

ABSTRACT
Ebola virus (EBOV), a member of the Filoviridae family, is a highly pathogenic virus that causes severe hemorrhagic fever in humans and is responsible for epidemics throughout sub-Saharan, central, and West Africa. The EBOV genome encodes VP35, an important viral protein involved in virus replication by acting as an essential cofactor of the viral polymerase as well as a potent antagonist of the host antiviral type I interferon (IFN-I) system. By using mass spectrometry analysis and coimmunoprecipitation assays, we show here that VP35 is ubiquitinated on lysine 309 (K309), a residue located on its IFN antagonist domain. We also found that VP35 interacts with TRIM6, a member of the E3-ubiquitin ligase tripartite motif (TRIM) family. We recently reported that TRIM6 promotes the synthesis of unanchored K48-linked polyubiquitin chains, which are not covalently attached to any protein, to induce efficient antiviral IFN-I-mediated responses. Consistent with this notion, VP35 also associated noncovalently with polyubiquitin chains and inhibited TRIM6-mediated IFN-I induction. Intriguingly, we also found that TRIM6 enhances EBOV polymerase activity in a minigenome assay and TRIM6 knockout cells have reduced replication of infectious EBOV, suggesting that VP35 hijacks TRIM6 to promote EBOV replication through ubiquitination. Our work provides evidence that TRIM6 is an important host cellular factor that promotes EBOV replication, and future studies will focus on whether TRIM6 could be targeted for therapeutic intervention against EBOV infection.
IMPORTANCE EBOV belongs to a family of highly pathogenic viruses that cause severe hemorrhagic fever in humans and other mammals with high mortality rates (40 to 90%). Because of its high pathogenicity and lack of licensed antivirals and vaccines, EBOV is listed as a tier 1 select-agent risk group 4 pathogen. An important mechanism for the severity of EBOV infection is its suppression of innate immune responses. The EBOV VP35 protein contributes to pathogenesis, because it serves as an essential cofactor of the viral polymerase as well as a potent antagonist of innate immunity. However, how VP35 function is regulated by host cellular factors is poorly understood. Here, we report that the host E3-ubiquitin ligase TRIM6 promotes VP35 ubiquitination and is important for efficient virus replication. Therefore, our study identifies a new host factor, TRIM6, as a potential target in the development of antiviral drugs against EBOV.
KEYWORDS: TRIM6, tripartite motif (TRIM) protein, VP35, viral RNA polymerase, Ebola virus, innate immunity, ubiquitination, unanchored ubiquitin, virus-host interactions
INTRODUCTION
Ebola virus (EBOV) belongs to the Filoviridae family of highly pathogenic viruses that cause severe hemorrhagic fever in humans and other mammals with high mortality rates (40 to 90%). EBOV is a nonsegmented, negative-sense RNA virus from the genus Ebolavirus. There are five species in this genus: Zaire ebolavirus (EBOV), Bundibugyo (BDBV), Sudan (SUDV), Reston (RESTV), and Tai Forest (TAFV) (1). Another member of the Filoviridae family, the Marburg virus (MARV; species Marburg marburgvirus), also causes severe hemorrhagic fever (2). In contrast to the highly pathogenic EBOV, RESTV is the only member of the Ebolavirus genus that does not appear to cause disease in humans, although it can cause lethal disease in nonhuman primates (3). The RNA genome of EBOV encodes seven structural proteins. Replication and transcription of the viral genome requires formation of a ribonucleoprotein complex, comprising the viral genome encapsidated by the nucleoprotein (NP) in association with the RNA-dependent RNA polymerase (RDRP) complex and a viral transcription factor, VP30. The RDRP complex consists of the catalytic subunit of the polymerase L and the polymerase cofactor VP35 (4–6). VP35 interacts with L and NP and regulates NP assembly and viral genome binding (7, 8), and it also controls viral RNA synthesis by modulating NP-viral RNA interactions (9).
VP35 is composed of an N-terminal coiled-coil domain and a C-terminal interferon (IFN) inhibitory domain (IID). The coiled-coil domain promotes VP35 oligomerization and is important for different functions, including polymerase activity (10–14). In addition to VP35's critical role in virus replication as a cofactor of the viral polymerase, it has also been extensively studied for its function in inhibition of innate signaling pathways and expression of antiviral type I interferons (IFN-I) (10, 11, 14–20). VP35 also inhibits the maturation of dendritic cells (DCs) and prevents efficient adaptive immune responses (21–25).
Innate immune signaling is initiated when pathogen-associated molecular patterns (PAMPs) are recognized by host pattern recognition receptors (PRRs), including Toll-like receptors (TLRs) and retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs). Virus recognition by PRRs triggers downstream signaling resulting in the induction of proinflammatory cytokines and IFN-I (26, 27). IFN-I production requires activation of the IKK-related kinases (TBK1 and IKKε), which phosphorylate the transcription factors IRF3 and IRF7 (28, 29). IFN-I then elicits its antiviral activity through the upregulation of IFN-I-stimulated genes (ISGs) (30). VP35 inhibits IFN-I production by multiple mechanisms. It sequesters viral RNA from the innate immune sensor RIG-I by directly interacting with double-stranded RNA (dsRNA) via its IID domain. Residues critical for RNA binding are also important for IFN-I inhibition but not for viral polymerase cofactor function of VP35 (31). VP35 also binds and inhibits RIG-I, PACT, and the IKKε and TBK-1 kinases to inhibit IFN-I production (18, 20, 32). Additionally, VP35 also antagonizes IFN-I induction by promoting sumoylation of IRF7 (33). VP35 is phosphorylated by the IKKε and TBK-1 kinases, acting as a decoy substrate (20); however, whether VP35 undergoes other posttranslational modifications is not known.
Ubiquitination of proteins is a conserved posttranslational modification important in many cellular functions, including immune signaling (34, 35). However, viruses have adapted to evade ubiquitin (Ub)-dependent antiviral responses and in many cases hijack the Ub system to enhance their own replication (36). Ub contains seven lysines, each of which can be conjugated by another Ub to form poly-Ub chains. Ub conjugation requires an E1 activating enzyme, E2 conjugating enzyme, and E3-ligase. The tripartite motif (TRIM) proteins belong to a large family of E3-Ub ligases, which are characterized by the presence of RING, B box, and coiled-coil domains (collectively called RBCC) and have been implicated in innate immunity (37–43). We recently showed that a large number of TRIMs positively regulate innate immune signaling pathways (42, 43). We further demonstrated that TRIM6 catalyzes the synthesis of unanchored K48-linked polyubiquitin chains, which activate the IKKε kinase for induction of IFN-I responses (44). As evidence of the critical role of TRIM6 in these antiviral responses, we recently showed that the pathogenic Nipah virus (family Paramyxoviridae) can inhibit IKKε and downstream signaling by targeting TRIM6 (45).
Recent studies suggest that host factors modulate VP35 function (18, 46, 47). However, the mechanisms by which the host influences VP35 activity are poorly defined. It is currently unknown whether VP35 undergoes posttranslational modifications and, if so, how it may regulate its function. Here, we show that TRIM6 interacts with the EBOV VP35 protein and promotes its ubiquitination, which stimulates virus polymerase activity and virus replication.
RESULTS
The host E3-ubiquitin ligase TRIM6 interacts with Ebola virus VP35 protein.
Our recent work has shown that TRIM6 activates the IKKε kinase for IFN-I production (44). Since EBOV VP35 previously has been shown to interact with and inhibit IKKε (20), we hypothesized that this inhibition occurs by interfering with TRIM6 function. In line with this hypothesis, TRIM6 efficiently interacted with VP35 in coimmunoprecipitation (co-IP) assays (Fig. 1A). In contrast, the NP protein from the lymphocytic choriomeningitis virus (LCMV), which also inhibits IKKε function (48), did not interact with TRIM6 (Fig. 1A). In addition, TRIM6 did not interact with the VP40 protein of the related Marburg virus, which is known to inhibit the activation of JAK1 kinase and IFN-I signaling (49, 50). To map the region of TRIM6 binding to VP35, we used deletion mutants of TRIM6 expressing the RING domain, B box domain, or the C-terminal SPRY domain (Fig. 1B) and tested interaction with VP35 by co-IP. The C-terminal SPRY domain of TRIM6 specifically interacted with VP35, whereas a mutant lacking only the SPRY domain (RBCC) lost the ability to interact with VP35 (Fig. 1C). Additional evidence of TRIM6-VP35 interaction was obtained in colocalization confocal microscopy assays. As we recently reported (44) and in line with previous reports (51), TRIM6 localized in distinct cytoplasmic dots (Fig. 1D, TRIM6 alone). In contrast, VP35 showed a diffuse, slightly granular cytoplasmic localization, and in some cases it localized in apparent inclusion bodies (Fig. 1D, VP35 alone). When VP35 and TRIM6 were coexpressed, VP35 was recruited to the TRIM6 cytoplasmic bodies where they colocalize (Fig. 1D). Consistent with our hypothesis and the binding assays, VP35 inhibited TRIM6-dependent IFN-β production when cells were stimulated through the RIG-I pathway (Fig. 1E); however, a K319A/R322A (KRA) VP35 mutant, which is unable to bind dsRNA and is known to have reduced IFN antagonist activity (11, 31), partially recovered IFN induction. Collectively, these data show that VP35 specifically interacts with the C-terminal SPRY domain of TRIM6 in cytoplasmic bodies, and VP35 is able to inhibit TRIM6-mediated IFN-I induction.
FIG 1.

EBOV VP35 interacts with the C-terminal SPRY region of TRIM6 and is recruited to TRIM6 cytoplasmic dots. (A) TRIM6 interacts with EBOV-VP35 but not with LCMV-NP or Marbug-VP40. HEK293T cells were transfected with plasmids encoding VP35, NP, VP40, and HA-TRIM6. Cells were harvested and lysed, and whole-cell extracts (WCE) were used for FLAG immunoprecipitation (IP) (VP35, NP, or VP40) using anti-FLAG beads. Immunobloting (IB) was performed with the indicated antibodies. (B and C) The C-terminal SPRY domain of TRIM6 interacts with VP35. (B) TRIM6 deletion mutants used for co-IP (GST tagged). (C) HEK293T cells were transfected with plasmids encoding the indicated GST-TRIM6 deletion mutants together with VP35. Cells were harvested and lysed, and WCE were used for TRIM6 pulldown using GST beads. (D) VP35 colocalizes with TRIM6 in cytoplasmic bodies. HeLa cells were transfected with plasmids encoding HA-TRIM6 and VP35. Cells were fixed and stained with the indicated primary antibodies (anti-HA and anti-Flag) and secondary antibodies (488 and 555) as described in Materials and Methods, followed by confocal microscopy. (E) VP35 inhibits RIG-I-induced TRIM6-mediated IFN-β. HEK293T cells were transfected with a plasmid encoding firefly luciferase under the control of the IFN-β promoter and a control plasmid encoding Renilla luciferase in the presence or absence of plasmids encoding the constitutively active RIG-I (2CARD), HA-TRIM6, and VP35 WT or an RNA binding-defective mutant of VP35 (KRA). Empty vector was used to ensure that the same amounts of plasmids were transfected. Cells were lysed after 30 h for luciferase assay. Data were normalized by the nonstimulated sample to obtain fold induction. The luciferase reporter assay was performed using triplicate samples, and results are depicted as the means ± SE (n = 3). All experiments were repeated at least two times, and representatives of independent experiments are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; NS, not significant.
VP35 is ubiquitinated on K309.
Since TRIM6 acts as an E3-Ub ligase and it interacts with VP35, we reasoned that VP35 might be subject to covalent ubiquitination. To test this, we performed mass spectrometry (MS) analysis from samples enriched for ubiquitinated proteins by immunoprecipitation of ectopically expressed hemagglutinin (HA)-tagged Ub in the presence of VP35, followed by trypsin digestion. This MS analysis identified a peptide of VP35 containing lysine 309 (K309), which was linked to diglycine residues, typically found on ubiquitinated peptides after trypsin digestion (Fig. 2A; peptides identified by MS are shown in red, and ubiquitinated lysine is shown in blue). To corroborate VP35 modification by Ub, we performed co-IP in which we pulled down ectopically expressed HA-tagged Ub in the presence of wild-type VP35 (VP35 WT) and a lysine-to-alanine change on residue 309 of VP35 (VP35 K309A). In support of the MS data, robust ubiquitination of VP35 WT was detected under these conditions (Fig. 2B). In contrast, reduced levels of ubiquitination were observed in the VP35 K309A mutant, suggesting that VP35 undergoes covalent ubiquitination on K309 (Fig. 2B). However, based on the predicted molecular masses of VP35 and the Ub protein and the different migrating forms of WT and K309A VP35 detected by immunoblotting (IB), our data suggest that in addition to K309 there is at least one other ubiquitination site on VP35. In addition, we also detected noncovalent interactions of VP35 with poly-Ub chains, and these interactions are not affected by the K309A mutation (Fig. 2B, the first band of VP35 corresponding to its monomer at approximately 37 kDa). According to these analyses, we could detect poly-Ub chains on VP35 WT of up to six Ub molecules (Hexa-Ub-VP35), from which only two Ub molecules are detectable on the VP35 K309 mutant. This observation suggests that the additional ubiquitination site is linked to two Ub molecules (di-Ub-VP35 on VP35 K309A) or, alternatively, two independent ubiquitination sites, each linked to mono-Ub. This observation is further substantiated by quantification of the different ubiquitinated forms of VP35, which indicates that levels of diubiquitinated VP35 and VP35 linked to longer poly-Ub chains are significantly decreased in VP35-K309A (Fig. 2C). Although we cannot conclusively exclude that VP35-K309A can be linked to chains longer than the di-Ub chains, our data suggest the presence of multiple potential ubiquitinated forms of VP35, which are schematically represented in Fig. 2D. Additional co-IP studies suggest that only the C-terminal region of VP35 (amino acids [aa] 219 to 340), which harbors the IID domain of VP35, associates with poly-Ub chains (Fig. 2E). Taken together, these data demonstrate that VP35 is covalently linked to poly-Ub chains on K309 and suggest that an additional site, probably on the C-terminal IID domain of VP35, also becomes covalently attached to Ub.
FIG 2.

EBOV-VP35 is ubiquitinated on K309. (A) Mass spectrometry analysis of ubiquitinated VP35. HEK293T cells were transfected with plasmids encoding VP35 and HA-Ub. After 30 h, cells were harvested and lysed, and whole-cell extracts (WCE) were used for HA immunoprecipitation (IP) using anti-HA beads. Ubiquitinated proteins were eluted with HA peptide. Eluted fractions were digested with trypsin and analyzed by MS. Peptides corresponding to VP35 are shown in red. GlyGly at residue 4 indicates ubiquitination (blue). (B) VP35 is ubiquitinated on K309. HEK293T cells were transfected with plasmids encoding VP35 WT or a VP35 K309A mutant and HA-Ub. Cells were harvested and lysed, and WCE were used for IP using anti-HA beads. Immunoblotting (IB) was performed with the indicated antibodies. The bands corresponding to the different ubiquitinated forms of VP35 are indicated according to the predicted molecular mass of Ub (∼8.5 kDa) and VP35 (37 kDa). (C) Quantification of the different ubiquitinated forms of VP35 WT and K309A. Densitometry of each separate band representing the different forms of ubiquitinated VP35 shown in panel B was done using ImageJ software. To normalize by the efficiency of HA-Ub pulldown and the levels of VP35 expression, the values were normalized first by the total amount of HA-Ub (shown in the IP and IB rows for HA-Ub in panel B) and then by the amounts of VP35 (from the WCE). The values obtained from 3 independent experiments were used to obtain statistics shown in panel C (*, P < 0.05; n = 3). (D) Schematic representation of the possible ubiquitinated forms of VP35 detected in the co-IP shown in panel B. Poly-Ub chains of up to 6 ubiquitin molecules attached to VP35 were detected based on molecular mass. The VP35 K309A mutant retains at least one di-Ub chain or two monoubiquitinated sites. (D) The C-terminal region of VP35 associates with ubiquitin. HEK293T cells were transfected with vectors encoding the N-terminal (1 to 218 aa) or C-terminal (219 to 340 aa) regions of VP35 (Flag tagged). Cells were harvested and WCE were used for IP using anti-FLAG beads. Immunoblotting was performed using anti-Ub antibodies.
VP35 interacts through covalent and noncovalent interactions with poly-Ub chains.
We have shown that VP35 is covalently linked to poly-Ub chains; however, these co-IP assays also suggested that VP35 could associate, by noncovalent interactions, with poly-Ub chains (Fig. 2B, the first band of VP35 corresponding to its monomer at approximately 37 kDa). Since TRIM6 interacts with VP35 and we recently showed that TRIM6 promotes the synthesis of unanchored poly-Ub chains (44), it was of interest to determine whether VP35 associates noncovalently with poly-Ub chains. To distinguish between VP35 covalently modified with Ub or VP35 interacting noncovalently with Ub or ubiquitinated proteins, a denaturing pulldown assay of His-tagged Ub coexpressed with VP35 was performed. These experiments show that denaturing washes with urea almost completely remove monomeric, noncovalently ubiquitinated VP35 (Fig. 3, first band from the bottom in the radioimmunoprecipitation assay [RIPA] wash), while slower-migrating ubiquitinated forms of VP35 corresponding to mono- and diubiquitinated VP35 were easily detectable after denaturing pulldown. It is worth noting that under these experimental conditions the proportion of VP35 that is covalently attached to Ub must be relatively low, because higher-migrating bands corresponding to polyubiquitinated VP35 are not detected in input samples and are only enriched upon Ub pulldown. Together, these data suggest that VP35 associates with poly-Ub chains via both covalent and noncovalent interactions.
FIG 3.
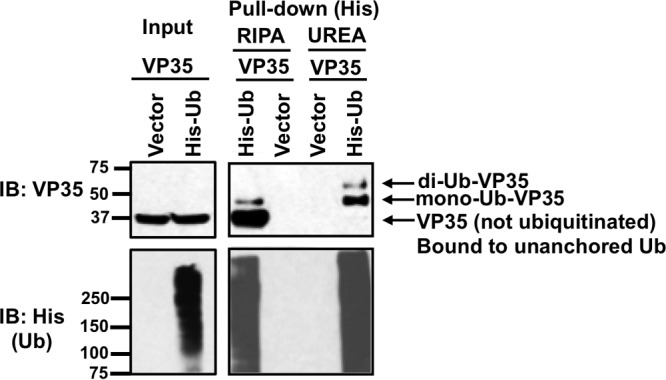
VP35 interacts with poly-Ub chains by covalent and noncovalent interactions. HEK293T cells, transfected with VP35 and His-tagged Ub, were subjected to His pulldown under denaturing conditions (urea washes) or nondenaturing conditions (RIPA washes). The amounts of His-Ub (lower left) and VP35 (upper left) used for the pulldown are shown (input samples). The upper right panel shows the amount of VP35 that associates with Ub by covalent interactions (urea wash) or noncovalent interactions (RIPA wash). The bottom right panel shows the efficiency of the ubiquitin pulldown.
TRIM6 enhances ubiquitination of VP35.
Since VP35 interacts with TRIM6, a known E3-ubiquitin ligase, we tested whether TRIM6 is involved in VP35 ubiquitination. Ectopic expression of TRIM6 resulted in enhanced association of VP35 with endogenous poly-Ub chains (Fig. 4A). Importantly, our experiments do not show evidence that ubiquitination of VP35 leads to its degradation because overexpression of TRIM6 does not affect VP35 protein levels (Fig. 4A, IP FLAG), suggesting that ubiquitination of VP35 regulates its function by nondegradative mechanisms. Additional co-IP assays using ectopically expressed HA-Ub to enrich for the different ubiquitinated forms of VP35 indicate that TRIM6 overexpression strongly enhances VP35 ubiquitination, especially by increasing linkage of 2 or more Ub molecules (di-Ub VP35 and longer) (Fig. 4B). These data suggest that TRIM6 is involved in polyubiquitination of VP35.
FIG 4.

TRIM6 overexpression enhances VP35 ubiquitination. (A) HEK293T cells were transfected with plasmids encoding FLAG-VP35 with or without plasmids encoding HA-TRIM6. Cells were harvested and whole-cell extracts (WCE) were used for FLAG IP using anti-FLAG beads. Ubiquitinated VP35 was detected by immunoblotting (IB) using a specific antibody against endogenous ubiquitin. (B) HEK293T cells were transfected with a plasmid encoding untagged VP35 with or without GST-TRIM6 and HA-Ub. Cells were harvested and whole-cell extracts (WCE) were used for HA IP using anti-HA beads, and the levels of ubiquitinated VP35 were assessed by IB with anti-VP35. Different exposures for the VP35 blots are shown. The bands corresponding to ubiquitinated VP35 are indicated based on the molecular masses of VP35 (approximately 37 kDa) and ubiquitin (8.5 kDa). Polyubiquitinated VP35 with more than 25 ubiquitin molecules can be detected (over 250 kDa).
TRIM6 enhances Ebola minigenome activity.
In addition to its IFN antagonist function, VP35 also plays an important role as a cofactor of the viral RNA polymerase. Therefore, we asked whether TRIM6 regulates VP35-mediated polymerase activity in a minigenome assay. To this end, the viral polymerase components of EBOV (NP, VP30, and L proteins) were ectopically expressed in the presence or absence of VP35 and a concentration gradient of TRIM6, and the polymerase activity was measured in an EBOV minigenome Renilla luciferase reporter assay. We found that TRIM6 enhanced EBOV polymerase activity in a dose-dependent manner when limiting amounts of VP35 WT were used (Fig. 5A, compare lanes 3 to 5 to lane 2, from left to right). In contrast, a catalytically inactive TRIM6 mutant that lacks E3-ligase activity (C15A) (44) did not significantly affect polymerase activity (Fig. 5A, compare lanes 6 to 8 to lane 2), suggesting that ubiquitination of VP35 by TRIM6 enhances VP35-mediated viral polymerase activity. We next asked whether the K309 residue on VP35 is involved in the enhanced polymerase activity induced by TRIM6. This was of particular interest because previous studies suggested that the K309 residue on VP35 is not critical for its polymerase function, since the VP35 K309A mutant is active in minigenome assays (31). As previously reported, the VP35 K309A mutant showed activity comparable to that of VP35 WT in this minigenome assay (Fig. 5A, compare lane 2 to lane 9). Surprisingly, ectopic expression of TRIM6 did not enhance VP35 K309-mediated polymerase activity or even decreased it (Fig. 5A, compare lanes 10 to 12 to lane 9). Similar results were observed in the presence of the catalytically inactive TRIM6 C15A mutant (Fig. 5A, compare lanes 13 to 15 to lane 9). Importantly, overexpression of TRIM6 in the presence of optimal amounts of VP35 WT decreases VP35 activity in this minigenome assay (Fig. 5B, high VP35), suggesting that TRIM6 positively regulates VP35 activity only at low concentrations of VP35 and that ubiquitination of VP35 plays regulatory roles that depend on the levels of VP35. Control immunoblot analysis showed similar levels of TRIM6 and VP35 WT and mutant proteins (Fig. 5C). These results indicate that TRIM6 enhances the activity of VP35 as a cofactor of the polymerase at limiting VP35 concentrations and suggest that ubiquitination of VP35 on the K309 residue by TRIM6 is important for efficient EBOV polymerase function.
FIG 5.

TRIM6 enhances VP35-dependent polymerase activity at limiting amounts of VP35. (A) An Ebola minigenome assay was performed in the presence of different amounts of transfected vectors expressing TRIM6 or C15A (10, 50, and 100 ng) and vectors expressing either VP35 WT or VP35 K309A at limiting amounts (25 ng). The fold increase was determined by setting the value for empty vector (no VP35) as 1. (B) TRIM6 expression decreases the VP35-mediated minigenome activity at optimal amounts of VP35. The minigenome assay was performed as described for panel A but using an optimal amount of VP35 vectors of 125 ng. These experiments are representative of 2 independent experiments, each performed in triplicates. The error bars indicate means ± SE (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; NS, not significant. (C) Representative samples from the minigenome assay shown in panels A and B (with TRIM6 at 100 ng only) were analyzed by immunoblotting. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
TRIM6 is important for efficient Ebola virus replication.
We have shown that VP35 is ubiquitinated on the K309 residue and that TRIM6 enhances EBOV viral polymerase activity. These results suggest that TRIM6 is important for efficient EBOV replication. However, TRIM6 also plays an important role in the establishment of an IFN-mediated antiviral state (44), which raises the question of which of the TRIM6 functions dominate during virus infection. To evaluate the balance between these two functions, we generated two TRIM6 knockout (KO) A549 cell lines by using CRISPR/Cas9 technology (KO cell lines named KO3 and KO6) (Fig. 6A). With evidence that ubiquitination of VP35 by TRIM6 enhances its activity as the cofactor of the viral polymerase, we predicted that infectious EBOV would replicate less efficiently even in the absence of an effective antiviral response. To test this, we first performed studies with recombinant EBOV expressing green fluorescent protein (GFP) in our TRIM6-KO cells. Consistent with a role of TRIM6 in promoting virus replication, TRIM6-KO cells showed lower levels of EBOV-GFP expression than WT cells, especially at earlier time points postinfection (p.i.) (Fig. 6B). Furthermore, visually reduced cytopathic effects correlated with reduced virus replication in TRIM6 KO cells compared to controls (Fig. 6B, bright fields at 48 and 72 h p.i.). Although quantification of virus replication by plaque assays showed little to no difference at 24 h p.i. between WT and TRIM6 KO cells, EBOV replicated less efficiently in TRIM6 KO cells at 48 and 72 h p.i., and larger differences were observed when cells were infected at lower multiplicities of infection (MOI) (at an MOI of 5, an approximately 7-fold difference; at an MOI of 0.01, a more than 100-fold difference at 48 h p.i.) (Fig. 6C). Consistent with the plaque assay data, the levels of EBOV RNA were also significantly reduced in TRIM6 KO cells, as measured by quantitative PCR (qPCR) (Fig. 7A, MOI of 0.5, left). Importantly, EBOV infection did not induce significant levels of IFN-β mRNA expression at early time points p.i., which is consistent with previous observations and with the notion that EBOV is a poor inducer of innate immune responses (15, 52, 53). However, virus accumulation at later time points postinfection (72 to 96 h p.i.) correlated with a significant increase in IFN-β mRNA expression in WT cells but not in TRIM6 KO cells (Fig. 7A, middle), consistent with a role of TRIM6 in IFN-β production, as we recently reported for other viruses and stimulations (44). The reduced viral load observed in TRIM6 KO cells could also explain the reduction in IFN-β induction. However, in contrast to the results obtained for IFN-β, the levels of the proinflammatory cytokine interleukin-6 (IL-6) were increased in TRIM6-KO-infected cells compared to those of the WT controls (Fig. 7A, right), suggesting that TRIM6 KO cells are able to recognize virus infection but cytokine production is deregulated. In support of this notion and as we previously reported (44), infection with Sendai virus (SeV), which is known to activate the RIG-I pathway, also resulted in reduced IFN-β induction in TRIM6 KO cells compared to WT cells (Fig. 7B, left). However, in contrast to our results obtained for EBOV infections, the levels of SeV replication were not affected in TRIM6 KO cells (Fig. 7B, right). These data suggest that TRIM6 is specifically required for optimal EBOV replication but not for other viruses.
FIG 6.

TRIM6 is required for efficient EBOV replication. (A to C) TRIM6 A549 knockout cell lines were infected with EBOV-GFP at different MOIs (5, 0.5, and 0.01). Immunoblotting of TRIM6 (A) and GFP images at different time points for an MOI of 5 (bright-field [BF] and GFP images for EBOV) (B) are shown. (C) Plaque assays of WT A549 and TRIM6 knockout cell lines infected at different MOIs. Each experiment was performed using triplicate samples, and results are depicted as the means ± standard errors (SE) (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Experiments shown are representative of at least 2 independent experiments. The limit of detection for the plaque assay was 1 × 102 PFU/ml.
FIG 7.

EBOV RNA and IFN-β but not IL-6 are reduced in TRIM6 knockout EBOV-infected cells and correlate with reduced levels of ubiquitinated VP35 in TRIM6 KO-transfected cells. (A) WT and TRIM6 knockout A549 cells were infected with EBOV-GFP at an MOI of 0.5. Cells were lysed at the indicated time points and RNA was extracted for qPCR analysis. (B) WT and TRIM6 knockout A549 cells were infected with Sendai virus (SeV; 100 HA units), and cells were harvested at 8 h p.i. for RNA extraction and qPCR analysis. (C) VP35 ubiquitination is reduced in TRIM6 KO cells. A549 WT and TRIM6 KO (KO6) cells were transfected with empty vector or a vector expressing FLAG-VP35. Forty-eight hours posttransfection, cells were lysed and whole-cell extracts (WCE) were used for FLAG (VP35) immunoprecipitation (IP) using anti-FLAG beads. Immunoblots with antiubiquitin and anti-VP35 are shown. The ubiquitin bands of VP35 correlate to the smear observed with VP35 antibody. (D) WT and TRIM6 KO cells were stimulated with IFN-β (100 U), and after 4 h cells were lysed for RNA extraction and qPCR analysis. Samples were normalized by the 18S housekeeping gene. Data for IFN-β, IL-6, ISG54, and STAT1 were further normalized by mock-treated samples to give fold induction. The qPCR was performed using triplicate samples, and results are depicted as the means ± SE (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To verify that TRIM6 is indeed the E3-ubiquitin ligase that is responsible for VP35 ubiquitination, we perform co-IP assays of VP35 in WT and TRIM6 KO cells. As predicted, the levels of endogenous poly-Ub chains associated with VP35 were strongly reduced in TRIM6 KO cells compared to WT controls (Fig. 7C, IB: Ub) and correlated with a reduction in the higher-molecular-mass forms of VP35 (Fig. 7C, IB: FLAG, ubiquitinated VP35; molecular mass, 50 to 75 kDa). Of note, long film exposures showed detectable ubiquitination on VP35 in TRIM6 KO cells, suggesting that TRIM6 is not the only E3-ubiquitin ligase that ubiquitinates VP35. These results show that TRIM6 is important for VP35 ubiquitination and correlate with the reduction of EBOV replication observed in TRIM6 KO cells (Fig. 6).
Finally, to further validate these TRIM6 knockout cells, we tested whether they reproduce the previously reported phenotype upon IFN-β treatments (44). Using different gene knockdown approaches, we previously reported that TRIM6 is required for efficient induction of a subset of IFN-I-induced IKKε-dependent ISGs (44). As expected, induction of ISG54 mRNA (an IKKε-dependent ISG) was significantly reduced in these TRIM6 KO cells, whereas STAT1 (an IKKε-independent ISG) expression levels were not affected (Fig. 7D). These data confirm that TRIM6 is required for optimal activation of IFN signaling via the IKKε branch of the pathway (44).
Taken together, our data show that the host E3-ubiquitin ligase TRIM6 is important for efficient EBOV replication and that this function of TRIM6 in promoting EBOV replication prevails over its role as an antiviral factor through the IFN-I system. Our data also suggest that VP35 ubiquitination on K309 by TRIM6 plays an important role in promoting efficient EBOV replication.
DISCUSSION
In this study, we demonstrate that the VP35 protein of EBOV is ubiquitinated on K309, a lysine residue located on its IFN antagonist domain. We further demonstrate that the E3-ubiquitin ligase TRIM6 promotes VP35 ubiquitination, and TRIM6 enhances VP35-mediated polymerase activity and virus replication. Our data suggest that TRIM6 is a host factor important for efficient EBOV replication by promoting VP35 ubiquitination. This is supported by the following lines of evidence: (i) identification of ubiquitinated K309 by MS and EBOV VP35 K309 mutant has reduced covalently linked poly-Ub chains, (ii) ectopic expression of TRIM6 enhances VP35 ubiquitination, (iii) ubiquitination of VP35 is reduced in TRIM6 knockout cells, (iv) ectopic expression of TRIM6 enhances VP35 WT but not VP35 K309 polymerase activity in minigenome assays, (v) expression of a catalytically inactive point mutant of TRIM6 fails to enhance VP35 polymerase activity, (vi) TRIM6 knockout cells have reduced EBOV replication compared to controls, and (vii) IFN-β production is reduced in TRIM6 knockout-infected cells.
Our MS analysis shows that VP35 is ubiquitinated at K309; however, this analysis did not provide the full spectrum of lysines present in VP35, because the peptides detected in the MS covered only approximately 50% of the VP35 sequence. Moreover, our co-IP studies suggest that K309 is not the only ubiquitinated residue, because the K309A mutant showed reduced but still detectable levels of ubiquitinated VP35. Although our data suggest that TRIM6 and K309 are at least in part responsible for the results observed, at this point we cannot rule out the possibility that TRIM6 ubiquitinates other lysines on VP35 in addition to or instead of K309, including the N-terminal region of VP35, and that other ubiquitination sites may be responsible for promoting polymerase activity. The roles of other ubiquitination sites in VP35 are under investigation. Importantly, it appears that the ubiquitination levels of the C-terminal region of VP35 are low when the truncated form of VP35 is used, suggesting that full-length VP35 is required for optimal VP35 ubiquitination. Although at the moment we cannot determine the levels of VP35 ubiquitination during infection, our data suggest that not all VP35 is ubiquitinated at any given time point, suggesting that this VP35 covalent modification is not required for its function but rather is a regulatory mechanism to control levels of virus replication.
The C-terminal IID region of VP35, which contains a basic patch of amino acid residues (R305, K309, R312, K319, and R322), is known to be required for inhibition of IFN-β production (11, 31). VP35 binds dsRNA, and mutants in the IID domain, which lack IFN antagonism activity, do not bind dsRNA (11, 19, 31, 54). Structural studies indicate that the basic amino acids in the IID domain directly contact dsRNA (31); therefore, ubiquitination of lysine residues could alter dsRNA binding and IFN antagonism. However, it is intriguing that while the polymerase activity of the VP35 K309A mutant is not significantly affected when expressed in the context of the minigenome assay (Fig. 5) (31), it is only upon overexpression of TRIM6 that the differences between VP35 WT and VP35 K309A become evident. Although these effects are consistent with the infectious virus experiments in TRIM6 KO cells, in which virus replication is reduced but not completely abrogated, they suggest that ubiquitination of VP35 is a regulatory mechanism but is not essential for virus viability. A recent study showed that the interaction of VP35 with the EBOV transcription factor VP30 depends on the dsRNA-binding capacity of VP35, and mutations in the VP35 central basic patch (R305A, K309A, and R312A) reduced its ability to bind VP30 (55). Therefore, VP35 ubiquitination on K309 could affect VP35-VP30 interactions, thereby regulating the switch between virus transcription and replication. Importantly, the K309 residue is conserved in the five members of the Ebolavirus genus (EBOV, BDBV, SUDV, RESTV, and TAFV) as well as in the related MARV, suggesting that indeed this region is important for virus replication. Furthermore, it has been proposed that the structural organization and dsRNA binding ability of EBOV versus RESTV VP35 contributes to their differential pathogenicity in humans (56, 57). Therefore, the K309 residue may play multiple roles in IFN antagonism as well as polymerase function.
It is important to point out that TRIM6 enhances VP35-mediated polymerase activity in the minigenome assay only when limiting amounts of VP35 are used (25 ng), and at optimal VP35 concentrations TRIM6 inhibits the minigenome activity (Fig. 5A and B). These effects are consistent with previous reports using titrations of VP35 plasmids, which have shown a bell curve effect for minigenome activity when the amounts of VP35 plasmid DNA were increased over the optimal amounts (6, 58). One possible explanation is that ubiquitination of VP35 increases its affinity with the polymerase complex and enhance interactions with L and/or NP, which may shift the bell curve, causing the effects previously observed by others (6, 58) to occur at lower concentrations of VP35. An alternative possibility is that TRIM6 promotes aggregation of VP35 at high concentrations, which in turn would decrease minigenome activity. In fact, this is the case for many TRIMs, because these proteins tend to oligomerize at high concentrations and can form high-molecular-weight complexes that aggregate (51). We have found that many TRIMs can enhance the signaling activity of multiple innate immune factors, including RIG-I, TBK-1, and IKKε, only when these factors are used at limiting concentrations (43, 44).
Our ubiquitination experiments using TRIM6 KO cells indicate that VP35 ubiquitination is significantly reduced in TRIM6 KO cells compared to controls, but it is not completely eliminated (Fig. 7C), suggesting that there are other E3-ubiquitin ligases that compensate for the loss of TRIM6 or play redundant roles. Alternatively, while TRIM6 may be responsible for ubiquitination on K309, it could be that other E3-ligases ubiquitinate other sites. Since the TRIM family is highly conserved, it would not be surprising if other members of this family of E3-ligases also are able to bind and ubiquitinate VP35.
VP35 is a well-known IFN-I antagonist that acts by targeting different components of the signaling pathway to produce IFN-I. These include RIG-I, PACT, IKKε, TBK, and IRF7 (18, 20, 32, 33). Here, we show that VP35 can also inhibit RIG-I-induced TRIM6-mediated IFN-I production. TRIM6 catalyzes the synthesis of unanchored poly-Ub chains required for optimal activation of IKKε (44). Since we show that VP35 can interact noncovalently with poly-Ub chains, it could be that VP35 sequesters these chains from IKKε, preventing its proper activation. An alternative possibility is that VP35 blocks TRIM6 activity similar to our recent report of TRIM6 antagonism by the matrix protein of Nipah virus (45). Although we did not find evidence that VP35 expression results in TRIM6 degradation, VP35 could compete with IKKε for TRIM6 binding, since both bind the C-terminal SPRY domain of TRIM6 (Fig. 1) (44). Interestingly, we found that the proinflammatory cytokine IL-6 is increased in EBOV-infected TRIM6 KO cells. This is consistent with previous observations in TRIM6 knockdown cells that produce higher levels of tumor necrosis factor alpha (TNF-α) and IL-6 in lipopolysaccharide (LPS)-stimulated dendritic cells (44). Together, these observations suggest that in addition to the positive function of TRIM6 in the IFN-I pathway, TRIM6 also acts as a negative regulator of proinflammatory cytokines, probably downstream of the adaptor protein MAVS. EBOV-VP35 is a potent inhibitor of RIG-I, which is critical for induction of both the IFN-I and NF-κB pathways (Fig. 8 depicts a schematic representation of the pathways), and even low levels of EBOV replication are enough to trigger higher levels of IL-6 (Fig. 7A).
FIG 8.

Proposed model of regulation of VP35 function by ubiquitination. Upon virus recognition of EBOV infection, pattern recognition receptor (PRR) signaling promotes the synthesis of unanchored K48-linked polyubiquitin chains by the E3-ubiquitin ligase TRIM6. These polyubiquitin chains interact with IKKε and induce its oligomerization and downstream signaling to produce IFN-β (44). VP35 inhibits IFN-β at the level of RIG-I and the kinases TBK1 and IKKε (20). Ubiquitination of VP35 by TRIM6 promotes VP35-mediated polymerase activity and enhances virus replication.
In summary, here we report a new function for the host E3-ubqiuitin ligase TRIM6 in promoting Ebola virus replication. We propose that the VP35 protein of Ebola virus hijacks TRIM6 to promote virus replication via ubiquitination (Fig. 8). Therefore, TRIM6 is emerging as a critical host factor targeted by multiple viruses to enhance their replication.
MATERIALS AND METHODS
Cells and viruses.
HEK-293T, HeLa, A549, Vero E6 (CRL1586), and Vero (CCL-81) cells all were purchased from the American Type Culture Collection (ATCC). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 1% penicillin-streptomycin (Gibco-BRL). Wild-type Ebola virus (EBOV) strain Zaire and recombinant EBOV expressing GFP (kindly provided by the Special Pathogens Branch, CDC, Atlanta, GA) were propagated in Vero E6 cells. Stock virus titers were determined by plaque assay in Vero-CCL81 cells. All work with live virus was carried out under biosafety level 4 (BSL4) conditions in the Robert E. Shope biosafety level 4 laboratory, UTMB. Sendai virus (SeV; Cantell strain) was obtained from Charles River Laboratories and propagated in 10-day-old pathogen-free embryonated chicken eggs (Charles River Laboratories, North Franklin, CT).
Plasmids and reagents.
The VP35 WT (untagged or Flag-tagged), VP35 K309A mutant, and TRIM6 and ubiquitin expression constructs have been described before (11, 44). The RIG-I (2CARD) expression constructs as well as pIFNβ fLuc reporters were kindly provided by A. Garcia-Sastre (Icahn School of Medicine at Mount Sinai) and have been described previously (43, 44). The reporter plasmid carrying the Renilla luciferase gene (REN-Luc/pRL-TK) was purchased from Promega. The HA-TRIM6 plasmid was kindly provided by Andrea Ballabio (51). All sequences were confirmed by sequencing analysis at the UTMB molecular genomics core facility.
Transient transfections were performed with TransIT-LT1 (Mirus), Lipofectamine 2000, or RNAiMax (Invitrogen) according to the manufacturers' instructions. Mouse anti-VP35 antibody has been described previously (18, 46). Mouse and rabbit anti-FLAG antibodies, rabbit anti-HA antibody, mouse anti-β-tubulin, and mouse anti-β-actin antibodies were from Sigma. Rabbit monoclonal anti-ubiquitin lysine 48 (K48, clone Apu2) was purchased from Millipore. Rabbit anti-glutathione S-transferase (GST) antibody (OTI4G1) was from Bethyl Laboratories. Fluorescently labeled secondary antibodies for imaging Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 donkey anti-rabbit, Alexa Fluor 555 goat anti-mouse, Alexa Fluor 555 donkey anti-rabbit, and goat anti-mouse Alexa Fluor 633 were purchased from Thermo Scientific (Life Technologies). Directly conjugated antibodies toward Flag/DYKDDDK tag for rabbit (Alexa 555) and HA tag for mouse (Alexa 488) were from Thermo Scientific and Cell Signaling Technologies.
Ebola virus infections.
WT and KO A549 cells were seeded into 24-well plates 18 to 24 h prior to infection in complete medium (DMEM, 10% heat-inactivated FBS [HI-FBS], 1% penicillin-streptomycin [PS]). At 80% confluence, growth medium was removed and EBOV-GFP (or WT EBOV) was added at the indicated multiplicity of infection (MOI). Cells were returned to a 37°C, 5% CO2 humidified incubator for 1 h and rocked every 15 min. Following a 1-h incubation, the inoculum was removed and the cells washed with Dulbecco's phosphate-buffered saline (PBS) and returned to the incubator with viral growth medium (DMEM, 2% HI-FBS, 1% PS). Growth medium for titration was removed at the indicated time point and the monolayer lysed using TRIzol reagent for RNA extraction. Titers of samples were determined on Vero CCL-81 cells in 12-well plates by standard protocols. Briefly, log dilutions of virus were added to 80% confluent monolayers and rocked every 15 min for infection of A549. At 1 h postinfection, cells were overlaid with a semisolid overlay of MEM, 0.5% methylcellulose, 2% HI-FBS, and 1% PS. Ten days postinfection, the overlay was removed and monolayers were stained and fixed using 10% neutral buffered formalin with crystal violet. Plaques were enumerated and virus titers, as PFU per milliliter, were calculated.
Luciferase reporter assays.
HEK293T cells were transfected in 24-well (5 × 104 cells per well) or 96-well (1 × 104 cells per well) plates (Falcon, Becton Dickinson, NJ) with 10 to 50 ng of IFN-β reporter plasmid, 4 to 20 ng of Renilla luciferase, and 2 to 50 ng plasmids using TransIT-LT1 (Mirus) at a ratio of 1:3. Empty vector was used to ensure that the plasmid concentration in each well was the same. Twenty-four hours later, cells were lysed and Dual-Luciferase assay was performed according to the manufacturer's instructions (Promega, Madison, WI, USA). Values were obtained by normalizing the luciferase values by Renilla values (fLuc/rLuc) and then normalized to the nonstimulated sample to obtain fold induction.
Coimmunoprecipitation and Western blotting.
Transfected 293T cells were harvested in RIPA lysis buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% (vol/vol) NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail (Roche) and supplemented with 5 mM N-ethylmaleimide (NEM) and iodoacetamide as deubiquitinase inhibitors. Cell lysate was clarified by microcentrifugation at 14,000 rpm for 20 min. One-tenth of the aliquot from clarified lysate was taken and added to 2× Laemmli buffer with β-mercaptoethanol (β-ME) and stored at −20°C for Western blotting (whole-cell extracts [WCE]). Mouse anti-FLAG or anti-HA antibody cross-linked to agarose beads (EZview Red Anti-FLAG M2 or EZview Red Anti-HA affinity gel; Sigma) were added to the rest of the lysate, and the mixture was rotated on a benchtop shaker overnight at 4°C. Beads were extensively washed, and the bound proteins were eluted by boiling for 10 min in Laemmli loading buffer. For immunoblotting, proteins were resolved by SDS-PAGE (7.5% or 4 to 15%; Bio-Rad) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Immobilon; Bio-Rad Laboratories). The following primary antibodies were used: anti-Ub-K48 (1:1,000), anti-Flag (1:3,000) (Sigma), anti-HA (1:5,000) (Sigma), anti-GST (1:2,000) (Bethyl Laboratories), and anti-actin/tubulin (1:5,000).
Immunoblots (IB) were developed with the following secondary antibodies: ECL anti-rabbit IgG horseradish peroxidase-conjugated whole antibody from donkey and ECL anti-mouse IgG horseradish peroxidase-conjugated whole antibody from sheep (GE Healthcare, Buckinghamshire, England). The proteins were visualized by an enhanced chemiluminescence reagent (Pierce).
Denaturing pulldown of ubiquitinated VP35.
HEK293T cells were transfected with His-tagged Ub and VP35. After 30 h, cells were harvested in RIPA lysis buffer supplemented with protease inhibitor cocktail and 5 mM N-ethylmaleimide (NEM) and iodoacetamide as deubiquitinase inhibitors. Cell lysate was clarified by microcentrifugation at 14,000 rpm for 20 min. One-tenth of the aliquot from clarified lysate was taken and added to 2× Laemmli buffer with β-ME and stored at −20°C for Western blotting (input). The rest of the lysate was bound to nickel-nitrilotriacetic acid (Ni-NTA) beads (Qiagen) overnight at 4°C in a shaker. The next day, beads were extensively washed with either RIPA buffer (nondenaturing pulldown) or with a denaturing buffer containing 6 M urea, 350 mM NaCl, 0.5% NP-40, 40 mM imidazole in 50 mM Tris (pH 8.0). After 7 washes, the bound ubiquitinated proteins were eluted on the rotator in a cold room for 30 min in 50 mM Tris, pH 8.0, containing 300 mM imidazole. Protein eluates were mixed with Laemmli loading buffer and analyzed by immunoblotting.
Quantitative PCR.
qPCR was performed as previously described (44). In brief, total RNA was extracted from A549 cells with TRIzol reagent (Sigma) and purified using an RNeasy kit (Qiagen). cDNA was prepared by using the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) by following the manufacturer's instructions. Relative gene expression was determined by using SYBR green mix (Bio-Rad) with a Bio-Rad CFX384 instrument. CFX Manager software (Bio-Rad) was used to analyze the relative mRNA expression levels by the change in the threshold cycle (ΔCT) and normalized using the 18S gene as the housekeeping gene.
Immunofluorescence microscopy and image analysis.
For colocalization studies of HA-TRIM6 and Flag-VP35, HeLa cells were seeded into Lab-Tek II 8-well chamber slides (CC2 glass slide; Nunc, Rochester, NY). After 12 to 16 h, 300 to 700 ng of plasmids was transfected with Lipofectamine 2000 (Invitrogen) at a ratio of 1:1. Six hours later, medium was replaced, and 16 to 24 h later, cells were washed with PBS, fixed with 4% paraformaldehyde, permeabilized with 0.5% (vol/vol) NP-40 in PBS, and blocked with 0.5% bovine serum albumin (BSA) and 0.2% fish gelatin in PBS for 1 h (blocking solution). Staining was performed with anti-HA and anti-Flag antibodies (1:200) (prepared in blocking solution) overnight at 4°C. The next day, cells were washed 3 times with blocking solution, and the secondary antibodies donkey anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 555 (Invitrogen), diluted in blocking buffer along with 4′,6-diamidino-2-phenylindole (DAPI) (1:3,000), were used to visualize the proteins. The slides were imaged on a Zeiss LSM 510 confocal microscope in the UTMB optical imaging core at a magnification of ×63.
Ebola minigenome polymerase assay.
A functional polymerase complex was reconstituted by transfection of plasmids expressing L (500 ng), NP (250 ng), VP30 (100 ng), and model viral RNA encoding the Renilla reporter flanked by viral regulatory sequences (200 ng) with or without VP35 (25 ng or 125 ng) and T7 polymerase (200 ng) into HEK293T cells using Lipofectamine 2000 (Invitrogen) transfection reagent. A firefly plasmid (10 ng) is also coexpressed to determine transfection efficiency. Plasmids encoding HA-TRIM6 or HA-TRIM6 C15A were also cotransfected as indicated in the figure. The plasmid amounts are indicative of one well of a 12-well plate, and the reaction is divided into 4 wells of a 96-well plate with 100,000 cells/well, where three replicates were used for minigenome assay and one replicate was used for Western blotting. Forty-eight hours posttransfection, the luciferase activities were determined using Dual Glo reagent (Promega) and reading on an Envision plate reader (PerkinElmer). The Renilla activity was normalized with firefly activity, and fold increase was calculated by setting the no-VP35 value as 1. Note that TRIM6 and VP35 plasmid concentrations need to be carefully titrated to allow an experimental window for enhancement upon TRIM6 coexpression. A limited amount of VP35 (25 ng) was selected after titrations. The levels of protein expression were validated by immunoblotting. All of the minigenome helper plasmids are in pCAGGS and MG-Reporter reporter plasmid in pTM1.
Mass spectrometry analysis.
To identify ubiquitinated sites on EBOV VP35, we first enriched for ubiquitinated proteins by co-IP. HEK293T cells were transfected with 500 ng of HA-Ub and 500 ng of EBOV VP35 expression vector. Thirty hours posttransfection, cells were lysed in RIPA buffer and immunoprecipitation was performed using anti-HA beads. After extensive washes, ubiquitinated proteins, including VP35, were eluted using HA peptide (Sigma). Ubiquitinated protein eluates were separated by SDS-PAGE, and gel bands at or above the molecular mass of VP35 (35 to 82 kDa) were excised. Gel slices were digested in-gel with trypsin for liquid chromatography-tandem MS (LC-MS/MS) analysis (59). Following digestion, samples were concentrated using C18 ZipTips (Millipore) according to the manufacturer's specifications. Desalted samples were evaporated to dryness and resuspended in 0.1% formic acid for mass spectrometry analysis. Digested peptide mixtures were analyzed in technical duplicate on a Thermo Scientific LTQ Orbitrap Elite mass spectrometry system equipped with a Proxeon Easy nLC 1000 ultra-high-pressure liquid chromatography and autosampler system (laboratory of J. R. Johnson and N. J. Krogan at UCSF). Samples were injected onto a C18 column (25 cm by 75 μm inner diameter, packed with ReproSil Pur C18 AQ 1.9-μm particles) in 0.1% formic acid and then subjected to a 2-h gradient from 0.1% formic acid to 30% ACN–0.1% formic acid. The mass spectrometer collected data in a data-dependent fashion, collecting one full scan in the Orbitrap at 120,000 resolution followed by 20 collision-induced dissociation MS/MS scans in the dual linear ion trap for the 20 most intense peaks from the full scan. Dynamic exclusion was enabled for 30 s with a repeat count of 1. Charge state screening was employed to reject analysis of singly charged species or species for which a charge could not be assigned.
Raw mass spectrometry data were searched with the Protein Prospector algorithm (60) against the VP35 sequence with a precursor charge state range of 2 to 4, monoisotopic parent mass tolerance of ±20 ppm, fragment ion mass tolerance of ±0.8 Da, constant modification for cysteine carbamidomethylation, and variable modifications for N-terminal protein acetylation, methionine oxidation, peptide N-terminal glutamine-to-pyroglutamine conversion, protein N-terminal methionine cleavage, and lysine ubiquitin remnant (diGly) modification to identify trypsin-digested ubiquitination sites. Data were filtered with a minimum peptide score of 15.0 and a maximum peptide E value of 0.05.
Generation of A549 TRIM6 CRISPR/Cas9 knockout.
The TRIM6 knockout cell lines were generated using a CRISPR-CAS9 vector, (pSpCas9n)(BB)-2A-GFP (PX458) (Addgene plasmid 48138), with an single-guide RNA (sgRNA) targeting exon 2 of the human TRIM6 gene (target sequence, ACTGGTGGACATACGAGAAG). Sequences were designed using the following link: http://crispr.mit.edu:8079/. The sgRNA sequence was cloned into the CRISPR-Cas9 vector as described by Ran et al. (61). To generate the knockout cell line, 5 × 105 A549 cells (ATCC) were seeded in a 6-well plate and transfected with 2 μg of CRISPR-CAS9 vector in 500 μl of Opti-MEM and 1.5 μl of Lipofectamine 2000. Medium was changed 6 h later. Forty-eight hours posttransfection, cells were observed for GFP expression, trypsinized to a single-cell suspension, passed through a 70-μm strainer, and sorted by flow cytometry using a BD FACSAria2 (core facility at UTMB) for stable GFP expression. Cells were allowed to recover for 24 h. To generate stably modified clones, cells isolated by fluorescence-activated cell sorting according to a desired florescence level were diluted to 2.5 cells/ml, and 200 μl of cells was plated to 96-well plates and expanded to clonal cell lines. Knockout lines were verified by Western blotting. Transfections of these A549 WT and TRIM6 KO cells was performed in 6-well plates with 1 × 105 cells per well (in triplicate wells). Cells were allowed to attached to the plates overnight, and transfections were then performed using 1 μg of VP35 plasmid and Lipofectamine LTX (ThermoFisher) by following the manufacturer's instructions. Forty-eight hours posttransfection, cells were lysed with RIPA buffer and the triplicate wells were combined to obtain enough express protein for detection in co-IP assays.
Statistical analysis.
Statistical analysis was performed with Prism (version 5.0; GraphPad Software). Student's paired t test or one-way analysis of variance with Dunnett's multiple-comparison test was used: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; and NS (not significant).
ACKNOWLEDGMENTS
We thank Adolfo García-Sastre (Icahn School of Medicine at Mount Sinai) for his immense generosity in sharing reagents and advice.
This work was supported by funding from the Institute for Human Infections and Immunity (IHII) and University of Texas Medical Branch (UTMB) to R.R. and A.N.F.
REFERENCES
- 1.Kuhn JH, Becker S, Ebihara H, Geisbert TW, Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol ST, Palacios G, Peters CJ, Tenorio A, Volchkov VE, Jahrling PB. 2010. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch Virol 155:2083–2103. doi: 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahanty S, Bray M. 2004. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis 4:487–498. doi: 10.1016/S1473-3099(04)01103-X. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez AGT, Feldmann H. 2006. Filoviridae: Marburg and Ebola viruses, p 1409–1448. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Muhlberger E, Lotfering B, Klenk HD, Becker S. 1998. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J Virol 72:8756–8764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muhlberger E. 2007. Filovirus replication and transcription. Future Virol 2:205–215. doi: 10.2217/17460794.2.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muhlberger E, Weik M, Volchkov VE, Klenk HD, Becker S. 1999. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. J Virol 73:2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirchdoerfer RN, Abelson DM, Li S, Wood MR, Saphire EO. 2015. Assembly of the Ebola virus nucleoprotein from a chaperoned VP35 complex. Cell Rep 12:140–149. doi: 10.1016/j.celrep.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trunschke M, Conrad D, Enterlein S, Olejnik J, Brauburger K, Muhlberger E. 2013. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology 441:135–145. doi: 10.1016/j.virol.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leung DW, Borek D, Luthra P, Binning JM, Anantpadma M, Liu G, Harvey IB, Su Z, Endlich-Frazier A, Pan J, Shabman RS, Chiu W, Davey RA, Otwinowski Z, Basler CF, Amarasinghe GK. 2015. An intrinsically disordered peptide from Ebola virus VP35 controls viral RNA synthesis by modulating nucleoprotein-RNA interactions. Cell Rep 11:376–389. doi: 10.1016/j.celrep.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. 2003. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol 77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartman AL, Towner JS, Nichol ST. 2004. A C-terminal basic amino acid motif of Zaire ebolavirus VP35 is essential for type I interferon antagonism and displays high identity with the RNA-binding domain of another interferon antagonist, the NS1 protein of influenza A virus. Virology 328:177–184. doi: 10.1016/j.virol.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Messaoudi I, Amarasinghe GK, Basler CF. 2015. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 13:663–676. doi: 10.1038/nrmicro3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reid SP, Cardenas WB, Basler CF. 2005. Homo-oligomerization facilitates the interferon-antagonist activity of the ebolavirus VP35 protein. Virology 341:179–189. doi: 10.1016/j.virol.2005.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basler CF, Amarasinghe GK. 2009. Evasion of interferon responses by Ebola and Marburg viruses. J Interferon Cytokine Res 29:511–520. doi: 10.1089/jir.2009.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basler CF, Wang X, Muhlberger E, Volchkov V, Paragas J, Klenk HD, Garcia-Sastre A, Palese P. 2000. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci U S A 97:12289–12294. doi: 10.1073/pnas.220398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung LW, Park MS, Martinez O, Valmas C, Lopez CB, Basler CF. 2011. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol Cell Biol 89:792–802. doi: 10.1038/icb.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, Amarasinghe GK, Basler CF. 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF. 2010. Basic residues within the ebolavirus VP35 protein are required for its viral polymerase cofactor function. J Virol 84:10581–10591. doi: 10.1128/JVI.00925-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prins KC, Cardenas WB, Basler CF. 2009. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J Virol 83:3069–3077. doi: 10.1128/JVI.01875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin H, Yan Z, Prabhakar BS, Feng Z, Ma Y, Verpooten D, Ganesh B, He B. 2010. The VP35 protein of Ebola virus impairs dendritic cell maturation induced by virus and lipopolysaccharide. J Gen Virol 91:352–361. doi: 10.1099/vir.0.017343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yen B, Mulder LC, Martinez O, Basler CF. 2014. Molecular basis for ebolavirus VP35 suppression of human dendritic cell maturation. J Virol 88:12500–12510. doi: 10.1128/JVI.02163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ilinykh PA, Lubaki NM, Widen SG, Renn LA, Theisen TC, Rabin RL, Wood TG, Bukreyev A. 2015. Different temporal effects of Ebola virus VP35 and VP24 proteins on global gene expression in human dendritic cells. J Virol 89:7567–7583. doi: 10.1128/JVI.00924-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lubaki NM, Ilinykh P, Pietzsch C, Tigabu B, Freiberg AN, Koup RA, Bukreyev A. 2013. The lack of maturation of Ebola virus-infected dendritic cells results from the cooperative effect of at least two viral domains. J Virol 87:7471–7485. doi: 10.1128/JVI.03316-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lubaki NM, Younan P, Santos RI, Meyer M, Iampietro M, Koup RA, Bukreyev A. 2016. The Ebola interferon inhibiting domains attenuate and dysregulate cell-mediated immune responses. PLoS Pathog 12:e1006031. doi: 10.1371/journal.ppat.1006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Meylan E, Tschopp J, Karin M. 2006. Intracellular pattern recognition receptors in the host response. Nature 442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 28.Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S. 2004. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 199:1641–1650. doi: 10.1084/jem.20040520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma S, Tenover BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 30.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leung DW, Prins KC, Borek DM, Farahbakhsh M, Tufariello JM, Ramanan P, Nix JC, Helgeson LA, Otwinowski Z, Honzatko RB, Basler CF, Amarasinghe GK. 2010. Structural basis for dsRNA recognition and interferon antagonism by Ebola VP35. Nat Struct Mol Biol 17:165–172. doi: 10.1038/nsmb.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramanan P, Shabman RS, Brown CS, Amarasinghe GK, Basler CF, Leung DW. 2011. Filoviral immune evasion mechanisms. Viruses 3:1634–1649. doi: 10.3390/v3091634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. 2009. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog 5:e1000493. doi: 10.1371/journal.ppat.1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen ZJ, Sun LJ. 2009. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Oudshoorn D, Versteeg GA, Kikkert M. 2012. Regulation of the innate immune system by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor Rev 23:273–282. doi: 10.1016/j.cytogfr.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajsbaum R, Garcia-Sastre A. 2013. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol 21:421–429. doi: 10.1016/j.tim.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNab FW, Rajsbaum R, Stoye JP, O'Garra A. 2011. Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol 23:46–56. doi: 10.1016/j.coi.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 38.Ozato K, Shin DM, Chang TH, Morse HC III. 2008. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajsbaum R, Garcia-Sastre A, Versteeg GA. 2014. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol 426:1265–1284. doi: 10.1016/j.jmb.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajsbaum R, Stoye JP, O'Garra A. 2008. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol 38:619–630. doi: 10.1002/eji.200737916. [DOI] [PubMed] [Google Scholar]
- 41.Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W. 2013. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol 87:257–272. doi: 10.1128/JVI.01804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Versteeg GA, Benke S, Garcia-Sastre A, Rajsbaum R. 2014. InTRIMsic immunity: positive and negative regulation of immune signaling by tripartite motif proteins. Cytokine Growth Factor Rev 25:563–576. doi: 10.1016/j.cytogfr.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Versteeg GA, Rajsbaum R, Sanchez-Aparicio MT, Maestre AM, Valdiviezo J, Shi M, Inn KS, Fernandez-Sesma A, Jung J, Garcia-Sastre A. 2013. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38:384–398. doi: 10.1016/j.immuni.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajsbaum R, Versteeg GA, Schmid S, Maestre AM, Belicha-Villanueva A, Martinez-Romero C, Patel JR, Morrison J, Pisanelli G, Miorin L, Laurent-Rolle M, Moulton HM, Stein DA, Fernandez-Sesma A, Tenover BR, Garcia-Sastre A. 2014. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKepsilon kinase-mediated antiviral response. Immunity 40:880–895. doi: 10.1016/j.immuni.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, Basler CF, Freiberg AN, Lee B, Rajsbaum R. 2016. The matrix protein of Nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKepsilon kinase-mediated type-I IFN antiviral response. PLoS Pathog 12:e1005880. doi: 10.1371/journal.ppat.1005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shabman RS, Leung DW, Johnson J, Glennon N, Gulcicek EE, Stone KL, Leung L, Hensley L, Amarasinghe GK, Basler CF. 2011. DRBP76 associates with Ebola virus VP35 and suppresses viral polymerase function. J Infect Dis 204(Suppl 3):S911–S918. doi: 10.1093/infdis/jir343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luthra P, Jordan DS, Leung DW, Amarasinghe GK, Basler CF. 2015. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J Virol 89:5148–5153. doi: 10.1128/JVI.03652-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pythoud C, Rodrigo WW, Pasqual G, Rothenberger S, Martinez-Sobrido L, de la Torre JC, Kunz S. 2012. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKepsilon. J Virol 86:7728–7738. doi: 10.1128/JVI.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valmas C, Basler CF. 2011. Marburg virus VP40 antagonizes interferon signaling in a species-specific manner. J Virol 85:4309–4317. doi: 10.1128/JVI.02575-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valmas C, Grosch MN, Schumann M, Olejnik J, Martinez O, Best SM, Krahling V, Basler CF, Muhlberger E. 2010. Marburg virus evades interferon responses by a mechanism distinct from Ebola virus. PLoS Pathog 6:e1000721. doi: 10.1371/journal.ppat.1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. 2001. The tripartite motif family identifies cell compartments. EMBO J 20:2140–2151. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartman AL, Ling L, Nichol ST, Hibberd ML. 2008. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J Virol 82:5348–5358. doi: 10.1128/JVI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kash JC, Muhlberger E, Carter V, Grosch M, Perwitasari O, Proll SC, Thomas MJ, Weber F, Klenk HD, Katze MG. 2006. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J Virol 80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prins KC, Delpeut S, Leung DW, Reynard O, Volchkova VA, Reid SP, Ramanan P, Cardenas WB, Amarasinghe GK, Volchkov VE, Basler CF. 2010. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J Virol 84:3004–3015. doi: 10.1128/JVI.02459-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Biedenkopf N, Schlereth J, Grunweller A, Becker S, Hartmann RK. 2016. RNA binding of Ebola virus VP30 is essential for activating viral transcription. J Virol 90:7481–7496. doi: 10.1128/JVI.00271-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimberlin CR, Bornholdt ZA, Li S, Woods VL Jr, MacRae IJ, Saphire EO. 2010. Ebolavirus VP35 uses a bimodal strategy to bind dsRNA for innate immune suppression. Proc Natl Acad Sci U S A 107:314–319. doi: 10.1073/pnas.0910547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leung DW, Shabman RS, Farahbakhsh M, Prins KC, Borek DM, Wang T, Muhlberger E, Basler CF, Amarasinghe GK. 2010. Structural and functional characterization of Reston Ebola virus VP35 interferon inhibitory domain. J Mol Biol 399:347–357. doi: 10.1016/j.jmb.2010.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boehmann Y, Enterlein S, Randolf A, Muhlberger E. 2005. A reconstituted replication and transcription system for Ebola virus Reston and comparison with Ebola virus Zaire. Virology 332:406–417. doi: 10.1016/j.virol.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 59.Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. 1992. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem 203:173–179. doi: 10.1016/0003-2697(92)90061-B. [DOI] [PubMed] [Google Scholar]
- 60.Clauser KR, Baker P, Burlingame AL. 1999. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 61.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]