

ABSTRACT
The Western Reserve (WR) strain of mature vaccinia virus contains an A26 envelope protein that mediates virus binding to cell surface laminin and subsequent endocytic entry into HeLa cells. Removal of the A26 protein from the WR strain mature virus generates a mutant, WRΔA26, that enters HeLa cells through plasma membrane fusion. Here, we infected murine bone marrow-derived macrophages (BMDM) with wild-type strain WR and the WRΔA26 mutant and analyzed viral gene expression and cellular innate immune signaling. In contrast to previous studies, in which both HeLa cells infected with WR and HeLa cells infected with WRΔA26 expressed abundant viral late proteins, we found that WR expressed much less viral late protein than WRΔA26 in BMDM. Microarray analysis of the cellular transcripts in BMDM induced by virus infection revealed that WR preferentially activated type 1 interferon receptor (IFNAR)-dependent signaling but WRΔA26 did not. We consistently detected a higher level of soluble beta interferon secretion and phosphorylation of the STAT1 protein in BMDM infected with WR than in BMDM infected with WRΔA26. When IFNAR-knockout BMDM were infected with WR, late viral protein expression increased, confirming that IFNAR-dependent signaling was differentially induced by WR and, in turn, restricted viral late gene expression. Finally, wild-type C57BL/6 mice were more susceptible to mortality from WRΔA26 infection than to that from WR infection, whereas IFNAR-knockout mice were equally susceptible to WR and WRΔA26 infection, demonstrating that the ability of WRΔA26 to evade IFNAR signaling has an important influence on viral pathogenesis in vivo.
IMPORTANCE The vaccinia virus A26 protein was previously shown to mediate virus attachment and to regulate viral endocytosis. Here, we show that infection with strain WR induces a robust innate immune response that activates type 1 interferon receptor (IFNAR)-dependent cellular genes in BMDM, whereas infection with the WRΔA26 mutant does not. We further demonstrated that the differential activation of IFNAR-dependent cellular signaling between WR and WRΔA26 not only is important for differential host restriction in BMDM but also is important for viral virulence in vivo. Our study reveals a new property of WRΔA26, which is in regulating host antiviral innate immunity in vitro and in vivo.
KEYWORDS: BMDM, interferon signaling, microarray, vaccinia virus
INTRODUCTION
Vaccinia virus is an Orthopoxvirus of the family Poxviridae. It is characterized by its large 190-kb double-stranded DNA genome that contains more than 200 open reading frames (ORFs) (1). Vaccinia virus replicates in defined areas in the host's cytoplasm called viral factories. Viral gene expression is regulated through a cascade involving early, intermediate, and late genes, followed by complex virion morphogenesis to produce three forms of virus particles, namely, mature virus (MV), wrapped virus, and extracellular virus (EV) (2). MV particles have a single membrane and are the most abundant infectious particles produced in infected cells (3). Proteomic analyses have revealed that MVs are composed of 76 to 80 viral proteins, implying highly complex viral protein-protein interactions at multiple stages of the vaccinia virus life cycle (4–6).
When vaccinia MV infects cells, it binds to surface glycosaminoglycans and laminin through four attachment proteins: A27, D8, H3, and A26 (7–12). Previous data have shown that after attaching to HeLa cells, MV subsequently clusters at cell surface lipid rafts and interacts with the cellular surface receptors integrin β1 (13) and CD98 (14), which triggers downstream Akt and mitogen-activated protein kinase signaling for fluid-phase endocytosis (15). Mercer and Helenius, on the other hand, reported that apoptotic mimicry drives macropinocytosis of vaccinia MV (16), and a role for the phosphatidylserine (PS) receptor Axl has been implicated (17). To date, it has been clear that anionic lipids are important MV membrane components (18, 19), but whether a PS receptor plays a role in vaccinia MV entry remains controversial (19–21). After being internalized into endosomal vesicles (22, 23), acidic environments trigger a conformational change in the A26 protein so that it dissociates from the viral entry fusion complex (EFC) and activates membrane fusion to release viral cores into the cytoplasm (24, 25).
These above-described studies were mainly performed in HeLa cells with the Western Reserve (WR) strain of vaccinia MV. However, it is known that the specificity of the vaccinia MV entry pathway is dependent on the cell type (26–28), as well as the viral strain (29–31). By comparing the MV entry pathways of several different vaccinia virus strains, we previously showed that removal of the viral A26 protein from MV particles of the WR strain, resulting in the WRΔA26 mutant, induces the preferential entry of WRΔA26 particles into HeLa cells via the plasma membrane route (24). We have proposed that the viral A26 protein functions as an MV fusion suppressor (25). MV of several vaccinia virus strains (such as IHD-W and MVA), which lack the A26 protein, consistently became fusion competent at neutral pH and entered cells through plasma membrane fusion (24). Studies of the entry of WR and WRΔA26 into cells of the CHO-K1 and L cell lines also support these findings (24). Although the virus entry pathways differ, the WR and WRΔA26 viruses replicate equally well in these cell lines, exhibiting similar MV and EV yields (12, 24), demonstrating that the A26 protein does not affect postentry viral growth in these cell lines.
In this study, we extended vaccinia virus infections into macrophages. Macrophages are important in innate immune defense, removing pathogens and secreting cytokines that recruit neutrophils and that activate B and T cells during vaccinia virus or other respiratory virus infections (32–34). Early reports showed that vaccinia virus infections in primary and immortalized murine macrophages induced apoptosis and that the virus life cycle became abortive (35–38). Furthermore, infections of macrophages by the MVA strain of vaccinia virus were shown to trigger strong innate immunity and cytokine expression through interferon regulatory factor 3 (IRF3) and NF-κB activation (37, 39, 40). In contrast, WR virus infections did not induce substantial changes in immunomodulatory genes (40), possibly because the genome of the strain encodes viral genes that antagonize host immune responses (41, 42). Here, we infected bone marrow-derived macrophages (BMDM) with WR and WRΔA26 and monitored cellular gene responses after virus infection in order to investigate whether WR and WRΔA26 induce different innate immune signaling that, in turn, differentially regulates viral gene expression in BMDM. We also investigated whether any differential cellular signaling induced by WR and WRΔA26 contributes to viral pathogenesis in vivo.
RESULTS
Differential viral late gene expression in BMDM infected with WR-A4mCherry or WRΔA26-A4mCherry.
We infected BSC40 cells and BMDM with two recombinant vaccinia viruses, WR-A4mCherry or WRΔA26-A4mCherry, and monitored fluorescent A4mCherry expression using live imaging analyses (see Video S1 in the supplemental material). Strong signals for the fluorescent A4mCherry protein were readily detected at 8 to 12 h postinfection (p.i.) in BSC40 cells infected with WR-A4mCherry or WRΔA26-A4mCherry. However, strong signals for fluorescent A4mCherry expression were detected only in BMDM infected with WRΔA26-A4mCherry and not in BMDM infected with WR-A4mCherry (Fig. 1A). We quantified A4mCherry fluorescence in the infected cells by fluorescence-activated cells sorting (FACS) (Fig. 1B). In BSC40 cells infected with WR-A4mCherry or WRΔA26-A4mCherry, similarly high levels of A4mCherry (91.1% and 74.5%, respectively) were detected, whereas BMDM infected with WR exhibited a reduced level of A4mCherry compared to WRΔA26-infected BMDM (8.79% and 63.3%, respectively). This is a novel finding, since WR and WRΔA26 grew equally well and formed plaques of similar size in several previously tested cell lines, such as BSC40, HeLa, and L cells and mouse embryonic fibroblasts (12, 24; data not shown). We then infected cells of a mouse macrophage cell line, RAW 264.7, with each virus, and, like BMDM, at 8 h p.i. WR-A4mCherry expressed less A4mCherry than WRΔA26-A4mCherry (data not shown), raising the possibility that innate immune cells such as BMDM respond differently to WR and WRΔA26 infection and can impose differential restriction on viral late gene expression through activation of host antiviral signaling. Despite the differential late A4mCherry expression between WR-A4mCherry and WRΔA26-A4mCherry described above, neither WR-A4mCherry nor WRΔA26-A4mCherry grew well in BMDM (data not shown).
FIG 1.

Differential vaccinia viral gene expression in BMDM infected with WR-A4mCherry and WRΔA26-A4mCherry viruses. (A) WR-A4mCherry and WRΔA26-A4mCherry were used to infect BSC40 cells (MOI = 5 PFU/cell) and BMDM (MOI = 20 PFU/cell), and fluorescent expression of the A4mCherry protein from the viral late promoter was recorded by live imaging analysis using a Zeiss LSM510 META-NLO confocal microscope. Images were photographed at 16 h p.i. (B) Flow cytometry analyses of A4mCherry protein expression from infected BSC40 cells and BMDM, as described in the legend to panel A. Contours represent cell populations positive for A4mCherry fluorescence. The blue outline marks A4mCherry-expressing cells, and the numbers above the outline represent the percentages of fluorescence-positive cells. The x axis represents fluorescence intensity, and the y axis represents propidium iodide (PI) staining.
WR preferentially upregulated interferon receptor-dependent host gene expression, but WRΔA26 did not.
To investigate host cell signaling responses after WR or WRΔA26 infection in BMDM, we performed microarray analyses using total RNA isolated from mock-infected or virus-infected BMDM at 1, 2, 4, and 8 h p.i. Microarray data were compiled from three independent infection experiments covering 31,612 probes representing 22,707 genes on an Agilent G3 mouse array. Cellular transcript levels in WR- and WRΔA26-infected cells were normalized to those in mock-infected cells at each time point, as described in Materials and Methods. We marked genes as upregulated if the increase in the fold change (FC) in expression was more than 2 (FC > 2) or as downregulated if the decrease in the fold change in expression was more than 2 (FC < −2). As shown in Fig. 2A, most changes in cellular transcript levels were observed at later time points, with maximal differences occurring at 8 h p.i., represented by 5.3% and 10.7% of the genes being upregulated in BMDM infected with WR or WRΔA26, respectively, and 13.0% and 19.4% of the genes being downregulated in BMDM infected with WR or WRΔA26, respectively (Fig. 2A). Overall, more cellular transcripts were globally affected in BMDM infected with WRΔA26 than in BMDM infected with WR at each time point (Fig. 2A and B).
FIG 2.

Microarray analysis and IPA of cellular transcripts regulated by WR and WRΔA26 infection in BMDM. (A) Numbers of cellular transcripts that are upregulated more than 2-fold (FC > 2) or downregulated more than 2-fold (FC < −2) at 1, 2, 4, and 8 h p.i. in BMDM infected with WR or WRΔA26. (B) Overlap of upregulated (FC > 2) or downregulated (FC < −2) genes at 1, 2, 4, and 8 h p.i. in BMDM infected with WR or WRΔA26. Numbers in parentheses represent numbers of genes. (C) IPA of canonical pathways of cellular transcripts that are upregulated (FC > 2) at 8 h p.i. in BMDM infected with WR and WRΔA26. The 10 most significant canonical pathways are shown. The complete description of the canonical pathway is provided in Table 1. (D) IPA of canonical pathways of cellular transcripts in 284 ORFs, representing genes upregulated more than 2-fold after WR infection relative to their levels of expression after WRΔA26 infection. The 10 most significant canonical pathways are shown. The complete description of the canonical pathway is provided in Table 2. For panels C and D, the number of genes in each pathway is shown at the end of the black bars; the percentage of genes in each pathway is shown in parentheses.
We then focused on the 8 h p.i. time point to analyze the cellular transcripts that are commonly induced after WR and WRΔA26 infections. We selected cellular genes whose transcript levels fulfilled two criteria for further analyses. First, each cellular transcript level had to be above the background level with a P value of ≤0.05 (43). Second, the gene expression level had to be upregulated more than 2-fold (FC > 2) in both WR- and WRΔA26-infected cells compared with its expression level in mock-infected control cells. Using these criteria, we identified 806 candidate open reading frames (ORFs) that represent genes whose expression was upregulated under both WR and WRΔA26 infection. These 806 ORFs were subjected to Qiagen's Ingenuity pathway analysis (IPA) to provide a comprehensive overview of common host signaling induced after WR and WRΔA26 infection in BMDM (Fig. 2C and Table 1). The 10 most significant canonical pathways involve genes responsible for intrinsic and extrinsic prothrombin activation, as well as Wnt/GSK-3β and G-protein-coupled receptor signaling, including cyclic AMP (cAMP)-mediated signaling, among other functions (Fig. 2C). Currently, little information regarding whether these signaling pathways regulate the vaccinia virus life cycle is known. We searched the literature and found one microarray study by Guerra et al. showing activation of the adenosine A2a receptor by WR infection in HeLa cells (44), whereas another study, by Leao-Ferreira et al., described that activation of the G-protein-coupled receptor inhibited vaccinia virus growth in BSC40 cells (45). Literature searches also found a study by Jia et al. showing that vaccinia virus infections inhibits the Wnt signaling pathway in tumor cells (46).
TABLE 1.
Ten most significant canonical pathways of 806 ORFs
| Canonical pathway | Genes | P valuea |
|---|---|---|
| Intrinsic prothrombin activation pathway | COL3A1, KLK3, COL11A2, F5, SERPINC1 | 1.45E−03 |
| Role of Wnt/GSK-3β signaling in the pathogenesis of influenza virus | CSNK1E, WNT5A, DVL2, IFNB1, APC2, IFNA4, WNT9A | 1.78E−03 |
| G-protein-coupled receptor signaling | ADORA2A, VIPR2, GABBR1, PDE1B, PIK3C2B, GNAL, PRKAR1A, DRD1, GABBR2, GNAS, MAP3K8, DRD4, HTR2C, GRM1, CHRM4, ADRA2A | 7.24E−03 |
| Role of hypercytokinemia/hyperchemokinemia in the pathogenesis of influenza virus | CXCL10, IL-6, IFNB1, IFNA4 | 8.32E03 |
| CDK5 signaling | DRD1, GNAS, CDK5R1, LAMB1, PPP1CB, GNAL, PPP2CA, PRKAR1A | 8.91E−03 |
| Retinoic acid-mediated apoptosis signaling | TIPARP, CRABP2, IFNB1, RARB, IFNA4 | 9.12E−03 |
| Extrinsic prothrombin activation pathway | F3, F5, SERPINC1 | 1.07E−02 |
| Axonal guidance signaling | MYL4, SEMA3E, WNT5A, PGF, SEMA3B, PIK3C2B, EPHB2, SRGAP1, RGS3, GNAL, PRKAR1A, ABLIM3, GNAS, SLIT2, SEMA6C, BMP4, SEMA3A, NFAT5, SRGAP2, EFNA2, PLXNA, WNT9A | 1.45E−02 |
| cAMP-mediated signaling | ADORA2A, VIPR2, GABBR1, PDE1B, GNAL, PRKAR1A, DRD1, GABBR2, GNAS, DRD4, CHRM4, ADRA2A, PKIB | 1.55E−02 |
| Role of lipids/lipid rafts in the pathogenesis of influenza virus | IFNB1, IFNA4 | 2.57E−02 |
A canonical pathway was considered significant if P was <0.05. Data were analyzed using Qiagen's Ingenuity pathway analysis (IPA).
We hypothesized that WR infection but not WRΔA26 infection of BMDM preferentially activates cellular antiviral signaling to inhibit A4mCherry expression in BMDM at 8 h p.i. (Fig. 1), so we were more interested in identifying those cellular transcripts differentially upregulated in BMDM infected with WR compared to their regulation in BMDM infected with WRΔA26. Thus, we selected cellular genes whose transcript levels fulfilled three criteria for further analyses. First, each gene expression level had to be above the background level with a P value of ≤0.05 (43). Second, the gene expression level in WR-infected BMDM had to be upregulated compared to its expression level in mock-infected control cells at 8 h p.i. Third, the gene expression level had to be 2-fold higher (FC > 2) in BMDM infected with WR than in BMDM infected with WRΔA26. Using these criteria, we identified 284 candidate genes, named “284 ORFs” (see Supplemental Material S2). When these 284 ORFs were subjected to IPA, we found that several canonical cellular pathways were activated in WR-infected BMDM but not in BMDM infected with WRΔA26, including pathways involved in cytosolic pattern recognition, as well as TREM1, death receptor, and interferon signaling, among others (Fig. 2D and Table 2).
TABLE 2.
Ten most significant canonical pathways of 284 ORFs
| Canonical pathway | Genes | P valuea |
|---|---|---|
| Activation of IRF by cytosolic pattern recognition receptors | DHX58, IRF7, STAT1, STAT2, IFIH1, DDX58, IL-10, ZBP1, IFIT2, CD40, IL-6, IKBKE | 2.51E−12 |
| TREM1 signaling | JAK2, TLR3, CCL2, NLRC5, NOD1, IL-10, CD40, IL-6, ICAM1, CD86 | 8.13E−09 |
| Dendritic cell maturation | IL-15, JAK2, FCGR1A, STAT2, CD40, IL-6, IL1RN, TLR3, STAT1, IL-10, ICAM1, CD86, FCGR3A/FCGR3B, IKBKE | 2.14E−08 |
| Communication between innate and adaptive immune cells | IL-15, IL1RN, TLR3, CXCL10, IL-10, CCL4, CD40, IL-6, CCL3L3, CD86 | 4.37E−08 |
| Death receptor signaling | PARP12, PARP10, PARP11, TNFSF15, TNFSF10, PARP9, PARP14, FAS, CYCS, IKBKE | 6.03E−08 |
| Interferon signaling | OAS1, JAK2, STAT1, IFIT3, STAT2, IFI35, TAP1 | 1.02E−07 |
| Role of pattern recognition receptors in recognition of bacteria and viruses | EIF2AK2, OAS1, IRF7, C3, TLR3, IFIH1, NOD1, OAS2, DDX58, IL-10, IL-6 | 3.02E−07 |
| Altered T-cell and B-cell signaling in rheumatoid arthritis | IL-15, IL1RN, TLR3, CSF1, IL-10, FAS, CD40, IL-6, CD86 | 5.75E−07 |
| Role of macrophages, fibroblasts, and endothelial cells in rheumatoid arthritis | IL-15, JAK2, FCGR1A, IL-6, IL1RN, TLR3, CCND1, CCL, CSF1, Prss1, IL-10, TRAF1, ICAM1, FCGR3A/FCGR3B, IKBKE | 1.51E−06 |
| Allograft rejection signaling | H2-M2, H2-T9, H2-T10, IL-10, H2-M5, FAS, CD40, CD86 | 3.55E−06 |
A canonical pathway was considered significant if P was <0.001. Data were analyzed using Qiagen's Ingenuity pathway analysis (IPA).
We compared our 284 ORFs with a gene list derived from a study by Tong et al. (47) that investigated the innate immune gene signaling network in mouse BMDM stimulated with lipid A (a lipid component of lipopolysaccharide) and found that 47 of our 284 ORFs were type 1 interferon receptor (IFNAR)-dependent secondary response genes, 72.3% of the genes reported by Tong et al. (47) (Fig. 3A and Table 3). We also found that 20 of our 284 ORFs were primary response genes involved in inflammation and functions of blood cells (Fig. 3B and Table 3). Eight of our 284 ORFs are involved in the secondary IFNAR-independent response that is mostly involved in T-cell activation (Fig. 3C and Table 3). Taken together, our results revealed that the IFNAR-dependent secondary response pathway is preferentially activated in WR-infected BMDM at 8 h p.i. and not in WRΔA26-infected cells.
FIG 3.

Heatmaps of cellular genes that are preferentially upregulated in BMDM infected with WR and not in BMDM infected with WRΔA26. (A) Heatmap showing the 47 genes out of the 284 ORFs that are IFNAR-dependent secondary response genes. Genes in the dark green panel are also present in the cluster 19 group described in Fig. 4A. (B) Heatmap showing the 20 genes out of the 284 ORFs that are primary response genes involved in inflammation and blood cell functions. (C) Heatmap showing the 8 genes out of 284 ORFs that are IFNAR-independent secondary response genes. W1, W2, W4, and W8, expression of cellular genes at 1, 2, 4, and 8 h p.i. in BMDM infected with WR, respectively; A1, A2, A4, and A8, expression of cellular genes at 1, 2, 4, and 8 h p.i. in BMDM infected with WRΔA26, respectively.
TABLE 3.
Comparison of gene classes defined by Tong et al. (47) with the 284 ORFs identified in this study
| Group | Gene class | Function | No. of genes found by Tong et al. (47) | No. of genes in 284 ORFs (% genes found by Tong et al. [47]). |
|---|---|---|---|---|
| 1 | IFNAR-dependent secondary response | Antiviral response | 65 | 47 (72.3) |
| 2 | Primary response | Inflammation and functions of blood cells | 132 | 20 (15.1) |
| 3 | IFNAR-independent secondary response | T-cell proliferation, differentiation, and activation | 29 | 8 (27.6) |
We then took a different approach by performing k-means analyses of the total microarray data obtained with 31,612 probes to sort gene groups into different clusters on the basis of characteristic cellular gene expression patterns from 1 to 8 h p.i. in BMDM infected with WR and WRΔA26. As shown in Fig. 4A, k-means analyses identified 25 gene clusters (C1 to C25). Each cluster is represented by two plots, with one representing the relative expression levels of a group of genes at 1, 2, 4, and 8 h p.i. in BMDM infected with WR (after normalization against the level of expression in mock-infected cells) and the second plot representing the relative expression levels of the same group of genes at the same four time points in BMDM infected with WRΔA26. Of the 25 clusters, only cluster 19 (C19) represented 168 genes whose expression was preferentially induced by WR in BMDM at 8 h p.i. but not by WRΔA26 in BMDM (Fig. 4A and Supplemental Material S3). We then performed scatter plot analyses (Fig. 4B) for each cluster by plotting the normalized gene expression levels in WR-infected cells at each time point (on the x axis) against those in WRΔA26-infected cells (on the y axis). If gene expression within a given cluster is equally up- or downregulated by WR and WRΔA26, we expected to observe a positive linear relationship covering all four time points (Fig. 4B). Most of the clusters fitted our prediction, with positive and high correlation coefficients (r) of 0.71 to 0.99. Only C19 exhibited a deviant correlation coefficient of −0.2 (Fig. 4C). Therefore, cluster 19 represents cellular genes whose expression levels are preferentially upregulated in BMDM infected with WR but not those infected with WRΔA26.
FIG 4.

Characteristic expression patterns represented in clusters 1 to 25. (A) k-means clustering (C1 to C25, for a total of 25 clusters) of genes based on Euclidean distance metrics of expression over time for WR- and WRΔA26-infected BMDM compared to the levels of expression in mock-infected control cells and plotted as the relative expression level for each gene. Each cluster has eight data points per gene: data for WR-infected cells at 1, 2, 4, and 8 h p.i. (W1, W2, W4, and W8 in the first plot), followed by data for WRΔA26-infected cells at 1, 2, 4, and 8 h p.i. (A1, A2, A4, and A8 in the second plot). (B) Scatter plot of log2 ratios of normalized gene expression (at 1, 2, 4, and 8 h p.i.) for WR-infected cells on the x axis and WRΔA26-infected cells on the y axis for each of the 25 clusters. (C) Correlation coefficients (r) for each cluster in the scatter plot. The weak correlation coefficient of cluster 19 (r = −0.2079) is highlighted in red.
Finally, we compared the genes of cluster 19 with our previously identified 284 ORFs and with 65 IFNAR-dependent secondary response genes identified by Tong et al. (47) and found significant overlap (Fig. 5). One hundred forty (∼83%) of the cluster 19 genes were also represented in our 284 ORFs, corresponding to almost half (∼49%) of the 284 ORFs. Furthermore, 38 (∼23%) of the cluster 19 genes were also identified to be IFNAR-dependent secondary response genes in the study by Tong et al. (47). In total, 49 (∼75%) of the IFNAR-dependent response genes were identified in the 284 ORFs and cluster 19. Taken together, our microarray analyses have revealed that multiple signaling pathways are activated by WR and WRΔA26 infection in BMDM. Despite the complexity of gene regulatory pathways, we can conclude that genes involved in the IFNAR-dependent secondary response are the major transcriptional units that are upregulated in BMDM infected with WR but not those infected with WRΔA26.
FIG 5.
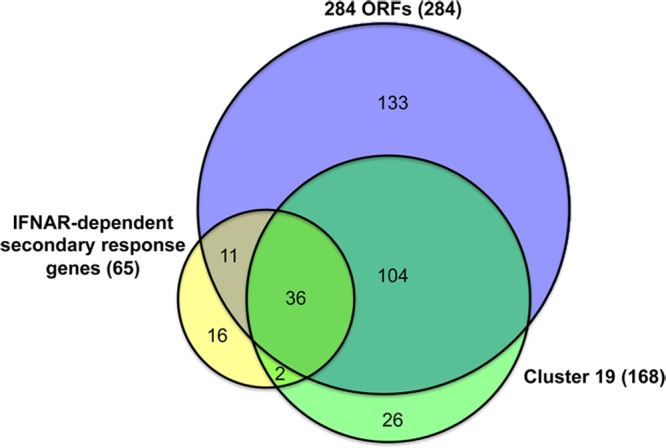
Gene overlap among three different data sets. Colored circles represent different gene lists. Purple, 284 ORFs; green, genes in cluster 19; yellow, the 65 IFNAR-dependent secondary response genes described by Tong et al. (47).
Activation of IFNAR-dependent signaling in BMDM infected with WR MV.
We then performed quantitative real-time reverse transcription-PCR (qRT-PCR) to validate our microarray analyses, focusing on three genes (IFIT2, IRF7, and ZBP1) that are known to be IFNAR-dependent secondary targets (47). As shown in Fig. 6A, the transcript levels of these three genes were at least 2-fold higher in WR-infected BMDM than in WRΔA26-infected cells at 8 h p.i., supporting the suggestion that WR infection of BMDM preferentially induces IFNAR-dependent secondary response genes. At 8 h p.i., we also collected supernatants from BMDM infected with WR and WRΔA26 for enzyme-linked immunosorbent assay (ELISA) detection of beta interferon (IFN-β) (Fig. 6B) and CXCL10 (a downstream IFNAR-signaling molecule) (Fig. 6C). Indeed, the levels of soluble IFN-β and CXCL10 were higher in the culture medium of BMDM infected with WR than in the culture medium of WRΔA26-infected cells (Fig. 6B and C). The reduced level of soluble IFN-β detected in WRΔA26-infected BMDM was not simply a delayed response, since the amounts of IFN-β did not increase significantly even after overnight culture (data not shown). We analyzed IFN-β-induced IFNAR-dependent signaling by monitoring STAT1 phosphorylation in immunoblots (Fig. 6D). WR infection of BMDM induced STAT1 phosphorylation at residue Y701 at 4 and 8 h p.i., but WRΔA26 infection did not (Fig. 6D). Thus, consistent with our microarray data, these findings showed that WR infection preferentially triggered an IFNAR-dependent secondary response but WRΔA26 infection did not.
FIG 6.

The IFN signaling pathway is activated in BMDM infected with WR but not those infected with WRΔA26. (A) qRT-PCR analyses of cellular transcripts (IFIT2, IRF7, and ZBP1) that are upregulated in WR-infected BMDM at 1, 2, 4, and 8 h p.i. after their levels are normalized to the levels of the same transcripts in WRΔA26-infected BMDM. Data from three independent experiments were averaged, with error bars representing the standard deviations. Data were analyzed using a one-tailed Student's t test. *, P < 0.05. (B and C) Soluble IFN-β (B) and CXCL10 (C) levels at 8 h p.i. in supernatants of BMDM that were mock infected or infected with WR or WRΔA26. Data from three independent experiments were averaged, with error bars representing the standard deviations. Data were analyzed using a two-tailed unpaired Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) Immunoblot analysis of STAT1 phosphorylation in WR- or WRΔA26-infected BMDM at 1, 2, 4, and 8 h p.i. Blots were probed with anti-phospho-STAT1 (Y701) (1:1,000) and anti-STAT1 (1:1,000) antibodies. Anti-β-actin antibody (1:2,000) was included as the loading control. pSTAT1, phosphorylated STAT1.
Rescue of WR late A4mCherry expression in IFNAR-knockout BMDM.
Our results led us to investigate whether the preferential activation of IFN-β signaling suppresses A4mCherry expression in BMDM infected with WR. Therefore, we isolated BMDM from WT and IFNAR-knockout (IFNAR-KO) mice (48) for further analyses. As a control experiment, BMDM from WT and IFNAR-KO mice were treated with poly(I·C) (10 μg/ml) or IFN-β (10 U/ml) for 12 and 4 h, respectively, to induce IFNAR-dependent signaling, as previously described (49–52) (Fig. 7A). We anticipated that poly(I·C) would trigger the activation of the IFN-β–STAT1 axis to increase the level of expression of the IFNAR target gene CXCL10, similar to what we found for exogenous IFN-β (Fig. 6C). Supernatants from BMDM of WT and IFNAR-KO mice were assayed for CXCL10 by ELISA, which confirmed that CXCL10 was specifically induced in WT BMDM but not in IFNAR-KO BMDM (Fig. 7A). WT and IFNAR-KO BMDM were then infected with WR or WRΔA26 and subjected to live imaging analyses, and we found that A4mCherry was detected in BMDM infected with either WR or WRΔA26 (Fig. 7B). Consistently, STAT1 phosphorylation was not observed in IFNAR-KO BMDM infected with either virus (Fig. 7C), indicating that IFNAR-STAT1 signaling was specifically induced in BMDM upon infection with WR virus and consequently restricted viral late A4mCherry gene expression.
FIG 7.

IFNAR KO rescues viral late gene expression in BMDM infected with WR-A4mCherry. (A) Soluble CXCL10 levels in supernatants of WT and IFNAR-KO BMDM pretreated with poly I·C (10 μg/ml) or IFN-β (10 U/ml) for 12 and 4 h, respectively. Data from three independent experiments were averaged, with error bars representing the standard deviations. NA, nontreated samples. (B) WR-A4mCherry and WRΔA26-A4mCherry were used to infect BSC40 cells (MOI = 5 PFU/cell) and IFNAR-KO BMDM (MOI = 20 PFU/cell), and the fluorescent expression of the A4mCherry protein from the viral late promoter was recorded by live imaging analysis using a Zeiss LSM510 META-NLO confocal microscope. The images shown here were photographed at 16 h p.i. (C) Immunoblot analyses of STAT1 phosphorylation at 1, 2, 4, and 8 h p.i. in WT and IFNAR-KO BMDM infected with WR or WRΔA26. The blots were probed with anti-phospho-STAT1 (Y701) (1:1,000) and anti-STAT1 (1:1,000) antibodies. Anti-β-actin antibody (1:2,000) was included as the loading control.
IFNAR-dependent signaling contributes to the differential pathogenesis between WR and WRΔA26 infection in vivo.
We were surprised to observe that only WR MV infection and not WRΔA26 infection triggered IFNAR-dependent signaling in BMDM in vitro. Therefore, we intranasally inoculated C57BL/6 mice with 104 PFU per mouse of WR or WRΔA26 and monitored the mice for signs of infection over a 2-week period. We also examined a previously reported recombinant A26 revertant virus (REV-A26) (25) that had a Flag-tagged A26L ORF reinserted into the WRΔA26 genome. REV-A26 allowed us to assess if any differences in the in vivo pathogenesis between WR and WRΔA26 were due to other mutations in the WRΔA26 genome. As shown in Fig. 8A, WRΔA26 infection caused a more rapid and significant loss of body weight at 4 to 6 days p.i than either WR or REV-A26 infection. Furthermore, WRΔA26 infection induced higher mortality (90% over 2 weeks; Fig. 8B) in mice than WR and REV-A26 infection, showing that deletion of the A26L ORF specifically contributed to enhanced viral virulence in mice. Since the rate of in vivo mortality after REV-A26 infection was comparable to that after WR infection, we considered it appropriate to use only WR and WRΔA26 infections for comparisons in subsequent experiments and thereby reduce the number of mice that had to be used.
FIG 8.

WRΔA26 is more pathogenic than WR during the intranasal route of infection in C57BL/6 mice. (A) C57/BL6 mice (n = 15) were infected intranasally with 104 PFU of WR, WRΔA26, or REV-A26 per mouse and monitored daily for body weight change. Data were analyzed using a two-tailed unpaired Student's t test. **, P < 0.01; ***, P < 0.001. (B) The same mice described in the legend to panel A were monitored for mortality for a period of 12 days. Survival curves were analyzed using the log-rank (Mantel-Cox) test. ***, P < 0.001. (C) Scatter plot representation of vaccinia MV titers in nasal turbinates, lungs, brains, and ovaries of the infected mice. Organs were removed from infected mice (n = 10) at days 1, 2, 4, 6, 8, and 10 (D1, D2, D4, D6, D8, and D10, respectively) and homogenized, and MV titers were determined by plaque assays on BSC40 cells. Dashed black lines, limit of sensitivity of plaque detection (≤10 PFU per organ). Data were analyzed using a two-tailed unpaired Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) H&E staining of nasal cavity tissue obtained at day 4 p.i. from mice infected with WR or WRΔA26. Magnifications, ×100; bars, 450 μm.
We repeated intranasal infections of mice with WR or WRΔA26 and collected organs of primary infection, such as nasal turbinate and lung, as well as organs of secondary infection (brain and ovary), to monitor virus dissemination in vivo. The virus titers in each organ were determined by plaque assays on BSC40 cells (Fig. 8C). WRΔA26 infection induced higher viral titers on days 2, 4, and 6 p.i. in nasal turbinate than WR infections did. WRΔA26 infection also produced more virus in lung tissue than WR at the same time. Both WR and WRΔA26 produced much less virus in the brain and ovary than in the primary infection sites. Also, viral titers in the brain and ovary of WRΔA26-infected animals did not differ from those in the brain and ovary of WR-infected animals, suggesting that the WRΔA26-induced pathogenesis took effect before virus dissemination into secondary organs. We dissected one of the primary infection sites, i.e., the nasal turbinate, at day 4 p.i. for histopathological examination using hematoxylin and eosin (H&E) staining. As shown in Fig. 8D, mock-infected control mice showed intact olfactory epithelium (OE) layers and no sign of inflammation. Mice infected with the WR strain exhibited an altered OE, reflected in changes to the spongiform layer; mild to moderate disruption of the underlying lamina propria and Bowman's glands; as well as eosinophilic edematous material. Nasal lesions were more extensive in mice infected with WRΔA26, and these mice exhibited severe necrosis of the OE and a buildup of necrotic neutrophil and epithelial cell debris. To extend our in vitro finding that IFNAR-dependent signaling is differentially induced by WR and WRΔA26, we sought to determine whether this difference also modulates the varied in vivo virulence between them. Therefore, we performed intranasal infections of age-matched and gender-matched IFNAR+/+ and IFNAR−/− sibling mice. As shown in Fig. 9, inoculation of control IFNAR+/+ mice with 104 PFU WRΔA26 resulted in more rapid weight loss (Fig. 9A) and higher mortality compared to the weight loss and mortality seen in mice inoculated with the same titer of WR (Fig. 9B), consistent with the data presented in Fig. 8. In contrast, weight loss did not differ between IFNAR-KO mice infected with WR and those infected with WRΔA26 (Fig. 9C), and all mice died by day 7 p.i. (Fig. 9D). These results show that host IFNAR-dependent signaling contributes to the differential in vivo virulence between WR and WRΔA26 infection.
FIG 9.

IFNAR-mediated antiviral signaling protects mice infected with WR but not those infected with WRΔA26. WT mice (IFNAR+/+ sibling control; n = 5 in panels A and B) and IFNAR−/− mice (n = 5 in panels C and D) were infected intranasally with 104 PFU of WR or WRΔA26 per mouse and monitored daily for body weight loss (A and C) and mortality (B and D). Statistical analyses of body weight change (A and C) and survival curves (B and D) were performed as described in the legend to Fig. 8A and B. *, P < 0.05.
DISCUSSION
In our previous investigations, we showed that strains WR and WRΔA26 enter cells of different cell lines, such as HeLa, CHO, and L cells, through different entry pathways; WR virus enters through fluid-phase endocytosis, whereas WRΔA26 virus enters through plasma membrane fusion (24, 25). Apart from the difference in their entry pathways, both WR and WRΔA26 viruses exhibit full replication and complete packaging in these cell lines, with no sign of differential host susceptibility being seen (12, 24). In contrast, here we found that infections with WR and WRΔA26 triggered differential innate immune responses, resulting in differential late gene expression in BMDM in vitro and altered virus pathogenicity in mice in vivo. Although previous studies have already shown that vaccinia virus growth in macrophages is abortive (35, 36, 53), we endeavored to investigate the cause of differential viral late gene expression in BMDM under WR or WRΔA26 infection. It is worth mentioning that, despite robust late A4mChery expression, the growth of WRΔA26 remained restricted in BMDM, similar to that of WR virus (data not shown). We still consider this an important issue not only because innate immune cells, such as macrophages, are an important first line of defense for hosts to induce an efficient antiviral response but also because deletion of the A26 protein had such a dramatic consequence that it may lead us to uncover an important role for this protein in innate immune regulation. Although we did not investigate the primary innate immune-sensing mechanism upstream of IFNAR-mediated signaling in BMDM, our data uncover a critical role of the IFNAR signaling pathway that contributes to the differential response to WR or WRΔA26 infection in BMDM. We also showed that WR and WRΔA26 exhibit different in vivo virulences in mice, and by comparing virus infections in WT and IFNAR-KO mice, we concluded that the phenotypic difference is mediated by IFNAR signaling, which represents an interesting correlation between macrophage restriction in vitro and host antipoxvirus innate immunity in vivo. This finding corroborates the findings of a previous report that demonstrated a critical role for macrophages in the clearance of vaccinia virus infection from mice, with the depletion of alveolar macrophages using liposome clodronate resulting in increased vaccinia virus virulence following intranasal infections (32).
Vaccinia virus has been used as an effective vaccine vector to eradicate smallpox disease, so its potency in the regulation of immune responses has been the center of poxvirus research for many years. It has been well documented that to elicit protective immunity in hosts, the early innate immune responses induced by vaccinia virus infection through dendritic cells (DCs), macrophages, and cytokines are required to activate subsequent T and B immune cells to clear the virus and prevent further dissemination (54–57). Therefore, studies of infection of primary innate immune cells with attenuated vaccine vectors (such as the MVA and NYVAC strains of vaccinia virus) have further demonstrated that both IRF3 and NF-κB, together with interferon signaling (37, 39, 43, 58–60), are critical for the activation of DCs and macrophages and for leukocyte recruitment to the infected areas (38, 40), followed by subsequent B-cell and CD4 and CD8 T-cell activation for virus clearance (61, 62).
In contrast to the findings obtained with the MVA strain, many studies have shown that WR virus infections do not induce substantial expression of immunomodulatory genes in various cell lines (44, 63, 64), primary fibroblasts (65), and human/mouse monocyte cells (37, 40, 63). Indeed, the WR genome encodes multiple viral proteins that are known to antagonize the IRF3-IFN type 1 signaling pathway (41, 42), such as E3 (66), C16 (67), A46 (68), C6 (69), K7 (70), N2 (71), and B18R (72–74). Despite the presence of these known viral antagonists, our BMDM study revealed several common canonical pathways induced by WR and WRΔA26, such as prothrombin activation, Wnt signaling, and G-protein-coupled receptor signaling. Most importantly, our microarray analyses showed strong activation of IFNAR signaling preferentially by the WR strain and not by WRΔA26. We compared our microarray data with those from other poxvirus microarray analyses presented in the literature and found differences in IFNAR signaling pathways (39, 43, 44, 59, 63, 64). Additional differences between our data and data from the literature on the G-protein-coupled receptor signaling pathway (44, 45) and the Wnt signaling pathway (46) were also found. The discrepancies in the results may be due to the use of different viral strains, host species, cell types, and data analysis methods and the complexity of the temporal regulation of cellular transcription during the course of infection. We did notice that the multiplicity of infection (MOI) of 20 PFU per cell that we used for BMDM infections is higher than the MOIs (MOI, 5 to 10 PFU per cell) described in the studies mentioned above. It remains to be determined whether infection with WR virus at such a high MOI results in such a strong interferon response that viral immunomodulatory proteins fail to suppress in BMDM. Nevertheless, our microarray data are consistent with our experimental results showing that WR infections of BMDM readily induce IFN-β production and STAT1 phosphorylation. Tellingly, the WRΔA26 mutant virus did not induce IFNAR signaling or STAT1 phosphorylation in BMDM at 2 to 8 h p.i. How does WRΔA26 escape immune detection at the time of entry into BMDM? We suspected that WRΔA26 may enter BMDM via the plasma membrane and thereby bypass endosomal innate immune detection. However, even though WRΔA26 entry appears to be less sensitive to bafilomycin inhibition than WR entry, we have not been able to induce cell-cell fusion from without on BMDM with WRΔA26 MV at neutral pH (unpublished results), unlike for HeLa, L, and CHO-K1 cells, in which WRΔA26 has been shown to be much more bafilomycin resistant and in which robust cell-cell fusion has been induced from without at neutral pH (24, 75). These preliminary data suggest that WRΔA26 may enter BMDM through an aberrant endocytic process that is different from the one used by WR. Clearly, previous studies already showed that enveloped virus entry into cells could readily disarm the downstream innate immune response. For example, Ebola virus, Marburg virus, varicella-zoster virus, and amphotropic murine leukemia virus bind to the TAM receptor during virus entry to downregulate the innate immune response in DCs (76). HIV binds to dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin and Toll-like receptor 8 to exploit innate immune signaling for virus production in DCs (77). In the future, we will compare the entry processes of WR and WRΔA26 in BMDM. We also intend to investigate whether different upstream innate immune sensing pathways are induced by WR and WRΔA26, contributing to the differential IFNAR signaling reported in this study.
Previous studies have shown that macrophages are important immune cells that restrict vaccinia virus infection (32, 36), raising the possibility that WR and WRΔA26 may exhibit different virulences in vivo. We infected mice intranasally with WR, WRΔA26, or the REV-A26 revertant virus to assess pathogenic phenotypes in vivo. Based on multiple criteria (weight loss, host mortality, viral titers in different organs, nasal tissue histology), we concluded that WRΔA26 is more severely pathogenic than WR. IFNAR-knockout mice infected with WR succumbed to an infection as severe as that in WRΔA26-infected mice (both groups exhibited 100% mortality by day 7 p.i.), implying a role for A26 in modulating innate immunity and viral virulence. Although we do not claim BMDM to be the only cell type contributing to host resistance to vaccinia virus, our study raises an interesting research avenue to determine how and why the A26 protein on MV regulates IFN signaling induction in vitro and in vivo.
MATERIALS AND METHODS
Cells, viruses, and reagents.
BSC40 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco). Anti-phospho-STAT1 Y701 (catalog number 9167S) and anti-total STAT1 (catalog number 9172S) antibodies were purchased from Cell Signaling Inc. Anti-β-actin antibody (clone BA3R) was purchased from BioVision Inc. WR-A4mCherry and WRΔA26-A4mCherry—two recombinant viruses containing an ORF encoding the A4mCherry fusion protein—were generated from WR and WRΔA26, respectively, as previously described (15). For in vitro BMDM infections, we used purified vaccinia viruses as infection inocula, whereas for in vivo infections, we used virus-infected lysates as infection inocula. When necessary, WR and WRΔA26 MVs were purified by the CsCl gradient centrifugation method as previously described (78, 79). Wild-type C57BL/6 mice were purchased from BioLasco Inc. Type 1 interferon receptor-knockout (IFNAR-KO) mice, which have the C57BL/6 mouse genetic background, were obtained from Chien-Kuo Lee at National Taiwan University (48). Offspring mice of IFNAR+/− heterozygous genotypes were intercrossed to obtain sibling IFNAR−/− and IFNAR+/+ mice that were gender and age matched for experimental use.
To establish cultured BMDM, we followed established protocols as previously described (80). In brief, bone marrow cells were flushed out of the femurs and tibias of 6- to 10-week-old C57BL/6 mice and treated with ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) to remove red blood cells. These cells were then plated onto 100-mm non-tissue culture petri dishes containing DMEM supplemented with 10% heat-inactivated fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Gibco), 2 mM l-glutamine (Gibco), 1 mM sodium pyruvate (Sigma), and 10% L929-conditioned medium. Cell cultures were replenished with fresh medium every 3 days, and BMDM purity was determined (>95%) at day 7 by staining with phycoerythrin (PE)-Cy7-conjugated anti-F4/80 (BioLegend) and allophycocyanin (APC)-conjugated anti-CD11b (BioLegend), before flow cytometry analyses as previously described (81). For the experiments, BMDM were reseeded at day 7 in BMDM growth medium plus 10 ng/ml recombinant macrophage colony-stimulating factor (Peprotech Inc.) and incubated overnight before use.
RNA preparation, labeling, and hybridization for microarray analyses.
BMDM were infected with CsCl-purified MV of WR and WRΔA26 at an MOI of 20 PFU/cell at 37°C for 60 min and washed, and incubation in growth medium was continued for 1, 2, 4, and 8 h before harvesting. Total RNA was extracted from these virus-infected BMDM as previously described (82). In brief, cells were lysed in 4 M guanidine thiocyanate and subjected to high-speed centrifugation in an SW60 Ti rotor (Beckman) at 14,000 × g through a dense cushion of cesium chloride (5.7 M) for 12 h at 25°C. Following centrifugation, the RNA pellets were then resuspended in 3 M sodium acetate and absolute ethanol and precipitated for 1 h at −80°C. The RNA pellets were then centrifuged and resuspended in diethyl pyrocarbonate-treated water. RNA quality was monitored with an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA). Total RNA (10 μg) was reverse transcribed with aminoallyl-modified dUTP (aa-dUTP) using a Superscript Plus indirect cDNA labeling system (Invitrogen) according to the manufacturer's protocol. The cDNA was then purified by use of a Qiagen column (Qiagen, Valencia, CA), followed by coupling with Alexa/CyDye. Control samples were labeled with Alexa Fluor 555, and test samples were labeled with Alexa Fluor 647. Alexa/CyDye-labeled aminoallyl-modified cDNA was then hybridized to an Agilent SurePrint G3 Mouse GE 8 × 60K microarray (G4852A) using Agilent gene expression hybridization and wash kits according to the manufacturer's protocols. The microarrays were scanned on an Agilent DNA microarray scanner (catalog number US9230696) using the two-color scan setting for 8 × 60K array slides.
Microarray data and pathway analyses.
The 55,821 microarray probes and the gene information from NCBI (data downloaded from ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene_info.gz; download date, 3 December 2015) were compared. The results showed that the microarray contained 31,612 probes (representing 22,707 total genes), from which labeled noncoding RNAs or predicted genes from NCBI were excluded, as were probes not identified from Mouse Genome Informatics (MGI; http://www.informatics.jax.org/). The microarray experiments were independently repeated three times. Mean values were obtained as the data set used for gene expression and k-means clustering analyses by the use of GeneSpring (version 12.6.1) software. k-means clustering divides genes into 25 groups on the basis of their expression patterns by using Euclidean distance metrics. The levels of gene expression by BMDM infected with WR and WRΔA26 were compared to those by mock-infected BMDM. We considered genes to be upregulated relative to their levels of expression in mock-infected BMDM if the fold change (FC) was >2 or downregulated if FC was <−2. Canonical pathways and functional analysis of the regulated genes were evaluated using Ingenuity pathway analysis (IPA; Qiagen Inc.).
Statistical analysis.
For microarray analysis, raw data were imported into GeneSpring (version 12.6.1) software, and signal intensities significantly above the background (P ≤ 0.05) were retained. Gene expression ratios (log2) were calculated using the values for mock-infected cells as the baseline. Expression ratios were subjected to k-means clustering based on a Euclidian distance metric performed in GeneSpring software with the criterion k equal 25. A scatter plot was used to show gene expression patterns based on a Cartesian coordinate system. Pearson correlation coefficients were used to measure the strength of a linear relationship between the gene expression values for WR- and WRΔA26-infected cells. Statistical analyses of the survival curves for mice were performed using the log-rank (Mantel-Cox) test, whereas the results of all other experiments were analyzed using Student's t test in Prism (version 5) software (GraphPad). Statistical significance is represented as P values of <0.05, <0.01, and <0.001.
Quantitative real-time RT-PCR.
Total RNA was isolated from WR-infected BMDM and WRΔA26-infected BMDM at 1, 2, 4, and 8 h p.i. as described above. RNA (1 μg) was treated with DNase I (Invitrogen) and reverse transcribed using SuperScript III reverse transcriptase (Invitrogen) with oligo(dT) and a random hexamer according to the manufacturer's protocol. Quantitative PCR was subsequently performed with cDNA using iQ SYBR green supermix (Bio-Rad) in a CFX-96 Touch system (Bio-Rad). Results were averaged from those from three independent experiments. The primer sequences used in qRT-PCR were as follows: for IFIT2, forward primer 5′-GCAGTCATGAGTACAACGAGTAAGG-3′ and reverse primer 5′-TATGTTGCACATGGTGGCTTTAA-3′; for ZBP1, forward primer 5′-GATGCTCACCCAAAGGCAAA-3′ and reverse primer 5′-GCAAGGTCGGTTCCACTTCTTA-3′; and for IRF7, forward primer 5′-CACACCCCCATCTTCGACTT-3′ and reverse primer 5′-TCACCAGGATCAGGGTCTTCTC-3′.
Flow cytometry analyses.
Flow cytometry of BMDM was performed using fluorescence-activated cell sorting (FACS) as previously described (81) After day 7 of culture, the purity of BMDM was determined to be >95% by staining with PE-Cy7-conjugated anti-F4/80 (BioLegend) and APC-conjugated anti-CD11b (BioLegend) antibodies, and BMDM were analyzed using an LSRII BD Bioscience system. Data were further analyzed and quantified with FlowJo (version 8.7) software.
Cytokine ELISAs.
BMDM were seeded onto 12-well dishes (4 × 105 cells/well) and incubated overnight in growth medium. On the next day, the BMDM were stimulated with poly(I·C) (10 μg/ml; InvivoGen) or IFN-β (10 U/ml; PBL Assay Science) for 12 h and 4 h, respectively. Alternatively, BMDM were infected with vaccinia viruses and incubated for 8 h. Supernatants were then collected from the cells and subjected to ELISA analyses for detection of CXCL10 (R&D) and IFN-β (PBL Assay Science) according to the manufacturers' protocols.
Live imaging of vaccinia virus infections in BMDM cultures.
BMDM were infected with recombinant viruses WR-A4mCherry and WRΔA26-A4mCherry at an MOI of 20 PFU per cell. These cells were washed and replated in BMDM growth medium, and live cell images were recorded for 24 h using a Carl Zeiss LSM510 META-NLO confocal microscope under 5% CO2 supplementation in a 37°C incubator. Cells were visualized using an LD Achroplan 20× (numerical aperture, 0.4) Korr Ph2 objective lens. Fluorescent images were recorded by exciting mCherry with a 561-nm diode-pumped solid-state laser (Melles Griot) and collected using a band-pass filter of between 576 and 615 IR nm. Time-lapse images were acquired every 15 min and recorded using Zen 2009 imaging software (Carl Zeiss).
Immunoblot analysis.
BMDM infected with WR and WRΔA26 were harvested at 1, 2, 4, and 8 h p.i. Cells were lysed using protein lysis buffer containing 50 mM Tris-HCl pH 7.5, 250 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100, 0.5% NP-40, 10% glycerol, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 0.7 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride, and a PhosStop tablet (Roche). Cells were lysed for 5 min on ice, followed by centrifugation at 16,000 × g for 15 min at 4°C. Protein concentrations in soluble lysates were determined using a Pierce bicinchoninic acid protein assay kit (Thermo Fischer Scientific). Equal amounts (10 μg) of lysates were loaded, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose membranes (PerkinElmer) using a wet transfer apparatus (Bio-Rad). Membranes were blocked in 5% bovine serum albumin (BSA) and incubated with primary anti-phospho-STAT1 (9167S) antibody or anti-total STAT1 (9172S) antibody overnight at 4°C or anti-β-actin control antibody for 1 h at room temperature. The blots were then probed with horseradish peroxide-conjugated goat anti-rabbit or goat anti-mouse immunoglobulin secondary antibody (Jackson ImmunoResearch Inc.) and developed using a Western lightning enhanced chemiluminescence kit (PerkinElmer) as previously described (24).
In vivo vaccinia virus infection in mice.
All animal protocols were approved by the Institutional Animal Care and Utilization Committee of Academia Sinica in strict accordance with the guidelines on animal use and care of the U.S. National Institutes of Health (84). BSC40 cells infected with the WR or WRΔA26 virus were harvested, freeze-thawed, and sonicated to be used as virus inocula. Age-matched C57BL/6 control and IFNAR-KO mice 6 to 8 weeks old were anesthetized with isoflurane and inoculated intranasally with 10 μl WR or WRΔA26 stocks at 104 PFU per mouse. For mock-infected controls, mice were inoculated with 10 μl phosphate-buffered saline plus 0.05% BSA and 10 mM magnesium chloride (PBS-AM). The titers of the virus inocula on BSC40 cells were immediately redetermined after mouse infections to ensure the accuracy of the virus titers. Mice were individually weighed daily and monitored for signs of illness. Changes in the body weights of the mice in each group were averaged. In some experiments, mice were sacrificed and their nasal turbinates, lungs, brains, and ovaries were homogenized and frozen and thawed three times, followed by sonication. Virus titers were then determined by plaque assays on BSC40 cells. For pathological tissue examination, the mice were anesthetized at day 4 p.i. with tribromoethanol (Avertin; 0.018 ml/g body weight), and the hearts were perfused with 30 ml PBS, as previously described (83). Nasal turbinates were removed postmortem and fixed in 10% neutral buffered formalin for at least 36 h before they were embedded in paraffin wax. Tissue sections in paraffin were stained with hematoxylin and eosin (H&E) for histopathological examination (National Laboratory Animal Center, National Applied Research Laboratories, Taipei, Taiwan, Republic of China).
Accession number(s).
The microarray data have been submitted to the Gene Expression Omnibus (GEO) database under accession number GSE97705.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sue-Ping Lee of the Imaging Core Facility of the Institute of Molecular Biology at Academia Sinica for help with cell imaging analyses. We also thank the FACS Core Facility, Genomics Core Facility, and Bioinformatics Core Facility of the Institute of Molecular Biology, Academia Sinica.
This work was supported by grants from Academia Sinica and the Ministry of Science and Technology (MOST 106-2321-B-001-014, MOST 105-2321-B-001-052).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00767-17.
REFERENCES
- 1.Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. 1990. The complete DNA sequence of vaccinia virus. Virology 179:247–266, 517–563. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- 2.Moss B. 2015. Poxvirus membrane biogenesis. Virology 479-480:619–626. doi: 10.1016/j.virol.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Condit RC, Moussatche N, Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 4.Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J Virol 80:2127–2140. doi: 10.1128/JVI.80.5.2127-2140.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoder JD, Chen TS, Gagnier CR, Vemulapalli S, Maier CS, Hruby DE. 2006. Pox proteomics: mass spectrometry analysis and identification of vaccinia virion proteins. Virol J 3:10. doi: 10.1186/1743-422X-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Resch W, Hixson KK, Moore RJ, Lipton MS, Moss B. 2007. Protein composition of the vaccinia virus mature virion. Virology 358:233–247. doi: 10.1016/j.virol.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol 72:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsiao JC, Chung CS, Chang W. 1998. Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J Virol 72:8374–8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsiao JC, Chung CS, Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J Virol 73:8750–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin CL, Chung CS, Heine HG, Chang W. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J Virol 74:3353–3365. doi: 10.1128/JVI.74.7.3353-3365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter GC, Law M, Hollinshead M, Smith GL. 2005. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol 86:1279–1290. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- 12.Chiu WL, Lin CL, Yang MH, Tzou DL, Chang W. 2007. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J Virol 81:2149–2157. doi: 10.1128/JVI.02302-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PW, Liao CL, Chang W. 2012. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J Virol 86:6677–6687. doi: 10.1128/JVI.06860-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroeder N, Chung CS, Chen CH, Liao CL, Chang W. 2012. The lipid raft-associated protein CD98 is required for vaccinia virus endocytosis. J Virol 86:4868–4882. doi: 10.1128/JVI.06610-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang CY, Lu TY, Bair CH, Chang YS, Jwo JK, Chang W. 2008. A novel cellular protein, VPEF, facilitates vaccinia virus penetration into HeLa cells through fluid phase endocytosis. J Virol 82:7988–7999. doi: 10.1128/JVI.00894-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 17.Frei AP, Jeon OY, Kilcher S, Moest H, Henning LM, Jost C, Pluckthun A, Mercer J, Aebersold R, Carreira EM, Wollscheid B. 2012. Direct identification of ligand-receptor interactions on living cells and tissues. Nat Biotechnol 30:997–1001. doi: 10.1038/nbt.2354. [DOI] [PubMed] [Google Scholar]
- 18.Oie M. 1985. Reversible inactivation and reactivation of vaccinia virus by manipulation of viral lipid composition. Virology 142:299–306. doi: 10.1016/0042-6822(85)90338-1. [DOI] [PubMed] [Google Scholar]
- 19.Laliberte JP, Moss B. 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc Natl Acad Sci U S A 106:17517–17521. doi: 10.1073/pnas.0909376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morizono K, Xie Y, Olafsen T, Lee B, Dasgupta A, Wu AM, Chen IS. 2011. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 9:286–298. doi: 10.1016/j.chom.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moss B. 2016. Membrane fusion during poxvirus entry. Semin Cell Dev Biol 60:89–96. doi: 10.1016/j.semcdb.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsiao JC, Chu LW, Lo YT, Lee SP, Chen TJ, Huang CY, Ping YH, Chang W. 2015. Intracellular transport of vaccinia virus in HeLa cells requires WASH-VPEF/FAM21-retromer complexes and recycling molecules Rab11 and Rab22. J Virol 89:8365–8382. doi: 10.1128/JVI.00209-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rizopoulos Z, Balistreri G, Kilcher S, Martin CK, Syedbasha M, Helenius A, Mercer J. 2015. Vaccinia virus infection requires maturation of macropinosomes. Traffic 16:814–831. doi: 10.1111/tra.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang SJ, Chang YX, Izmailyan R, Tang YL, Chang W. 2010. Vaccinia virus A25 and A26 proteins are fusion suppressors for mature virions and determine strain-specific virus entry pathways into HeLa, CHO-K1, and L cells. J Virol 84:8422–8432. doi: 10.1128/JVI.00599-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang SJ, Shih AC, Tang YL, Chang W. 2012. Vaccinia mature virus fusion regulator A26 protein binds to A16 and G9 proteins of the viral entry fusion complex and dissociates from mature virions at low pH. J Virol 86:3809–3818. doi: 10.1128/JVI.06081-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Locker JK, Kuehn A, Schleich S, Rutter G, Hohenberg H, Wepf R, Griffiths G. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membrane is signaling-dependent for the IMV but not the EEV. Mol Biol Cell 11:2497–2511. doi: 10.1091/mbc.11.7.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Townsley AC, Weisberg AS, Wagenaar TR, Moss B. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J Virol 80:8899–8908. doi: 10.1128/JVI.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitbeck JC, Foo CH, Ponce de Leon M, Eisenberg RJ, Cohen GH. 2009. Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology 385:383–391. doi: 10.1016/j.virol.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armstrong JA, Metz DH, Young MR. 1973. The mode of entry of vaccinia virus into L cells. J Gen Virol 21:533–537. doi: 10.1099/0022-1317-21-3-533. [DOI] [PubMed] [Google Scholar]
- 30.Chang A, Metz DH. 1976. Further investigations on the mode of entry of vaccinia virus into cells. J Gen Virol 32:275–282. doi: 10.1099/0022-1317-32-2-275. [DOI] [PubMed] [Google Scholar]
- 31.Bengali Z, Townsley AC, Moss B. 2009. Vaccinia virus strain differences in cell attachment and entry. Virology 389:132–140. doi: 10.1016/j.virol.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera R, Hutchens M, Luker KE, Sonstein J, Curtis JL, Luker GD. 2007. Murine alveolar macrophages limit replication of vaccinia virus. Virology 363:48–58. doi: 10.1016/j.virol.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider C, Nobs SP, Heer AK, Kurrer M, Klinke G, van Rooijen N, Vogel J, Kopf M. 2014. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog 10:e1004053. doi: 10.1371/journal.ppat.1004053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sang Y, Miller LC, Blecha F. 2015. Macrophage polarization in virus-host interactions. J Clin Cell Immunol 2015:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Humlova Z, Vokurka M, Esteban M, Melkova Z. 2002. Vaccinia virus induces apoptosis of infected macrophages. J Gen Virol 83:2821–2832. doi: 10.1099/0022-1317-83-11-2821. [DOI] [PubMed] [Google Scholar]
- 36.Yanguez E, Garcia-Culebras A, Frau A, Llompart C, Knobeloch KP, Gutierrez-Erlandsson S, Garcia-Sastre A, Esteban M, Nieto A, Guerra S. 2013. ISG15 regulates peritoneal macrophages functionality against viral infection. PLoS Pathog 9:e1003632. doi: 10.1371/journal.ppat.1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Royo S, Sainz B Jr, Hernandez-Jimenez E, Reyburn H, Lopez-Collazo E, Guerra S. 2014. Differential induction of apoptosis, interferon signaling, and phagocytosis in macrophages infected with a panel of attenuated and nonattenuated poxviruses. J Virol 88:5511–5523. doi: 10.1128/JVI.00468-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price PJ, Luckow B, Torres-Dominguez LE, Brandmuller C, Zorn J, Kirschning CJ, Sutter G, Lehmann MH. 2014. Chemokine (C-C motif) receptor 1 is required for efficient recruitment of neutrophils during respiratory infection with modified vaccinia virus Ankara. J Virol 88:10840–10850. doi: 10.1128/JVI.01524-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guerra S, Lopez-Fernandez LA, Conde R, Pascual-Montano A, Harshman K, Esteban M. 2004. Microarray analysis reveals characteristic changes of host cell gene expression in response to attenuated modified vaccinia virus Ankara infection of human HeLa cells. J Virol 78:5820–5834. doi: 10.1128/JVI.78.11.5820-5834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann MH, Price PJ, Brandmuller C, Sutter G. 2015. Modified vaccinia virus Ankara but not vaccinia virus induces chemokine expression in cells of the monocyte/macrophage lineage. Virol J 12:21. doi: 10.1186/s12985-015-0252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perdiguero B, Esteban M. 2009. The interferon system and vaccinia virus evasion mechanisms. J Interferon Cytokine Res 29:581–598. doi: 10.1089/jir.2009.0073. [DOI] [PubMed] [Google Scholar]
- 42.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 43.Offerman K, Deffur A, Carulei O, Wilkinson R, Douglass N, Williamson AL. 2015. Six host-range restricted poxviruses from three genera induce distinct gene expression profiles in an in vivo mouse model. BMC Genomics 16:510. doi: 10.1186/s12864-015-1659-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerra S, Lopez-Fernandez LA, Pascual-Montano A, Munoz M, Harshman K, Esteban M. 2003. Cellular gene expression survey of vaccinia virus infection of human HeLa cells. J Virol 77:6493–6506. doi: 10.1128/JVI.77.11.6493-6506.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leao-Ferreira LR, Paes-De-Carvalho R, De Mello FG, Moussatche N. 2002. Inhibition of vaccinia virus replication by adenosine in BSC-40 cells: involvement of A(2) receptor-mediated PKA activation. Arch Virol 147:1407–1423. doi: 10.1007/s00705-002-0795-5. [DOI] [PubMed] [Google Scholar]
- 46.Jia X, Chen Y, Zhao X, Lv C, Yan J. 2016. Oncolytic vaccinia virus inhibits human hepatocellular carcinoma MHCC97-H cell proliferation via endoplasmic reticulum stress, autophagy and Wnt pathways. J Gene Med 18:211–219. doi: 10.1002/jgm.2893. [DOI] [PubMed] [Google Scholar]
- 47.Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, Allred AL, Barish GD, Smale ST. 2016. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell 165:165–179. doi: 10.1016/j.cell.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen YL, Chen TT, Pai LM, Wesoly J, Bluyssen HA, Lee CK. 2013. A type I IFN-Flt3 ligand axis augments plasmacytoid dendritic cell development from common lymphoid progenitors. J Exp Med 210:2515–2522. doi: 10.1084/jem.20130536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reimer T, Brcic M, Schweizer M, Jungi TW. 2008. Poly(I:C) and LPS induce distinct IRF3 and NF-kappaB signaling during type-I IFN and TNF responses in human macrophages. J Leukoc Biol 83:1249–1257. doi: 10.1189/jlb.0607412. [DOI] [PubMed] [Google Scholar]
- 50.Park K, Scott AL. 2010. Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J Leukoc Biol 88:1081–1087. doi: 10.1189/jlb.0610318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schroder K, Spille M, Pilz A, Lattin J, Bode KA, Irvine KM, Burrows AD, Ravasi T, Weighardt H, Stacey KJ, Decker T, Hume DA, Dalpke AH, Sweet MJ. 2007. Differential effects of CpG DNA on IFN-beta induction and STAT1 activation in murine macrophages versus dendritic cells: alternatively activated STAT1 negatively regulates TLR signaling in macrophages. J Immunol 179:3495–3503. doi: 10.4049/jimmunol.179.6.3495. [DOI] [PubMed] [Google Scholar]
- 52.Fujimura Y, Hotokezaka H, Ohara N, Naito M, Sakai E, Yoshimura M, Narita Y, Kitaura H, Yoshida N, Nakayama K. 2006. The hemoglobin receptor protein of Porphyromonas gingivalis inhibits receptor activator NF-kappaB ligand-induced osteoclastogenesis from bone marrow macrophages. Infect Immun 74:2544–2551. doi: 10.1128/IAI.74.5.2544-2551.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broder CC, Kennedy PE, Michaels F, Berger EA. 1994. Expression of foreign genes in cultured human primary macrophages using recombinant vaccinia virus vectors. Gene 142:167–174. doi: 10.1016/0378-1119(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 54.Xu R, Johnson AJ, Liggitt D, Bevan MJ. 2004. Cellular and humoral immunity against vaccinia virus infection of mice. J Immunol 172:6265–6271. doi: 10.4049/jimmunol.172.10.6265. [DOI] [PubMed] [Google Scholar]
- 55.Bonduelle O, Duffy D, Verrier B, Combadiere C, Combadiere B. 2012. Cutting edge: protective effect of CX3CR1+ dendritic cells in a vaccinia virus pulmonary infection model. J Immunol 188:952–956. doi: 10.4049/jimmunol.1004164. [DOI] [PubMed] [Google Scholar]
- 56.Goulding J, Bogue R, Tahiliani V, Croft M, Salek-Ardakani S. 2012. CD8 T cells are essential for recovery from a respiratory vaccinia virus infection. J Immunol 189:2432–2440. doi: 10.4049/jimmunol.1200799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu J, Martinez J, Huang X, Yang Y. 2007. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood 109:619–625. doi: 10.1182/blood-2006-06-027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waibler Z, Anzaghe M, Ludwig H, Akira S, Weiss S, Sutter G, Kalinke U. 2007. Modified vaccinia virus Ankara induces Toll-like receptor-independent type I interferon responses. J Virol 81:12102–12110. doi: 10.1128/JVI.01190-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guerra S, Najera JL, Gonzalez JM, Lopez-Fernandez LA, Climent N, Gatell JM, Gallart T, Esteban M. 2007. Distinct gene expression profiling after infection of immature human monocyte-derived dendritic cells by the attenuated poxvirus vectors MVA and NYVAC. J Virol 81:8707–8721. doi: 10.1128/JVI.00444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dai P, Wang W, Cao H, Avogadri F, Dai L, Drexler I, Joyce JA, Li XD, Chen Z, Merghoub T, Shuman S, Deng L. 2014. Modified vaccinia virus Ankara triggers type I IFN production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog 10:e1003989. doi: 10.1371/journal.ppat.1003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramirez JC, Gherardi MM, Esteban M. 2000. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J Virol 74:923–933. doi: 10.1128/JVI.74.2.923-933.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L, Chavan R, Feinberg MB. 2008. Dendritic cells are preferentially targeted among hematolymphocytes by modified vaccinia virus Ankara and play a key role in the induction of virus-specific T cell responses in vivo. BMC Immunol 9:15. doi: 10.1186/1471-2172-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rubins KH, Hensley LE, Relman DA, Brown PO. 2011. Stunned silence: gene expression programs in human cells infected with monkeypox or vaccinia virus. PLoS One 6:e15615. doi: 10.1371/journal.pone.0015615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bourquain D, Dabrowski PW, Nitsche A. 2013. Comparison of host cell gene expression in cowpox, monkeypox or vaccinia virus-infected cells reveals virus-specific regulation of immune response genes. Virol J 10:61. doi: 10.1186/1743-422X-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hernaez B, Alonso G, Alonso-Lobo JM, Rastrojo A, Fischer C, Sauer S, Aguado B, Alcami A. 2017. RNA-Seq based transcriptome analysis of the type I interferon host response upon vaccinia virus infection of mouse cells. J Immunol Res 2017:5157626. doi: 10.1155/2017/5157626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A 89:4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fahy AS, Clark RH, Glyde EF, Smith GL. 2008. Vaccinia virus protein C16 acts intracellularly to modulate the host response and promote virulence. J Gen Virol 89:2377–2387. doi: 10.1099/vir.0.2008/004895-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. 2005. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med 201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Unterholzner L, Sumner RP, Baran M, Ren H, Mansur DS, Bourke NM, Randow F, Smith GL, Bowie AG. 2011. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog 7:e1002247. doi: 10.1371/journal.ppat.1002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schroder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ferguson BJ, Benfield CT, Ren H, Lee VH, Frazer GL, Strnadova P, Sumner RP, Smith GL. 2013. Vaccinia virus protein N2 is a nuclear IRF3 inhibitor that promotes virulence. J Gen Virol 94:2070–2081. doi: 10.1099/vir.0.054114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Symons JA, Alcami A, Smith GL. 1995. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81:551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 73.Alcami A, Symons JA, Smith GL. 2000. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol 74:11230–11239. doi: 10.1128/JVI.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montanuy I, Alejo A, Alcami A. 2011. Glycosaminoglycans mediate retention of the poxvirus type I interferon binding protein at the cell surface to locally block interferon antiviral responses. FASEB J 25:1960–1971. doi: 10.1096/fj.10-177188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang TH, Chang SJ, Hsieh FL, Ko TP, Lin CT, Ho MR, Wang I, Hsu ST, Guo RT, Chang W, Wang AH. 2013. Crystal structure of vaccinia viral A27 protein reveals a novel structure critical for its function and complex formation with A26 protein. PLoS Pathog 9:e1003563. doi: 10.1371/journal.ppat.1003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bhattacharyya S, Zagorska A, Lew ED, Shrestha B, Rothlin CV, Naughton J, Diamond MS, Lemke G, Young JA. 2013. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 14:136–147. doi: 10.1016/j.chom.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gringhuis SI, van der Vlist M, van den Berg LM, den Dunnen J, Litjens M, Geijtenbeek TB. 2010. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat Immunol 11:419–426. doi: 10.1038/ni.1858. [DOI] [PubMed] [Google Scholar]
- 78.Payne L. 1978. Polypeptide composition of extracellular enveloped vaccinia virus. J Virol 27:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engelstad M, Smith GL. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology 194:627–637. doi: 10.1006/viro.1993.1302. [DOI] [PubMed] [Google Scholar]
- 80.Hsieh WC, Chuang YT, Chiang IH, Hsu SC, Miaw SC, Lai MZ. 2014. Inability to resolve specific infection generates innate immunodeficiency syndrome in Xiap−/− mice. Blood 124:2847–2857. doi: 10.1182/blood-2014-03-564609. [DOI] [PubMed] [Google Scholar]
- 81.Zhang X, Goncalves R, Mosser DM. 2008. The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14:Unit 14.11. doi: 10.1002/0471142735.im1411s90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. 1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 83.Nagy A. 2003. Manipulating the mouse embryo: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 84.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.