

ABSTRACT
Diarrhea is the major cause of non-research-associated morbidity and mortality affecting the supply of rhesus macaques and, potentially, their responses to experimental treatments. Idiopathic chronic diarrhea (ICD) in rhesus macaques also resembles ulcerative colitis, one form of human inflammatory bowel disease. To test for viral etiologies, we characterized and compared the fecal viromes from 32 healthy animals, 31 animals with acute diarrhea, and 29 animals with ICD. The overall fractions of eukaryotic viral reads were 0.063% for the healthy group, 0.131% for the acute-diarrhea group, and 0.297% for the chronic-diarrhea group. Eukaryotic viruses belonging to 6 viral families, as well as numerous circular Rep-encoding single-stranded DNA (CRESS DNA) viral genomes, were identified. The most commonly detected sequences were from picornaviruses, making up 59 to 88% of all viral reads, followed by 9 to 17% for CRESS DNA virus sequences. The remaining 5 virus families, Adenoviridae, Astroviridae, Anelloviridae, Picobirnaviridae, and Parvoviridae, collectively made up 1 to 3% of the viral reads, except for parvoviruses, which made up 23% of the viral reads in the healthy group. Detected members of the families Picornaviridae and Parvoviridae were highly diverse, consisting of multiple genera, species, and genotypes. Coinfections with members of up to six viral families were detected. Complete and partial viral genomes were assembled and used to measure the number of matching short sequence reads in feces from the 92 animals in the two clinical and the healthy control groups. Several enterovirus genotypes and CRESS DNA genomes were associated with ICD relative to healthy animals. Conversely, higher read numbers from different parvoviruses were associated with healthy animals. Our study reveals a high level of enteric coinfections with diverse viruses in a captive rhesus macaque colony and identifies several viruses positively or negatively associated with ICD.
KEYWORDS: enterovirus, sapelovirus, parvovirus, CRESS-DNA, idiopathic diarrhea, rhesus macaques, enteric disease etiology
INTRODUCTION
We compared the enteric viromes of captive rhesus macaques (Macaca mulatta) with acute and idiopathic chronic diarrhea (ICD) to those of healthy animals using a case-control study. The genomes of several known and new viruses were characterized using metagenomics. We showed that some specific enterovirus genotypes and some small circular DNA genomes are associated with chronic idiopathic diarrhea, providing candidates for further testing of their pathogenicity. Other viruses, including some parvoviruses, were found at significantly higher levels in healthy animals.
ICD in captive macaques is the most common cause of non-medical-research mortality in primate research centers, reducing the availability of healthy rhesus macaques and increasing the cost of nonhuman primate (NHP) research (1, 2). Clinically, animals with ICD exhibit persistent or recurring nonbloody diarrhea with microscopic ulcers in the colon (2–5). The mucus layer of the colon is also thickened, with shortened crypts showing a very high level of lymphocyte and plasma cell infiltration, mimicking signs of a response to viral infection (6). This condition is akin to human ulcerative colitis, a subset of inflammatory bowel disease (IBD) in which small ulcers often begin in and extend throughout the colon (distinct from the other major form of IBD, Crohn's disease, which affects the entire gastrointestinal tract). In rhesus macaques, ICD cases are often diagnosed by 1 year of age. Their peripheral and gut inflammation is biased toward TH1 inflammation, and symptoms are always accompanied by dysbiosis in the gut microbial communities (7–10).
Bacterial infections with Shigella, Campylobacter, Yersinia, Salmonella, or Clostridium difficile and with parasites, including Cryptosporidium, are also common causes of diarrhea in macaque colonies (2, 11). However, in a subset of persistent and recurrent diarrhea cases, no pathogenic bacteria, parasites, or other etiological agents are identified, and are therefore defined as ICD (8).
At the California National Primate Research Center (CNPRC), approximately 20% of the animals are hospitalized annually due to acute diarrhea. Up to 25% of these acute-diarrhea cases are diagnosed as ICD (up to 5% annual incidence). The annual incidence of ICD cases has been reported to be as high as 10 to 15% of the population in other breeding colonies (5, 6). Affected animals with ICD respond poorly to medical management (including corticosteroids and antibiotics) and are frequently hospitalized due to dehydration and weight loss. Animals that are nonresponsive to treatment regimens are euthanized for ethical considerations (6).
Using various virus-specific reagents and methods, feces from rhesus macaques with diarrhea on a Chinese monkey farm were shown to contain enteric adenoviuses, enteroviruses, coronaviruses, rotaviruses, and picobirnaviruses, while those at the Yerkes National Primate Research Center (Yerkes, GA) contained adenovirus and picorbinaviruses (12). Using cytopathic effects in tissue culture as read out, adenoviruses were found to be associated with diarrhea in captivity-raised rhesus macaques, while rotaviruses were not considered a frequent cause of gastroenteritis (13). Experimental inoculation with primate caliciviruses was able to induce colitis in juvenile rhesus macaques (14). More recently, simian immunodeficiency virus (SIV)-induced enteropathy was associated with a greater diversity of enteric viruses, mucosal adenovirus, and parvovirus viremia than in an uninfected group of rhesus macaques (15). African green monkeys, which, unlike rhesus macaques, do not develop SIV-associated enteropathy, showed a less diverse enteric virome in both SIV-free and SIV-infected animals (15).
Numerous metagenomics virome analyses have shown that humans, as well as wild and domesticated animals, both healthy and with diarrhea, can shed multiple viruses in their feces (16–30). For animals weakened by other infections, immunodeficiency, age, or stress, generally innocuous infections may result in overt disease. Therefore, a major problem in uncovering the etiology of enteric animal diseases, such as ICD, is the presence of many largely innocuous viruses, some of which may be pathogenic in only the most susceptible hosts.
Here, we used viral metagenomic sequencing to characterize the enteric virome of rhesus macaques with acute and chronic diarrhea and compared its composition to that of healthy animals to identify viruses associated with acute diarrhea or ICD.
RESULTS
Detection of multiple viral families.
Viral metagenomic libraries were prepared from cryopreserved fecal samples from animals with acute diarrhea (n = 31) or idiopathic chronic diarrhea (n = 29) and healthy animals (n = 32). Samples were individually sequenced using two Illumina MiSeq runs, generating a total of 51 million reads. Viral reads from 6 viral families plus circular Rep-encoding single-stranded DNA (CRESS DNA) viruses were detected, and their distribution in each animal group is shown in Fig. 1A. The overall percentage of eukaryotic virus reads in these virus families was 0.063% in the healthy group, 0.131% in the acute-diarrhea group, and 0.297% in the chronic-diarrhea group, indicating a higher overall viral burden in diarrheic animals. The following virus families were identified in decreasing percentages of reads: Picornaviridae, CRESS DNA viruses, Parvoviridae, Adenoviridae, Picobirnaviridae, Anelloviridae, and Astroviridae. The dominant viral reads in all groups were from the family Picornaviridae, making up 59% of the viral reads in the healthy group and 86% in the chronic-diarrhea and 88% in the acute-diarrhea groups. The CRESS DNA viral sequences made up the second most common viral reads, accounting for 17% of viral reads in healthy animals, 9% in the acute-diarrhea group, and 13% in animals with chronic diarrhea. The most prominent difference among the three groups can be seen with members of the family Parvoviridae, which made up only 0.2% and 1.6% of the reads in the chronic- and acute-diarrhea groups, respectively, but 23% in the healthy group. Collectively, adenovirus, picobirnavirus, anellovirus, and astrovirus sequences were detected as a small fraction (1 to 3%) of the total viral reads in every group (Fig. 1A).
FIG 1.

(A) Distribution of sequence reads matching different eukaryotic virus families. The numbers in the black boxes are the percentages of eukaryotic viral reads relative to the total number of reads. The pie charts indicate percentages of viral sequence reads from different viral families. “Others” consists of Adenoviridae (purple), Picobirnaviridae (turquoise), Anelloviridae (orange), and Astroviridae (light blue). (B) Distribution of sequence reads from different viral families shown by individual rhesus macaque using percent viral reads divided by total reads. Horizonntal lines reflect mean values.
A comparison of percentages of hits for different viral families in each animal of the three clinical groups is shown in Fig. 1B. The mean values were greater for picornaviruses and picobirnaviruses in acute and chronic diarrhea than for healthy animals, and vice versa for parvoviruses, but after adjusting the P values derived by t tests for multiple comparisons (different viral families), the false-discovery rate (FDR) calculation yielded no differences of <0.05.
The number of coinfections with different viral families and CRESS DNA viruses in each individual animal group, irrespective of read numbers, ranged from 1 to 6. The level of coinfections between groups was not statistically different using the one-way analysis of variance (ANOVA) method.
Complete and partial viral genomes. (i) Picornaviridae. (a) Enterovirus.
A total of 22 contigs ranging in size from 1,076 nucleotides (nt) to 7,096 nt were generated (see Table S2 in the supplemental material). Members of two enterovirus species were identified, based on a protein similarity search (BLASTx): Enterovirus A (EV-A) (15 contigs) and Enterovirus J (EV-J) (7 contigs). A complete or nearly complete coding DNA sequence (CDS) was generated in 4 contigs from Enterovirus A species and 2 contigs from Enterovirus J species (Fig. 2A). Three shorter partial genomes were also generated (see Table S2 in the supplemental material). The sequences were subjected to genotype evaluation over the VP1 region using Enterovirus Genotyping Tool v. 01 (31). Four EV-A contigs were classified as serotype SV19, two as SV46, and three as Enterovirus J. These sequences, along with closely related genomes from this and other NHP enterovirus species, were used for phylogenetic analysis based on complete VP1 capsid sequences (Fig. 2B). Three main groups of enteroviruses related to EV-A SV19/WUHARV2 and -3, EV-A SV46, and EV-J POo-1 were identified. The EV-A SV19-like sequences (cg4006, cg4644, cg5250, and cg9811) showed 77 to 92% nucleotide identity to one another and 80 to 83% to SV19. These viruses also showed a close relationship to WUHARV enterovirus 2 (GenBank accession no. JX627571.1) and WUHARV enterovirus 3 (GenBank accession no. JX627572), with 79 to 82% and 78 to 86% nucleotide similarity, respectively. The SV46 sequences (cg4119 and cg5400) showed 82% nucleotide similarity in VP1 to simian enterovirus 46 strain RNM5 (GenBank accession no. EF667343.1) from rhesus macaque. Enterovirus J contigs (cg5275, cg8227, and cg3697) showed 86 to 91% nucleotide similarity to one another and 76% to enterovirus J strain POo-1 (GenBank accession no. FJ007373), which belongs to the EV-J103 genotype isolated from pig-tailed macaque (Macaca nemestrina).
FIG 2.

Enterovirus and sapelovirus genomes and phylogenetic analyses. (A) Genome coverage of the longest enterovirus contigs, shown in dark shading. (B) Phylogenetic analysis of enterovirus VP1. (C) Phylogenetic analysis of sapelovirus VP1.
In order to generate reference sequences to count reads to different enterovirus genotypes and variants, the diverse EV-A SV19-related sequences were further split into two groups (see Table S2 in the supplemental material) more closely related to either SV19/WUHARV3 or WUHRAV2. Another group of shorter enterovirus contigs most closely related to EV-A SV92/WUHARV1 (missing VP1 and therefore not included in Fig. 2A and B) were also selected. A total of 5 groups of different enterovirus sequences were therefore assembled for measuring read counts to different enterovirus variants (four EV-A and one EV-J) (see Table S2 in the supplemental material).
(b) Sapelovirus.
Sapelovirus is a distinct genus of the family Picornaviridae (32). Three contigs (cg10192, cg1921, and cg7136) contained a 5′ untranslated region (UTR), leader sequence, and capsid-encoding region (VP4 to VP1), respectively, of a sapelovirus genome. The contig locations relative to the closest reference of simian sapelovirus 1 (GenBank accession no. NP_758809.1) are shown in Fig. 2C. Two of these contigs (cg10192 and cg1921 [see Table S2 in the supplemental material]) included the first half of the sapelovirus genome from the 5′ UTR to VP3, with a 481-bp-long overlap (Fig. 2C). The third contig (cg7136), encoding VP1 capsid protein, shared 91% amino acid identity with simian sapelovirus 1 (GenBank accession no. NP_758809.1) and clustered together with group B simian sapeloviruses (Fig. 2C).
(ii) Parvoviridae. (a) Erythroparvovirus.
The complete CDS of an erythroparvovirus genome (cg5877), with a length of 5,041 nt, was generated (Fig. 3A). The virus showed 83% nucleotide similarity over the entire genome and 93% amino acid identity in the capsid protein to simian erythroparvovirus 2 (GenBank accession no. U26342) (see Table S2 in the supplemental material).
FIG 3.

Parvovirus genomes and phylogenetic analyses. (A) ORF maps of parvovirus contigs. (B) Phylogenetic analysis of parvovirus NS1. (C) Phylogenetic analysis of parvovirus VP1.
(b) Dependoparvovirus.
A complete CDS genome sequence of a dependovirus (cg34) with a length of 4,502 nt was generated (Fig. 3A), with 97% nucleotide similarity to adeno-associated virus 1 (GenBank accession no. AF063497) (99% capsid protein identity) originally isolated from a tissue culture of rhesus macaque cells (see Table S2 in the supplemental material).
(c) Bocaparvovirus.
A complete NS1 (649 amino acids [aa]) and partial middle gene NP1 (188 aa) of a novel bocaparvovirus (cg8092) were generated. The genetic map of this partial genome (2,815 nt) with 2 identified open reading frames (ORFs) is depicted in Fig. 3A. The closest GenBank accession number reference based on NS1 protein homology is canine bocavirus (GenBank accession no. AKG54784.1), with 49% amino acid identity to the novel bocavirus (Fig. 3A).
(d) Protoparvovirus.
A 1,707-nt-long contig (cg4720) of a simian parvovirus, encoding a partial NS1 (128 aa), a complete middle protein NP1 (105 aa), and a partial capsid protein (75 aa), was generated (Fig. 3A). The amino acid identities of cg4720 were as follows: 57% to NS1 of sea otter parvovirus 1 (GenBank accession no. YP_009272690.1), 52% to a putative middle protein of bat parvovirus BtBV_V3 (GenBank accession no. AKM21311.1), and 82% to capsid protein VP1 of porcine bufavirus (GenBank accession no. AMY15606.1), all of which belong to the genus Protoparvovirus.
A separate partial VP1 contig (cg3346) was also generated (Fig. 3A), which did not overlap a previously described sequence and encoded a 452-aa sequence with 80% identity to WUHARV parvovirus 1 (GenBank accession no. AFV48070.1), another protoparvovirus (see Table S2 in the supplemental material).
(e) Chapparvovirus.
Three contigs with homology to parvovirus NS1 (simian parvo-like virus 1 or Mopa1), simian parvo-like virus 2 (cg9738), and simian parvo-like virus 3 (cg5864) were generated, with sequence lengths of 1,605 nt, 1,272 nt, and 2,699 nt, respectively (Fig. 3A), including partial or complete ORFs for NS1 proteins of different lengths: Mopa1 (347 aa), cg9738 (359 aa), and cg5864 (712 aa). The closest relative was the NS1 of porcine parvovirus 7 (GenBank accession no. KU563733.1), with 52%, 48%, and 49% amino acid identity (BLASTp), which has been included in a genus recently labeled Chapparvovirus (33). The NS1 amino acid identities among these three chapparvovirus-like NS1 sequences were 54% to 69%.
The phylogenetic analyses of the parvovirus NS1 and VP1 described above are shown in Fig. 3B and C, supporting their genus assignments.
(iii) Picobirnaviridae.
Two contigs of picobirnavirus were generated: cg10460, encoding RdRp, and cg10459, including capsid genes (Fig. 4A and B). The partial RdRp (352 aa) showed 81% amino acid identity to a human picobirnavirus (GenBank accession no. ALL29321.1) and clustered together with group II picobirnaviruses. The partial capsid protein was 366 aa long and showed 33% identity to the capsid of a dromedary picobirnavirus (GenBank accession no. AIY31277.1). Phylogenetic analysis confirmed the close relationship of the RdRp segments to that of a human picobirnavirus (Fig. 4A) and more distant relationship of its capsid segment to that of a fox picobirnavirus (Fig. 4B).
FIG 4.

Picobirnavirus genome coverage and phylogenetic analyses. (A) Phylogenetic analysis of RdRp. (B) Phylogenetic analysis of capsid protein. Bootstrap values are shown at nodes.
(iv) Adenoviridae.
Adenovirus contigs (see Table S2 in the supplemental material) that mapped to simian adenovirus 13 (GenBank accession no. KP329563) with an average of 87% nucleotide similarity (see Table S2 in the supplemental material) were generated. Sequence analysis of the hexon protein, the region traditionally used for classification, confirmed its close relationship to that of simian adenovirus 13 (data not shown).
(v) Astroviridae.
Astrovirus reads were present only at low read number levels (2 to 16 reads) in one animal with acute diarrhea, three with chronic diarrhea, and two healthy animals. All the reads mapped to human astrovirus MLB1 strain BtnMLB1-86 (GenBank accession no. AB823732.1), with 92% and 63% amino acid identity to capsid and serine proteinase genes, respectively. Too few reads were detected to generate a contig of >1,000 bases.
(vi) Unclassified virus family: CRESS DNA viruses.
Numerous contigs (n = 63) of >1,000 bases encoded a Rep protein involved in rolling-circle replication of small circular single-stranded DNA (ssDNA) genomes (see Table S2 in the supplemental material). Twenty-one complete circular genomes encoding a Rep and a capsid protein were assembled de novo or completed by inverse PCR and Sanger sequencing. The characteristics of the genomes, which varied in size from 2,129 to 3,665 nt, are shown in Fig. 5. Eighteen viruses had bidirectional Rep and Cap genes, and for four genomes, both ORFs were on the same strand.
FIG 5.

Genome ORF maps of CRESS DNA viruses.
These genomes are part of the rapidly growing group of viruses recently described as CRESS-DNA viruses, known to infect plants, fungi, and vertebrates and found in multiple environments, as well as insect bodies (34). These 21 Rep sequences plus another 5 complete Rep ORFs from contigs (see Table S2 in the supplemental material) were selected for phylogenetic analysis. Ten Rep sequences clustered with a diverse group of genomes recently identified in mammalian fecal samples and labeled smacoviruses (35), but other taxa were widely dispersed throughout the tree (Fig. 6). A high degree of Rep genetic diversity was seen, but none clustered closely with any of the few CRESS DNA viruses with known cellular hosts. The tropism of these feces-derived CRESS DNA genomes therefore remains unknown.
FIG 6.

Phylogenetic analysis of CRESS DNA Rep proteins. Rep proteins from complete genomes are labeled with black circles; the open circles indicate complete Rep proteins from partial genomes.
Enterovirus load estimated by real-time (RT) PCR.
Using a panenterovirus 5′ UTR real-time PCR, the enterovirus loads were compared using cycle threshold (CT) values. The group mean CT values were lower in the acute group (P = 0.01; average CT = 17) and the chronic group (P = 0.004; average CT = 16) than they were in healthy animals (average CT = 19) (Fig. 7).
FIG 7.
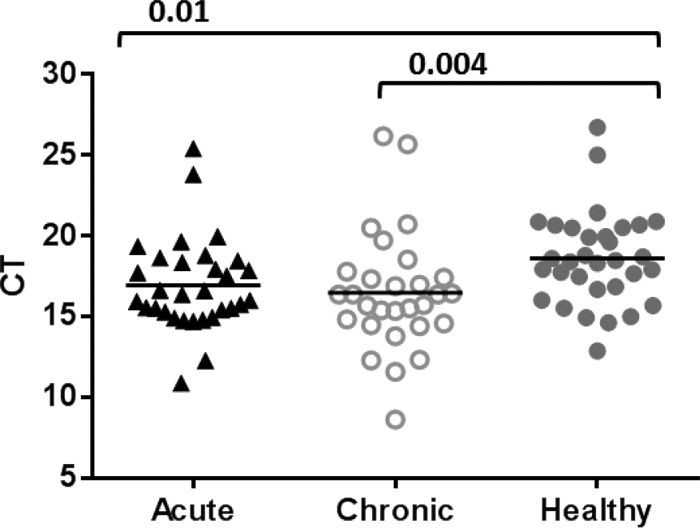
Panenterovirus real-time PCR signal cutoff values. Comparison between groups was performed using the Mann-Whitney test. P values (brackets) of ≤0.05 were considered statistically significant.
Disease associations of different viral genetic clusters.
To quantify the sequence reads to specific viruses (rather than viral families), viral contigs >1,000 bases long were used to enumerate matching reads in the feces from each animal. Contig lengths and percent translated amino acid identities and E values to their closest homolog in GenBank are shown in Table S2 in the supplemental material. When nearly complete genomes of genetically distinct viruses (with no close relative detected) were available, these contigs were used directly to enumerate matching raw reads from each animal. When contigs to different regions or segments of the same genome (closest to the same reference genome in GenBank) were present, concatenates of these contigs were used to increase the number of matching raw reads. In order to measure matching reads to multiple variants of the same virus genotypes, we also concatenated very closely related contigs based on the best sequence similarities to the same viral genomes in GenBank. The distribution of matching read numbers to each viral contig and concatenate was then measured in animals from the two clinical groups and the healthy group. To visualize the number of reads matching different enterovirus genotypes and parvovirus genera, these results are shown on a log scale (Fig. 8).
FIG 8.

Raw read numbers matching different enterovirus genotypes and parvovirus genera plotted on a log10 scale. Raw read numbers were adjusted for total reads in each library.
The distributions of viral reads matching the different viruses of mammalian origin were then compared between the three clinical groups using ANOVA and Kolmogorov-Smirnov (KS) statistical tests to test for a positive or negative association with health or disease (Fig. 9; see Table S2 in the supplemental material).
FIG 9.

Statistical comparisons of read number distributions matching different mammalian viruses generated from healthy animals and those with acute diarrhea or ICD. The numbers reflect the mean difference (the average of the first group minus the average of the second group) in different clinical groups. The colored bars are group mean differences (after log transformation). The red (blue) bars represent higher (lower) read numbers in animals with acute diarrhea or ICD versus healthy animals or in animals with ICD versus animals with acute diarrhea. The ANOVA and KS P values indicate whether the red or blue values are significantly different. P values below 0.05 are shaded.
Enteroviruses.
When comparing both clinical groups to the healthy group, higher percentages of viral reads were found in sick animals for all 5 genotypes (Fig. 10, red bars comparing acute- and chronic-diarrhea groups to the healthy group). After adjusting for multiple comparisons using the false-discovery rate, the EV-A92/WUHARV1 and SV46 genotypes were found to be significantly associated with ICD relative to healthy controls using both statistical methods (Fig. 9).
FIG 10.

Statistical comparisons of read number distributions matching different CRESS-DNA viruses of unknown cellular origin generated from healthy animals and those with acute diarrhea or ICD. The numbers reflect the mean difference (the average of the first group minus the average of the second group) in different clinical groups. The colored bars are group mean differences (after log transformation). The red (blue) bars represent higher (lower) read numbers in animals with acute diarrhea or ICD versus healthy animals or in animals with ICD versus animals with acute diarrhea. The ANOVA and KS P values indicate whether the red or blue values are significantly different. P values below 0.05 are shaded.
Sapelovirus.
Sapelovirus reads were more frequent in the healthy group than either of the two clinical groups (blue bars comparing the acute- and chronic-diarrhea groups to the healthy group in Fig. 9). The presence of sapelovirus reads was associated with healthy animals compared to ICD animals using ANOVA only (Fig. 9).
Adenoviruses.
Adenovirus reads were detected in numbers greater than 5 in only a single acute-diarrhea animal (see Table S2, sample 10A, in the supplemental material), and no association was therefore found between clinical state and adenovirus reads using both statistical tests (Fig. 9).
Parvoviruses.
For parvovirus contigs, a greater percentage of reads were consistently seen in the healthy animals than in the animals with either acute or chronic diarrhea (Fig. 9, blue bars comparing the acute- and chronic-diarrhea groups to the healthy group). Reads matching a new bocaparvovirus and erythroparvovirus were associated with healthy animals relative to acute- and chronic-diarrhea cases using ANOVA after adjustment for multiple comparisons (Fig. 9). The KS analysis yielded similar results, except for the erythroparvovirus acute-healthy comparison, with a P value of only 0.158. Differences between clinical groups and healthy animals did not reach statistical significance for the dependovirus (adeno-associated virus), protoparvovirus (related to bufavirus and the WUHARV-1 parvovirus), and chapparvovirus (simian parvo-like) sequences, except for the comparison of animals with chronic diarrhea versus healthy animals using ANOVA only (Fig. 9). The numbers of reads from each animal for all five parvoviruses are also plotted on a log scale to visualize their greater numbers in healthy animals than in clinical cases (Fig. 8).
Picobirnaviruses.
Four animals in the chronic-diarrhea group were positive, with both RdRp and capsid contigs matching reads (see Table S2 in the supplemental material). This concordance in the detection of reads from both segments indicated that the RdRp and capsid contigs belong to the same bipartite genome. While all 5 animals with >5 total picobirnavirus reads were found in the chronic group, this distribution of reads did not reach statistical significance using either ANOVA or the KS test (Fig. 9).
CRESS-DNA viruses.
The CRESS-DNA sequences as a group were the second most frequently detected viral reads (Fig. 1). The distributions of reads to these genomes of unknown cellular origin were also compared between clinical groups (Fig. 10). The majority of the 21 CRESS-DNA genomes used detected more matching reads in healthy animals than in both acute- and chronic-diarrheic animals, with five of the genomes reaching statistically significantly higher levels in healthy animals using ANOVA (Fig. 10). In the KS analysis, these five genomes reached statistically significant levels only in the acute-diarrhea versus healthy group comparison. A single CRESS-DNA genome was positively associated with acute diarrhea only using ANOVA and the KS test.
DISCUSSION
Diarrhea in captive rhesus macaques is a significant health problem in primate research facilities (36) whose study may provide vaccine candidates and an NHP model system for similar conditions in humans. In a subset of initially acute diarrhea cases, the condition does not resolve despite treatment, and pathogenic bacteria and parasites are not detected. Clinically, these monkeys show watery, nonbloody diarrhea, leading to weight loss and dehydration, without response to common therapies, and ultimately, these animals are euthanized for ethical reasons. Here, we describe the partial and complete viral genomes belonging to multiple viral families generated from the feces of 92 captive rhesus macaques. Differences in viral read numbers matching the different viruses circulating in that population were compared between healthy animals and those with acute diarrhea or ICD.
Genomes could be classified into 6 known viral families, plus a large group of unclassified viruses defined as CRESS DNA viruses (34). The overall percentage of eukaryotic virus hits was highest in chronic diarrhea, followed by acute diarrhea and the healthy group of rhesus macaques. Picornavidae family members were predominant in all three groups, but the acute- and chronic-diarrhea groups showed higher percentages of picornaviral reads (88% and 86%) than the healthy group (59%). From EV-A species, we identified three genotypes: A92/WUHARV1, SV46, and SV19 (which was further divided into SV19/WUHARV3 and WUHRAV2). We also characterized enterovirus species J viruses related to the EV-J103 genotype. These viruses were initially described from NHPs using tissue culture and metagenomics sequencing and were found in cases of captive primates with diarrhea or SIV-induced enteropathy (15, 32, 37, 38). Based on matching read numbers measured, enteroviruses EV-A92/WUHARV1 and SV46 were both significantly associated with ICD, using two different statistical tests after adjusting for multiple comparisons. While the other enterovirus genotypes (EV-A SV19/WUHARV3 and WUHRAV2 plus the EV-J103 genotypes) also showed higher levels in diarrheic animals (Fig. 9), this difference did not reach statistical significance after adjustment for multiple comparisons. SV19 was reported by RT-PCR in 41% of tested rhesus macaque fecal specimens collected from a zoo in Dhaka, Bangladesh (39). Using a panenterovirus real-time PCR, we also showed significantly lower CT values in the acute- and chronic-diarrhea groups of animals, supporting generally higher picornavirus RNA loads in diarrheic cases than in the healthy group. The notion that the percentage of sequence read numbers generally reflects the viral load is also supported by studies reporting a correlation between the viral reads and real-time PCR-estimated viral loads (40–42). A related virome study of SIV-infected rhesus macaques also showed a greater percentage of picornavirus reads (all genotypes combined) in the feces of animals with SIV enteropathy than in a non-SIV-infected control group (15).
Reads from sapeloviruses, members of a Picornaviridae genus distinct from enteroviruses, were reduced in ICD versus healthy animals by one statistical measure only. Whether such a negative association with diarrhea reflects interference between sapeloviruses and potentially pathogenic picornaviruses, such as enteroviruses EV-A92/WUHARV1 and SV46, is unknown.
Numerous parvoviruses were detected, which could be classified into 5 different Parvoviridae genera. Simian erythroparvovirus was originally discovered in cynomolgus macaques (Macaca fascicularis) with severe anemia and abnormal erythroid morphology similar to those seen in human B19 infection (43–45). Previous studies measured the prevalence of related simian parvoviruses (SPV-2; GenBank accession no. AAA74974) in monkeys, with approximately 50% of cynomolgus macaques and 35% of rhesus macaques showing antibodies to VP2 capsid protein (46). In immunocompetent animals, primary infection is typically clinically silent, and animals with anti-SPV-2 antibody are resistant to reinfection (46, 47). Another parvovirus detected here was closely related to a recently described member of the genus Protoparvovirus found in fecal samples of rhesus macaques with SIV enteropathy and in the sera of animals with advanced simian AIDS (SAIDS) (15). Other parvoviruses were less closely related to genomes already described, including a bocavirus and several members of a newly proposed Parvoviridae genus named Chapparvovirus (33, 48–50). Unexpectedly the distribution of reads for bocavirus and erythroparvovirus were positively associated with healthy animals relative to diarrheic animals.
A prior study found a greater proportion of SIV-immunosuppressed than SIV-negative rhesus macaques shedding adenoviruses (15). Simian adenovirus 13 (51) was detected here, but in high read numbers (>5) only in a single animal with acute diarrhea (see Table S2, sample 10A, in the supplemental material). Adenoviruses therefore do not seem to be a frequent cause of diarrhea in the animals from the different NHP facility analyzed here. Frequent calicivirus infections were also reported in SIV-infected rhesus macaques (15), but that viral family was not detected in the animals tested here, likely reflecting differences between primate colonies.
Picobirnavirus sequences yielded two long contigs corresponding to the RdRp- and capsid-encoding segments. Matching reads showed picobirnavirus to be distributed in 5 ICD animals, each at low read numbers (see Table S2 in the supplemental material). Despite this uneven distribution, the two statistical tools used did not show a statistical correlation between picobirnavirus reads and ICD. The picobirnavirus RdRp belonged to group II picobirnaviruses and clustered closely with human picobirnavirus isolate ChXz-4 (52). Genogroup I picobirnaviruses have been described previously from NHPs (cynomolgus and pigtailed macaques) with diarrhea (53). Picobirnavirus reads (both groups I and II) were also the most common viruses detected by metagenomics analysis in the feces of wild, presumably healthy rhesus macaques from Bangladesh (54). The role of picobirnavirus in diarrhea remains uncertain, as it is also a common infection in healthy rhesus macaques, as well as in other primates (55, 56), including humans (57), and other mammals (53, 58, 59). The intermingling of picobirnaviruses sequence from different hosts seen during phylogenetic analyses indicates that cross-species transmission may be occurring frequently (60, 61). The large amount of genetic diversity within picobirnaviruses leaves open the possibility that, as is the case for enteroviruses in humans, only a subset of the numerous genotypes circulating have pathogenic potential.
The CRESS DNA viral reads were the most common after the picornavirus reads (Fig. 1). While the tropism of some CRESS DNA viruses, such as circoviruses (infecting vertebrates) and geminivirus (infecting plants), has been clearly defined, the large majority of such genomes have been characterized using metagenomics from environmental or nonsterile samples, such as feces (13, 24, 26, 35, 62–65). The tropism of the large majority of CRESS DNA viruses is therefore still unknown (66). The diverse CRESS DNA virus genomes in fecal samples could reflect infection of macaque cells or protozoan residents of the gut or originate from plants or other components of the diet. Given the high diversity of the CRESS DNA viruses reported here, it is also possible that collectively these viruses infect multiple cellular hosts. The disease association of the complete CRESS DNA virus genomes generated here showed a minority of them (5/21) positively associated with healthy relative to diarrheic animals. Only one such CRESS-DNA virus genome was positively associated with acute diarrhea relative to healthy animals. Most CRESS DNA virus genomes showed no statistically significant difference in their distribution among the 3 animal groups.
The association of a subset of enteroviruses detected here with ICD is compatible with the wide range of clinical signs that have been associated with different member of this highly diverse genus (http://www.picornaviridae.com). Enterovirus infections are common and include numerous human and animal pathogens, as well as highly prevalent genotypes with low or rare pathogenic outcomes (67–72). Disease induction by enteroviruses is likely to be influenced by viral genotypes, host genetics, and immunological factors.
While the association of simian sapelovirus 1, some parvoviruses, and some CRESS DNA viruses with healthy animals is unexplained, several factors may be influential. By removing target cells or inducing other changes in the enteric environment, acute or chronic diarrhea may result in an environment less conducive to replication by generally innocuous viruses. Pathogenic viruses may also share receptors or intracellular proteins/pathways also required by other viruses, thereby reducing their availability for viral replication. Innate antiviral responses may also be induced by potentially “protective” viruses that increase resistance to high-level replication by pathogenic viruses in the same (sapelovirus versus enterovirus) or different (parvovirus versus enterovirus) viral families (73–78).
MATERIALS AND METHODS
Animals and biological samples.
The rhesus macaques (Macaca mulatta) assigned to this study were all housed in outdoor colonies at some point in their lives. They all received the same food biscuits (LabDiet, St. Louis, MO), which contain approximately 25% protein, 11% fat (ether extract and acid hydrolysis), and 5% fiber (crude). The diet is also balanced to contain the required vitamins and minerals for the species. Animals housed outdoors are commonly housed in half-acre corrals. These animals supplement their food by foraging on grass; however, the main source of their diet is commercially available biscuits supplemented weekly by fresh seasonal vegetables, fruits, and nuts. Animals with acute diarrhea were selected based on their first admission to the hospital for having liquid stool. Healthy controls were randomly selected during semiannual physical examinations of animals housed outdoors. The feces from ICD cases were randomly selected from animals with a long history of diarrhea and at least two negative cultures (Shigella, Yersinia, Salmonella, and Campylobacter) and parasitology tests. These animals received antibiotic therapy for treatment of diarrhea with no improvement and were returned to the hospital with recurrent watery diarrhea.
In total, 92 animals housed at the CNPRC were assigned to the study. The animals were divided into 3 groups based on their health status: acute diarrhea (n = 31), ICD (n = 29), and healthy animals (n = 32). The male/female ratio was 2:1 in the acute-diarrhea group, 1:1 in the chronic-diarrhea group, and 7:1 in the healthy group. The median ages of the animals at the time of sampling were 3 years (acute), 2 years (chronic), and 2 years (healthy). Fecal samples (sick animals) were collected directly from cage pans of hospitalized animals, and rectal swabs (healthy animals) were collected by insertion about 1 in. into the rectum. Both sample types were kept in empty sterile tubes and immediately stored and maintained at −80°C until processed. The sample identifier (ID), date of collection, and number of reads derived from each sample are included in Table S1 in the supplemental material. Whether the concentration or relative stability of nucleic acids from different enteric viral families consistently differs between anal swabs and fecal samples is unknown.
Ethics statement.
This study was conducted in accordance with Institutional Animal Care and Use Committee (IACUC) policy and was approved by the IACUC at the University of California at Davis (UC Davis) (protocol 18031). All sample collection procedures conformed to the requirements of the Animal Welfare Act, were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council, and followed the CNPRC standard operating protocols. The CNPRC houses approximately 5,000 nonhuman primates and is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), which is a private, nonprofit organization that promotes the humane treatment of animals in science through voluntary accreditation.
Sample processing.
Samples were handled in the order one acute, one ICD, and one healthy-animal sample, repeated until all samples were processed, and then sequenced in two Illumina MiSeq runs. Diarrhea samples (500 μl) and rectal swabs were mixed with 0.75 ml of phosphate-buffered saline (PBS) and 0.2 g of zirconia beads, vortexed, and spun at 12,000 rpm in a tabletop microcentrifuge for 10 min, and the supernatants were transferred into Eppendorf tubes. Two hundred microliters of fecal supernatant was then filtered through a 0.45-μm-pore-size filter (Millipore) to exclude cells and large particles. The filtrates were then digested with a combination of DNase and RNase nucleases to reduce the background of host and bacterial genetic material and to enrich for viral nucleic acids protected from nuclease digestion within their capsids (79, 80).
Nucleic acid extraction was performed using the Qiagen viral RNA minikit according to the manufacturer's instructions. Viral cDNA synthesis was performed using 10 μl extracted viral nucleic acids with 100 pmol of random hexamer (IDT) at 72°C for 2 min; 200 U SuperScript III reverse transcriptase (Invitrogen), 0.5 mM each deoxynucleoside triphosphate (dNTP), 10 mM dithiothreitol, and 1× first-strand extension buffer were added to the mixture and incubated at 25°C for 10 min, followed by 50°C incubation for 1 h and 70°C for 15 min. The second-strand cDNA synthesis was performed by incubation of reverse transcribed products with 5 U of Klenow fragment DNA polymerase (New England BioLabs) at 37°C for 1 h, followed by 75°C for 20 min. The resulting double-stranded cDNA served as input for viral-library construction using the transposon-based Nextera XT DNA sample preparation kit (Illumina). Multiplexing of individual samples was achieved by applying unique dual-index (barcode) primer combinations. The quantity of the final library was assessed with a Kapa Library Quant kit (Kapa Biosystems), following the manufacturer's instructions, with sequencing on the MiSeq instrument (Illumina) using 250 paired-end sequences.
Bioinformatics pipeline.
Next-generation sequencing (NGS) reads of 250 bp generated by MiSeq were debarcoded with Illumina vendor software. Using an in-house analysis pipeline running on a 36-node Linux cluster, bacterial reads were subtracted by mapping to human and bacterial nucleotide sequences from the GenBank nucleotide database using bowtie2 (81). Reads were considered duplicates if base positions 5 to 55 were identical. One random copy of duplicates was kept. Low-sequencing-quality tails were trimmed using a Phred quality score of 20 as the threshold. Adaptor and primer sequences were trimmed using the default parameters of VecScreen (82). The cleaned reads were then de novo assembled using Ensemble Assembler (83). The assembled contigs, along with the remaining singlets, were aligned with an in-house viral proteome database using BLASTx. Matches to virus proteins were then aligned to an in-house nonvirus-nonredundant (NVNR) universal proteome database using BLASTx.
The human and bacterial nucleotide database was compiled as follows. A human reference genome sequence and mRNA sequences (hg38) were concatenated. Bacterial nucleotide sequences were extracted from the NCBI nt fasta file (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/ [30 November 2015]) based on NCBI taxonomy (ftp://ftp.ncbi.nih.gov/pub/taxonomy [30 November 2015]). Human and bacterial nucleotide sequences were compiled into the bowtie2 (version 2.2.4) databases (81) for cellular sequence subtraction. Two databases were constructed: (i) a virus BLASTx database compiled using the NCBI virus reference proteome (ftp://ftp.ncbi.nih.gov/refseq/release/viral/ [30 November 2015]), to which was added viral protein sequences from the NCBI nr fasta file (based on annotation taxonomy in Virus Kingdom), and (ii) the NVNR database, compiled using nonviral protein sequences extracted from the NCBI nr fasta file (based on annotation taxonomy excluding Virus Kingdom). Repeats and low-complexity regions were masked using segmasker from the BLAST+ suite (version 2.2.7) (81, 82, 84–88). Hits with more significant (lower) adjusted E values to NVNR than to viral proteins were removed.
Assessment of virus abundance.
For quantification of viral read hits at the family level, sequence reads with the lowest (best) E score to viral proteins with values of <10−2 were counted and classified into 6 known virus families and a group of diverse CRESS DNA genomes. The virus family-specific reads from individual samples were then divided by the total number of reads generated from each sample, with the resulting adjusted read numbers expressed as a percentage of the virus reads. Significant differences among the animal groups were assessed using t tests (GraphPad Prism version 7.00 for Windows; GraphPad Software, La Jolla, CA, USA).
The numbers of raw reads matching selected viral sequences (contigs and concatenates) were measured using the bowtie2 program with all raw data output reads (81) in local-fast mode. The seed length parameter -L was set at 30, and an alignment is considered a hit if the alignment identity to the reference genome was >95%.
Comparing the distribution of sequence reads matching viral contigs in both clinical groups and healthy animals was done using one-way ANOVA to determine whether the mean values of the groups were equal. To achieve normality of skewed data, viral match counts from bowtie analysis were incremented by 1, divided by the total number of reads, and log2 transformed. The mean differences between groups after log transformation were visualized in Excel (Microsoft Office 2010 for Windows) (red and blue bars) using the conditional-formatting option. ANOVA was followed by a post hoc Tukey test to generate a pairwise comparison of P values (R Core Team, 2014 [http://www.R-project.org/]). The post hoc Tukey range test is used in conjunction with ANOVA for pairwise comparisons to find means that are significantly different from each other (89).
The KS test was also used to compare the overall matching read number distributions (rather than the mean) of each group (90). The two-sample KS test is a nonparametric method for comparing two samples, as it is sensitive to differences in both the location and shape of the empirical cumulative distribution functions of the two samples. The test statistic of the KS test is the maximum-probability distance along the two cumulative probability distributions.
Adjustment of P values for multiple comparisons was performed with the FDR correction available online at http://www.sdmproject.com/utilities/.
For comparison of CT values, results were generated using one-way ANOVA. Comparison between groups was performed using the Mann-Whitney test (GraphPad Prism version 7.00 for Windows; GraphPad Software, La Jolla, CA, USA).
Phylogenetic analyses and pairwise genetic distance comparisons.
Sequence alignments were performed using MUSCLE with default settings, and phylogenies were generated by the neighbor-joining method (Kimura 2 parameter for nucleotide sequences and p distance model for amino acid sequences) integrated in the MEGA package, version 6.0 (91). The statistical significance of tree topologies was evaluated by 1,000 bootstrap resampling iterations, and a bootstrap value of 50% was used as the cutoff point for cluster analysis. The nucleotide/amino acid similarity/identity plots were generated in BioEdit v1 (92).
Acquisition of complete genomes of CRESS DNA viruses.
Major ORFs were identified using ORF finder with a minimum window size of 200 nt (Geneious R7 v. 7.1.9; Biomatters). Following de novo assembly, the whole genomes of circular DNA viruses encoding a Rep protein were obtained using direct repeats at the ends of linear contigs to close the circular genomes after deleting one repeat. In some cases, inverse PCR and Sanger sequencing were used to close the gaps of circular DNA genomes. Genome annotation and visualization of the circular genomes were performed in Geneious R7. MFOLD software was used to identify the putative replication origins of newly acquired genomes and to generate their stem-loop structures (http://unafold.rna.albany.edu).
Real-time PCR to measure enterovirus relative viral loads detected by NGS.
Total nucleic acids were directly extracted from 140 μl of fecal supernatant samples using the Qiagen viral RNA extraction kit. Ten microliters of nucleic acids was combined with 1 μl of 100 mM random hexamer (IDT) and incubated at 72°C for 2 min. The cDNA was generated by using 4 μl of 1× Protoscript II reaction buffer, 2 μl 10 nM dNTP, 1 μl of 10 mM dithiothreitol (DTT), 1 μl RiboLock RNase inhibitor (Thermo Fisher Scientific), and 1 μl ProtoScript II reverse transcriptase (New England BioLabs). The incubation was carried out at 25°C for 10 min, 42°C for 50 min, and 65°C for 20 min. Five microliters of cDNA was added to the reaction mixture, which consisted of 15 mM KCl, 40 mM Tris HCl, 25 μg bovine serum albumin (BSA), 5 mM Mg2+, 20 mM dNTP, 0.5 μM each primer, 0.75 U of Fastart Taq polymerase (Roche), and 0.15 μl of SYBR green dye (Invitrogen). Real-time PCR amplification was performed in a Light Cycler II 480 (Roche) instrument. The panenterovirus genus-specific primers targeted the conserved region of the enterovirus 5′ UTR: Entero F1 (5′-TAG TAG TCC TCC GGC CCC TGA ATG C-3′), with positions nt 440 to 464, and Entero R1 (5′-ACA CGG ACA CCC AAA GTA GTC GGT T-3′), with positions nt 538 to 562 relative to the reference sequence (GenBank accession no. EF667343), with an amplified product size of 123 bp.
The amplification profile was as follows: 1-min initial denaturation at 95°C, followed by 45 cycles of 30 s of denaturation at 95°C, 30 s of annealing at 60°C, and 30 s of extension at 72°C. Data collection was enabled at the extension step. A CT value was used to estimate the relative viral load of each sample, which then was compared among the three animal groups using one-way ANOVA.
Accession number(s).
All raw data have been submitted to GenBank under SRA number SRP065074. The GenBank accession numbers of 64 complete and partial genomes are listed in Table S2 in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff of the California Research Primate Center for sample collection and Lani Montalvo at BSRI for assistance with real-time PCR.
This work was supported by a National Institute of Allergy and Infectious Diseases (NIAID) grant (R01AI123376) and a CNPRC Base Grant, Office of Research Infrastructure Programs/OD (P51OD011107).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00952-17.
REFERENCES
- 1.Prongay K, Park B, Murphy SJ. 2013. Risk factor analysis may provide clues to diarrhea prevention in outdoor-housed rhesus macaques (Macaca mulatta). Am J Primatol 75:872–882. doi: 10.1002/ajp.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sestak K, Merritt CK, Borda J, Saylor E, Schwamberger SR, Cogswell F, Didier ES, Didier PJ, Plauche G, Bohm RP, Aye PP, Alexa P, Ward RL, Lackner AA. 2003. Infectious agent and immune response characteristics of chronic enterocolitis in captive rhesus macaques. Infect Immun 71:4079–4086. doi: 10.1128/IAI.71.7.4079-4086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingle SB, Adgaonkar BD, Ingle CRH. 2014. Microscopic colitis: common cause of unexplained nonbloody diarrhea. World J Gastrointest Pathophysiol 5:48–53. doi: 10.4291/wjgp.v5.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elmore DB, Anderson JH, Hird DW, Sanders KD, Lerche NW. 1992. Diarrhea rates and risk factors for developing chronic diarrhea in infant and juvenile rhesus monkeys. Lab Anim Sci 42:356–359. [PubMed] [Google Scholar]
- 5.Blackwood RS, Tarara RP, Christe KL, Spinner A, Lerche NW. 2008. Effects of the macrolide drug tylosin on chronic diarrhea in rhesus macaques (Macaca mulatta). Comp Med 58:81–87. [PMC free article] [PubMed] [Google Scholar]
- 6.Ardeshir A, Oslund KL, Ventimiglia F, Yee J, Lerche NW, Hyde DM. 2013. Idiopathic microscopic colitis of rhesus macaques: quantitative assessment of colonic mucosa. Anat Rec (Hoboken) 296:1169–1179. doi: 10.1002/ar.22727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ardeshir A, Sankaran S, Oslund KL, Hartigan-O'Connor DJ, Lerche NW, Hyde DM, Dandekar S. 2014. Inulin treatment leads to changes in intestinal microbiota and resolution of idiopathic chronic diarrhea in rhesus macaques. Ann Am Thorac Soc 11:Suppl 1. doi: 10.1513/AnnalsATS.201306-208MG. [DOI] [Google Scholar]
- 8.Broadhurst MJ, Ardeshir A, Kanwar B, Mirpuri J, Gundra UM, Leung JM, Wiens KE, Vujkovic-Cvijin I, Kim CC, Yarovinsky F, Lerche NW, McCune JM, Loke P. 2012. Therapeutic helminth infection of macaques with idiopathic chronic diarrhea alters the inflammatory signature and mucosal microbiota of the colon. PLoS Pathog 8:e1003000. doi: 10.1371/journal.ppat.1003000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamboli CP, Neut C, Desreumaux P, Colombel JF. 2004. Dysbiosis in inflammatory bowel disease. Gut 53:1–4. doi: 10.1136/gut.53.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Round JL, Mazmanian SK. 2009. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Russell RG, Rosenkranz SL, Lee LA, Howard H, DiGiacomo RF, Bronsdon MA, Blakley GA, Tsai CC, Morton WR. 1987. Epidemiology and etiology of diarrhea in colony-born Macaca nemestrina. Lab Anim Sci 37:309–316. [PubMed] [Google Scholar]
- 12.Wang Y, Tu X, Humphrey C, McClure H, Jiang X, Qin C, Glass RI, Jiang B. 2007. Detection of viral agents in fecal specimens of monkeys with diarrhea. J Med Primatol 36:101–107. doi: 10.1111/j.1600-0684.2006.00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stuker G, Oshiro LS, Schmidt NJ, Holmberg CA, Anderson JH, Glaser CA, Henrickson RV. 1979. Virus detection in monkeys with diarrhea: the association of adenoviruses with diarrhea and the possible role of rotaviruses. Lab Anim Sci 29:610–616. [PubMed] [Google Scholar]
- 14.Sestak K, Feely S, Fey B, Dufour J, Hargitt E, Alvarez X, Pahar B, Gregoricus N, Vinje J, Farkas T. 2012. Experimental inoculation of juvenile rhesus macaques with primate enteric caliciviruses. PLoS One 7:e37973. doi: 10.1371/journal.pone.0037973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, Abbink P, Maxfield LF, Kambal A, Duan E, Stanley K, Kramer J, Macri SC, Permar SR, Schmitz JE, Mansfield K, Brenchley JM, Veazey RS, Stappenbeck TS, Wang D, Barouch DH, Virgin HW. 2012. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng TF, Kondov NO, Deng X, Van Eenennaam A, Neibergs HL, Delwart E. 2015. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J Virol 89:5340–5349. doi: 10.1128/JVI.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Li L, Deng X, Kapusinszky B, Pesavento PA, Delwart E. 2014. Faecal virome of cats in an animal shelter. J Gen Virol 95:2553–2564. doi: 10.1099/vir.0.069674-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sachsenroder J, Braun A, Machnowska P, Ng TF, Deng X, Guenther S, Bernstein S, Ulrich RG, Delwart E, Johne R. 2014. Metagenomic identification of novel enteric viruses in urban wild rats and genome characterization of a group A rotavirus. J Gen Virol 95:2734–2747. doi: 10.1099/vir.0.070029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng TF, Mesquita JR, Nascimento MS, Kondov NO, Wong W, Reuter G, Knowles NJ, Vega E, Esona MD, Deng X, Vinje J, Delwart E. 2014. Feline fecal virome reveals novel and prevalent enteric viruses. Vet Microbiol 171:102–111. doi: 10.1016/j.vetmic.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phan TG, Vo NP, Boros A, Pankovics P, Reuter G, Li OT, Wang C, Deng X, Poon LL, Delwart E. 2013. The viruses of wild pigeon droppings. PLoS One 8:e72787. doi: 10.1371/journal.pone.0072787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Diab S, McGraw S, Barr B, Traslavina R, Higgins R, Talbot T, Blanchard P, Rimoldi G, Fahsbender E, Page B, Phan TG, Wang C, Deng X, Pesavento P, Delwart E. 2013. Divergent astrovirus associated with neurologic disease in cattle. Emerg Infect Dis 19:1385–1392. doi: 10.3201/eid1909.130682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L, Shan T, Wang C, Cote C, Kolman J, Onions D, Gulland FM, Delwart E. 2011. The fecal viral flora of California sea lions. J Virol 85:9909–9917. doi: 10.1128/JVI.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shan TL, Wang CM, Cui L, Delwart E, Yuan CL, Zhao W, Guo W, Dai XQ, Yu Y, Hua XG. 2010. Human parechovirus infections in monkeys with diarrhea, China. Emerg Infect Dis 16:1168–1169. doi: 10.3201/eid1607.091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phan TG, Kapusinszky B, Wang C, Rose RK, Lipton HL, Delwart EL. 2011. The fecal viral flora of wild rodents. PLoS Pathog 7:e1002218. doi: 10.1371/journal.ppat.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blinkova O, Victoria J, Li Y, Keele BF, Sanz C, Ndjango JB, Peeters M, Travis D, Lonsdorf EV, Wilson ML, Pusey AE, Hahn BH, Delwart EL. 2010. Novel circular DNA viruses in stool samples of wild-living chimpanzees. J Gen Virol 91:74–86. doi: 10.1099/vir.0.015446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Victoria JG, Wang C, Jones M, Fellers GM, Kunz TH, Delwart E. 2010. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol 84:6955–6965. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneeberger PH, Becker SL, Pothier JF, Duffy B, N′Goran EK, Beuret C, Frey JE, Utzinger J. 2016. Metagenomic diagnostics for the simultaneous detection of multiple pathogens in human stool specimens from Cote d'Ivoire: a proof-of-concept study. Infect Genet Evol 40:389–397. doi: 10.1016/j.meegid.2015.08.044. [DOI] [PubMed] [Google Scholar]
- 28.Phan TG, Nordgren J, Ouermi D, Simpore J, Nitiema LW, Deng X, Delwart E. 2014. New astrovirus in human feces from Burkina Faso. J Clin Virol 60:161–164. doi: 10.1016/j.jcv.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shan T, Li L, Simmonds P, Wang C, Moeser A, Delwart E. 2011. The fecal virome of pigs on a high-density farm. J Virol 85:11697–11708. doi: 10.1128/JVI.05217-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Pesavento PA, Shan T, Leutenegger CM, Wang C, Delwart E. 2011. Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. J Gen Virol 92:2534–2541. doi: 10.1099/vir.0.034611-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroneman A, Vennema H, Deforche K, van den Avoort H, Penaranda S, Oberste MS, Vinje J, Koopmans M. 2011. An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol 51:121–125. doi: 10.1016/j.jcv.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Oberste MS, Maher K, Pallansch MA. 2002. Molecular phylogeny and proposed classification of the simian picornaviruses. J Virol 76:1244–1251. doi: 10.1128/JVI.76.3.1244-1251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang S, Liu Z, Wang Y, Li W, Fu X, Lin Y, Shen Q, Wang X, Wang H, Zhang W. 2016. A novel rodent Chapparvovirus in feces of wild rats. Virol J 13:133. doi: 10.1186/s12985-016-0589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosario K, Dayaram A, Marinov M, Ware J, Kraberger S, Stainton D, Breitbart M, Varsani A. 2012. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J Gen Virol 93:2668–2681. doi: 10.1099/vir.0.045948-0. [DOI] [PubMed] [Google Scholar]
- 35.Ng TF, Zhang W, Sachsenroder J, Kondov NO, da Costa AC, Vega E, Holtz LR, Wu G, Wang D, Stine CO, Antonio M, Mulvaney US, Muench MO, Deng X, Ambert-Balay K, Pothier P, Vinje J, Delwart E. 2015. A diverse group of small circular ssDNA viral genomes in human and non-human primate stools. Virus Evol 1:vev017. doi: 10.1093/ve/vev017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hird DW, Anderson JH, Bielitzki JT. 1984. Diarrhea in nonhuman primates: a survey of primate colonies for incidence rates and clinical opinion. Lab Anim Sci 34:465–470. [PubMed] [Google Scholar]
- 37.Nix WA, Jiang B, Maher K, Strobert E, Oberste MS. 2008. Identification of enteroviruses in naturally infected captive primates. J Clin Microbiol 46:2874–2878. doi: 10.1128/JCM.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberste MS, Jiang X, Maher K, Nix WA, Jiang B. 2008. The complete genome sequences for three simian enteroviruses isolated from captive primates. Arch Virol 153:2117–2122. doi: 10.1007/s00705-008-0225-4. [DOI] [PubMed] [Google Scholar]
- 39.Oberste MS, Feeroz MM, Maher K, Nix WA, Engel GA, Begum S, Hasan KM, Oh G, Pallansch MA, Jones-Engel L. 2013. Naturally acquired picornavirus infections in primates at the Dhaka zoo. J Virol 87:572–580. doi: 10.1128/JVI.00838-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graf EH, Simmon KE, Tardif KD, Hymas W, Flygare S, Eilbeck K, Yandell M, Schlaberg R. 2016. Unbiased detection of respiratory viruses by use of RNA sequencing-based metagenomics: a systematic comparison to a commercial PCR panel. J Clin Microbiol 54:1000–1007. doi: 10.1128/JCM.03060-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monaco CL, Gootenberg DB, Zhao G, Handley SA, Ghebremichael MS, Lim ES, Lankowski A, Baldridge MT, Wilen CB, Flagg M, Norman JM, Keller BC, Luevano JM, Wang D, Boum Y, Martin JN, Hunt PW, Bangsberg DR, Siedner MJ, Kwon DS, Virgin HW. 2016. Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 19:311–322. doi: 10.1016/j.chom.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Deng X, Mee ET, Collot-Teixeira S, Anderson R, Schepelmann S, Minor PD, Delwart E. 2015. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J Virol Methods 213:139–146. doi: 10.1016/j.jviromet.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Sullivan MG, Anderson DK, Goodrich JA, Tulli H, Green SW, Young NS, Brown KE. 1997. Experimental infection of cynomolgus monkeys with simian parvovirus. J Virol 71:4517–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown KE, Young NS. 1995. Parvovirus B19 infection and hematopoiesis. Blood Rev 9:176–182. doi: 10.1016/0268-960X(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 45.O'Sullivan MG, Anderson DC, Fikes JD, Bain FT, Carlson CS, Green SW, Young NS, Brown KE. 1994. Identification of a novel simian parvovirus in cynomolgus monkeys with severe anemia. A paradigm of human B19 parvovirus infection. J Clin Invest 93:1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown, Young. 1997. The simian parvoviruses. Rev Med Virol 7:211–218. [DOI] [PubMed] [Google Scholar]
- 47.Simon MA. 2008. Simian parvoviruses: biology and implications for research. Comp Med 58:47–50. [PMC free article] [PubMed] [Google Scholar]
- 48.Palinski RM, Mitra N, Hause BM. 2016. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 52:564–567. doi: 10.1007/s11262-016-1322-1. [DOI] [PubMed] [Google Scholar]
- 49.Reuter G, Boros A, Delwart E, Pankovics P. 2014. Novel circular single-stranded DNA virus from turkey faeces. Arch Virol 159:2161–2164. doi: 10.1007/s00705-014-2025-3. [DOI] [PubMed] [Google Scholar]
- 50.Baker KS, Leggett RM, Bexfield NH, Alston M, Daly G, Todd S, Tachedjian M, Holmes CE, Crameri S, Wang LF, Heeney JL, Suu-Ire R, Kellam P, Cunningham AA, Wood JL, Caccamo M, Murcia PR. 2013. Metagenomic study of the viruses of African straw-coloured fruit bats: detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 441:95–106. doi: 10.1016/j.virol.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. 1999. Gene therapy vectors based on adeno-associated virus type 1. J Virol 73:3994–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun G, Zang Q, Gu Y, Niu G, Ding C, Zhang P. 2016. Viral metagenomics analysis of picobirnavirus-positive feces from children with sporadic diarrhea in China. Arch Virol 161:971–975. doi: 10.1007/s00705-015-2726-2. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Banyai K, Tu X, Jiang B. 2012. Simian genogroup I picobirnaviruses: prevalence, genetic diversity, and zoonotic potential. J Clin Microbiol 50:2779–2782. doi: 10.1128/JCM.00634-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anthony SJ, Islam A, Johnson C, Navarrete-Macias I, Liang E, Jain K, Hitchens PL, Che X, Soloyvov A, Hicks AL, Ojeda-Flores R, Zambrana-Torrelio C, Ulrich W, Rostal MK, Petrosov A, Garcia J, Haider N, Wolfe N, Goldstein T, Morse SS, Rahman M, Epstein JH, Mazet JK, Daszak P, Lipkin WI. 2015. Non-random patterns in viral diversity. Nat Commun 6:8147. doi: 10.1038/ncomms9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kapusinszky B, Minor P, Delwart E. 2012. Nearly constant shedding of diverse enteric viruses by two healthy infants. J Clin Microbiol 50:3427–3434. doi: 10.1128/JCM.01589-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masachessi G, Ganesh B, Martinez LC, Giordano MO, Barril PA, Isa MB, Pavan GV, Mateos CA, Nates SV. 2015. Maintenance of picobirnavirus (PBV) infection in an adult orangutan (Pongo pygmaeus) and genetic diversity of excreted viral strains during a three-year period. Infect Genet Evol 29:196–202. doi: 10.1016/j.meegid.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 57.Smits SL, Schapendonk CM, van Beek J, Vennema H, Schurch AC, Schipper D, Bodewes R, Haagmans BL, Osterhaus AD, Koopmans MP. 2014. New viruses in idiopathic human diarrhea cases, the Netherlands. Emerg Infect Dis 20:1218–1222. doi: 10.3201/eid2007.140190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malik YS, Kumar N, Sharma K, Dhama K, Shabbir MZ, Ganesh B, Kobayashi N, Banyai K. 2014. Epidemiology, phylogeny, and evolution of emerging enteric Picobirnaviruses of animal origin and their relationship to human strains. Biomed Res Int 2014:780752. doi: 10.1155/2014/780752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ganesh B, Masachessi G, Mladenova Z. 2014. Animal picobirnavirus. Virusdisease 25:223–238. doi: 10.1007/s13337-014-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giordano MO, Martinez LC, Masachessi G, Barril PA, Ferreyra LJ, Isa MB, Valle MC, Massari PU, Nates SV. 2011. Evidence of closely related picobirnavirus strains circulating in humans and pigs in Argentina. J Infect 62:45–51. doi: 10.1016/j.jinf.2010.09.031. [DOI] [PubMed] [Google Scholar]
- 61.Ganesh B, Nataraju SM, Rajendran K, Ramamurthy T, Kanungo S, Manna B, Nagashima S, Sur D, Kobayashi N, Krishnan T. 2010. Detection of closely related Picobirnaviruses among diarrhoeic children in Kolkata: evidence of zoonoses? Infect Genet Evol 10:511–516. doi: 10.1016/j.meegid.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 62.Phan TG, da Costa AC, Del Valle Mendoza J, Bucardo-Rivera F, Nordgren J, O'Ryan M, Deng X, Delwart E. 2016. The fecal virome of South and Central American children with diarrhea includes small circular DNA viral genomes of unknown origin. Arch Virol 161:959–966. doi: 10.1007/s00705-016-2756-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phan TG, Mori D, Deng X, Rajindrajith S, Ranawaka U, Fan Ng TF, Bucardo-Rivera F, Orlandi P, Ahmed K, Delwart E. 2015. Small circular single stranded DNA viral genomes in unexplained cases of human encephalitis, diarrhea, and in untreated sewage. Virology 482:98–104. doi: 10.1016/j.virol.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Male MF, Kraberger S, Stainton D, Kami V, Varsani A. 2016. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect Genet Evol 39:279–292. doi: 10.1016/j.meegid.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 65.Cheung AK, Ng TF, Lager KM, Bayles DO, Alt DP, Delwart EL, Pogranichniy RM, Kehrli ME Jr. 2013. A divergent clade of circular single-stranded DNA viruses from pig feces. Arch Virol 158:2157–2162. doi: 10.1007/s00705-013-1701-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tijssen P, Penzes JJ, Yu Q, Pham HT, Bergoin M. 2016. Diversity of small, single-stranded DNA viruses of invertebrates and their chaotic evolutionary past. J Invertebr Pathol 140:83–96. doi: 10.1016/j.jip.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 67.Pons-Salort M, Parker EP, Grassly NC. 2015. The epidemiology of non-polio enteroviruses: recent advances and outstanding questions. Curr Opin Infect Dis 28:479–487. doi: 10.1097/QCO.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rao DC, Ananda Babu M, Raghavendra A, Dhananjaya D, Kumar S, Maiya PP. 2013. Non-polio enteroviruses and their association with acute diarrhea in children in India. Infect Genet Evol 17:153–161. doi: 10.1016/j.meegid.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 69.Phan TG, Nguyen TA, Shimizu H, Yagyu F, Okitsu S, Muller WE, Ushijima H. 2005. Identification of enteroviral infection among infants and children admitted to hospital with acute gastroenteritis in Ho Chi Minh City, Vietnam. J Med Virol 77:257–264. doi: 10.1002/jmv.20445. [DOI] [PubMed] [Google Scholar]
- 70.Holtz LR, Cao S, Zhao G, Bauer IK, Denno DM, Klein EJ, Antonio M, Stine OC, Snelling TL, Kirkwood CD, Wang D. 2014. Geographic variation in the eukaryotic virome of human diarrhea. Virology 468-470:556–564. doi: 10.1016/j.virol.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rao DC, Reddy H, Sudheendra K, Raghavendra A, Varadharaj V, Edula S, Goparaju R, Ratnakar B, Srinivasa Rao AS, Maiya PP, Ananda Babu M. 2014. Non-polio enterovirus association with persistent diarrhea in children as revealed by a follow-up study of an Indian cohort during the first two years of life. J Clin Virol 61:125–131. doi: 10.1016/j.jcv.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 72.Palacios G, Oberste MS. 2005. Enteroviruses as agents of emerging infectious diseases. J Neurovirol 11:424–433. doi: 10.1080/13550280591002531. [DOI] [PubMed] [Google Scholar]
- 73.Painter MM, Morrison JH, Zoecklein LJ, Rinkoski TA, Watzlawik JO, Papke LM, Warrington AE, Bieber AJ, Matchett WE, Turkowski KL, Poeschla EM, Rodriguez M. 2015. Antiviral protection via RdRP-mediated stable activation of innate immunity. PLoS Pathog 11:e1005311. doi: 10.1371/journal.ppat.1005311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lorenzo G, Rodriguez-Pulido M, Lopez-Gil E, Sobrino F, Borrego B, Saiz M, Brun A. 2014. Protection against Rift Valley fever virus infection in mice upon administration of interferon-inducing RNA transcripts from the FMDV genome. Antiviral Res 109:64–67. doi: 10.1016/j.antiviral.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 75.Watkinson RE, McEwan WA, Tam JC, Vaysburd M, James LC. 2015. TRIM21 promotes cGAS and RIG-I sensing of viral genomes during infection by antibody-opsonized virus. PLoS Pathog 11:e1005253. doi: 10.1371/journal.ppat.1005253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A 113:2241–2246. doi: 10.1073/pnas.1519657113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lei X, Xiao X, Wang J. 2016. Innate immunity evasion by enteroviruses: insights into virus-host interaction. Viruses 8:E22. doi: 10.3390/v8010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grekova S, Zawatzky R, Horlein R, Cziepluch C, Mincberg M, Davis C, Rommelaere J, Daeffler L. 2010. Activation of an antiviral response in normal but not transformed mouse cells: a new determinant of minute virus of mice oncotropism. J Virol 84:516–531. doi: 10.1128/JVI.01618-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Allander T, Emerson SU, Engle RE, Purcell RH, Bukh J. 2001. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc Natl Acad Sci U S A 98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, Wang C, Naeem A, Zaidi S, Delwart E. 2009. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol 83:4642–4651. doi: 10.1128/JVI.02301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ye J, McGinnis S, Madden TL. 2006. BLAST: improvements for better sequence analysis. Nucleic Acids Res 34:W6–W9. doi: 10.1093/nar/gkl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL. 2015. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res 43:e46. doi: 10.1093/nar/gkv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Skewes-Cox P, Sharpton TJ, Pollard KS, DeRisi JL. 2014. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS One 9:e105067. doi: 10.1371/journal.pone.0105067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eddy SR. 2009. A new generation of homology search tools based on probabilistic inference. Genome Inform 23:205–211. [PubMed] [Google Scholar]
- 86.Johnson LS, Eddy SR, Portugaly E. 2010. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Magoc T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tukey JW. 1949. Comparing individual means in the analysis of variance. Biometrics 5:99–114. doi: 10.2307/3001913. [DOI] [PubMed] [Google Scholar]
- 90.Massey FJ. 1951. The Kolmogorov-Smirnov test for goodness of fit. J Am Stat Assoc 46:68–78. doi: 10.1080/01621459.1951.10500769. [DOI] [Google Scholar]
- 91.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.