

ABSTRACT
The mumps virus (MuV) small hydrophobic protein (SH) is a type I membrane protein expressed in infected cells. SH has been reported to interfere with innate immunity by inhibiting tumor necrosis factor alpha (TNF-α)-mediated apoptosis and NF-κB activation. To elucidate the underlying mechanism, we generated recombinant MuVs (rMuVs) expressing the SH protein with an N-terminal FLAG epitope or lacking SH expression due to the insertion of three stop codons into the SH gene. Using these viruses, we were able to show that SH reduces the phosphorylation of IKKβ, IκBα, and p65 as well as the translocation of p65 into the nucleus of infected A549 cells. Reporter gene assays revealed that SH interferes not only with TNF-α-mediated NF-κB activation but also with IL-1β- and poly(I·C)-mediated NF-κB activation, and that this inhibition occurs upstream of the NF-κB pathway components TRAF2, TRAF6, and TAK1. Since SH coimmunoprecipitated with tumor necrosis factor receptor 1 (TNFR1), RIP1, and IRAK1, we hypothesize that SH exerts its inhibitory function by interacting with TNFR1, interleukin-1 receptor type 1 (IL-1R1), and TLR3 complexes in the plasma membrane of infected cells.
IMPORTANCE The MuV SH has been shown to impede TNF-α-mediated NF-κB activation and is therefore thought to contribute to viral immune evasion. However, the mechanisms by which SH mediates NF-κB inhibition remained largely unknown. In this study, we show that SH interacts with TNFR1, IL-1R1, and TLR3 complexes in infected cells. We thereby not only shed light on the mechanisms of SH-mediated NF-κB inhibition but also reveal that SH interferes with NF-κB activation induced by interleukin-1β (IL-1β) and double-stranded RNA.
KEYWORDS: NF-κB, mumps, paramyxovirus
INTRODUCTION
Mumps virus (MuV) causes acute infections in humans which are characterized by parotitis, fever, headache, and fatigue (1). MuV is highly neurotropic with involvement of the central nervous system (CNS) in half of all cases, where it can induce aseptic meningitis or encephalitis. Other complications include orchitis, oophoritis, pancreatitis, and deafness (2). Mumps is a vaccine-preventable disease, and worldwide vaccination campaigns have led to a considerable reduction of incidence (3–5). However, in recent years, an increasing number of mumps outbreaks have been reported worldwide within vaccinated populations (6–11).
MuV, a member of the Paramyxoviridae family, is an enveloped virus with a nonsegmented negative-stranded RNA genome encoding nine proteins in seven tandemly linked transcription units (2). The gene encoding the small hydrophobic protein (SH) is located in a highly variable region of the MuV genome and is therefore used for genotyping according to a WHO protocol (12–15).
Previous results provided evidence that the SH protein is a type I membrane protein expressed in the membrane of infected cells (16). Like the SH proteins of related paramyxoviruses, MuV SH is not essential for virus growth in tissue culture (17–21). Nevertheless, it is suspected to be a viral antagonist to the host's innate immune system, since it has been shown to interfere with the release of tumor necrosis factor α (TNF-α) from infected cells and to impede with the activation of TNF-α-mediated nuclear factor κB (NF-κB) in transiently transfected cells (21, 22). The precise mechanism of SH-mediated NF-κB inhibition, however, remained to be uncovered.
NF-κB transcription factors play a crucial role in the activation of the innate immune system. They are activated by the binding of cytokines like TNF-α to tumor necrosis factor receptor 1 (TNFR1) or interleukin-1β (IL-1β) to interleukin-1 receptor type 1 (IL-1R1) (23). Furthermore, the activation of Toll-like receptors (TLRs) promotes NF-κB-regulated gene expression through the detection of pathogen-associated molecular patterns (PAMPs), including bacterial products like triacylated lipoproteins, recognized by TLR2 in combination with TLR1 (24), and lipopolysaccharides (LPS), which activate NF-κB via binding to TLR4 (25). In contrast, TLR3 recognizes double-stranded RNA (dsRNA) molecules derived from viruses and their replication intermediates (26).
NF-κB is a mono- or heterodimer of two Rel proteins. In the canonical pathway, NF-κB consists primarily of the proteins p50 and p65 (23). Inactive NF-κB is associated with its inhibitor, IκBα (inhibitor of κBα), and is located in the cytoplasm. Receptor stimulation leads to the recruitment of adaptor proteins like receptor-interacting serine/threonine-protein kinase 1 (RIP1), interleukin-1 receptor-associated kinase 1 (IRAK-1), or myeloid differentiation primary response gene 88 (MyD88) to specific receptors. The recruitment of these adaptor proteins leads to the activation of kinase cascades that ultimately converge in the activation of the kinase complex IKKα/β. Once activated, the IKKβ subunit phosphorylates IκBα at position serine 32 (Ser32), resulting in further ubiquitination and proteasome-mediated degradation of IκBα, thereby unmasking the nuclear localization signal (NLS) sequence of NF-κB subunit p65 (27). Consequently, p65 is transported into the nucleus and binds to its specific target DNA sequences (28). In addition to the phosphorylation of IκBα, IKKβ also directly phosphorylates p65 at position Ser536, which enhances the transcriptional activation of NF-κB target genes (29).
In order to elucidate the mechanism of SH-mediated NF-κB inhibition, we generated SH-expressing and, by inserting three stop codons, SH-deficient rMuVs, each with a FLAG epitope fused to the SH gene. These modifications enabled us to detect SH protein expression or deficiency in infected cells while maintaining the viral genomic structure important for proper virus replication. Using these viruses, we revealed that the SH protein reduces NF-κB activation by decreasing IKKβ, IκBα, and p65 phosphorylation as well as p65 translocation into the nucleus of infected cells. In addition, we demonstrated for the first time that SH impedes TNF-α- as well as IL-1β- and poly(I·C)-mediated NF-κB activation. Since we found that SH coimmunoprecipitated with TNFR1, RIP1, and IRAK1, we propose that SH exerts its inhibitory effect in the membrane of MuV-infected cells by interacting with receptor complexes involved in NF-κB activation.
RESULTS
Rescue of recombinant mumps viruses.
To characterize the function of the SH protein in the context of MuV infection, SH-expressing and SH-deficient recombinant mumps viruses were generated. In each virus, a FLAG epitope was fused to the N terminus or the C terminus of the SH protein (rMuV-SH, rMuV-SHstop, rMuV-SH-C, and rMuV-SHstop-C) (Fig. 1A). The FLAG epitope was introduced due to the absence of an antibody against the SH protein to allow the detection of SH. The successful generation of the rMuVs was indicated by observing syncytium formation in transfected BSR-T7 cells after 7 days (Fig. 1B) and by detection of F protein expression in Vero76 cells infected with the rescued rMuVs by immunofluorescence (Fig. 1C). Integrity of the rMuVs was verified by sequencing. The expression of the SH protein in rMuV-SH- and rMuV-SH-C-infected cells was confirmed by Western blot analysis. Furthermore, the SH protein was not detected in cells infected with SH-deficient viruses rMuV-SHstop and rMuV-SHstop-C (Fig. 1D).
FIG 1.

Generation of rMuV with mutated SH genes. (A) Scheme of the MuV genome and the modified SH fragments of the generated rMuVs. For the SH-deficient viruses, the positions of introduced stop codons starting at the ATG of SH (rMuV-SHstop-C) and the FLAG epitope sequence (rMuV-SHstop) are stated. (B) BSR-T7 cells were transfected with pG-MuV-FL_SH-N-flag, pG-MuV-FL_SH-3stop-N-flag, pG-MuV-FL_SH-C-flag, or pG-MuV-FL_SH-3stop-C-flag plus pMuV-L, pMuV-N, and pMuV-P. As a negative control (mock), cells were transfected with pMuV-EGFP. Pictures of syncytia were taken at 7 days posttransfection. (C) Vero76 cells were infected with rMuV-SH, rMuV-SHstop, rMuV-SH-C, or rMuV-SHstop-C at an MOI of 0.01 or left untreated (mock). At 2 days p.i., cells were fixed and stained using an MuV F-specific Cy3-coupled antibody and DAPI. Images were generated using a Zeiss cLSM780 confocal laser scanning microscope. (D) At 3 days p.i., rMuV-infected Vero76 cells (MOI, 0.01) were lysed and subjected to Western blot analysis. MuV N and GAPDH were detected by specific monoclonal antibodies, and MuV SH was detected by an anti-FLAG antibody.
Characterization of rMuV growth and SH membrane orientation.
To analyze if the lack of SH protein expression influences viral replication, we investigated extracellular rMuV titers after infection of Vero76 cells. As shown in Fig. 2A, all rMuVs replicated to comparable titers, indicating no pronounced effect of the SH protein on virus replication. Additionally, extracellular titers of an rMuV with an unchanged genome were compared to rMuV-SH, revealing no noticeable effect of the insertion of the FLAG epitope on virus replication (data not shown).
FIG 2.

rMuV replication and SH protein expression. (A) Vero76 cells were infected with rMuV-SH, rMuV-SHstop, rMuV-SH-C, or rMuV-SHstop-C at an MOI of 0.01. Supernatants were collected at the indicated time points and titrated in triplicate on Vero76 cells. The detection limit due to assay procedure is 102 (dashed line). The graph depicts means ± standard deviations (SD) from one representative experiment for which three independent infections were performed at the same time. A549 (B and D) and Vero76 (C and E) cells were infected with rMuVs at an MOI of 5. Cells were harvested at the indicated time points and analyzed by flow cytometry. Total SH protein (B and C) was detected using an APC-conjugated FLAG-specific antibody. The values of rMuV-SHstop or rMuV-SHstop-C were subtracted from the values of rMuV-SH or rMuV-SH-C. Total N protein (D and E) was detected using an N-specific primary antibody and an Alexa Fluor 488-conjugated secondary antibody. The values of mock-infected cells were subtracted from the values of rMuV-infected cells. Graphs depict means ± SD from three individual experiments. *, P < 0.05; results were calculated by unpaired t tests with a two-tailed P value. (F) A549 cells were infected with rMuVs at an MOI of 5 and harvested at 20 h postinfection. Cells were treated with 0.5% saponin (permeabilized) for intra- and extracellular SH detection or not treated (nonpermeabilized) for extracellular SH detection by flow cytometry. Staining and calculation for total SH protein were carried out as described for panels B and C. The graph depicts means ± SD from three individual experiments. ***, P < 0.001; results were calculated by unpaired t tests with a two-tailed P value.
The expression kinetics of the SH and the N protein in rMuV-infected cells were analyzed next. We used Vero76 cells, since this cell line is routinely used for MuV propagation, and also A549 cells, because epithelial cells are assumed to be a primary target for MuV in vivo (2). Syncytium formation was similar for all viruses at all time points (data not shown). Flow cytometry analysis revealed a significantly earlier expression of the SH protein in A549 cells infected with rMuV-SH than with rMuV-SH-C (Fig. 2B, 10 and 12 h postinfection [p.i.]). At the later time points, similar numbers of cells expressed SH regardless which virus was used for infection, with approximately 60% of cells being positive for SH expression at 20 h p.i. (Fig. 2B). Vero76 cells infected with rMuV-SH or rMuV-SH-C showed no statistically significant differences in the level of SH-positive cells at all time points. At 20 h p.i., approximately 50% of rMuV-infected Vero76 cells expressed the SH protein (Fig. 2C). To determine infection rates, rMuV-infected A549 and Vero76 cells were analyzed for the expression of the viral N protein. Independently of the expression of the SH protein or the location of the FLAG epitope fused to the SH gene, similar numbers of cells expressed the N protein, suggesting a similar rate of infection for all rMuVs, and differences were found not to be statistically significant (Fig. 2D and E).
The SH protein has been described as a membrane protein with its N terminus located at the cell surface and its C terminus orientated toward the cytoplasm (16). To corroborate these data in the context of an MuV infection, permeabilized and nonpermeabilized rMuV-infected A549 cells were examined for SH expression by staining the SH protein with an anti-FLAG antibody followed by flow cytometry analysis. In nonpermeabilized cells, the SH protein was detected in rMuV-SH- but not in rMuV-SH-C-infected cells (Fig. 2F, gray bars), confirming that the N terminus is oriented toward the extracellular space and classifying SH as a type I membrane protein. We therefore assume that the C-terminal domain of SH may interfere with cellular signaling pathways, which might be influenced by a C-terminal FLAG tag. Because of this, we decided to perform all further studies with rMuV-SH and rMuV-SHstop exclusively.
The SH protein reduces phosphorylation of IKKβ, IκBα, and p65.
To investigate if the SH protein influences NF-κB activation in the context of MuV infection, phosphorylation of IKKβ and its substrates, IκBα and p65, was analyzed by Western blotting. A549 cells were lysed at 1, 15, and 20 h p.i. and analyzed with respect to the amounts of total IKKβ, IκBα, and p65 and of their phosphorylated forms. MuV N and SH proteins were detected as a control for successful infection. Uninfected cells, either nonstimulated or stimulated with TNF-α, served as controls for NF-κB activation. At 1 h p.i., levels of phosphorylated IKKβ in rMuV-SH- and rMuV-SHstop-infected cells were comparable to those of stimulated control cells (Fig. 3A), possibly due to the large amount of virus particles and serum components in the suspension used for infection. In contrast, at later time points of infection, the levels of phosphorylated IKKβ were lower in all infected cells. However, the phosphorylation was even more reduced in the presence of the SH protein in rMuV-SH-infected cells compared to cells infected with rMuV-SHstop which lack SH expression. The amount of total IKKβ was not affected by SH. This indicates that SH does not influence IKKβ expression but rather reduces IKKβ phosphorylation (Fig. 3A). The analysis of IκBα (Fig. 3B) and p65 (Fig. 3C) phosphorylation also revealed lower levels of phosphorylation in rMuV-SH-infected compared to rMuV-SHstop-infected cells at 15 h and 20 h p.i., demonstrating that SH also leads to reduced IκBα and p65 phosphorylation.
FIG 3.
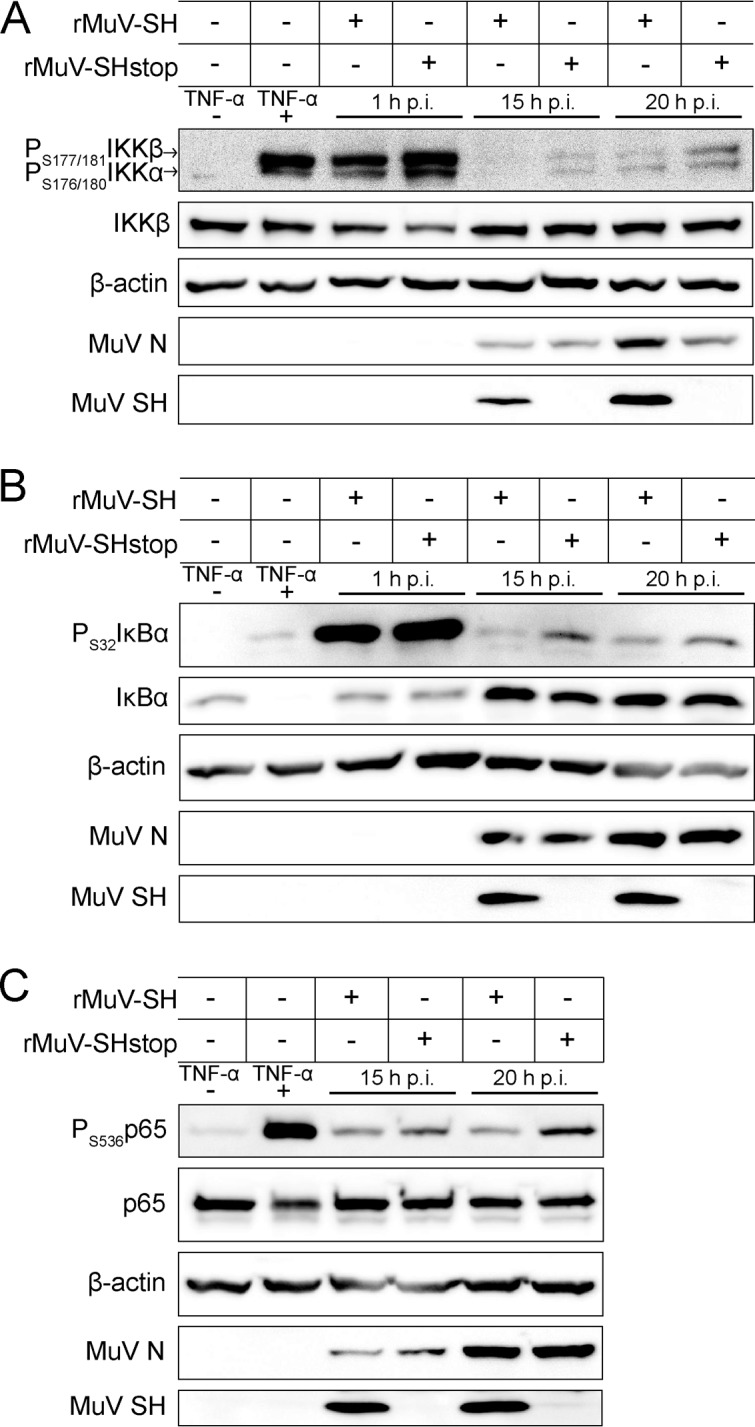
Effect of SH on the phosphorylation of NF-κB pathway proteins. A549 cells were infected with rMuV-SH or rMuV-SHstop at an MOI of 5. As a positive control, cells were stimulated with TNF-α (100 ng/ml) for 15 min (+). Untreated cells served as a negative control (−). Cells were lysed at time points indicated and analyzed by Western blotting for phosphorylation of IKKβ at Ser177/181 (A), IκBα at Ser32 (B), and p65 at Ser536 (C) using antibodies specific for the phosphorylated form of the respective protein. Detection of total levels of IKKβ, p65, IκBα, β-actin, MuV N, and MuV SH served as controls. The data shown are representative of three independent experiments.
The SH protein decreases p65 translocation into the nucleus.
The reduced levels of IKKβ and IκBα phosphorylation in the presence of SH suggested that the translocation of p65 into the nucleus might also be diminished by SH in infected cells. This assumption was investigated by immunofluorescence analysis. Therefore, rMuV-infected A549 cells were fixed at 15 h and 20 h p.i. and stained for p65 and MuV N protein. At both time points, syncytia as well as the N protein were detected; no difference in infection rate between cells infected with rMuV-SH or rMuV-SHstop was observed (Fig. 4A; data for 15 h p.i. are not shown). In contrast, at both time points the number of cells with nuclear localization of p65 was significantly reduced by about 84% in cells infected with rMuV-SH compared to levels in cells infected with rMuV-SHstop (1.4% versus 9.8% of cells were positive for p65 nuclear localization at 15 h p.i. and 4.8% versus 28.2% at 20 h p.i.) (Fig. 4A and B). The closeup of representative syncytia at 20 h p.i. highlights that p65 is distributed evenly across the cytoplasm in most of the cells infected with rMuV-SH, whereas in rMuV-SHstop-infected cells, p65 is almost entirely localized in the nuclei. These data support the assumption that SH interferes with the translocation of p65 into the nucleus of infected cells.
FIG 4.

p65 translocation in rMuV-infected cells. A549 cells were infected with rMuV-SH or rMuV-SHstop at an MOI of 5. (A) At 20 h p.i., cells were fixed and stained for p65 using a specific Cy3-coupled antibody, the viral N protein was stained using a specific Alexa 488-coupled antibody, and the nuclei were DAPI stained (not shown). Images were generated using a Zeiss cLSM780 confocal laser scanning microscope. Representative syncytia are shown in a closeup. (B) A total of 1.6 × 104 cells from three individual experiments were analyzed for p65 translocation in infected cells at 15 and 20 h p.i. The graph depicts means ± SD. ****, P < 0.0001; results were calculated by unpaired t tests with a two-tailed P value.
The SH protein interferes with NF-κB activation at the level of receptor complexes.
The impact of SH on NF-κB activation for a number of different stimuli and receptors was investigated using a firefly luciferase reporter gene assay. 293G and A549 cells were transfected with the luciferase reporter plasmid and a plasmid encoding SH or an empty vector as a control. NF-κB activation was stimulated by the treatment of the cells with either TNF-α (ligand of TNFR1), the dsRNA analogue poly(I·C) (ligand of TLR3), IL-1β (ligand of IL-1R1), the synthetic triacylated lipopeptide Pam3CSK4 (ligand of TLR1/2), or LPS (ligand of TLR4). In 293G cells, TNF-α and poly(I·C) induced a 15- or 20-fold NF-κB activation, respectively (Fig. 5A, black bars). Treatment with IL-1β, Pam3CSK4, or LPS resulted in less than a 2-fold activation of NF-κB. In the presence of SH, NF-κB activation was significantly reduced by about 60% when induced by TNF-α or poly(I·C) (Fig. 5A, gray bars). Due to the low level of activation following stimulation with IL-1β, Pam3CSK4, or LPS, the effect of SH was not investigated after treatment with those stimuli. In A549 cells, TNF-α and IL-1β induced a 140- or 130-fold activation, respectively, which was also reduced significantly in the presence of the SH protein by about 60% (Fig. 5B). Treatment with Pam3CSK4, LPS, and poly(I·C) resulted in a 20-, 6-, and 2-fold NF-κB activation, respectively, which was reduced in the presence of SH by 60% in cells treated with Pam3CSK4 and by 30% in cells treated with LPS, albeit not significantly. Reduction upon poly(I·C) stimulation was not investigated due to the low level of NF-κB activation. Taken together, these findings indicate that the SH protein hinders activation of NF-κB via stimulation of not only TNFR1 but also of IL-1R1 and TLR3.
FIG 5.

Effect of SH on NF-κB activation after stimulation with TNF-α, IL-1β, and poly(I·C). 293G (A) and A549 (B) cells were transfected with p(PRDII)5tkΔ(−39)lucter, pCMV-βGal, and pCMV-N-HA-SH or pCMV-HA as a control. At 24 h p.t., cells were stimulated for 4 h with 100 ng/ml TNF-α, 20 μg/ml poly(I·C), 10 ng/ml IL-1β, 5 μg/ml Pam3CSK4, or 1 μg/ml LPS. Afterwards, cells were lysed and analyzed for luciferase and β-galactosidase activity. Graphs depict means ± SD for two (A) or three (B) individual experiments. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001; results were calculated by unpaired t tests with a two-tailed P value. (C) A549 cells were transfected with luciferase and β-galactosidase reporter plasmids. At 24 h p.t., cells were infected with rMuV-SH or rMuV-SHstop at an MOI of 0.5. At 16 h p.i., cells were stimulated for 4 h with 100 ng/ml TNF-α or 10 ng/ml IL-1β. Afterwards, cells were lysed and analyzed for luciferase and β-galactosidase activity. The values for rMuV-SH-infected cells were normalized to those for rMuV-SHstop-infected cells. Graphs depict means ± SD from at least three individual experiments. (D) 293G cells were transfected with luciferase and β-galactosidase reporter plasmids and a plasmid for SH protein expression or the empty vector. In addition, cells were transfected with pUNO.MyD88, pFLAG-CMV2-TRAF2, pME18SFLAG-TRAF6, pCMV-TAK1, or pcDNA3-IKKβ. At 28 h p.t., cells were lysed and analyzed for luciferase and β-galactosidase activity. The values for pCMV-N-HA-SH-transfected cells were normalized to those for pCMV-HA-transfected cells. Graph depicts means ± SD from three individual experiments. **, P < 0.01; results were calculated by unpaired t tests with a two-tailed P value.
Additionally, the impact of the SH protein on NF-κB activation was investigated in infected A549 cells. NF-κB activation following stimulation with TNF-α and IL-1β was reduced by about 60% in rMuV-SH-infected cells compared to rMuV-SHstop-infected cells, confirming the impact of SH on NF-κB activation via the stimulation of TNFR1 and IL-1R1 also in the context of an MuV infection (Fig. 5C).
To narrow down at which point in the respective signaling pathway the SH protein interferes with NF-κB activation, cells were transfected with plasmids encoding the proteins MyD88, TRAF2, TRAF6, TAK1, and IKKβ. Overexpression of these proteins results in the activation of the NF-κB pathway downstream of the respective protein. 293G cells were cotransfected with the luciferase reporter plasmid and a plasmid encoding SH or the empty vector as a control. As shown in Fig. 5D, the SH protein reduced NF-κB activation by 60% in cells overexpressing MyD88. In contrast, NF-κB activation due to overexpression of TRAF2, TRAF6, TAK1, and IKKβ was not affected by the presence of SH. Our findings indicate that the SH protein impacts NF-κB signaling downstream of or at the level of MyD88 and upstream of TRAF2, TRAF6, TAK1, and IKKβ in the respective pathways (see Fig. 7). We therefore hypothesize that SH interacts with proteins of the receptor complex of TNFR1, IL-1R1, and TLR3.
FIG 7.

Model of MuV SH interference with NF-κB activation. SH expression leads to a reduction of MuV-induced IKKβ, IκBα, and p65 phosphorylation and p65 translocation into the nucleus, resulting in decreased activation of NF-κB-controlled genes. Luciferase reporter gene assays revealed that SH (red) impacts NF-κB activation downstream of or at the level of MyD88 (blue) and upstream of TRAF2, TRAF6, TAK1, and IKKβ (orange) in the respective pathways. As SH was shown to interact with TNFR1, RIP1, and IRAK1 (green), we assume that SH inhibits NF-κB activation by interacting with the receptor complexes of TNFR1, IL-1R1, and TLR3 in the plasma membranes of infected cells.
The SH protein interacts with the TNFR1, IL-1R1, and TLR3 receptor complexes.
To verify the interaction of SH with these receptor complexes, coimmunoprecipitation assays were performed. 293G or A549 cells were transfected with a plasmid encoding FLAG-tagged SH or an empty vector as a control. Proteins were precipitated by the use of FLAG-specific antibodies and analyzed by Western blotting. The SH protein was only detected in lysates and precipitates of cells transfected with pCMV-N-flag-SH (Fig. 6A). Endogenous expression of TNFR1, RIP1, IRAK1, MyD88, and IKKβ was similar in the cell lysates of all samples, independent of SH expression. Most importantly, TNFR1, RIP1, and IRAK1 coimmunoprecipitated with the SH protein, while IKKβ and MyD88 did not (Fig. 6A). To verify the interaction of SH with the receptor complexes in the context of an MuV infection, A549 or 293G cells were infected with rMuV-SH and rMuV-SHstop and their lysates were subjected to coimmunoprecipitation and Western blot analysis. The SH protein was detected in the precipitates of rMuV-SH-infected cells but not in all cell lysates, presumably due to the low expression levels of SH in infected cells. Endogenous TNFR1, RIP1, IRAK1, and MyD88 were detected at similar levels in all cell lysates. TNFR1, RIP1, and IRAK1 coimmunoprecipitated with SH, whereas MyD88 did not (Fig. 6B). These findings support the assumption that the SH protein interacts with the receptor complexes of TNFR1, IL-1R1, and TLR3.
FIG 6.

Coimmunoprecipitation of SH with proteins of the TNFR1, IL-1R, and TLR3 receptor complexes. (A) 293G cells or A549 cells (for MyD88) were transfected with a plasmid for expression of an N-terminally flagged SH protein (pCMV-N-flag-SH) or the empty vector (pCMV-HA) as a control. At 20 h p.t., the cells were lysed and incubated overnight with agarose beads covalently attached to an anti-FLAG antibody. Subsequently, bound proteins were eluted and separated by SDS-PAGE and analyzed via Western blotting using an anti-FLAG antibody and specific monoclonal antibodies against TNFR1, RIP1, IKKβ, MyD88, and IKAK1. GAPDH served as a control. The data shown are representative of three independent experiments. IP, immunoprecipitation. (B) A549 or 293G cells (for RIP1) were infected with rMuV-SH or rMuV-SHstop at an MOI of 5 or 10, respectively. At 20 h p.i., cells were lysed and analyzed as described for panel A. The data shown are representative of three independent experiments.
DISCUSSION
Small RNA viruses encode proteins interfering with innate immunity to promote their successful replication and dissemination (30). Here, we show that MuV SH impedes NF-κB activation in infected cells by decreasing the phosphorylation of IKKβ, IκBα, and p65 and the translocation of p65 into the nucleus. SH-mediated inhibition was observed not only after activation of NF-κB by TNF-α but also after stimulation of cells with IL-1β and poly(I·C). Because we found SH coimmunoprecipitated with TNFR1, RIP1, and IRAK1, we conclude that the SH protein reduces NF-κB activation by interacting with TNFR1, IL-1R1, and TLR3 complexes in the membranes of infected cells.
Efficient paramyxovirus replication is dependent on a well-balanced gradient of viral mRNAs, provided by the order and the number of viral genes on the single-stranded RNA genome (31), as well as on a hexameric genome length (rule of six) for promoter recognition and viral editing (32). In order to generate rMuV deficient in SH with the least possible effect on virus replication, we introduced three stop codons into the SH gene (similar to Malik et al. [33]). To allow the detection of SH in infected cells, we additionally fused a FLAG epitope coding sequence to the SH gene while complying with the rule of six. Earlier studies usually investigated the function of paramyxoviral SH proteins using viruses lacking the complete SH gene. Cytopathic effect (CPE) and apoptosis induction was enhanced in cells infected with those viruses, which was mainly attributed to an increase of NF-κB-dependent secretion of TNF-α (19, 21, 22, 34–36). Similarly, we found NF-κB activation to be enhanced in cells infected with rMuV deficient in SH, confirming that the observed effect was due to the interference of MuV SH with NF-κB activation and not a consequence of the disruption of the genomic structure.
Manipulation of the NF-κB pathway is a common principle used by a plethora of viruses to impede the host's innate immune system. The intervention can occur at various levels, ranging from the interaction with cellular receptors to the modulation of downstream enzyme activities. We were able to show that MuV SH leads to a reduction of the phosphorylation of IκBα (Ser32), IKKβ (Ser177/181), and p65 (Ser536) as well as of the translocation of p65 when SH is present in infected cells, corroborating the results observed in studies using viruses with deleted SH genes (19, 21, 22, 34, 35, 37). Most importantly, we found that SH not only inhibits NF-κB activation mediated by TNF-α but also interferes with NF-κB activation induced by IL-1β and poly(I·C). These findings show that SH interferes with the signaling of TNFR1 as well as IL-1R and TLR3, thereby extending the knowledge of the pathways that SH influences. SH reduced NF-κB activation via these receptors to approximately the same extent (60%) (Fig. 5A, B, and C), leading to the hypothesis that the mechanism of SH-mediated NF-κB repression is similar. In fact, we found SH to coimmunoprecipitate with TNFR1, RIP1, and IRAK1. RIP1 functions as a key regulatory element in TNFR1- and TLR3-mediated activation of NF-κB and is recruited to the activated receptors (38); IRAK1 is described as an adaptor molecule of activated IL-1R (39). Whether SH interacts with TNFR1, RIP1, or IRAK1 directly or whether the interaction is mediated by SH binding to other proteins of the receptor complexes has not yet been determined. We hypothesize that SH interacts with the receptor complexes of TNFR1, IL-1R1, and TLR3 in the plasma membrane, thereby inhibiting downstream signaling and NF-κB activation (Fig. 7). This assumption is supported by detection of the SH protein in the plasma membrane of infected cells (flow cytometry) (Fig. 2F) and the specification of the pathway level at which SH impedes NF-κB activation (luciferase reporter assay) (Fig. 5D).
Two cell lines were employed which differed in their response to the various stimuli, possibly as a result of cell- or strain-specific defects in signaling pathways and/or receptor expression. A549 cells have already been reported not to respond to poly(I·C) when it was added to the culture medium without other supplementation (40); 293G cells have been shown to need IL-1R, TLR2, or TLR4 supplementation in order to respond efficiently to IL-1β, bacterial lipoproteins, or LPS (41–43). Older studies described 293G cells to be deficient in TLR3 expression, while more recent publications showed that they express TLR3 mRNA and that they are positive for intracellular TLR3 staining (44–46). Similarly, we proved the cells to be positive for TLR3 (Western blotting; data not shown) and were able to induce NF-κB activation by adding poly(I·C) to the medium.
Although paramyxoviral SH proteins exert similar functions, they exhibit no sequence homology except for their transmembrane region. They differ in size (from 183 amino acids in human metapneumovirus to 44 amino acids in parainfluenza virus type 5) and in membrane orientation (16, 20, 35, 47–50). Flow cytometry analysis revealed the N terminus of MuV SH to be directed toward the extracellular space and the C terminus toward the intracellular space in infected cells, classifying MuV SH as a type I membrane protein and confirming previous findings by Takeuchi et al. (16). The 8 amino acids at the N terminus of MuV SH are highly conserved among different MuV strains. In contrast, the 26-amino-acid C terminus is highly variable (51, 52). Nevertheless, it contains several polar and charged amino acids, including one conserved threonine and serine residue. The contribution of those amino acids to SH-mediated NF-κB inhibition in correlation with strain variation will be the subject of future studies.
Interference with NF-κB activation often results in the reduction of apoptosis and a reduced expression of proinflammatory cytokines such as TNF-α or IL-1β, providing more time for virus replication (21, 36, 53). We did not observe MuV SH to have a severe impact on virus replication in cell culture, which is in line with previous findings by Malik et al. (33). In a neurotoxicity test involving intracerebral injection of MuV into the brain of newborn rats (54), rMuV lacking the entire SH gene was shown to be attenuated, while rMuV deficient in SH due to additional stop codons was not (33). However, effects on NF-κB signaling might become evident only in a more physiological setting, i.e., during MuV infection via the respiratory tract.
In conclusion, we report that MuV SH protein interferes with NF-κB activation by interacting with the complexes of TNFR1, IL-1R1, and TLR3 in the plasma membrane of infected cells, thereby possibly serving as a virulence factor for MuV replication in vivo.
MATERIALS AND METHODS
Cell culture.
293G (kindly provided by C. Uhlenhaut, Berlin, Germany), Vero76, and A549 cells (both from ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 5% fetal bovine serum (FBS; PAN Laboratories GmbH), 2 mM l-glutamine (Lonza), 100 U/ml penicillin (Gibco), and 100 μg/ml streptomycin (Gibco). BSR-T7 cells (kindly provided by K. K. Conzelmann, Munich, Germany) were cultured in Eagle's minimum essential medium (EMEM; Gibco) supplemented with 5% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Geneticin (G-418; PAA Laboratories GmbH) was added to every second passage at a concentration of 1 mg/ml. All cell lines were cultured in a humidified atmosphere at 37°C and 5% CO2.
Plasmid construction and rescue of recombinant mumps viruses.
To generate recombinant viruses with a FLAG epitope fused to the SH gene and recombinant viruses deficient in SH protein translation, the plasmid pG-MuV-FL (kindly provided by S. A. Udem, New York, USA) was used, which carries the MuV genome of the major component of the vaccine strain Jeryl Lynn (JL5). A fragment consisting of part of the F gene, the SH gene, and part of the intergenic region between the SH and HN genes was amplified via PCR (primers GCAACCTCCCTAGGATTATACCT and CGGAACACAGTTGTGATAGCAG) and cloned into the vector pCR2.1-TOPO (Invitrogen). To enable the reinsertion of the modified fragment into pG-MuV-FL, the AvrII restriction site in the intergenic region between the F and SH genes was modified by site-directed mutagenesis. For further modification, the SH fragment was inserted into the shuttle vector pUC19 (Clontech) using EcoRI. Three translational stop codons were introduced into the SH gene at positions 13 (CAA mutated to TAA), 75 (TAT mutated to TAA), and 115 (CGA mutated to TGA) using site-directed mutagenesis. In addition, a FLAG epitope coding sequence was fused to either the 5′ terminus or the 3′ terminus of SH. The modified SH fragments were reinserted into pG-MuV-FL using AvrII. Infectious viruses from the mutated MuV genomes were generated by cotransfecting the modified pG-MuV-FL plasmids (5 μg) with pMuV-L (200 ng), pMuV-N (300 ng), and pMuV-P (50 ng) (all kindly provided by S. A. Udem, New York, USA) into BSR-T7 cells, which were seeded in 60-mm dishes, using 2 μl polyethylenimine (PEI) (Sigma-Aldrich). The virus-containing supernatants were harvested when 80 to 90% of cells showed cytopathic effects (CPE). The identity of the rescued viruses, rMuV-SH-N-flag (henceforth referred to as rMuV-SH), rMuV-SH-3stop-N-flag (rMuV-SHstop), rMuV-SH-C-flag (rMuV-SH-C), and rMuV-SH-3stop-C-flag (rMuV-SHstop-C), was confirmed by reverse transcription-PCR followed by dideoxy sequencing.
Virus propagation.
Vero76 cells were infected with rMuV at a multiplicity of infection (MOI) of 0.003 and incubated in DMEM containing 2% FBS. Supernatants were harvested when 90 to 100% of cells showed CPE. Cell debris was pelleted by centrifugation at 2,000 × g for 10 min. rMuV was enriched in the supernatant using Vivaspin 20 columns (Sartorius) and centrifugation at 3,000 × g.
Virus growth analysis.
To analyze virus growth, 1.6 × 105 Vero76 cells were infected at an MOI of 0.01. After virus adsorption for 1 h, the cells were washed three times with phosphate-buffered saline (PBS) to remove unbound viruses and further incubated in DMEM containing 2% FBS. Every 24 h, supernatants were collected and cell debris was removed by low-speed centrifugation. Virus titers were determined by immunocolorimetric plaque assay, which involved incubating Vero76 cells with virus dilutions for 1 h and then overlaying the cells with DMEM, 0.5% carboxymethyl cellulose (CMC; Sigma-Aldrich), 6% 10× minimum essential medium (MEM), 0.165% sodium bicarbonate, 100 U/ml penicillin, and 100 μg/ml streptomycin. At 6 days postinfection, the cells were fixed with 2% paraformaldehyde (PFA; Thermo Fisher Scientific), permeabilized with 100% methanol (Roth) at −20°C, and blocked in PBS containing 0.1% Tween 20 (Roth), 0.6% bovine serum albumin (BSA; GE Healthcare), and 1% FBS. Plaques were visualized on the monolayer by staining with mouse monoclonal anti-mumps fusion protein antibody (Mab846; Merck Millipore), goat-anti-mouse IgG conjugated with horseradish peroxidase (HRP; Merck Millipore), and 3,3′,5,5′-tetramethylbenzidine substrate (TMB; Mikrogen Diagnostik).
Immunoblot analysis.
Vero76 or A549 cells (6 × 105) were infected with the different rMuVs at an MOI of 0.01 or 5, respectively. Virus suspension was removed after 1 h and cells were washed two times with PBS and cultured in DMEM containing 5% FBS. Cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) containing protease inhibitor cocktail (P8340; Sigma-Aldrich) 1 h, 15 h, 20 h, or 72 h postinfection and incubated with 0.125 U/μl Benzonase nuclease (Merck Millipore) for 5 min at room temperature (RT). The protein concentration was determined using the Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher) according to the manufacturer's recommendations. Fifty to 60 μg of protein was subjected to SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked with PBS containing 0.1% Tween 20 and 5% nonfat milk powder for 1 h at RT and incubated with primary antibodies according to the manufacturer's recommendations. The following primary antibodies were used: rabbit polyclonal anti-phospho-IKKα/β (Ser176/180) (no. 2697), rabbit polyclonal anti-IKKβ (no. 2684), rabbit monoclonal anti-phospho-NF-κB p65 (Ser536) (no. 3033), rabbit monoclonal anti-NF-κB p65 (no. 8242), rabbit polyclonal anti-phospho-IκBα (Ser32) (no. 2859), mouse monoclonal anti-IκBα (no. 4814), rabbit monoclonal anti-β-actin (no. 4970), rabbit monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (no. 5174), rabbit monoclonal anti-TNFR1 (no. 3736), rabbit monoclonal anti-RIP1 (no. 3493), rabbit monoclonal anti-IRAK1 (no. 4504), rabbit monoclonal anti-MyD88 (no. 4283) (all Cell Signaling Technology), mouse monoclonal anti-mumps NP (sc-57919; Santa Cruz Biotechnology), and mouse monoclonal anti-FLAG M2 (no. F1804; Sigma-Aldrich) to detect the SH protein. Primary antibodies were detected with anti-rabbit or anti-mouse IgG conjugated with HRP (no. 7074 and 7076, respectively; Cell Signaling Technology) and applied according to the manufacturer's recommendations. Antibody binding was visualized using Lumi-Light Plus Western blotting substrate (Sigma-Aldrich) and detected by an Intas Advanced Imager.
Flow cytometry.
A total of 6 × 105 Vero76 or A549 cells were infected with the different rMuVs at an MOI of 5. After incubation for 1 h, the cells were washed two times with PBS and cultured in DMEM containing 5% FBS. At various time points, cells were detached with trypsin 0.05%–EDTA 0.02% (PAN Laboratories GmbH), fixed with 2% PFA in PBS for 20 min at RT, and permeabilized with 0.5% saponin (Sigma-Aldrich) in PBS containing 2.5% FBS and 0.05% sodium azide (Merck Millipore). Cells were stained with rat monoclonal anti-FLAG antibody coupled to allophycocyanin (APC; no. 637307; BioLegend) or with mouse monoclonal anti-mumps NP (sc-57922; Santa Cruz Biotechnology) diluted in PBS containing 0.5% saponin, 2.5% FBS, and 0.05% sodium azide for 20 min on ice. For detection of mumps NP protein, cells were additionally stained with goat Fab anti-mouse IgG (H+L)-Alexa Fluor 488 (no. 115-547-003; Dianova) for 15 min on ice. Data were generated using a FACSCalibur flow cytometer (BD Biosciences) and Cell Quest Pro software (BD Biosciences). Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, Inc.). For calculations of significance, an unpaired t test with a two-tailed P value was used.
Immunofluorescence assay.
Vero76 and A549 cells grown on coverslips in 24-well tissue culture plates were infected for 1 h with the different rMuVs at an MOI of 0.01 (Vero76) or 5 (A549). The cells were washed two times with PBS before cultivating them in DMEM containing 5% FBS. At 15 h, 20 h, or 48 h postinfection, the cells were fixed with 4% PFA in PBS for 20 min at RT, permeabilized with 0.1% Triton X-100 (Merck Millipore) in PBS for 10 min at RT, and blocked with 1% BSA in PBS containing 0.05% Tween 20 for 1 h at RT. To couple the primary antibodies to Fab fragments fused with fluorescence dyes, mouse monoclonal anti-mumps fusion protein (Mab846; Merck Millipore), mouse monoclonal anti-mumps NP (sc-57919; Santa Cruz Biotechnology), and rabbit monoclonal anti-NF-κB p65 (no. 8242; Cell Signaling Technology) were incubated for 20 min with goat Fab anti-mouse IgG (H+L)-Cy3 (no. 115-167-003), goat Fab anti-mouse IgG (H+L)-Alexa Fluor 488 (no. 115-547-003), and goat Fab anti-rabbit IgG (H+L)-Cy3 (no. 111-167-003) (all Dianova), respectively. The coupled antibodies were incubated for 20 min with unspecific IgG against mouse or rabbit (Dianova) before the antibodies were mixed, diluted in blocking buffer, and incubated with the cells for 1 h at RT. Following staining with 4′,6-diamidino-2-phenylindole (DAPI) (Roth) for 10 min at RT, the cells were mounted using Mowiol (Merck Millipore). Images were obtained using a Zeiss cLSM780 confocal laser scanning microscope and Zen 2012 software. To quantify the p65 translocation into the nucleus, three to four images per virus and time point of three individual experiments, each image containing 200 to 500 cells, were analyzed. Each cell was assessed with respect to MuV N expression and p65 being distributed in the cytoplasm or in the nucleus. The percentage of MuV N-positive cells with p65 translocated to the nucleus in relation to the total number of N-positive cells was calculated for each time point and both rMuVs. Statistical analyses were performed using GraphPad Prism 5. For calculations of significance, an unpaired t test with a two-tailed P value was used.
Luciferase reporter assay.
A total of 3 × 105 293G or A549 cells were transfected with the luciferase reporter plasmid p(PRDII)5tkΔ(−39)lucter, which contains the luciferase gene under the control of NF-κB-binding elements of the beta interferon promoter (55) (293G cells, 240 ng; A549 cells, 480 ng) and the β-galactosidase reporter plasmid pCMV-βGal (293G cells, 35 ng; A549 cells, 70 ng) (both plasmids kindly provided by S. Goodbourn, London, United Kingdom). The cells were additionally transfected with pCMV-N-HA-SH (56) for SH protein expression or the empty vector pCMV-HA (Clontech) alone or in combination with pUNO.MyD88 (Invivogen), pFLAG-CMV2-TRAF2, pME18SFLAG-TRAF6 (57) (kindly provided by W. Brune, Hamburg, Germany), pCMV-TAK1 (kindly provided by K. Matsumoto, Nagoya, Japan), or pcDNA3-IKKβ (kindly provided by S. Goodbourn, London, United Kingdom) (293G cells, 725 ng; A549 cells, 1,450 ng) using X-tremeGENE HP DNA transfection reagent (Roche) according to the manufacturer's recommendations. In a different approach, 3 × 105 A549 cells were transfected with p(PRDII)5tkΔ(−39)lucter (480 ng), pCMV-βGal (70 ng), and pCMV-HA (1,450 ng). Cells were additionally infected 24 h posttransfection (p.t.) with rMuVs at an MOI of 0.5 for 1 h, washed two times with PBS, and incubated in DMEM containing 5% FBS. Cells were stimulated using 100 ng/ml TNF-α (PromoKine), 10 ng/ml IL-1β (InvivoGen), 5 μg/ml Pam3CSK4 (InvivoGen), 1 µg/ml LPS-EK ultrapure Escherichia coli strain K-12 (InvivoGen), or 20 μg/ml poly(I·C) (InvivoGen) or were left untreated. Cell lysis was carried out using reporter lysis buffer (Promega), and luciferase and β-galactosidase activities were analyzed using the ONE-Glo luciferase assay system and β-galactosidase enzyme assay system with reporter lysis buffer according to the manufacturer's recommendations (both from Promega). Data were compiled with a FLUOstar Omega photometer (BMG Labtech) using MARS data analysis software (BMG Labtech). Expression of β-galactosidase was used as an internal control of transfection. Fold induction was calculated in relation to unstimulated cells. Statistical analyses were performed using GraphPad Prism 5. For calculations of significance, an unpaired t test with a two-tailed P value was used.
Coimmunoprecipitation assay.
A total of 6 × 105 293G or A549 cells were transfected with pCMV-N-flag-SH or pCMV-HA using X-tremeGENE HP DNA transfection reagent (Roche) according to the manufacturer's recommendations. In a different approach, 6 × 105 A549 or 293G cells were infected with rMuV-SH or rMuV-SHstop at an MOI of 5 or 10, respectively, for 1 h, washed two times with PBS, and incubated in DMEM containing 5% FBS. Twenty hours after transfection or infection, the coimmunoprecipitation assay was performed using anti-FLAG M2 affinity gel (Sigma-Aldrich) according to the manufacturer's recommendations. Briefly, the cells were lysed on ice and cell debris was removed by centrifugation. Lysis buffer contained 50 mM Tris (Roth), pH 7.4 (HCl), 150 mM NaCl (Merck Millipore), 1 mM EDTA (Roth), 1% Triton X-100, and protease inhibitor cocktail. A fraction of the lysates, 3.3%, was stored separately for analysis of total protein expression. The remaining fraction of the lysates was incubated for 3 to 6 h with nProtein A Sepharose 4 Fast Flow (GE Healthcare) prior to addition to the resin. The resin was washed with Tris-buffered saline (TBS) and incubated for 3 to 6 h with 1% BSA (GE Healthcare), washed again, and added to the lysates. After overnight incubation, the resin was washed with lysis buffer and the proteins were eluted with 2× RotiLoad 1 (Roth). SDS-PAGE and Western blot analysis were performed as described for immunoblot analysis.
ACKNOWLEDGMENTS
We kindly thank Christine Uhlenhaut for proving 293G cells and Karl-Klaus Conzelmann for providing BSR-T7 cells. We are grateful to Stephen A. Udem, Steve Goodbourn, Wolfram Brune, and Kunihiro Matsumoto for providing plasmids. We also thank Ana Reis for helpful discussion, Anne Wolbert for her help writing the manuscript, and Jennifer Kaiser for experimental assistance.
The Sonnenfeld Foundation and the Georg and Agnes Blumenthal Foundation provided funding to Stephanie Franz.
REFERENCES
- 1.Hviid A, Rubin S, Muhlemann K. 2008. Mumps. Lancet 371:932–944. doi: 10.1016/S0140-6736(08)60419-5. [DOI] [PubMed] [Google Scholar]
- 2.Rubin S, Eckhaus M, Rennick LJ, Bamford CG, Duprex WP. 2015. Molecular biology, pathogenesis and pathology of mumps virus. J Pathol 235:242–252. doi: 10.1002/path.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galazka AM, Robertson SE, Kraigher A. 1999. Mumps and mumps vaccine: a global review. Bull World Health Organ 77:3–14. [PMC free article] [PubMed] [Google Scholar]
- 4.van Loon FP, Holmes SJ, Sirotkin BI, Williams WW, Cochi SL, Hadler SC, Lindegren ML. 1995. Mumps surveillance–United States, 1988-1993. MMWR CDC Surveill Summ 44:1–14. [PubMed] [Google Scholar]
- 5.Davidkin I, Kontio M, Paunio M, Peltola H. 2010. MMR vaccination and disease elimination: the Finnish experience. Expert Rev Vaccines 9:1045–1053. doi: 10.1586/erv.10.99. [DOI] [PubMed] [Google Scholar]
- 6.Borgmann S, Schwab F, Santibanez S, Mankertz A. 2014. Mumps virus infection in vaccinated patients can be detected by an increase in specific IgG antibodies to high titres: a retrospective study. Epidemiol Infect 142:2388–2396. doi: 10.1017/S0950268813003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eriksen J, Davidkin I, Kafatos G, Andrews N, Barbara C, Cohen D, Duks A, Griskevicius A, Johansen K, Bartha K, Kriz B, Mitis G, Mossong J, Nardone A, O'Flanagan D, De Ory F, Pistol A, Theeten H, Prosenc K, Slacikova M, Pebody R. 2013. Seroepidemiology of mumps in Europe (1996-2008): why do outbreaks occur in highly vaccinated populations? Epidemiol Infect 141:651–666. doi: 10.1017/S0950268812001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mankertz A, Beutel U, Schmidt FJ, Borgmann S, Wenzel JJ, Ziegler P, Weissbrich B, Santibanez S. 2015. Laboratory-based investigation of suspected mumps cases submitted to the German National Reference Centre for Measles, Mumps, and Rubella, 2008 to 2013. Int J Med Microbiol 305:619–626. doi: 10.1016/j.ijmm.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 9.Okajima K, Iseki K, Koyano S, Kato A, Azuma H. 2013. Virological analysis of a regional mumps outbreak in the northern island of Japan-mumps virus genotyping and clinical description. Jpn J Infect Dis 66:561–563. doi: 10.7883/yoken.66.561. [DOI] [PubMed] [Google Scholar]
- 10.Otto W, Mankertz A, Santibanez S, Saygili H, Wenzel J, Jilg W, Wieland W, Borgmann S. 2010. Ongoing outbreak of mumps affecting adolescents and young adults in Bavaria, Germany, August to October 2010. Euro Surveill 15:19748. [PubMed] [Google Scholar]
- 11.Watson-Creed G, Saunders A, Scott J, Lowe L, Pettipas J, Hatchette TF. 2006. Two successive outbreaks of mumps in Nova Scotia among vaccinated adolescents and young adults. CMAJ 175:483–488. doi: 10.1503/cmaj.060660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeuchi K, Tanabayashi K, Hishiyama M, Yamada A, Sugiura A. 1991. Variations of nucleotide sequences and transcription of the SH gene among mumps virus strains. Virology 181:364–366. doi: 10.1016/0042-6822(91)90504-5. [DOI] [PubMed] [Google Scholar]
- 13.Johansson B, Tecle T, Orvell C. 2002. Proposed criteria for classification of new genotypes of mumps virus. Scand J Infect Dis 34:355–357. doi: 10.1080/00365540110080043. [DOI] [PubMed] [Google Scholar]
- 14.WHO. 2012. Mumps virus nomenclature update: 2012. Wkly Epidemiol Rec 87:217–224. [PubMed] [Google Scholar]
- 15.Jin L, Orvell C, Myers R, Rota PA, Nakayama T, Forcic D, Hiebert J, Brown KE. 2015. Genomic diversity of mumps virus and global distribution of the 12 genotypes. Rev Med Virol 25:85–101. doi: 10.1002/rmv.1819. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi K, Tanabayashi K, Hishiyama M, Yamada A. 1996. The mumps virus SH protein is a membrane protein and not essential for virus growth. Virology 225:156–162. doi: 10.1006/viro.1996.0583. [DOI] [PubMed] [Google Scholar]
- 17.Bukreyev A, Whitehead SS, Murphy BR, Collins PL. 1997. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J Virol 71:8973–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He B, Leser GP, Paterson RG, Lamb RA. 1998. The paramyxovirus SV5 small hydrophobic (SH) protein is not essential for virus growth in tissue culture cells. Virology 250:30–40. doi: 10.1006/viro.1998.9354. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y, Bright AC, Rothermel TA, He B. 2003. Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic gene. J Virol 77:3371–3383. doi: 10.1128/JVI.77.6.3371-3383.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR, Collins PL, Buchholz UJ. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J Virol 78:12877–12887. doi: 10.1128/JVI.78.23.12877-12887.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu P, Li Z, Sun D, Lin Y, Wu J, Rota PA, He B. 2011. Rescue of wild-type mumps virus from a strain associated with recent outbreaks helps to define the role of the SH ORF in the pathogenesis of mumps virus. Virology 417:126–136. doi: 10.1016/j.virol.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson RL, Fuentes SM, Wang P, Taddeo EC, Klatt A, Henderson AJ, He B. 2006. Function of small hydrophobic proteins of paramyxovirus. J Virol 80:1700–1709. doi: 10.1128/JVI.80.4.1700-1709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verstrepen L, Bekaert T, Chau TL, Tavernier J, Chariot A, Beyaert R. 2008. TLR-4, IL-1R and TNF-R signaling to NF-kappaB: variations on a common theme. Cell Mol Life Sci 65:2964–2978. doi: 10.1007/s00018-008-8064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doyle SL, O'Neill LA. 2006. Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol 72:1102–1113. doi: 10.1016/j.bcp.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. 1999. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162:3749–3752. [PubMed] [Google Scholar]
- 26.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 27.Gilmore TD. 2006. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 28.Karin M, Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol 18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 29.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. 1999. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 30.Katze MG, Fornek JL, Palermo RE, Walters KA, Korth MJ. 2008. Innate immune modulation by RNA viruses: emerging insights from functional genomics. Nat Rev Immunol 8:644–654. doi: 10.1038/nri2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee AK, Barik S, De BP. 1991. Gene expression of nonsegmented negative strand RNA viruses. Pharmacol Ther 51:47–70. doi: 10.1016/0163-7258(91)90041-J. [DOI] [PubMed] [Google Scholar]
- 32.Kolakofsky D, Pelet T, Garcin D, Hausmann S, Curran J, Roux L. 1998. Paramyxovirus RNA synthesis and the requirement for hexamer genome length: the rule of six revisited. J Virol 72:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malik T, Shegogue CW, Werner K, Ngo L, Sauder C, Zhang C, Duprex WP, Rubin S. 2011. Discrimination of mumps virus small hydrophobic gene deletion effects from gene translation effects on virus virulence. J Virol 85:6082–6085. doi: 10.1128/JVI.02686-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuentes S, Tran KC, Luthra P, Teng MN, He B. 2007. Function of the respiratory syncytial virus small hydrophobic protein. J Virol 81:8361–8366. doi: 10.1128/JVI.02717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Z, Xu J, Patel J, Fuentes S, Lin Y, Anderson D, Sakamoto K, Wang LF, He B. 2011. Function of the small hydrophobic protein of J paramyxovirus. J Virol 85:32–42. doi: 10.1128/JVI.01673-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor G, Wyld S, Valarcher JF, Guzman E, Thom M, Widdison S, Buchholz UJ. 2014. Recombinant bovine respiratory syncytial virus with deletion of the SH gene induces increased apoptosis and pro-inflammatory cytokines in vitro, and is attenuated and induces protective immunity in calves. J Gen Virol 95:1244–1254. doi: 10.1099/vir.0.064931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bao X, Kolli D, Liu T, Shan Y, Garofalo RP, Casola A. 2008. Human metapneumovirus small hydrophobic protein inhibits NF-kappaB transcriptional activity. J Virol 82:8224–8229. doi: 10.1128/JVI.02584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ofengeim D, Yuan J. 2013. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 14:727–736. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- 39.Cao Z, Henzel WJ, Gao X. 1996. IRAK: a kinase associated with the interleukin-1 receptor. Science 271:1128–1131. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- 40.Tanabe M, Kurita-Taniguchi M, Takeuchi K, Takeda M, Ayata M, Ogura H, Matsumoto M, Seya T. 2003. Mechanism of up-regulation of human Toll-like receptor 3 secondary to infection of measles virus-attenuated strains. Biochem Biophys Res Commun 311:39–48. doi: 10.1016/j.bbrc.2003.09.159. [DOI] [PubMed] [Google Scholar]
- 41.Croston GE, Cao Z, Goeddel DV. 1995. NF-kappa B activation by interleukin-1 (IL-1) requires an IL-1 receptor-associated protein kinase activity. J Biol Chem 270:16514–16517. doi: 10.1074/jbc.270.28.16514. [DOI] [PubMed] [Google Scholar]
- 42.Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 274:10689–10692. doi: 10.1074/jbc.274.16.10689. [DOI] [PubMed] [Google Scholar]
- 43.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. 1999. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 44.Sarkar SN, Smith HL, Rowe TM, Sen GC. 2003. Double-stranded RNA signaling by Toll-like receptor 3 requires specific tyrosine residues in its cytoplasmic domain. J Biol Chem 278:4393–4396. doi: 10.1074/jbc.C200655200. [DOI] [PubMed] [Google Scholar]
- 45.Rudd BD, Burstein E, Duckett CS, Li X, Lukacs NW. 2005. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J Virol 79:3350–3357. doi: 10.1128/JVI.79.6.3350-3357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weber C, Muller C, Podszuweit A, Montino C, Vollmer J, Forsbach A. 2012. Toll-like receptor (TLR) 3 immune modulation by unformulated small interfering RNA or DNA and the role of CD14 (in TLR-mediated effects). Immunology 136:64–77. doi: 10.1111/j.1365-2567.2012.03559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hiebert SW, Paterson RG, Lamb RA. 1985. Identification and predicted sequence of a previously unrecognized small hydrophobic protein, SH, of the paramyxovirus simian virus 5. J Virol 55:744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hiebert SW, Richardson CD, Lamb RA. 1988. Cell surface expression and orientation in membranes of the 44-amino-acid SH protein of simian virus 5. J Virol 62:2347–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olmsted RA, Collins PL. 1989. The 1A protein of respiratory syncytial virus is an integral membrane protein present as multiple, structurally distinct species. J Virol 63:2019–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van den Hoogen BG, Bestebroer TM, Osterhaus AD, Fouchier RA. 2002. Analysis of the genomic sequence of a human metapneumovirus. Virology 295:119–132. doi: 10.1006/viro.2001.1355. [DOI] [PubMed] [Google Scholar]
- 51.Kyte J, Doolittle RF. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol 157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 52.Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, Grosdidier A, Hernandez C, Ioannidis V, Kuznetsov D, Liechti R, Moretti S, Mostaguir K, Redaschi N, Rossier G, Xenarios I, Stockinger H. 2012. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40:W597–W603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russell RF, McDonald JU, Ivanova M, Zhong Z, Bukreyev A, Tregoning JS. 2015. Partial attenuation of respiratory syncytial virus with a deletion of a small hydrophobic gene is associated with elevated interleukin-1beta responses. J Virol 89:8974–8981. doi: 10.1128/JVI.01070-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rubin SA, Pletnikov M, Carbone KM. 1998. Comparison of the neurovirulence of a vaccine and a wild-type mumps virus strain in the developing rat brain. J Virol 72:8037–8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Visvanathan KV, Goodbourn S. 1989. Double-stranded RNA activates binding of NF-kappa B to an inducible element in the human beta-interferon promoter. EMBO J 8:1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woznik M, Rodner C, Lemon K, Rima B, Mankertz A, Finsterbusch T. 2010. Mumps virus small hydrophobic protein targets ataxin-1 ubiquitin-like interacting protein (ubiquilin 4). J Gen Virol 91:2773–2781. doi: 10.1099/vir.0.024638-0. [DOI] [PubMed] [Google Scholar]
- 57.Ishida T, Mizushima S, Azuma S, Kobayashi N, Tojo T, Suzuki K, Aizawa S, Watanabe T, Mosialos G, Kieff E, Yamamoto T, Inoue J. 1996. Identification of TRAF6, a novel tumor necrosis factor receptor-associated factor protein that mediates signaling from an amino-terminal domain of the CD40 cytoplasmic region. J Biol Chem 271:28745–28748. doi: 10.1074/jbc.271.46.28745. [DOI] [PubMed] [Google Scholar]