B, Michelet M, Pecheur E-I, Parent R, Bartosch B, Zoulim F, Durantel D. 2013. Very-low-density lipoprotein (VLDL)-producing and hepatitis C virus-replicating HepG2 cells secrete no more lipoviroparticles than VLDL-deficient Huh7.5 cells. J Virol 87:5065–5080. doi: 10.1128/JVI.01405-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res 42:3858–3863. [PubMed] [Google Scholar]
- 33.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouillé Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. 2007. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol 88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 35.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fénéant L, Ghosn J, Fouquet B, Helle F, Belouzard S, Vausselin T, Séron K, Delfraissy J-F, Dubuisson J, Misrahi M, Cocquerel L. 2015. Claudin-6 and occludin natural variants found in a patient highly exposed but not infected with hepatitis C virus (HCV) do not confer HCV resistance in vitro. PLoS One 10:e0142539. doi: 10.1371/journal.pone.0142539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belouzard S, Delcroix D, Rouillé Y. 2004. Low levels of expression of leptin receptor at the cell surface result from constitutive endocytosis and intracellular retention in the biosynthetic pathway. J Biol Chem 279:28499–28508. doi: 10.1074/jbc.M400508200. [DOI] [PubMed] [Google Scholar]
- 38.Castelain S, Descamps V, Thibault V, François C, Bonte D, Morel V, Izopet J, Capron D, Zawadzki P, Duverlie G. 2004. TaqMan amplification system with an internal positive control for HCV RNA quantitation. J Clin Virol 31:227–234. doi: 10.1016/j.jcv.2004.03.009. [DOI] [PubMed] [Google Scholar]