

ABSTRACT
Antibiotic-resistant bacteria are an emerging threat to global public health. New classes of antibiotics and tools for antimicrobial discovery are urgently needed. Type III secretion systems (T3SS), which are required by dozens of Gram-negative bacteria for virulence but largely absent from nonpathogenic bacteria, are promising virulence blocker targets. The ability of mammalian cells to recognize the presence of a functional T3SS and trigger NF-κB activation provides a rapid and sensitive method for identifying chemical inhibitors of T3SS activity. In this study, we generated a HEK293 stable cell line expressing green fluorescent protein (GFP) driven by a promoter containing NF-κB enhancer elements to serve as a readout of T3SS function. We identified a family of synthetic cyclic peptide-peptoid hybrid molecules (peptomers) that exhibited dose-dependent inhibition of T3SS effector secretion in Yersinia pseudotuberculosis and Pseudomonas aeruginosa without affecting bacterial growth or motility. Among these inhibitors, EpD-3′N, EpD-1,2N, EpD-1,3′N, EpD-1,2,3′N, and EpD-1,2,4′N exhibited strong inhibitory effects on translocation of the Yersinia YopM effector protein into mammalian cells (>40% translocation inhibition at 7.5 μM) and showed no toxicity to mammalian cells at 240 μM. In addition, EpD-3′N and EpD-1,2,4′N reduced the rounding of HeLa cells caused by the activity of Yersinia effector proteins that target the actin cytoskeleton. In summary, we have discovered a family of novel cyclic peptomers that inhibit the injectisome T3SS but not the flagellar T3SS.
KEYWORDS: Yersinia, type III secretion system, T3SS, cyclic peptides, peptoids, peptomers, virulence blocker, Pseudomonas aeruginosa
INTRODUCTION
As currently available antibiotics become ineffective due to the rise in antibiotic resistance among pathogenic bacteria, development of completely new classes of antibiotics is critical (1). Classic antibiotics target pathogens and commensal bacteria indiscriminately; therefore, their use puts selective pressure on both populations. Because of the abundance of commensals within a mammalian host (1013 to 1014), antibiotic resistance is thought to arise more frequently in commensal bacteria and is horizontally transferred to pathogens (2–4). In contrast to classic antibiotics, virulence blockers are compounds that selectively inhibit the expression or function of a virulence factor in a pathogen or group of pathogens (5). Advantages of virulence blockers are twofold. For one, selective pressure on a limited number of microbes, i.e., only pathogens expressing the molecular target of the virulence blocker, should limit the evolution of resistance (6). Second, the decreased commensal killing by virulence blockers has the potential to preserve a healthy microbiota, which is critical for maintaining gut homeostasis and defending against opportunistic pathogens (7, 8).
Type III secretion systems (T3SS) are bacterial appendages required by dozens of pathogens to cause disease, including Salmonella, enteropathogenic Escherichia coli (EPEC), Shigella, Pseudomonas, and Yersinia, but they are largely absent in nonpathogenic bacteria (9). Bacteria use T3SS to inject bacterial effector proteins into target host cells to manipulate host processes for the benefit of the pathogen. Seven T3SS injectisome families have been identified (9) and share a number of homologous membrane-associated components with the flagellar basal body (10). While many aspects of T3SS structure are similar among different T3SS families, the repertoire of encoded T3SS effector proteins is unique for each genus or even species (11, 12). For example, pathogenic Yersinia species, which include the plague agent Yersinia pestis and the enteropathogens Yersinia enterocolitica and Yersinia pseudotuberculosis, and the opportunistic pathogen Pseudomonas aeruginosa encode a Ysc family T3SS that contributes to virulence. However, the Yersinia T3SS translocates the Yersinia outer proteins (Yops) YopH, YopE, YopM, YopO, YopJ, and YopT, whereas Pseudomonas secretes the proteins ExoS, ExoU, and ExoT (13, 14). Yersinia avoids phagocytosis by using effectors targeting the actin polymerization process and signaling pathways. YopE has RhoGAP activity and leads to depolymerization of actin stress fibers in eukaryotic cells (15, 16). YopH, a protein tyrosine phosphatase, antagonizes several signaling pathways associated with focal adhesions (17, 18). YopO, also known as YpkA, binds to actin monomers, blocking actin filament polymerization, and sequesters actin polymerization regulators, which disrupts phagocytosis (19).
P. aeruginosa is a major cause of hospital-acquired infections (20, 21), and its T3SS is important in infection establishment, dissemination, and survival in animals (14, 22). P. aeruginosa is resistant to a number of antibiotics because its outer membrane is highly restrictive (20, 21, 23); therefore, new drugs to treat P. aeruginosa are of high interest. The widespread importance and structural conservation of T3SS among bacterial pathogens make T3SS optimal targets for antimicrobial agents.
NF-κB (nuclear factor kappa light chain-enhancer of activated B cells) is a family of mammalian transcription factors involved in inflammation and development (24). Yersinia lacking the YopHEMOJT T3SS effector proteins but expressing an otherwise functional Ysc T3SS triggers NF-κB activation in HEK293T cells, while Yersinia lacking the YopB translocator protein essential for translocation of T3SS cargo inside host cells does not activate NF-κB (25). Previously, we used host NF-κB-driven luciferase in HEK293T cells as a readout for Yersinia T3SS activity in a pilot T3SS inhibitor screen and identified a family of T3SS inhibitors called piericidins (26). In order to improve our screening strategy, we generated a HEK293 stable cell line expressing an NF-κB-driven green fluorescent protein (GFP) reporter gene. Using this cell line, we identified a group of eight cyclic peptomers that inhibit type III secretion in Yersinia and Pseudomonas aeruginosa.
RESULTS
Development and use of an NF-κB-GFP stable cell line for identification of T3SS inhibitors.
To reduce the variability and batch effects from transient transfection of an NF-κB reporter, we improved upon our previous NF-κB-based screen for T3SS inhibitors by generating an NF-κB-driven GFP-expressing HEK293 stable cell line (HEK293-GFP). To validate this cell line, we infected HEK293-GFP cells with Y. pseudotuberculosis lacking the Yersinia YopHEMOJT effector proteins (ΔyopHEMOJ; this strain is referred to as the Δyop6 strain from this point forward), as several Yops modulate NF-κB signaling (13). Approximately 40% of the cells were positive for GFP after 5 h of infection with the Δyop6 strain at a multiplicity of infection (MOI) of 7 (Fig. 1). In contrast, Y. pseudotuberculosis carrying a nonfunctional T3SS (Δyop6 ΔyopB) induced low levels of GFP expression similar to those of uninfected samples. The known T3SS inhibitor piericidin A1 significantly reduced the percentage of GFP-expressing cells in response to the Δyop6 strain (Fig. 1). A Z-score, described in detail previously (27), is a single numerical value that accurately validates the reproducibility of high-throughput assays. Our stable cell line generated data with Z-scores ranging from 0.4 to 0.8, which is in the reliable range. These data suggest that our stable cell line can be used for high-throughput screens for T3SS inhibitors.
FIG 1.

A newly developed NF-κB-GFP reporter stable cell line can be used to identify chemical inhibitors of the Yersinia T3SS. A HEK293-based stable cell line developed for this study was infected with Y. pseudotuberculosis Δyop6 (T3SS+) or Δyop6 ΔyopB (T3SS−), and GFP fluorescence was quantified as a readout of T3SS-induced NF-κB activation in the presence or absence of a known T3SS inhibitor, piericidin A1. The results are representative of three independent replicates. Data were analyzed by one-way ANOVA with Dunnett's multiple comparison test. ****, P < 0.0001; ***, P < 0.001.
As part of a previous study (28), a library of 20 synthetic peptides and peptomers based on the structure of a natural product, phepropeptin D (29), were synthesized and placed in an internal screening library. These peptomers were not previously screened for any bioactivity and may have a high intrinsic potential to disrupt protein-protein interactions (30), such as those critical for assembly of a large proteinaceous structure such as the T3SS. The compounds were screened for the ability to inhibit NF-κB activation during Y. pseudotuberculosis Δyop6 infection of HEK293-GFP cells. We used 2 standard deviations below the mean for the dimethyl sulfoxide (DMSO) control as the threshold for hit calling (26). The following 8 of the 20 peptomers reduced NF-κB-driven GFP expression at the two highest concentrations tested: EpD-1N, EpD-1′N, EpD-2N, EpD-3N, EpD-3′N, EpD-4′N, EpD-6N, and EpD-6′N (Fig. 2A and B) (Z-score, ∼0.5). While the “hit rate” is high, each of these eight peptomers has a single peptide-to-peptoid substitution compared to the parent compound, epiphepropeptin D (EpD), a phepropeptin D derivative containing a stereo-inversion at the proline position, and are thus structurally similar to each other (Fig. 2A).
FIG 2.

First-generation cyclic peptomers inhibit NF-κB activation in infected host cells and secretion of Yersinia T3SS effector proteins. (A) Structures of the first-generation cyclic peptomers, each of which carries one side chain modification compared to the parental compound, epiphepropeptin D (EpD). (B) HEK293-GFP cells were infected with Y. pseudotuberculosis Δyop6 in the presence or absence of the first-generation cyclic peptomers, and GFP fluorescence was quantified. The fold reductions of the GFP signals in the presence of cyclic peptomers compared to the signal with DMSO are shown as averages for three independent experiments. The dotted lines represent 2 standard deviations above the DMSO control level. (C and D) WT Y. pseudotuberculosis was grown under T3SS-inducing conditions with 60 μM cyclic peptomer, 71 μM piericidin A1, or an equivalent volume of DMSO, and secretion of T3SS cargo into the culture supernatant was assessed by precipitating the secreted proteins, visualizing them with Coomassie blue (C), and quantifying the YopE effector band intensity (D). The data were analyzed by one-way ANOVA with Dunnett's multiple comparison test. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; ns, not significant. For panel C, the lanes containing the ladder and samples shown were separated by unrelated samples in the intervening lanes of the same gel and are therefore shown divided by white borders.
Cyclic peptomers inhibit type III secretion in Yersinia pseudotuberculosis.
In order to validate whether the cyclic peptomers we identified in our primary screen were indeed T3SS inhibitors, we evaluated the effects of our hits on secretion of Y. pseudotuberculosis effectors under T3SS-inducing conditions in vitro. The previously identified T3SS inhibitor piericidin A1 (26) was used as a positive control. Wild-type (WT) Y. pseudotuberculosis was grown in low-calcium medium in the presence or absence of one of the compounds at 60 μM, an equivalent volume of DMSO, or 71 μM piericidin A1, and secretion of the YopE effector protein was analyzed as a measure of general type III secretion. YopE was selected because it can easily be distinguished from other secreted proteins visible upon Coomassie blue staining (Fig. 2C; see Fig. S1 in the supplemental material) (26). Y. pseudotuberculosis lacking one of the T3SS structural operons (ΔyscNU) was used as a negative control. YopE secretion was reduced ≥70% by EpD-4′N and EpD-3′N treatments (P < 0.0001) and ≥45% by EpD-1N, EpD-1′N, EpD-2N, and EpD-3N treatments (P < 0.001) (Fig. 2D). Furthermore, the abundances of the other Yops visible upon Coomassie blue staining were also reduced upon compound treatment (Fig. 2C; Fig. S1). Given that each of the eight peptomers carries only one peptoid substitution compared to the parent compound, we hypothesized that a combination of peptoid substitutions may further improve the compounds' bioactivity. We synthesized second-generation peptomers with combinatorial changes at positions 1, 2, 3, and 4 on the cyclic peptide backbone (Fig. 3A). All five new compounds led to >70% reductions in secretion (P < 0.0001) (Fig. 3B).
FIG 3.

Second-generation cyclic peptomers inhibit secretion of Yersinia T3SS effector proteins. (A) Structures of the second-generation cyclic peptomers, each of which carries two, three, or four side chain modifications compared to EpD. (B) WT Y. pseudotuberculosis was grown under T3SS-inducing conditions with 60 μM cyclic peptomer, 71 μM piericidin A1, or an equivalent volume of DMSO, and secretion of T3SS cargo into the culture supernatant was assessed by precipitating secreted proteins and visualizing with Coomassie blue. Data were analyzed by one-way ANOVA with Dunnett's multiple comparison test. ****, P < 0.0001.
To further validate that the candidate T3SS inhibitors are active in a dose-dependent manner, the eight compounds showing the greatest inhibition at 60 μM were chosen for testing at 2-fold increasing concentrations from 1.875 μM to 120 μM (Fig. 4; Table 1). EpD-1N, EpD-3′N, EpD-1,2N, EpD-1,2,3′N, EpD-1,2,4′N, and EpD-1,2,3′,4′N showed a dose-response relationship and reduced secretion of YopE, to less than 40% of normal DMSO control levels, at 30 μM. Surprisingly, although the parental compound EpD did not decrease NF-κB activation in our primary screen (Fig. 2B), EpD did inhibit YopE secretion in a dose-dependent manner (Fig. 4B). However, the EpD-6N peptomer did not show dose-dependent inhibition of YopE secretion (Fig. 4B), consistent with the inability of this compound to inhibit YopE secretion at 60 μM (Fig. 2D). These data suggest that specific peptoid substitutions in the epiphepropeptin scaffold improve the bioactivity of peptomers as T3SS inhibitors.
FIG 4.

Cyclic peptomers inhibit type III secretion in Yersinia pseudotuberculosis in a dose-dependent manner. WT Y. pseudotuberculosis was grown under T3SS-inducing conditions with increasing concentrations of peptomers. Secretion of T3SS cargo into the culture supernatant was assessed by precipitating secreted proteins and visualizing them with Coomassie blue. YopE band intensities were quantified and normalized to that of the DMSO control. The results are from two independent experiments. Nonlinear curve fitting is shown to depict the trend of inhibition. (A) EpD-1N, EpD-3′N, EpD-4′N, EpD-1,2N, EpD-1,3′N, EpD-1,2,3′N, EpD-1,2,4′N, and EpD-1,2,3′,4′N. (B) EpD and EpD-6N.
TABLE 1.
Cyclic peptomer data summaryh
| Compound | YopE secretion (%)a |
ExoU secretion (%)b |
YopM-Bla translocation (%)c | Concn (μM) with cell roundingd | Concn (μM) with cell toxicitye | % reduction in motilityf | Growthg | ||
|---|---|---|---|---|---|---|---|---|---|
| 60 μM | 30 μM | 60 μM | 30 μM | ||||||
| EpD-1N | 30 | 18 | 17 | 21 | 62 | NE | All | NE | NE |
| EpD-1′N | 51 | ND | ND | ND | ND | ND | ND | ND | ND |
| EpD-2N | 43 | ND | ND | ND | ND | ND | ND | ND | ND |
| EpD-3N | 55 | ND | ND | ND | ND | ND | ND | ND | ND |
| EpD-3′N | 21 | 30 | 11 | 17 | 59 | 4 | NE | NE | NE |
| EpD-4′N | 28 | 82 | 13 | 84 | 75 | 2 | >102 | NE | NE |
| EpD-1,2N | 21 | 25 | 19 | 15 | 54 | NE | NE | NE | NE |
| EpD-1,3′N | 36 | 69 | 21 | 21 | 51 | 2 | NE | NE | NE |
| EpD-1,2,3′N | 26 | 31 | 18 | 14 | 51 | 2 | NE | 20 | NE |
| EpD-1,2,4′N | 17 | 14 | 18 | 12 | 56 | 2 | NE | NE | NE |
| EpD-1,2,3′,4′N | 16 | 27 | 14 | 12 | 65 | 2 | NE | NE | NE |
| EpD-6N | 94 | ND | ND | ND | ND | ND | ND | ND | ND |
| EpD-6′N | 76 | ND | ND | ND | ND | ND | ND | ND | ND |
Yersinia YopE secretion with 60 or 30 μM peptomers compared to that with DMSO (100%).
Pseudomonas ExoU secretion with 60 or 30 μM peptomers compared to that with DMSO (100%).
YopM-Bla translocation into CHO-K1 cells treated with 7.5 μM peptomers, normalized to that with DMSO (100%).
Lowest concentration of peptomers at which cell rounding in HeLa cells was statistically significantly reversed.
Concentration of compound showing cytotoxicity in HeLa cells by the MTT assay.
Percent reduction in Yersinia motility compared to that with DMSO (100%).
Yersinia pYV− growth at 37°C over 8 h compared to that of the DMSO control.
NE, no effect (not significantly different from the DMSO control); ND, not determined.
We observed that several of the cyclic peptomers formed aggregates over time when they were added to culture media. We set out to measure the amounts of monomeric peptomers in bacteriological and cell culture media. The solubility of peptomers able to inhibit type III secretion ranged from 1 to 84 μM (Fig. S4). To determine whether cyclic peptomer aggregates were responsible for the observed T3SS-inhibitory activity, we sought to disaggregate the compounds and test their activity by adding the detergent Triton X-100 to bacterial cultures during type III secretion. Concentrations of Triton X-100 of >0.005% (vol/vol), in the absence of cyclic peptomers, led to enhanced Yop release into the culture media (Fig. S5A), perhaps as a result of cell envelope leakiness or perturbations to normal T3SS regulation with high levels of detergent. Therefore, we chose to carry out our disaggregation experiment with 0.001% (vol/vol) Triton X-100, as this concentration of detergent did not cause enhanced Yop release but has been shown to be effective at inhibiting nonspecific activity due to aggregate formation (31) and to give a decreased absorbance at 600 nm when added to cyclic peptomers (data not shown). In the presence of 0.001% Triton X-100, EpD-1,2,4′N showed activity comparable to that of the no-detergent control (Fig. 4; Fig. S5B), suggesting that peptomer aggregates that can be disrupted by 0.001% Triton X-100 are not required for T3SS-inhibitory activity.
In order to rule out nonspecific interaction with protein as the cause of the T3SS-inhibitory activity observed for the cyclic peptomers, we carried out the Yop secretion assay in the presence of increasing concentrations of bovine serum albumin (BSA). We observed no effect of BSA on secretion inhibition by EpD-1,2N, and the presence of BSA did not shift the dose-response curve for EpD-1,2N (Fig. S6). These data indicate that nonspecific protein binding does not underlie the T3SS-inhibitory activity of the cyclic peptomers.
Cyclic peptomers do not affect Yersinia growth or flagellar motility.
To determine whether the peptomers affect type III secretion specifically or have a broader impact on Yersinia physiology, we monitored bacterial growth and metabolic activity following drug treatment. Active type III secretion, induced under low-calcium conditions at 37°C in vitro, causes growth arrest, complicating analysis of bacterial growth. Therefore, we used a strain of Y. pseudotuberculosis lacking the T3SS-encoding virulence plasmid pYV (Yersinia pYV−). Kanamycin was used as a positive control because it is known to inhibit Yersinia growth. In the presence of 60 μM cyclic peptomers, Yersinia pYV− grew similarly to the DMSO-treated control, while kanamycin inhibited growth (Fig. 5A). Furthermore, in the presence of a high concentration of cyclic peptomers (120 μM), no significant difference in metabolic activity was detected compared to that of the DMSO-treated control, while kanamycin induced a significant reduction in metabolic activity (Fig. 5B). These results suggest that, unlike conventional antibiotics, peptomers do not inhibit bacterial replication or metabolic activity, yet they inhibit type III secretion in Yersinia.
FIG 5.
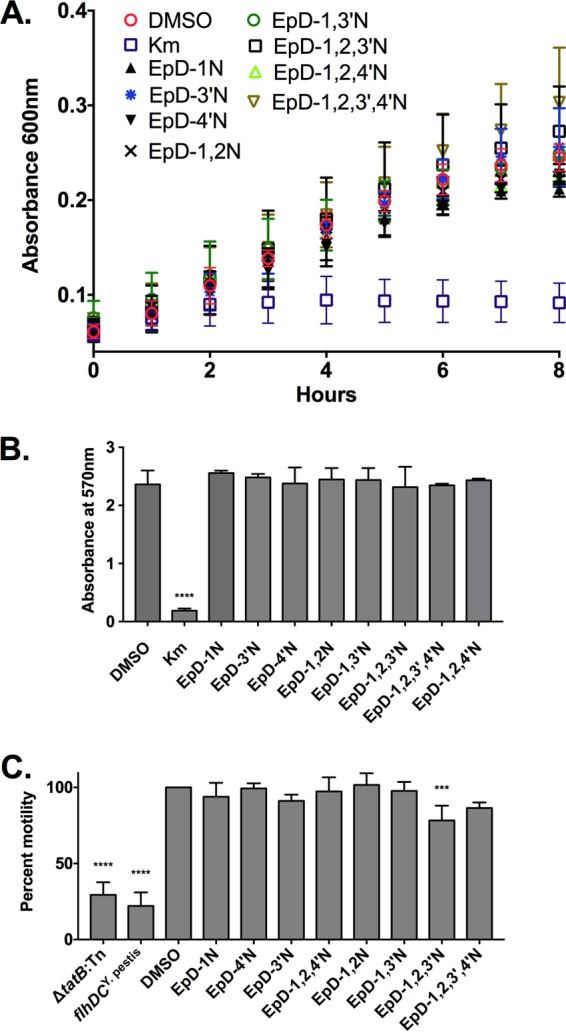
Cyclic peptomers do not affect Yersinia growth or motility. (A) Yersinia pYV− was grown in low-calcium medium at 37°C in the presence of 60 μM cyclic peptomer, an equivalent volume of DMSO, or kanamycin. The bacterial density over time was measured by determining the absorbance at 600 nm. (B) Yersinia pYV− was grown in low-calcium medium at 37°C in the presence of 120 μM cyclic peptomer, an equivalent volume of DMSO, or kanamycin for 24 h. MTT was added, and the absorbance at 570 nm was used as a readout of bacterial metabolic activity. (C) Cyclic peptomers were added to motility agar plates at 60 μM, and the diameter of WT Yersinia colonies was measured as a readout of flagellar motility. Averages for three independent experiments are shown. Data were analyzed by one-way ANOVA with Dunnett's multiple comparison test. ****, P < 0.0001; ***, P < 0.001.
The flagellar basal body, which is a T3SS, is structurally related to the injectisome T3SS (32). For Y. enterocolitica, the proton motive force was shown to be required for both type III secretion and motility (33). In addition, the twin-arginine translocation (Tat) secretion system is required for motility in Y. pseudotuberculosis and requires the proton motive force (34, 35). Therefore, compounds that affect the proton motive force or the Tat system should affect motility. To determine if the cyclic peptomers affect either of these processes, WT Y. pseudotuberculosis was spotted on soft motility agar with 60 μM cyclic peptomers or an equivalent volume of DMSO. A Y. pseudotuberculosis mutant carrying the inactive Y. pestis allele of the flhDC flagellar master regulator genes (flhDCY. pestis) and a Tat mutant (tatB::Tn) were used as negative controls. Colony diameters were measured and normalized to that of the DMSO control (100%). At 60 μM, the cyclic peptomers did not affect Yersinia motility compared to that with DMSO, except for EpD-1,2,3′N, which reduced motility by 20% (P = 0.0005) (Fig. 5C). These results suggest that cyclic peptomers do not inhibit type III secretion by targeting the Tat secretion system, the flagellar T3SS, or the proton motive force.
In order to assess the ability of the cyclic peptomers to disrupt flagellar T3SS activity in liquid culture over a shorter period more comparable to that of the T3SS secretion conditions, we assessed the motility of Salmonella and Pseudomonas in liquid culture after 2 to 3 h of growth in the presence or absence of cyclic peptomers. These bacteria were chosen because, unlike Yersinia, they express flagella at 37°C (36, 37). As the bacterial culture increased in density 32-fold during this time, new flagellar synthesis should have occurred in the presence of the cyclic peptomers. However, we observed no difference in swimming distance, speed, or percentage of motile bacteria (Fig. S7). These data suggest that cyclic peptomers do not nonspecifically block the assembly or activity of the flagellar T3SS or, more broadly, the functions of proteinaceous appendages in bacteria, other than the injectisome T3SS.
Cyclic peptomers inhibit type III secretion in Pseudomonas aeruginosa.
To determine whether the cyclic peptomers specifically blocked type III secretion in Yersinia or were active against the T3SS of other pathogens, we tested their bioactivity in P. aeruginosa. We found that the cyclic peptomers at 60 μM significantly reduced secretion of the effector protein ExoU compared to the DMSO control (Fig. 6A). In addition, these compounds showed a dose-response inhibitory effect (Fig. 6B; Table 1). At 30 μM, EpD-3′N, EpD-1,2N, EpD-1,2,3′N, EpD-1,2,4′N, and EpD-1,2,3′,4′N reduced secretion of ExoU to less than 20% of that with DSMO, which was normalized to 100%. These data suggest that five cyclic peptomers, EpD-3′N, EpD-1,2N, EpD-1,2,3′N, EpD-1,2,4′N, and EpD-1,2,3′,4′N, strongly inhibit type III secretion in both Yersinia and Pseudomonas.
FIG 6.

Cyclic peptomers inhibit type III secretion in Pseudomonas aeruginosa. WT Pseudomonas PA103 was grown under T3SS-inducing conditions with 60 μM cyclic peptomers (A), cyclic peptomers at concentrations ranging from 1.9 μM to 120 μM (B), or an equivalent volume of DMSO. Secretion of T3SS cargo into the culture supernatant was assessed by precipitating secreted proteins and visualizing them with Coomassie blue. ExoU band intensities were quantified and normalized to that of the DMSO control. Piericidin A1, which does not inhibit T3SS secretion in P. aeruginosa, was used as a negative control. (A) Data were analyzed by one-way ANOVA with Dunnett's multiple comparison test. ****, P < 0.0001; ***, P < 0.001. (B) The results are averages for at least two independent experiments. Nonlinear curve fitting is shown to depict the trend of inhibition.
Cyclic peptomers inhibit T3SS effector protein translocation and activity but are not toxic to host cells.
To test whether the cyclic peptomers affect mammalian cell physiology, we evaluated HeLa cell metabolic activity following exposure to the compounds (Fig. 7). TPEN, a known cytotoxic compound (38–40), was used as a positive control and showed toxicity at concentrations of >3.75 μM, in agreement with previous findings (38, 39). Most cyclic peptomers were not cytotoxic to HeLa cells, except for EpD-4′N, which showed toxicity at concentrations of >102 μM. In addition, EpD-1N-treated cells displayed low absorbance in the MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay at all concentrations, for reasons that are unclear but may reflect the compound's physical properties under these conditions. These data suggest that the majority of the cyclic peptomers are not toxic to mammalian cells.
FIG 7.
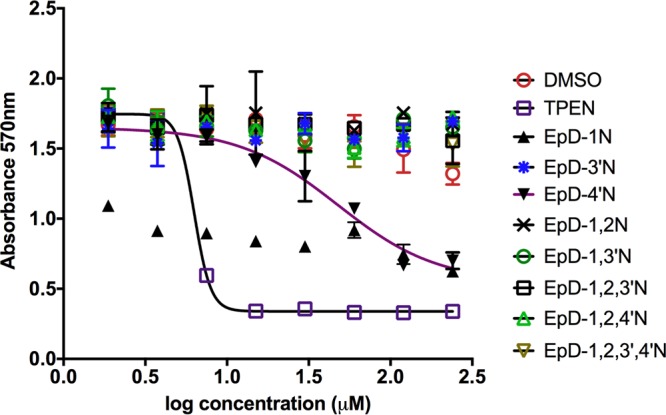
The majority of cyclic peptomers are not toxic to mammalian cells. HeLa cells were treated with increasing concentrations of cyclic peptomers or an equivalent volume of DMSO for 24 h. MTT was added to the culture, and the absorbance at 570 nm was used as a readout of cellular metabolism. TPEN, a known cytotoxic compound, was used as a control. Data shown are the averages for two independent biological replicates.
We tested the ability of the cyclic peptomers to inhibit translocation of the Y. pseudotuberculosis effector YopM, translationally fused to a reporter β-lactamase (YopM-Bla), into CHO-K1 cells. Infected cells were loaded with the fluorescent β-lactamase substrate CCF2-AM, and green (uncleaved) versus blue (cleaved) fluorescence was monitored. All eight peptomers significantly reduced YopM-Bla translocation into CHO-K1 cells at 7.5 μM (Fig. 8A). DMSO affected the percentage of blue cells at high concentrations (Fig. S2). Thus, we chose 7.5 μM because the volume of DMSO necessary to achieve this final concentration does not exceed the maximum tolerable dose of DMSO for this assay.
FIG 8.

Cyclic peptomers block translocation of a T3SS effector protein into host cells and protect host cells from T3SS effector activity. (A) Yersinia carrying a Yop effector–β-lactamase fusion (Δyop6/YopM-Bla) was used to infect CHO-K1 cells at an MOI of 5 in the presence of 7.5 μM peptomer or an equivalent volume of DMSO. CCF2-AM was added, and the percentage of cells with cleaved CCF2-AM (blue) among the total number of cells (green) was quantified. Data shown are averages for 3 independent experiments. (B) HeLa cells were infected with WT Yersinia at an MOI of 40 in the presence of increasing concentrations of peptomers for 3.5 h. Yersinia lacking a T3SS (ΔyscNU) was used as a negative control. Total cell area was quantified as a measure of cell rounding, which is a readout of host actin cytoskeleton modulation by the Yersinia T3SS YopEHO effector proteins. Median total area values for all imaged cells were plotted. Data shown are averages for two independent replicates with three technical replicates each. (C) Micrographs of HeLa cells infected with the ΔyscNU mutant or WT Yersinia and treated with EpD-3′N, EpD-1,2,4′N, or DMSO.
We next tested the ability of the cyclic peptomers to inhibit the ability of translocated Yersinia effector proteins to target the host actin cytoskeleton. As a result of YopEHO activity on the actin cytoskeleton, HeLa cells become rounded following Yersinia infection and YopEHO translocation. HeLa cells were treated with increasing concentrations of cyclic peptomers during WT Y. pseudotuberculosis infection at an MOI of 40, and the total cellular area was quantified as a measure of cell rounding (Fig. 8B). EpD-3′N and EpD-1,2,4′N reversed cell rounding induced by WT Yersinia with increasing concentrations of these compounds, starting at 3.75 μM and 1.875 μM, respectively (Fig. 8B and C). EpD-4′N, EpD-1,3′N, EpD-1,2,3′N, and EpD-1,2,3′,4′N reversed cell rounding to some degree at lower concentrations but enhanced rounding at higher concentrations, indicating a possible effect on the actin cytoskeleton at the higher concentrations. EpD-1N and EpD-1,2N enhanced rounding at almost all concentrations tested, indicating an even greater off-target effect. These data suggest that while some of the cyclic peptomers may affect mammalian cell physiology, EpD-3′N and EpD-1,2,4′N inhibit the activity of Yop effector proteins on the actin cytoskeleton. This is especially important because the activity of these effectors is critical for Yersinia to avoid important host defense mechanisms and cause disseminated disease (41).
DISCUSSION
Here we present a HEK293 stable cell line expressing an NF-κB-GFP reporter that enables high-throughput screening for inhibitors of the bacterial T3SS. We used this cell line to identify a novel set of T3SS inhibitors, i.e., synthetic peptomer analogs of the natural product phepropeptin. We found that a subset of these compounds inhibit type III secretion in both Yersinia and Pseudomonas as well as limit translocation of Yersinia effector proteins inside target host cells and, consequently, their activity on host cell processes. Importantly, the majority of these compounds do not affect bacterial growth, mammalian cell metabolism, or flagellar motility, indicating a specific effect on the T3SS.
Cyclic peptides may be particularly well suited for novel therapeutic strategies, including the targeting of virulence factors. The rich structural diversity of cyclic peptides and their larger size than that of traditional drugs may allow greater potency and specificity than those of typical small molecules (42). Oligomers of N-substituted glycine units (“peptoids”) were originally conceived as synthetic peptide derivatives that possess the modularity and protein binding characteristics of peptides but avoid the pitfalls associated with the notoriously poor metabolic stability of peptides (43, 44). Peptide-to-peptoid substitutions can increase or maintain the ability of cyclic peptides to permeate eukaryotic cells while enabling greater side chain diversity than that in the pool of commercially available amino acids (45). The phepropeptins are cyclic hexapeptides isolated from Streptomyces sp. and previously characterized for their activity as weak proteasome inhibitors (29). It was shown previously that stereo-inversion of the Pro residue to form epiphepropeptin (EpD) significantly altered the compound conformation as determined by nuclear magnetic resonance (NMR) analysis (28). Thus, it is unlikely that the compounds described here as possessing T3SS-inhibitory activity would retain the proteasomal inhibition of the natural product. In addition, Yersinia and Pseudomonas lack a proteasome. This suggests that phepropeptin compared to EpD and its cyclic peptomer analogs has distinct bioactivities and that proteasome inhibition does not underlie the T3SS-inhibitory activity of the EpD cyclic peptomers.
Peptides as a class have rich antimicrobial activities, such as that of antimicrobial peptides (AMPs), which are naturally produced by host cells to protect themselves from pathogens (46). AMPs kill a wide range of species, including fungi and viruses (46–48) as well as Gram-positive and Gram-negative bacteria, including Listeria monocytogenes, Staphylococcus aureus, Salmonella enterica serovar Typhimurium, Escherichia coli, and Pseudomonas aeruginosa (49). However, AMPs, which are ribosomally produced, exhibit unfavorable properties for use as antimicrobial therapies, such as susceptibility to degradation, cytotoxicity to host cells, instability with heat or enzymes, and a high cost of production (50, 51). It might be worth noting that most nonribosomal antimicrobial peptides, such as polymyxin, fusaricidin, and daptomycin, are “lipopeptides” which contain a mixture of charged residues and aliphatic tails and act by disrupting cell membrane or cell wall function (52, 53). In contrast, our peptides contain only aliphatic groups and are reminiscent of natural products that inhibit intracellular targets, such as griselimycin, which targets DnaN of Mycobacterium tuberculosis (54). Whether the peptomers can cross the bacterial membrane remains to be tested. Peptide analogues of a native autoinducing peptide signal in Staphylococcus aureus were shown to be potent at inhibiting quorum sensing and attenuating the virulence of the bacterium (55). Amide-bridged peptide analogues were more enzymatically stable than the peptides while still showing moderate quorum sensing inhibition (56). Our finding provides the first evidence suggesting that peptide-peptoid derivatives (peptomers) can act as T3SS inhibitors. Furthermore, our compounds contain peptoid positions that allow for facile diversification, making further optimization of these compounds synthetically tractable.
Our results suggest that the assay for T3SS inhibition with the largest dynamic range is the secretion assay, for both Yersinia and Pseudomonas T3SS effector proteins. In the absence of a standard measurement of compound activity, such as the MIC typically determined for classical antibiotics that inhibit bacterial growth or viability, we relied mainly on the secretion assay to determine the relative potencies of cyclic peptomers. When the single peptoid substitutions are compared to the parent compound, it appears that positions 1, 3, and 4 are the most important peptoid positions for activity. Although not every peptoid substitution was tested as both the N-peptoid and the chain-elongated N′-peptoid (Fig. 9), there appears to be a preference for one side chain over the other in all matched pairs, especially for position 3, with EpD-3′N inhibiting 79% of secretion while EpD-3N inhibits only 45% of secretion at 60 μM (Fig. 2 and Table 1), hinting at some specificity of interaction. While the peptoids with single substitutions at positions 1, 3, and 4 display high levels of YopE secretion inhibition at 60 μM, none of them show >50% inhibition when diluted to 15 μM. When both positions 1 and 2 are replaced with a peptoid, the potency is increased compared to those for both individual substitutions and the 1,3 combination (Fig. 4 and Table 1). The peptomers containing substitutions at both positions 1 and 2 all inhibit secretion >50% at 15 μM, with EpD-1,2,4′N and EpD-1,2,3′,4′N showing the most inhibition at this concentration. As several cyclic peptomers were found to perturb the host actin cytoskeleton and a subset of these disrupted mammalian cell metabolism, further derivatization is warranted to mitigate such off-target effects while improving activity. Importantly, the activities displayed by the cyclic peptomers are comparable to those of other known T3SS inhibitors, such as piericidin A1 (active at 71 μM) (26), N-hydroxybenzimidazole derivatives (50% inhibitory concentration [IC50] in the range of 3 to 70 μM) (57), various T3SS ATPase inhibitors (IC50 in the range of 17 to 70 μM) (58), and the salicylidene acylhydrazide INP0010 (active at 40 μM), for which three molecular targets have been identified (59).
FIG 9.

Structure-activity relationships and structural descriptions of peptoid analogs of epiphepropeptin D. N and N′ peptoids differ by a single -CH2 group in the peptoid chain. The YopE secretion assay was used to evaluate potency, with the first- and second-generation sets analyzed at 60 μM and 15 μM, respectively. Secretion inhibition was colored to highlight potency differences between compounds.
Aggregation effects have been attributed to nonspecific or promiscuous inhibitor activity (60). Most of the active cyclic peptomers have solubility values of <30 μM, while secretion inhibition was observed at higher concentrations. However, detergent disaggregation of compounds did not ablate T3SS-inhibitory activity, indicating that aggregate formation is not essential for T3SS inhibition. In addition, adding BSA to bacteria undergoing type III secretion in the presence of compounds did not abolish the compounds' activity, indicating that secretion inhibition was not due to nonspecific protein binding. Taken together, our data show that it is possible that both monomeric cyclic peptomers and peptomer aggregates are able to inhibit secretion. Alternatively, the active moiety, perhaps of a specific aggregate size, may be present at a range of cyclic peptomer concentrations as well as in the presence of Triton X-100. Without further testing (e.g., using dynamic light scattering), the observed T3SS inhibition in the presence of detergent and BSA does not formally rule out the possibility that some degree of aggregation plays a role in these compounds' activity. Future medicinal chemistry optimization will be aimed at improving both inhibitory potency and solubility, for example, by replacing one or more of the peptoid side chains with more polar groups.
The proton motive force is important for the Yersinia flagellar T3SS, as the proton motive force inhibitor carbonyl cyanide m-chlorophenylhydrazone (CCCP) prevents Yersinia motility (33, 61). Yet the majority of our compounds do not affect flagellar motility. This suggests that the cyclic peptomers do not inhibit type III secretion by perturbing the proton motive force. Since the Tat secretion system has been shown to be essential for Yersinia type III secretion (34), our data also indicate that the cyclic peptomers do not generally block bacterial secretion systems. Furthermore, the cyclic peptomers inhibit type III secretion in both Yersinia and Pseudomonas but do not block flagellar motility in Yersinia, Pseudomonas, or Salmonella. The flagellar and injectisome T3SS share eight conserved components, including an ATPase and several proteins that make up basal body membrane rings (62). However, over a dozen other T3SS components are conserved between Yersinia and Pseudomonas but are divergent or absent from the flagellar T3SS. Our data predict that the cyclic peptomers act on such a target, such as the SctF needle subunit. The activity of the cyclic peptomers is in contrast to that of the T3SS inhibitor piericidin A1, which inhibits the Yersinia Ysc T3SS but is inactive against the Pseudomonas T3SS (61). The Pseudomonas cell envelope is particularly exclusive to chemicals (63); the activity of the cyclic peptomers on the Pseudomonas T3SS suggests that these compounds either act on an extracellular T3SS component or can gain access to the Pseudomonas periplasm or cytoplasm. The specific mechanism of action of the cyclic peptomers will be the subject of another study.
In summary, the HEK293-GFP stable cell line and high-throughput screen reported here provide a rapid and reproducible platform for T3SS inhibitor discovery. Using this strategy, we have identified a new class of T3SS inhibitors, synthetic derivatives of the natural product phepropeptin, that significantly decrease type III secretion in Y. pseudotuberculosis and P. aeruginosa without affecting bacterial survival, motility, or other secretion systems, and without host cell toxicity. Future studies will focus on compound EpD-1,2,4′N, as this compound inhibited type III secretion in both Yersinia and Pseudomonas and blocked the native activity of Yersinia T3SS effector proteins on target host cells.
MATERIALS AND METHODS
Bacterial strains and mammalian cells used in this study.
The bacterial strains and cell lines used in this study are listed in Table 2. Y. pseudotuberculosis was grown in 2× YT (2× yeast extract and tryptone) at 26°C with shaking at 250 rpm overnight, unless otherwise noted. The cultures were back diluted to an optical density at 600 nm (OD600) of 0.2 in low-calcium medium (2× YT with 20 mM sodium oxalate and 20 mM MgCl2) or M9 medium supplemented with Casamino Acids to induce the T3SS (64). Cultures were grown for 1.5 h at 26°C with shaking at 250 rpm. Compounds or DMSO was added to bacterial cultures prior to shifting to 37°C to induce T3SS synthesis. Pseudomonas aeruginosa was grown in Luria-Bertani medium (LB) at 37°C with shaking at 250 rpm overnight. Low-calcium medium (LB with 20 mM EGTA and 20 mM MgCl2) was used to induce the T3SS.
TABLE 2.
Bacterial strains used in this study
| Strain | Description | Reference |
|---|---|---|
| Y. pseudotuberculosis strains | ||
| Wild type (WT) | Strain IP2666 | 68 |
| Δyop6 | ΔyopHEMOJ mutant | 25 |
| Δyop6 ΔyopB | ΔyopHEMOJB mutant | 25 |
| Δyop6/pYopM-Bla | ΔyopHEMOJ mutant carrying pYopM-Bla | 64 |
| Δyop6 ΔyopB/pYopM-Bla | ΔyopHEMOJB mutant carrying pYopM-Bla | 64 |
| ΔyscNU | Deletion of yscNU operon | 69 |
| pYV− | IP2666 cured of its virulence plasmid | 69 |
| tatB::Tn | Δyop6 with a tatB::TnHimar1 insertion | This study |
| flhDCY. pestis | Δyop6 with inactive Y. pestis flhDC | 64 |
| Pseudomonas aeruginosa strains | ||
| Wild type | Strain PA103 | 70 |
| ΔexoUT | ΔexoU ΔexoT mutant | 71 |
| PAO1 | PAO1 | 72 |
| Salmonella Typhimurium strains | 73 | |
| Wild type | Strain LT2 | |
| ΔflhDC | TH5736 (ΔflhDC-motAB-cheAW-tar) |
HeLa (ATCC), HEK293 (ATCC), and HEK293-GFP (described below) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). CHO-K1 cells (ATCC) were cultured in F-12K medium with 10% FBS. All cell lines were incubated at 37°C with 5% CO2.
Stable cell line generation.
A promoter containing five repeats of an NF-κB enhancer element (5′-TGGGGACTTTCCGC-3′) (26) was cloned upstream of the GFP gene, followed by integration of the construct into the HEK293 chromosome. The primers 5′-ATTAGAGCTCTGCAATTGTTGTTAACTTGTTTATT-3′ (forward primer; SacI restriction site is underlined) and 5′-ATTAAAGCTTTATATACCCTCTAGAGTCTCCGCG-3′ (reverse primer; HindIII restriction site is underlined) were used to clone the NF-κB reporter construct (26) into the multicloning site of peTurboGFP-PRL-dest1, containing a gene encoding a destabilized variant of green fluorescent protein (TurboGFP; Evrogen, Russia). The plasmid was linearized with BsaI and transfected into HEK293 cells by use of Lipofectamine 2000 (Invitrogen). Neomycin was used to select for cells carrying the plasmid, as previously described (65). Single cells were grown in DMEM supplemented with fresh neomycin every 2 weeks until they reached confluence in 96-well plates. Monoclonal NF-κB-driven GFP-expressing HEK293 cells were tested for their NF-κB response to the Yersinia T3SS, and positive clones were transferred to 25-cm2 and then 75-cm2 flasks and cryotubes. One clone, HEK293-GFP, was selected and used for the screen described below.
NF-κB screen for T3SS inhibitors.
HEK293-GFP cells were plated on poly-l-lysine-coated 384-well plates at a density of 1.2 × 104 cells/well in 25 μl FluoroBrite DMEM (Thermo Fisher Scientific). Y. pseudotuberculosis Δyop6 or Δyop6 ΔyopB (Table 2) overnight cultures were back diluted to an OD600 of 0.2 in M9 medium supplemented with Casamino Acids and grown for 1.5 h with shaking at 250 rpm and 26°C. Bacteria were transferred to 384-well plates. Compounds were added to bacteria prior to shifting the plate to 37°C, as well as to cell monolayers prior to infection by use of a pinning robot (Janus MDT robot; PerkinElmer). After 2 h of incubation at 37°C, bacteria were added to cell monolayers at an MOI of 7 (Janus MDT robot), and the infected cell plates were incubated for 5 h at 37°C and 5% CO2. Cells were stained with the nucleic acid dye Hoechst 33342 (Thermo Fisher Scientific) and fixed with 1% paraformaldehyde (PFA; Sigma-Aldrich) on ice for 10 min. Plates were washed with phosphate-buffered saline (PBS), and residual PFA was quenched by incubation with 0.1 M glycine for 30 min. Plates were washed in PBS four times and imaged with a Molecular Devices Image Express instrument within 24 h. T3SS function was quantified as the percentage of GFP-expressing cells among the total number of cells (total nuclei).
Cyclic peptide synthesis.
Peptides were synthesized using standard Fmoc solid-phase peptide synthesis, utilizing the submonomer approach for peptoid synthesis (66), either at room temperature or with microwave assistance. Cyclization was done in solution at a high dilution.
Loading of 2-chlorotrityl resin.
Fmoc-Xaa (10 mmol) was added to a flame-dried round-bottomed flask and dried in a vacuum desiccator with phosphorous pentoxide overnight. Fifty milliliters of dry dichloromethane (DCM) was cannula transferred into the flask, followed by 2.5 ml of N,N-diisopropylethylamine (DIPEA) transferred via syringe. After sonication for 10 min, 5 g of 2-chlorotrityl resin was added under a stream of N2 and allowed to shake for 4 h. The resin was capped with a 15-ml solution of 1:2:17 methanol (MeOH):DIPEA:dimethylformamide (DMF) (3 times for 15 min each). The resin was washed with DMF (3 times with 15 ml each) followed by DCM (3 times with 15 ml each). The loading value was calculated by determining the mass increase of dried, loaded resin.
Amino acid coupling at room temperature.
Four equivalents of Fmoc-Xaa, 5 eq of DIPEA, and 4 eq of O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) were added to the resin in DMF. The reaction mixture was agitated via shaking for 45 min and then drained. The resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each). The reaction was monitored by liquid chromatography-mass spectrometry (LC-MS) and repeated until the starting material was no longer observed. For microwave conditions, a solution of 4 eq of Fmoc-Xaa, 4 eq of HBTU, and 6 eq of DIPEA in DMF was allowed to prereact for 5 min. This solution was added to the deprotected peptide on-resin and allowed to react for 10 min at 50°C under microwave heating. The solution was drained, and the resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each). The reaction was monitored by LC-MS and repeated until the starting material was no longer observed.
Coupling of BrAcOH at room temperature.
A solution of 5 eq of bromoacetic acid (BrAcOH) and 5 eq of N,N′-diisopropylcarbodiimide (DIC) in DMF was allowed to prereact for 10 min. This solution was added to the deprotected peptide on-resin. The reaction mixture was agitated via shaking for 45 min and then drained. The resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each). The reaction was monitored by LC-MS and repeated until the starting material was no longer observed. For microwave conditions, a solution of 5 eq of BrAcOH and 5 eq of DIC in DMF was allowed to prereact for 10 min. This solution was added to the deprotected peptide on-resin and allowed to react for 10 min at 50°C under microwave heating. The solution was drained, and the resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each). The reaction was monitored by LC-MS and repeated until the starting material was no longer observed.
Installation of peptoid side chain.
A solution of 5 to 10 eq of the desired amine was prepared in a minimum volume of DMF. The resin containing the BrAc-peptide was swollen with DCM for 5 min prior to reaction. The amine was added, and the reaction mixture was agitated vigorously for 45 min. The solution was drained, and the resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each). The reaction was monitored by LC-MS and repeated until the starting material was no longer observed.
Removal of the N-Fmoc protection group at room temperature.
A solution of 2% piperidine and 2% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in DMF was added to the resin. The reaction mixture was agitated via shaking for 20 min and then drained. The resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each). For microwave conditions, a solution of 2% piperidine and 2% DBU in DMF was added to the resin. The reaction mixture was allowed to react for 5 min at 50°C under microwave heating and then drained. The resin was washed with DMF (3 times with 3 ml each) and DCM (3 times with 3 ml each).
Peptide cleavage.
Complete linear peptides were cleaved off the resin in 5 resin volumes of 2.5% trifluoroacetic acid (TFA) in DCM for 4 min three times, with a 5-resin-volume DCM wash between steps. Solvent was removed under N2, followed by dissolution in acetone or DCM and evaporation under reduced pressure. Residual TFA was removed in vacuo overnight.
Cyclization with COMU.
Linear peptides were dissolved in 20 ml of dry acetonitrile (ACN) with 4 eq of DIPEA and added dropwise (final concentration, 1 mg crude peptide per ml) to a solution of 1:1 tetrahydrofuran (THF)-ACN containing 2 eq of (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU). Reaction mixtures were stirred for 0.5 to 24 h, until complete cyclization was achieved as monitored by LC-MS. The reaction mixture was reduced in vacuo for purification via high-pressure liquid chromatography (HPLC).
Purification of peptides.
COMU by-products were removed after solution-phase cyclization on a Biotage Isolera Prime system equipped with a KP-C18-HS 12g column eluting with H2O-acetonitrile modified with 0.1% TFA. Peptides of insufficient purity were subsequently purified on a Waters mass-directed AutoPure system equipped with an X-Bridge BEH130 5-μm 19 × 150 C18 column eluting with H2O-acetonitrile, modified with 0.1% formic acid or 0.1% TFA. The mass spectra of all peptides are shown in Fig. S3 in the supplemental material.
Cyclic peptide manipulation.
Stock peptides were kept at 15 mM and were prediluted in DMSO prior to experiments. All the treatment and control pairs had the same DMSO volumes in all assays.
Secretion assay.
Visualization of proteins secreted into the culture medium was carried out as described previously (25). Briefly, Y. pseudotuberculosis or Pseudomonas aeruginosa was grown in T3SS-inducing medium (as described above) at 37°C in the presence of peptomers or an equivalent volume of DMSO carrier for 2 h. The cultures were normalized based on bacterial density (OD600) and then centrifuged at 13,200 rpm for 15 min. The supernatants were transferred to a new tube, mixed with 6.1 N trichloroacetic acid (TCA) to a final volume of 10%, and vortexed vigorously for 30 s. Samples were incubated on ice for 30 min and then spun down at 13,200 rpm for 15 min at 4°C. The pellet was resuspended in final sample buffer (FSB) plus 20% dithiothreitol (DTT) and boiled for 5 min prior to running in a 12.5% or 7.5% SDS-PAGE gel for Y. pseudotuberculosis or Pseudomonas aeruginosa, respectively. Quantification of the bands relative to those of DMSO-treated controls was carried out using Image Lab software (Bio-Rad). The WT Y. pseudotuberculosis YopE or Pseudomonas aeruginosa ExoU bands in DMSO control samples were set to 1.00. Bovine serum albumin (BSA) or Triton X-100 (Sigma-Aldrich) was added to the bacterial culture at the same time as the compounds in a subset of secretion assays, as indicated in Results. Note that the amount of BSA used in the assays (32 μg/ml) approached the maximum amount of BSA possible, as more BSA resulted in BSA aggregation following TCA addition (data not shown).
Translocation assay.
Translocation of T3SS effector proteins into mammalian cells was assayed as previously described (26). Briefly, 6,000 CHO-K1 cells were plated in each well of a 384-well plate in 50 μl of F-12K medium plus 10% FBS and incubated overnight at 37°C with 5% CO2. Overnight cultures of Y. pseudotuberculosis encoding a T3SS effector protein translocation reporter, YopM translationally fused with a β-lactamase (Bla), were back diluted to an OD600 of 0.2 and grown at 26°C for 1.5 h with shaking. The cultures were diluted to obtain a multiplicity of infection (MOI) of 5 and then transferred to a 384-well plate. Peptomers or DMSO alone was added to the plates containing the bacterial cultures and incubated for 1.5 h at 37°C. Immediately prior to infection, the peptomers or DMSO was added to the CHO-K1 cells. Bacteria were then transferred to cells by use of a pinning robot (Janus MDT; PerkinElmer) and incubated for 1 h at 37°C and 5% CO2. The β-lactamase substrate CCF2-AM (Invitrogen) was added to each well, and the plate was incubated in the dark for 30 min at room temperature. Cells were fixed with 4% PFA for 20 min and then stained with the DNA dye DRAQ5 (Cell Signaling Technology) in PBS for 10 min. Plates were washed once with PBS and imaged using an ImageXpress automated microscope, and the images were analyzed using MetaXpress analysis software (Molecular Devices). Translocation was quantified as the number of YopM-Bla-positive cells (blue cells containing cleaved CCF2) divided by the total number of cells (green cells containing uncleaved CCF2).
Growth curves.
Overnight cultures of Y. pseudotuberculosis pYV− (lacking the virulence plasmid that encodes the Ysc T3SS) were back diluted to an OD600 of 0.1 in low-calcium medium, and 100 μl was added to each well of 96-well plates. This T3SS-deficient strain was used because active type III secretion induces growth arrest in Yersinia. Peptomers were added to each well, to a final concentration of 60 μM, in three technical replicates. Equivalent volumes of DMSO were added to the positive-control wells, and kanamycin was used as a negative control (50 μg/ml). Bacteria were maintained at 37°C, and the OD600 values of the cultures were measured every hour for 8 h in a plate reader (VersaMax tunable microplate reader; Molecular Devices).
Motility agar assay.
Motility agar plates (1% tryptone, 0.25% agar) supplemented with peptomers at a final concentration of 60 μM or with an equivalent volume of DMSO were prepared in triplicate. Motility was analyzed by spotting 1-μl aliquots of either nonmotile Y. pseudotuberculosis (flhDCY. pestis or tatB::Tn) or motile bacteria (WT) onto the motility agar from overnight cultures standardized to an OD600 of 2.5 in PBS. Plates were incubated at room temperature for 1 day. Colonies were imaged by use of a Chemidoc system (Bio-Rad), and the diameters of the colonies were determined in Image Lab (Bio-Rad) and used to calculate the percent motility relative to the motility with DMSO, which was set to 100%.
Liquid motility assay.
Salmonella Typhimurium or Pseudomonas aeruginosa PAO1 was cultured in LB at 37°C with shaking at 250 rpm overnight. The cultures were then diluted to an OD of 0.025 in LB, and 30 μM peptomers or an equivalent volume of DMSO was added. An S. Typhimurium flagellar mutant (ΔflhDC) (Table 2) was used as a negative control for motility. S. Typhimurium and PAO1 were filmed by use of a Hamamatsu C4742-95 digital camera and a Nikon Eclipse E600 microscope after 2 and 3 h of incubation, respectively. Ten-microliter aliquots of culture were spotted onto coverslips, and videos were taken every 1 s for 60 frames. Five videos, from the four corners and the center of the slide, were recorded for each sample. The movies were analyzed in Imaris v.8 and subsequently processed in Matlab2016a and GraphPad Prism 7. The movement observed for nonmotile bacteria was due to drift of the culture on the slide during the course of the movie, as all bacteria in the negative-control culture showed drift in the same direction. Bacteria were considered motile if they traveled a distance 2 standard deviations greater than the average nonmotile bacterial drifting distance.
Cell viability.
The yellow tetrazolium MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] is reduced by metabolically active cells by the action of dehydrogenase enzymes, resulting in intracellular purple formazan. The MTT assay measures the cell proliferation rate, which correlates with formazan signals. To measure mammalian cell cytotoxicity in the presence of peptomers, HeLa cells were placed in 384-well plates at a density of 2,000 cells/well in triplicate and incubated at 37°C and 5% CO2 for 24 h. Compounds were added to each well in a 2-fold dilution series, starting at 240 μM, and the cultures were incubated at 37°C and 5% CO2 for 20 h. Ten microliters of MTT was added to each well, and plates were returned to the incubator for 4 h. Plates were washed once with PBS. Fifty microliters of ammonia-DMSO (67) was added to each well to solubilize the formazan crystals. Plates were incubated in the dark for 30 min at room temperature, and the absorbance at 570 nm was read in a plate reader (VersaMax tunable microplate reader; Molecular Devices).
To measure bacterial cell cytotoxicity in the presence of peptomers, Y. pseudotuberculosis pYV− overnight cultures were back diluted as described above for growth curves and then treated with compounds at a final concentration of 120 μM. The cultures were incubated at 37°C for 24 h. Ten microliters of MTT reagent was added to each well, and the plate was incubated for 2 h in the dark. One hundred microliters of 0.04 N HCl-DMSO (67) was added to each well and mixed thoroughly to solubilize the formazan crystals. Plates were read at 570 nm in a plate reader (VersaMax tunable microplate reader; Molecular Devices) within a few minutes.
Cell rounding.
HeLa cells were placed in 384-well plates at a density of 2,000 cells/well, centrifuged at 1,000 rpm for 5 min, and maintained at 37°C with 5% CO2. WT Yersinia pseudotuberculosis, which encodes the actin-targeting YopEHO effectors that cause cell rounding in host cells, was treated with peptomers in a 2-fold dilution series, starting at 120 μM, in low-calcium medium at 37°C for 2 h. HeLa cells were infected with WT Yersinia at an MOI of 40, and plates were spun at 750 rpm for 5 min and maintained at 37°C for 3 h. WT Yersinia treated with an equivalent amount of DMSO was used as a positive control, and the T3SS-deficient ΔyscNU mutant was used as a negative control for induction of cell rounding. Cells were stained with the plasma membrane stain CellMask deep red (Thermo Fisher Scientific) and the nuclear stain Hoechst 33342 (Thermo Fisher Scientific) for 20 min, fixed with 4% PFA (Sigma-Aldrich) for 10 min, washed with PBS, and imaged with a Molecular Devices Image Express system.
Thermodynamic solubility assay.
Stocks (15 mM) of the compounds were prepared in DMSO. Each stock solution was dispensed into a 96-well plate and evaporated overnight under reduced pressure with heat (50°C). To the evaporated film/solid, 125 μl of DMEM was added to make a 2 mM solution of the target compound. This solution was sonicated for 1 h and then shaken at 37°C for 16 h. The turbid solution was passed over a 0.7-μm glass-fiber filter. The solution was diluted 1:4 in acetonitrile. The solution was centrifuged at 1,000 × g for 10 min. The supernatant (10 μl) was injected onto a Thermo Fisher Orbitrap Velos Pro instrument. A 10 μM standard was used for comparison, and the assay was done in triplicate.
Kinetic solubility assay.
The kinetic solubility assay was done similarly to the thermodynamic solubility assay, except that the compounds were shaken for 2 h in the presence of 0.8% DMSO to mimic the conditions in most of our assays. The kinetic solubility assay mimics many of our assay conditions, in that compounds are predissolved in DMSO and tested after a short incubation time.
Statistical analysis.
GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA) was used to calculate the means, standard errors of the means, medians, standard errors of the medians, and one-way analysis of variance (ANOVA) values shown in the figures.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge National Institutes of Health grant R01AI106930 (to V.A.), NIH National Human Genome Research Institute grant R25HG006836 (to J.M.), and the Vietnam Education Foundation (to H.L.) for support.
We thank Joanne Engel and Susanne Häussler for the Pseudomonas aeruginosa strains, Melanie Marketon for the YopM-Bla plasmid, Karen Ottemann for Salmonella Typhimurium strains, Benjamin Abrams for helping with spot-tracking analysis, Matthew R. Naylor for helping with the Orbitrap instrument, and Hector Ramirez for characterization of the tatB::Tn strain.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00060-17.
REFERENCES
- 1.Coates AR, Halls G, Hu Y. 2011. Novel classes of antibiotics or more of the same? Br J Pharmacol 163:184–194. doi: 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 3.Schjorring S, Krogfelt KA. 2011. Assessment of bacterial antibiotic resistance transfer in the gut. Int J Microbiol 2011:312956. doi: 10.1155/2011/312956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schjorring S, Struve C, Krogfelt KA. 2008. Transfer of antimicrobial resistance plasmids from Klebsiella pneumoniae to Escherichia coli in the mouse intestine. J Antimicrob Chemother 62:1086–1093. doi: 10.1093/jac/dkn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duncan MC, Linington RG, Auerbuch V. 2012. Chemical inhibitors of the type three secretion system: disarming bacterial pathogens. Antimicrob Agents Chemother 56:5433–5441. doi: 10.1128/AAC.00975-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis K. 2013. Platforms for antibiotic discovery. Nat Rev Drug Discov 12:371–387. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 7.Kamada N, Chen GY, Inohara N, Nunez G. 2013. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumler AJ, Sperandio V. 2016. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535:85–93. doi: 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Troisfontaines P, Cornelis GR. 2005. Type III secretion: more systems than you think. Physiology 20:326–339. doi: 10.1152/physiol.00011.2005. [DOI] [PubMed] [Google Scholar]
- 10.Diepold A, Armitage JP. 2015. Type III secretion systems: the bacterial flagellum and the injectisome. Philos Trans R Soc Lond B Biol Sci 370:20150020. doi: 10.1098/rstb.2015.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Notti RQ, Stebbins CE. 12 February 2016. The structure and function of type III secretion systems. Microbiol Spectr 4:VMBF–0004–2015. doi: 10.1128/microbiolspec.VMBF-0004-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsumoto H, Young GM. 2009. Translocated effectors of Yersinia. Curr Opin Microbiol 12:94–100. doi: 10.1016/j.mib.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pha K, Navarro L. 2016. Yersinia type III effectors perturb host innate immune responses. World J Biol Chem 7:1–13. doi: 10.4331/wjbc.v7.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Von Pawel-Rammingen U, Telepnev MV, Schmidt G, Aktories K, Wolf-Watz H, Rosqvist R. 2000. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol Microbiol 36:737–748. doi: 10.1046/j.1365-2958.2000.01898.x. [DOI] [PubMed] [Google Scholar]
- 16.Black DS, Bliska JB. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol 37:515–527. doi: 10.1046/j.1365-2958.2000.02021.x. [DOI] [PubMed] [Google Scholar]
- 17.Guan KL, Dixon JE. 1990. Protein tyrosine phosphatase-activity of an essential virulence determinant in Yersinia. Science 249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- 18.Fallman M, Persson C, Wolf-Watz H. 1997. Yersinia proteins that target host cell signaling pathways. J Clin Invest 99:1153–1157. doi: 10.1172/JCI119270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee WL, Grimes JM, Robinson RC. 2015. Yersinia effector YopO uses actin as bait to phosphorylate proteins that regulate actin polymerization. Nat Struct Mol Biol 22:248–255. doi: 10.1038/nsmb.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mesaros N, Nordmann P, Plesiat P, Roussel-Delvallez M, Van Eldere J, Glupczynski Y, Van Laethem Y, Jacobs F, Lebecque P, Malfroot A, Tulkens PM, Van Bambeke F. 2007. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin Microbiol Infect 13:560–578. doi: 10.1111/j.1469-0691.2007.01681.x. [DOI] [PubMed] [Google Scholar]
- 21.Driscoll JA, Brody SL, Kollef MH. 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- 22.Schulert GS, Feltman H, Rabin SDP, Martin CG, Battle SE, Rello J, Hauser AR. 2003. Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J Infect Dis 188:1695–1706. doi: 10.1086/379372. [DOI] [PubMed] [Google Scholar]
- 23.Livermore DM. 2002. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis 34:634–640. doi: 10.1086/338782. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell S, Vargas J, Hoffmann A. 2016. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med 8:227–241. doi: 10.1002/wsbm.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auerbuch V, Golenbock DT, Isberg RR. 2009. Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog 5:e1000686. doi: 10.1371/journal.ppat.1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duncan MC, Wong WR, Dupzyk AJ, Bray WM, Linington RG, Auerbuch V. 2014. An NF-κB-based high-throughput screen identifies piericidins as inhibitors of the Yersinia pseudotuberculosis type III secretion system. Antimicrob Agents Chemother 58:1118–1126. doi: 10.1128/AAC.02025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J-H, Chung TDY, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Schwochert J, Lao Y, Pye CR, Naylor MR, Desai PV, Gonzalez Valcarcel IC, Barrett JA, Sawada G, Blanco MJ, Lokey RS. 2016. Stereochemistry balances cell permeability and solubility in the naturally derived phepropeptin cyclic peptides. ACS Med Chem Lett 7:757–761. doi: 10.1021/acsmedchemlett.6b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sekizawa R, Momose I, Kinoshita N, Naganawa H, Hamada M, Muraoka Y, Iinuma H, Takeuchi T. 2001. Isolation and structural determination of phepropeptins A, B, C, and D, new proteasome inhibitors, produced by Streptomyces sp. J Antibiot (Tokyo) 54:874–881. doi: 10.7164/antibiotics.54.874. [DOI] [PubMed] [Google Scholar]
- 30.Qian Z, Dougherty PG, Pei D. 2017. Targeting intracellular protein-protein interactions with cell-permeable cyclic peptides. Curr Opin Chem Biol 38:80–86. doi: 10.1016/j.cbpa.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGovern SL, Helfand BT, Feng B, Shoichet BK. 2003. A specific mechanism of nonspecific inhibition. J Med Chem 46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 32.Portaliou AG, Tsolis KC, Loos MS, Zorzini V, Economou A. 2016. Type III secretion: building and operating a remarkable nanomachine. Trends Biochem Sci 41:175–189. doi: 10.1016/j.tibs.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Wilharm G, Lehmann V, Krauss K, Lehnert B, Richter S, Ruckdeschel K, Heesemann J, Trulzsch K. 2004. Yersinia enterocolitica type III secretion depends on the proton motive force but not on the flagellar motor components MotA and MotB. Infect Immun 72:4004–4009. doi: 10.1128/IAI.72.7.4004-4009.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lavander M, Ericsson SK, Broms JE, Forsberg A. 2006. The twin arginine translocation system is essential for virulence of Yersinia pseudotuberculosis. Infect Immun 74:1768–1776. doi: 10.1128/IAI.74.3.1768-1776.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mould RM, Robinson C. 1991. A proton gradient is required for the transport of 2 lumenal oxygen-evolving proteins across the thylakoid membrane. J Biol Chem 266:12189–12193. [PubMed] [Google Scholar]
- 36.Wozniak CE, Chevance FF, Hughes KT. 2010. Multiple promoters contribute to swarming and the coordination of transcription with flagellar assembly in Salmonella. J Bacteriol 192:4752–4762. doi: 10.1128/JB.00093-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, Ramphal R. 2003. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol Microbiol 50:809–824. doi: 10.1046/j.1365-2958.2003.03740.x. [DOI] [PubMed] [Google Scholar]
- 38.Mendivil-Perez M, Velez-Pardo C, Jimenez-Del-Rio M. 2012. TPEN induces apoptosis independently of zinc chelator activity in a model of acute lymphoblastic leukemia and ex vivo acute leukemia cells through oxidative stress and mitochondria caspase-3- and AIF-dependent pathways. Oxid Med Cell Longev 2012:313275. doi: 10.1155/2012/313275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fatfat M, Merhi RA, Rahal O, Stoyanovsky DA, Zaki A, Haidar H, Kagan VE, Gali-Muhtasib H, Machaca K. 2014. Copper chelation selectively kills colon cancer cells through redox cycling and generation of reactive oxygen species. BMC Cancer 14:527. doi: 10.1186/1471-2407-14-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adler M, Shafer H, Hamilton T, Petrali JP. 1999. Cytotoxic actions of the heavy metal chelator TPEN on NG108-15 neuroblastoma-glioma cells. Neurotoxicology 20:571–582. [PubMed] [Google Scholar]
- 41.Viboud GI, Bliska JB. 2005. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol 59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 42.Craik DJ, Fairlie DP, Liras S, Price D. 2013. The future of peptide-based drugs. Chem Biol Drug Des 81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 43.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. 1994. Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg Med Chem Lett 4:2657–2662. doi: 10.1016/S0960-894X(01)80691-0. [DOI] [Google Scholar]
- 44.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Spellmeyer DC, Tan RY, Frankel AD, Santi DV, Cohen FE, Bartlett PA. 1992. Peptoids—a modular approach to drug discovery. Proc Natl Acad Sci U S A 89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwochert J, Turner R, Thang M, Berkeley RF, Ponkey AR, Rodriguez KM, Leung SSF, Khunte B, Goetz G, Limberakis C, Kalgutkar AS, Eng H, Shapiro MJ, Mathiowetz AM, Price DA, Liras S, Jacobson MP, Lokey RS. 2015. Peptide to peptoid substitutions increase cell permeability in cyclic hexapeptides. Org Lett 17:2928–2931. doi: 10.1021/acs.orglett.5b01162. [DOI] [PubMed] [Google Scholar]
- 46.Wassing GM, Bergman P, Lindbom L, van der Does AM. 2015. Complexity of antimicrobial peptide regulation during pathogen-host interactions. Int J Antimicrob Agents 45:447–454. doi: 10.1016/j.ijantimicag.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Wang G. 2015. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol Biol 1268:43–66. doi: 10.1007/978-1-4939-2285-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park SC, Park Y, Hahm KS. 2011. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int J Mol Sci 12:5971–5992. doi: 10.3390/ijms12095971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jang A, Jo C, Kang KS, Lee M. 2008. Antimicrobial and human cancer cell cytotoxic effect of synthetic angiotensin-converting enzyme (ACE) inhibitory peptides. Food Chem 107:327–336. doi: 10.1016/j.foodchem.2007.08.036. [DOI] [Google Scholar]
- 50.Ebbensgaard A, Mordhorst H, Overgaard MT, Nielsen CG, Aarestrup FM, Hansen EB. 2015. Comparative evaluation of the antimicrobial activity of different antimicrobial peptides against a range of pathogenic bacteria. PLoS One 10:e0144611. doi: 10.1371/journal.pone.0144611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mader JS, Hoskin DW. 2006. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin Invest Drugs 15:933–946. doi: 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- 52.Straus SK, Hancock REW. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta 1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 53.Horn JN, Romo TD, Pitman MC, Grossfield A. 2010. Binding of antimicrobial lipopeptides to lipid bilayers characterized by microsecond molecular dynamics simulations. Biophys J 98:81a. [Google Scholar]
- 54.Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, Konig C, Ammerman NC, Barrio MB, Borchers K, Bordon-Pallier F, Bronstrup M, Courtemanche G, Gerlitz M, Geslin M, Hammann P, Heinz DW, Hoffmann H, Klieber S, Kohlmann M, Kurz M, Lair C, Matter H, Nuermberger E, Tyagi S, Fraisse L, Grosset JH, Lagrange S, Muller R. 2015. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 348:1106–1112. doi: 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- 55.Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, Blackwell HE. 2013. Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J Am Chem Soc 135:7869–7882. doi: 10.1021/ja3112115. [DOI] [PubMed] [Google Scholar]
- 56.Tal-Gan Y, Ivancic M, Cornilescu G, Yang T, Blackwell HE. 2016. Highly stable, amide-bridged autoinducing peptide analogues that strongly inhibit the AgrC quorum sensing receptor in Staphylococcus aureus. Angew Chem Int Ed Engl 55:8913–8917. doi: 10.1002/anie.201602974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim OK, Garrity-Ryan LK, Bartlett VJ, Grier MC, Verma AK, Medjanis G, Donatelli JE, Macone AB, Tanaka SK, Levy SB, Alekshun MN. 2009. N-hydroxybenzimidazole inhibitors of the transcription factor LcrF in Yersinia: novel antivirulence agents. J Med Chem 52:5626–5634. doi: 10.1021/jm9006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swietnicki W, Carmany D, Retford M, Guelta M, Dorsey R, Bozue J, Lee MS, Olson MA. 2011. Identification of small-molecule inhibitors of Yersinia pestis type III secretion system YscN ATPase. PLoS One 6:e19716. doi: 10.1371/journal.pone.0019716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D, Zetterstrom CE, Gabrielsen M, Beckham KS, Tree JJ, Macdonald SE, Byron O, Mitchell TJ, Gally DL, Herzyk P, Mahajan A, Uvell H, Burchmore R, Smith BO, Elofsson M, Roe AJ. 2011. Identification of bacterial target proteins for the salicylidene acylhydrazide class of virulence-blocking compounds. J Biol Chem 286:29922–29931. doi: 10.1074/jbc.M111.233858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. 2002. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem 45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 61.Morgan JM, Duncan MC, Johnson KS, Diepold A, Lam H, Dupzyk AJ, Martin LR, Wong WR, Armitage JP, Linington RG, Auerbuch V. 2017. Piericidin A1 blocks Yersinia Ysc type III secretion system needle assembly. mSphere 2:e00030-17. doi: 10.1128/mSphere.00030-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cornelis GR. 2006. The type III secretion injectisome. Nat Rev Microbiol 4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 63.Zgurskaya HI, Lopez CA, Gnanakaran S. 2015. Permeability barrier of Gram-negative cell envelopes and approaches to bypass it. ACS Infect Dis 1:512–522. doi: 10.1021/acsinfecdis.5b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller HK, Kwuan L, Schwiesow L, Bernick DL, Mettert E, Ramirez HA, Ragle JM, Chan PP, Kiley PJ, Lowe TM, Auerbuch V. 2014. IscR is essential for Yersinia pseudotuberculosis type III secretion and virulence. PLoS Pathog 10:e1004194. doi: 10.1371/journal.ppat.1004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ludwig DL. May 2006. Mammalian expression cassette engineering for high-level protein production. BioProcess Int 2006(Suppl):14–23. [Google Scholar]
- 66.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. 1992. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J Am Chem Soc 114:10646–10647. doi: 10.1021/ja00052a076. [DOI] [Google Scholar]
- 67.Wang HW, Wang FQ, Tao XY, Cheng HR. 2012. Ammonia-containing dimethyl sulfoxide: an improved solvent for the dissolution of formazan crystals in the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Anal Biochem 421:324–326. doi: 10.1016/j.ab.2011.10.043. [DOI] [PubMed] [Google Scholar]
- 68.Bliska JB, Guan KL, Dixon JE, Falkow S. 1991. Tyrosine phosphate hydrolysis of host proteins by an essential Yersinia-virulence determinant. Proc Natl Acad Sci U S A 88:1187–1191. doi: 10.1073/pnas.88.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Balada-Llasat JM, Mecsas J. 2006. Yersinia has a tropism for B and T cell zones of lymph nodes that is independent of the type III secretion system. PLoS Pathog 2:816–828. doi: 10.1371/journal.ppat.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rabin SD, Hauser AR. 2005. Functional regions of the Pseudomonas aeruginosa cytotoxin ExoU. Infect Immun 73:573–582. doi: 10.1128/IAI.73.1.573-582.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rangel SM, Diaz MH, Knoten CA, Zhang A, Hauser AR. 2015. The role of ExoS in dissemination of Pseudomonas aeruginosa during pneumonia. PLoS Pathog 11:e1004945. doi: 10.1371/journal.ppat.1004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GKS, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 73.Frye J, Karlinsey JE, Felise HR, Marzolf B, Dowidar N, McClelland M, Hughes KT. 2006. Identification of new flagellar genes of Salmonella enterica serovar Typhimurium. J Bacteriol 188:2233–2243. doi: 10.1128/JB.188.6.2233-2243.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.