

ABSTRACT
The in vitro effects of 18 dinuclear thiolato-bridged arene ruthenium complexes (1 monohiolato compound, 4 dithiolato compounds, and 13 trithiolato compounds), originally designed as anticancer agents, on the apicomplexan parasite Toxoplasma gondii grown in human foreskin fibroblast (HFF) host cells were studied. Some trithiolato compounds exhibited antiparasitic efficacy at concentrations of 250 nM and below. Among those, complex 1 and complex 2 inhibited T. gondii proliferation with 50% inhibitory concentrations (IC50s) of 34 and 62 nM, respectively, and they did not affect HFFs at dosages of 200 μM or above, resulting in selectivity indices of >23,000. The IC50s of complex 9 were 1.2 nM for T. gondii and above 5 μM for HFFs. Transmission electron microscopy detected ultrastructural alterations in the matrix of the parasite mitochondria at the early stages of treatment, followed by a more pronounced destruction of tachyzoites. However, none of the three compounds applied at 250 nM for 15 days was parasiticidal. By affinity chromatography using complex 9 coupled to epoxy-activated Sepharose followed by mass spectrometry, T. gondii translation elongation factor 1α and two ribosomal proteins, RPS18 and RPL27, were identified to be potential binding proteins. In conclusion, organometallic ruthenium complexes exhibit promising activities against Toxoplasma, and the potential mechanisms of action of these compounds as well as their prospective applications for the treatment of toxoplasmosis are discussed.
KEYWORDS: Toxoplasma gondii, affinity chromatography, electron microscopy, in vitro culture, mitochondrion, ruthenium complex, toxoplasmosis, translation elongation factor 1α
INTRODUCTION
Organometallic compounds, especially platinum complexes, are widely applied as anticancer chemotherapeutics (1). However, due to their drawbacks (i.e., severe side effects, insurgence of tumor resistance, etc.), a variety of complexes of other transition metals, such as copper, gold, or ruthenium, have been investigated as potential alternative anticancer drug candidates (2–10). Among the different metal complexes studied, arene ruthenium complexes showed very promising anticancer properties with 50% inhibitory concentration (IC50) values in the low-micromolar range and certain selectivity for tumor cells over nontumorigenic cells (11–13). One such compound, namely, RAPTA-C (where RAPTA is ruthenium–arene complexes bearing the 1,3,5-triaza-7-phosphatricyclo-[3.3.1.1]decane ligand, and C is para-cymene), is currently in preclinical evaluation (14). Recently, some of us have shown that thiolato-bridged dinuclear arene ruthenium complexes, in particular, trithiolato dinuclear complexes of the type [(η6-p-Me-C6H4Pri)2Ru2(μ2-SR)3]+ (where Me is methyl) and [(η6-p-Me-C6H4Pri)2Ru2(μ2-SR1)(μ2-SR2)2]+ (where Pri is isopropyl and SR is a thiolato ligand), were among the most cytotoxic ruthenium complexes reported so far, with nanomolar IC50s against both A2780 human ovarian cancer cells and their cisplatin-resistant mutant variant, A2780cisR cells (15–21). Interestingly, treatment of mice in in vivo studies of one of these compounds, namely, [(η6-p-Me-C6H4Pri)2Ru2(μ2-SC6H4-p-But)3]+ (where But is tert-butyl) (diruthenium-1), demonstrated a significant increase in the survival of treated mice (22).
Arene ruthenium complexes were also shown to be effective against bacteria (23); against protozoan parasites, including the two closely related apicomplexans Neospora caninum and Toxoplasma gondii (24); and against helminths, such as Schistosoma mansoni (25, 26) and Echinococcus multilocularis (27). Interestingly, some ruthenium-clotrimazole (Ctz) complexes displayed high levels of in vitro activity against Leishmania major and Trypanosoma cruzi and low levels of toxicity when their activities were assessed in normal mammalian cells (28). In addition to ruthenium, other organometallic complexes have also been reported to display interesting antiparasitic and/or anti-infective activities (29–42). For instance, one manganese(I) tricarbonyl complex, [Mn(CO)3(bpyR,R)(Ctz)]PF6 (where bpyR,R is 2,2′-bipyridine), showed submicromolar activity against Staphylococcus aureus and Staphylococcus epidermidis with MICs of 0.625 μM. Moreover, the related complex [Mn(CO)3(bpyR,R)(Ktz)]PF6 (where Ktz is ketoconazole) was active against Trypanosoma brucei with an IC50 of 0.7 μM, while the IC50 for mammalian cells was more than 10 times higher (43).
Among the different above-mentioned pathogens, T. gondii is the most widespread parasite worldwide and infects approximately one-third of the human population (44). In general, T. gondii infestation remains without clinical symptoms in immunocompetent individuals, and no treatment is required. However, Toxoplasma infection has been linked to neuropsychiatric disease. Importantly, upon immunosuppression or primary infection during pregnancy, T. gondii can cause toxoplasmosis, a life-threatening disease affecting both humans and food and farm animals, which can lead to severe pathology, including fetal malformation and abortion. Current treatment options for toxoplasmosis include macrolide antibiotics and sulfonamides (45), which inhibit protein biosynthesis and intermediary metabolism in the apicoplast, a prokaryote-like organelle that is unique to apicomplexans (46). However, these treatments are often characterized by adverse side effects and do not eliminate the parasite; thus, these compounds do not act in a parasiticidal manner. It is therefore of high interest to investigate whether dinuclear thiolato-bridged arene ruthenium complexes exhibit selective toxicity and parasiticidal activity against T. gondii. Moreover, compounds with good efficacy against T. gondii have good chances of being active against related apicomplexan parasites of high medical and veterinary medical interest, such as the coccidians Cryptosporidium and Eimeria and the closely related Neospora caninum.
RESULTS
In vitro efficacy of Ru(II) complexes.
Trithiolato complexes 1 to 5 and the mixed complex 9 inhibited the proliferation of T. gondii with IC50s of approximately 500 nM or less (Fig. 1 and Table 1). Trithiolato complex 7 and mixed complexes 8 and 10 to 13 had no measurable antiparasitic activity or were already toxic for host cells at concentrations of 250 nM or 2,500 nM. The same was true for dithiolato complexes 14 to 17 and monothiolato complex 18. The activity of the complexes against T. gondii parallels to a certain extent the results previously found against several cancer cell lines: the IC50s of complex 7 were 2 orders of magnitude larger than those of the other complexes (20), and the mono- and dithiolato complexes were found to be only moderately cytotoxic in vitro against cancer cell lines (IC50s, between 0.2 and 2.5 μM) (47, 48).
FIG 1.

Structures of complexes 1 to 18 used in this study. Note that compounds 1, 2, and 9 were further characterized.
TABLE 1.
Efficacies of dinuclear thiolate-bridged arene ruthenium complexes against T. gondii beta-galactosidase-expressing tachyzoites, host cell (HFF) cytotoxicity, and physicochemical dataa
| Complex | IC50 (nM) for T. gondii beta-gal | IC50 (μM) for HFFs | LogP for RSH group |
|---|---|---|---|
| 1 | 34 ± 4 | 800 | 2.98 ± 0.28 |
| 2 | 62 ± 10 | >1,000 | 4.21 ± 0.29 |
| 3 | 540 ± 60 | ND | 2.38 ± 0.32 |
| 4 | 130 ± 20 | ND | 2.83 ± 0.42 |
| 5 | 120 ± 20 | ND | 1.68 ± 0.29 |
| 9 | 1.2 ± 0.5 | 5,129 | ND |
Chloride salts of the corresponding thiols of complexes 1 to 5 and 9 were used for all experiments. For the determination of efficacies, confluent HFF monolayers grown in a 96-well plate were treated with the complexes at various concentrations and were infected with T. gondii beta-gal tachyzoites (103 per well). After 3 days, beta-galactosidase activity or host cell viability was determined, and IC50s were calculated as described in the text. The logP values correspond to the values that were calculated for the thiol RSH groups (17). ND, not done.
Complexes 1, 2, and 9 appeared to be the most active, with IC50s of 34, 62, and 1.2 nM, respectively (Table 1). Accordingly, host cell toxicity was investigated for these three complexes. In the presence of complex 1, the vitality of human foreskin fibroblasts (HFFs) was decreased to 63% of the control value at a concentration of 250 μM, which was the highest concentration used in these assays. Thus, an extrapolated but purely theoretical IC50 of 800 μM was calculated for complex 1, since the solubility limit in water-based solutions was about 500 μM. Complex 2 did not affect the vitality of HFFs up to a concentration of 250 μM. Complex 9, exhibiting by far the lowest IC50s, had an IC50 for HFFs of approximately 5 μM. Thus, all three complexes affected T. gondii tachyzoites at low-nanomolar concentrations, and these effects were parasite specific with high selective toxicity indices: >23,000 for complex 1, >16,000 for complex 2, and >5,000 for complex 9. Interestingly, long-term treatment with complex 9 at 250 nM over a period of up to 15 days did not eliminate all parasites, since the regrowth of tachyzoites was observed 5 to 10 days after the release of drug pressure for compound 9. This indicates that these compounds acted in a parasitostatic rather than parasiticidal manner.
Ultrastructural changes induced by Ru(II) complexes show that one of the primary target organelles in T. gondii tachyzoites is the mitochondrion.
To obtain more detailed information on the subcellular effects of these 3 thiolato-bridged dinuclear arene ruthenium complexes, transmission electron microscopy (TEM) was performed on drug-treated HFFs infected with T. gondii (Fig. 2 and 3). Nontreated parasites, exemplified in Fig. 2, were located intracellularly and were undergoing proliferation by endodyogeny within a parasitophorous vacuole (PV) surrounded by a distinct PV membrane. These parasites exhibited the typical apicomplexan structural features, including rhoptries, dense granules, micronemes, and a conoid at the anterior part. The parasite mitochondrion, filled with a structured electron-dense matrix, could be readily identified in these nontreated parasites (Fig. 2C). In cultures exposed to complex 1, alterations within the mitochondria of T. gondii were already evident after 6 h of treatment, showing a progressive degeneration of the electron-dense intramitochondrial matrix (Fig. 3B and C). The interior ultrastructural organization of these mitochondria was largely distorted, and only membranous residues were present in some cases. However, the outer membrane of the mitochondria was still intact, and parasites maintained their overall shape. After 48 h of treatment with complex 1, T. gondii tachyzoites had lost their characteristic shape, and the parasites displayed a largely distorted morphology, no internal organelles were recognizable anymore, and the PV and its membrane were essentially lost. However, host cell mitochondria exhibited a normal morphology with clearly discernible cristae (Fig. 3D). Similar results were obtained in T. gondii-infected cultures treated with complex 2 (data not shown). For treatments with complex 9, mitochondrial changes were not noted in T. gondii tachyzoites after 6 h of treatment (data not shown), but alterations similar to those observed during treatments with complex 1 became evident after 24 to 48 h of exposure to complex 9 (Fig. 3E and F). However, intact parasites could also be observed in cultures treated with all three complexes. Overall, this suggested that these three ruthenium complexes induced largely similar ultrastructural changes by inducing distinct alterations in the mitochondria and could thus act with a similar or identical mechanism(s) of action.
FIG 2.

Ultrastructure of T. gondii tachyzoites grown in HFFs. (A) Low-magnification view of infected HFFs. The boxed area is shown at a higher magnification in panel B. Tachyzoites proliferate within a parasitophorous vacuole surrounded by a parasitophorous vacuole membrane. nuc, nucleus; dg, dense granules; mic, micronemes; rop, rhoptries; mito, mitochondrion; con, conoid. The boxed area in panel B shows the mitochondrial matrix and is enlarged in panel C. Bars = 1.8 μm (A), 0.3 μm (B), and 0.1 μm (C).
FIG 3.

Ultrastructure of T. gondii tachyzoites grown in HFFs and treated with ruthenium complexes 1 and 9. Treatments were carried out using 200 nM complex 1 (A to D) or complex 9 (E, F). (A) Low-magnification view of parasites treated with complex 1 for 6 h. The boxed areas are enlarged in panels B and C. (D) Parasites exposed to complex 1 for 48 h. (E, F) Parasites exposed to complex 9 for 24 h. Note the distinct alterations in the mitochondria (mito) in panels B, C, and E and the still intact host cell mitochondria (h-mito) in panel D. The boxed area in panel F is enlarged in panel E. Bars = 1 μm (A, F), 0.4 μm (B, C, E), and 0.8 μm (D).
Complex 9 affects extracellular parasites and interferes in adhesion, invasion, or intracellular establishment but does not act efficiently against T. gondii proliferation once parasites reside inside the host cell.
Since long-term treatment studies as well as TEM suggested that these ruthenium complexes did not act in a parasiticidal manner, we wanted to determine whether these compounds affected host cell invasion, intracellular proliferation, or both. For this, HFF monolayers were infected with T. gondii tachyzoites, and complex 9 (100 μM) was added either at the time of infection or at 1 h, 5 h, or 24 h postinfection (Fig. 4). Complex 9 efficiently inhibited tachyzoite proliferation when it was added at the time of infection and also when it was applied at 5 h postinfection but only partially when it was added at 24 h postinfection. Thus, complex 9 acted mainly during the first steps of the infection process (e.g., host cell invasion and intracellular establishment) and only with limited efficacy once parasites resided inside the host cell.
FIG 4.
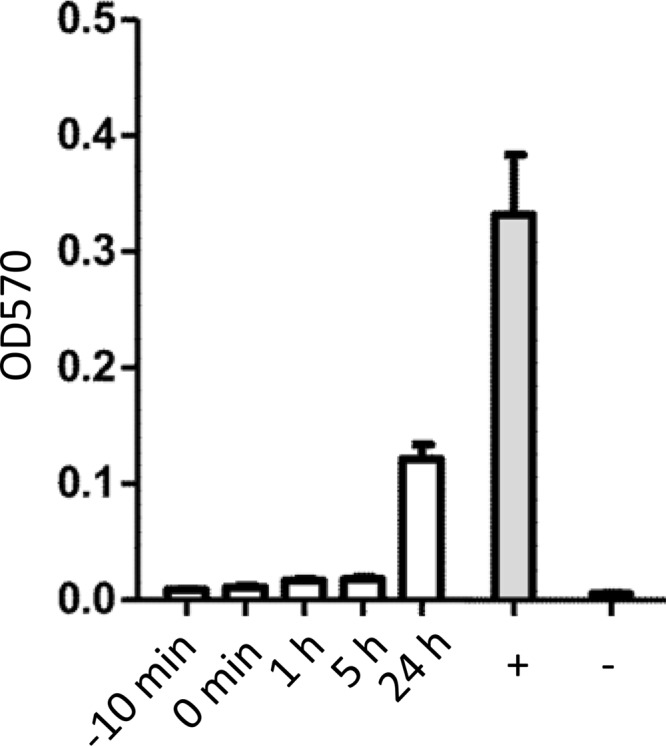
Compound 9 inhibits T. gondii tachyzoite proliferation only when it is applied early during infection. HFF monolayers grown in 96-well plates were treated with complex 9 (100 nM) either 10 min prior to infection or 1 h, 5 h, or 24 h after infection with T. gondii tachyzoites. The proliferation of tachyzoites was measured after 2 days of culture by beta-galactosidase assay, as described in Materials and Methods. OD570, optical density at 570 nm.
Complex 9 interacts with ribosomal proteins from T. gondii and from the host cell.
By affinity chromatography on complex 9–epoxy-Sepharose, two major bands of approximately 50 kDa and 20 kDa that were not present in the eluate of the mock epoxy-Sepharose column were identified (Fig. 5A). Mass spectrometry (MS) analysis identified ribosomal proteins of host and parasite origin as major components of the 20-kDa band (Table 2). The composition of the 50-kDa band was more heterogeneous. As quantified both via protein match score summation and via protein score, the major component of the 50-kDa band was T. gondii elongation factor 1α (TgEF1α; Table 2) with a unique peptide coverage of nearly 50% of the sequence (Fig. 5B). The second most abundant protein was its human homologue. Moreover, other proteins of human origin were identified in this fraction (Table 2).
FIG 5.

Identification of complex 9-binding proteins. (A) SDS-PAGE and silver staining of tandem (the column with mock epoxy-Sepharose and the column with compound 9-Sepharose) affinity chromatography of a protein extract prepared from T. gondii-infected HFFs. The soluble extract and the nonbinding fraction (flowthrough) are shown on the left, followed by wash and eluate fractions of the column with mock epoxy-Sepharose and the column with complex 9-Sepharose. The two arrows point to the two bands of 50 kDa and 20 kDa, which were cut out and analyzed by LC-MS/MS. (B) Amino acid sequence of the 50-kDa band identified as TgEF1α. The peptide sequences identified by LC-MS/MS are underlined.
TABLE 2.
| Band | UniProt accession no. | ID | PMSS | Protein score | No. of unique peptides | Coverage (%) | Protein mass (Da) | Description |
|---|---|---|---|---|---|---|---|---|
| 20 kDa | P46783 | RS10_HUMAN | 79 | 147 | 5 | 32.7 | 18,898 | 40S ribosomal protein S10 |
| P30050 | RL12_HUMAN | 72 | 161 | 5 | 43.0 | 17,819 | 60S ribosomal protein L12 | |
| S8FA78 | S8FA78_TOXGO | 46 | 86 | 3 | 25.5 | 17,821 | Ribosomal protein RPL12 | |
| V4YUP9 | V4YUP9_TOXGO | 30 | 69 | 3 | 21.8 | 16,331 | Ribosomal protein RPL27 | |
| P62269 | RS18_HUMAN | 29 | 67 | 3 | 19.1 | 17,719 | 40S ribosomal protein S18 | |
| S8EUB1 | S8EUB1_TOXGO | 26 | 61 | 3 | 21.8 | 17,723 | Ribosomal protein RPS18 | |
| P61254 | RL26_HUMAN | 25 | 55 | 3 | 16.6 | 17,258 | 60S ribosomal protein L26 | |
| P62851 | RS25_HUMAN | 24 | 35 | 2 | 13.6 | 13,742 | 40S ribosomal protein S25 | |
| Q5SGD8 | Q5SGD8_TOXGO | 18 | 37 | 2 | 11.1 | 19,983 | Tgd057 | |
| 50 kDa | S8GV85 | S8GV85_TOXGO | 323 | 518 | 17 | 47.5 | 49,006 | Elongation factor 1α |
| P68104 | EF1A1_HUMAN | 143 | 196 | 8 | 22.9 | 50,141 | Elongation factor 1α 1 | |
| O14773-2 | TPP1_HUMAN | 71 | 120 | 5 | 27.2 | 34,464 | Isoform 2 of tripeptidyl-peptidase 1 | |
| P63261 | ACTG_HUMAN | 36 | 77 | 4 | 14.7 | 41,793 | Actin, cytoplasmic 2 | |
| P06576 | ATPB_HUMAN | 32 | 61 | 3 | 8.3 | 56,560 | ATP synthase subunit beta, mitochondrial | |
| P22234 | PUR6_HUMAN | 26 | 47 | 2 | 6.1 | 47,079 | Multifunctional protein ADE2 | |
| P16989-2 | YBOX3_HUMAN | 22 | 42 | 2 | 8.3 | 31,947 | Isoform 2 of Y-box-binding protein 3 | |
| O75821 | EIF3G_HUMAN | 18 | 32 | 2 | 5.0 | 35,611 | Eukaryotic translation initiation factor 3 subunit G | |
| Q9Y6N5 | SQRD_HUMAN | 17 | 31 | 2 | 6.2 | 49,961 | Sulfide:quinone oxidoreductase, mitochondrial |
ID, UniProt identifier; PMSS, protein match score summation.
DISCUSSION
We report here on a series of 18 dinuclear thiophenolato-bridged arene ruthenium complexes, which exhibited highly promising in vitro activities against T. gondii tachyzoites. The organometallic complexes studied in this work have been previously described (15, 17, 47, 49). Very importantly, recent studies by some of us have shown that these dinuclear arene ruthenium complexes are inert to ligand substitutions and remain stable for long periods in water solutions or in organic solvents, like dimethyl sulfoxide (16, 21). These ruthenium complexes were originally generated for the treatment of cancer cells. Cancer cells and protozoan parasites, including Toxoplasma, share several features: they both live and multiply in a host organism and do not immediately kill their hosts, they have a potentially infinite proliferative capacity, and they escape in immunocompromised tissues. Cancer cells are largely resistant to apoptosis, while Toxoplasma and Neospora are known to interfere with the programmed cell death machinery of their host cell (50). Thus, we hypothesize that a potentially lucrative starting point for the discovery of novel drug candidates with activity against T. gondii and other protozoans is to examine compounds that are being developed against cancer.
Among the 18 compounds studied, the trithiolato complexes 1, 2, and 9 were highly efficacious against T. gondii, with IC50s ranging from 1.2 to 62 nM. In addition, these compounds exhibited a highly favorable selective toxicity index of up to 23,000. TEM demonstrated that one of the first organelles that exhibited ultrastructural alterations upon treatment with these compounds was the tachyzoite mitochondrion, which had already lost its interior membranous matrix and cristae after 6 to 24 h. More severe distortions, including a complete breakdown of other organelles within the parasite cytoplasm and a general disintegration of the tachzoites and the parasitophorous vacuole and its membrane, were observed after 48 h.
In comparison to the results obtained with other drugs, the in vitro results obtained with complexes 1, 2, and 9 are encouraging. Pyrimethamine, sulfadiazine, and atovaquone, compounds currently clinically used against toxoplasmosis, inhibited T. gondii beta-gal (transgenic T. gondii RH expressing the beta-galactosidase gene from Escherichia coli [51]) with IC50s of 1 mM, 80 μM, and 19 to 50 nM, respectively (51). The calcium-dependent protein kinase inhibitor BKI-1294, highly active against T. gondii and N. caninum infections in mice, inhibited T. gondii and N. caninum beta-galactosidase proliferation under identical conditions with IC50s of 137 and 40 nM, respectively (52). Two previously identified organometallic ruthenium complexes exhibited IC50s of 18 and 41 nM (24), however, with selective toxicity indices being below 100. As can be noticed from the calculated solubility (logP) values (Table 1) and as previously observed against cancer cells (17), the efficacy of inhibition is, to some extent, correlated to the lipophilicity of the complexes. Unlike the findings obtained with A2780 and A2780cisR cancer cells, the most lipophilic complex, complex 2, was not the complex that was the most potent against T. gondii, possibly suggesting that the different chemical natures of the cell and T. gondii outer membranes could influence the uptake of dinuclear thiolato-bridged arene ruthenium complexes.
While complexes 1, 2, and 9 were highly efficacious against T. gondii and exhibited excellent selective toxicity, we obtained evidence that these compounds did not act in a parasiticidal manner. Removal of the drugs after continuous treatment at 250 nM for up to 15 days did not result in the complete elimination of viable tachyzoites, and regrowth of the parasites was observed within 5 to 10 days after the release of the drug pressure. This was confirmed by TEM, where a small number of largely intact tachyzoites were still found after 48 h of continuous in vitro treatment. Similar results were previously reported for dicationic arylimidamides (53) and ruthenium phosphite complexes in T. gondii (24) and for buparvaquone, BKI-1294, as well as artemisinin derivatives in the closely related organism N. caninum (52, 54, 55). In some of these reports, rapid adaptation of T. gondii and N. caninum tachyzoites to the increased concentrations of drugs within a few days was documented (53, 54). This outstanding adaptive ability represents a major obstacle for the development of drugs efficacious against these parasites. Nevertheless, the lack of parasiticidal activity in vitro still allows excellent in vivo efficacy, as documented for BKI-1294 in models of N. caninum infection in pregnant mice (52, 55).
All three compounds had a profound impact on the ultrastructure of the parasite mitochondria, which lost their characteristic electron-dense matrix and cristae within 6 to 24 h after the initiation of drug treatments. After 48 h, this impacted the entire tachyzoites, leading, in most cases, to severe alterations and death. Of note, mitochondria are also targeted by other drugs currently used against apicomplexans, such as atovaquone, buparvaquone, and decoquinate, which have been shown to impair the cytochrome b/c1 complex in Toxoplasma, Plasmodium, and Theileria parasites (56–59).
The mitochondrion represents an attractive drug target. The disruption of mitochondria has recently been investigated as a potential novel chemotherapeutic mechanism for cancer treatment, because it circumvents upstream apoptotic pathways that may be mutated or lacking in cancer cells (60). Moreover, cancer cells have higher mitochondrial membrane potentials, rendering them more susceptible to mitochondrial perturbations than nonimmortalized cells (61). On the basis of these factors, numerous mitochondrion-targeting agents have been developed in order to disrupt the mitochondrial membrane potential and to further permeabilize the mitochondrial outer membrane. Some ruthenium(II) complexes can induce mitochondrion-mediated apoptosis in cancer cells (62–65). However, while in mammalian cells the mitochondrion represents the main ATP-generating organelle that allows complete oxidation of carbohydrates, lipids, and amino acids via the tricarboxylic acid (TCA) cycle and the electron transport chain, the situation in apicomplexans appears to be slightly different. Apicomplexans have a single tubular mitochondrial network that also hosts part of heme biosynthesis, iron-sulfur cluster assembly, and lipoic acid salvage, and the mitochondrion participates in the synthesis of many metabolic intermediates, including pyrimidines (66).
How exactly the mitochondrion is targeted by our ruthenium complexes is not known. Affinity chromatography using extracts from T. gondii-infected HFFs led to the identification of TgEF1α as well as its human homologue as major complex 9-binding partners. This is not surprising, since EF1α is expressed in all eukaryotic cells and is highly conserved (67). In eukaryotic cells, EF1α promotes the GTP-dependent transfer of aminoacylated tRNA to the ribosome A site and, hence, represents an essential component of protein synthesis. In addition, other activities have been attributed to EF1α in different eukaryotes, and these activities are associated with vital cellular functions, such as cell growth, motility, protein metabolism, signal transduction, DNA replication/repair protein networks, and apoptosis (68–70). In Trypanosoma brucei and T. gondii, EF1α mediates the specificity of mitochondrial tRNA import (71, 72), and disruption of this process could lead to the observed mitochondrial alterations.
In another apicomplexan parasite, Cryptosporidium parvum, C. parvum EF1α (CpEF1α) was shown to localize to the apical region of C. parvum sporozoites, and antibodies directed against CpEF1α inhibited host cell invasion (73). The same was shown for T. gondii (74). Our study also showed that complex 9 had profound efficacy when it was applied at the early stages of host cell infection, namely, either during or 1 to 5 h after exposure of T. gondii tachyzoites to host cells, but more limited efficacy was noted when it was added 24 h after infection. This would be consistent with a mode of action that is relevant for invasion or early host cell establishment. In addition, vaccination of mice with recombinant TgEF1α and a DNA vaccine coding for TgEF1α led to significantly prolonged survival times in T. gondii-infected mice (74, 75), underlining the importance of TgEF1α for the infection process.
As outlined in Table 1, two other ribosomal proteins of both host and parasite origin and various other host proteins were found to bind to complex 9 as well. This may explain the low, but still detectable, host cell toxicity of complex 9.
In conclusion, we have identified three promising dinuclear thiolato-bridged arene ruthenium complexes with promising and highly specific antiparasitic activity, as assessed against T. gondii. These complexes induce severe mitochondrial alterations within 6 to 24 h of drug treatment and efficiently inhibit proliferation but do not act in a parasiticidal manner. One of these complexes, complex 9, interacts with TgTEF1α and other parasite and host ribosomal proteins. Further studies will focus on the interactions of complex 9 and other promising ruthenium complexes with putative apicomplexan drug targets and on the use of these drugs in vivo.
MATERIALS AND METHODS
Chemicals and synthesis of ruthenium complexes.
All reagents were commercially available and were used as received. The complexes assessed in this study are shown in Fig. 1. The symmetrical trithiolato complexes 1 to 7 were synthesized following a protocol slightly modified from that published previously (17). The dinuclear complex [(η6-p-Me-C6H4Pri)Ru2(μ-Cl)Cl2] was first dissolved and heated in refluxing technical-grade ethanol, and a solution of 6 equivalents of the corresponding thiol SR in 5 ml technical-grade ethanol (EtOH) was added dropwise [R = 4-C6H4CH3 (complex 1), 4-C6H4But (complex 2), 4-C6H4OH (complex 3), 3,4-C6H3(OMe)2 (where OMe is a methoxy group; complex 4) 4-methylcoumarinyl (complex 5), 3-C6H4Cl (complex 6), and 3-C6H4NH2 (complex 7)]. The resulting mixture was refluxed for 18 h. After cooling to room temperature, the solvent was removed under reduced pressure. The oil obtained was purified by column chromatography on silica gel using a mixture of dichloromethane and ethanol (5:1) as the eluent. Mixed trithiolato complexes 8 to 13 were synthesized in two steps, as previously described (19, 49). First, the neutral dichlorido dithiolato intermediates [(η6-p-Me-C6H4Pri)2Ru2(μ2-SCH2-C6H4-R)2Cl2] were obtained from the reaction of the p-cymene ruthenium dichloride dimer [(η6-p-Me-C6H4Pri)Ru2(μ-Cl)Cl2] with 2 equivalents of the respective thiol SCH2R (R = C6H5 [complex 8], 4-C6H4CH3 [complex 9], 4-C6H4OMe [complex 10], 4-C6H4F [complex 11], 4-C6H4Cl [complex 12], and 4-C6H4Br [complex 13]) in ethanol at 0°C, according to the published method (48). These intermediates reacted in refluxing ethanol for 15 h with 6 equivalents of 4-mercaptophenol (4-HS-C6H4-OH) to give the corresponding mixed trithiolato complexes [(η6-p-Me-C6H4Pri)2Ru2(μ2-S-C6H4-OH)(μ2-SR)2]+ 8 to 13. Dithiolato complexes 14 to 17 and monothiolato complex 18 were synthesized according to published methods (47, 48). The resulting complexes 1 to 18 (Fig. 1), which were isolated as chloride or tetrafluoroborate salts, were air-stable, orange to red solids and were dried under a vacuum. The analytical data matched those previously reported in the literature (15, 17, 47, 49).
Host cell cultivation and parasite cultures.
If not stated otherwise, all tissue culture media were purchased from Gibco-BRL (Zurich, Switzerland) and biochemical reagents were from Sigma (St. Louis, MO). Human foreskin fibroblasts (HFFs) and Vero cells (green monkey kidney epithelial cells) were maintained in RPMI medium containing 10% fetal calf serum (FCS) (Gibco-BRL, Zurich, Switzerland) and antibiotics as described earlier (24). T. gondii beta-gal (transgenic T. gondii RH expressing the beta-galactosidase gene from E. coli [51]) was maintained in Vero cells and was isolated and separated from the host cells as described previously (24).
In vitro assessment of drug efficacy.
To study the effects of the complexes against T. gondii tachyzoites in vitro, 0.5 mM stock solutions of the complexes were prepared in water, sterile filtered, and stored at 4°C.
For assessment of drug efficacy against T. gondii tachyzoites, parasites were isolated (24) and assays were performed using HFFs as host cells (24). In short, 5 × 103 HFFs/well were grown to confluence in a 96-well plate in phenol-red free culture medium at 37°C with 5% CO2. Cultures were infected with freshly isolated T. gondii beta-gal tachyzoites (1 × 103/well), and drugs were added at the time of infection. Initial assessments of drug efficacy were done by exposing parasite cultures to 2,500 nM, 250 nM, 25 nM, or 2.5 nM each compound for a period of 3 days, or water was added as a control. For IC50 determinations, 6 selected complexes (complexes 1 to 5 and complex 9) were added at concentrations ranging from 0 to 2,000 nM. After 3 days at 37°C with 5% CO2, the plates were centrifuged at 500 × g, the medium was removed, and the cells in the cultures were lysed in phosphate-buffered saline (PBS) containing 0.05% Triton X-100. After addition of 10 μl of 5 mM chlorophenol red–β-d-galactopyranoside (CPRG; Roche Diagnostics, Rotkreuz, Switzerland) dissolved in PBS, the absorption shift was measured at a 570-nm wavelength at various time points on a VersaMax multiplate reader (Bucher Biotec, Basel, Switzerland). The activity, measured as the amount of chlorophenol red released over time, was proportional to the number of live parasites down to 50 per well, as determined in pilot assays. IC50s were calculated after the logit log transformation of relative growth and subsequent regression analysis by use of the corresponding software tool contained in the Excel software package (Microsoft, Seattle, WA).
In one time course experiment, complex 9 (100 nM) was added to HFF monolayers either 10 min prior to infection or 1 h, 5 h, or 24 h after infection with T. gondii tachyzoites. The proliferation of tachyzoites was measured after 2 days of culture as described above.
For long-term treatment assays, T. gondii-infected HFFs grown in T25 culture flasks were exposed to 250 nM complex 1, 2, or 9 for a period of 15 days, after which the cultures were washed with medium and were further maintained in medium devoid of drugs. Regrowth of the parasites was monitored on a daily basis by light microscopy.
Assays for cytotoxicity for noninfected confluent HFFs were also performed in 96-well plates by exposing HFFs to concentrations of 2.5 nM, 25 nM, 250 nM, and 2.5 μM each complex and assessment of the viability by an alamarBlue assay, as described previously (76).
TEM.
HFFs (5 × 104 per inoculum) cultured in T25 tissue culture flasks for 24 h were infected with 105 T. gondii beta-gal tachyzoites, and 200 nM complex 1, 2, or 9 was added at 24 h postinfection. After 6, 24, or 48 h, cells were harvested using a cell scraper, and they were placed into the primary fixation solution (2.5% glutaraldehyde in 100 mM sodium cacodylate buffer, pH 7.3) for 2 h. Specimens were then washed 2 times in cacodylate buffer and were postfixed in 2% OsO4 in cacodylate buffer for 2 h, followed by washing in water, prestaining in saturated uranyl acetate solution, and stepwise dehydration in ethanol. They were then embedded in Epon 812 resin and processed for transmission electron microscopy (TEM) as described previously (24). Specimens were viewed on a Phillips 400 transmission electron microscope operating at 80 kV.
Coupling of complex 9 to epoxy-activated Sepharose, affinity chromatography, and identification of a drug-binding protein by LC-MS/MS analysis.
To prepare a complex 9-Sepharose matrix, 20 mg of complex 9 was added to 0.5 mg of epoxy-Sepharose suspended in 2 ml of coupling buffer (0.1 M NaCO3, pH 9.5) followed by incubation for 2 days at 37°C on a shaker. Furthermore, a mock epoxy-Sepharose column was prepared by treatment with coupling buffer without complex 9 and blocking with ethanolamine. Prior to the runs, both columns were combined in tandem (the mock epoxy-Sepharose column first and then the column with complex 9) and washed with 25 ml of PBS equilibrated at 20°C.
To identify potential binding proteins both from T. gondii and from the host cell, three T75 flasks containing HFF monolayers were infected with 2 × 107 T. gondii tachyzoites and incubated for 3 to 4 days. Then, the cells were harvested by scraping and pelleted (1,000 × g, 10 min, 4°C). For protein extraction, frozen pellets were resuspended in 1 ml ice-cold PBS containing 1% Triton X-100 and 1 mM phenylmethylsulfonyl fluoride. The suspensions were thoroughly vortexed and centrifuged (15,200 × g, 10 min, 4°C). Extraction of pellets was repeated twice. The supernatants were combined (5 to 10 mg of total protein) and subjected to affinity chromatography by loading them onto the column tandem at a flow rate of 0.25 ml/min. The columns were washed with PBS until a flat baseline was detected (which was after washing with ca. 20 ml PBS). The columns were separated, and proteins binding to the columns were eluted with a pH shift (100 mM glycine Cl−, pH 2.9). Fractions (3 ml) were taken before, during, and after elution and precipitated overnight with 80% acetone at −20°C. The precipitates were solubilized in 30 μl of Laemmli buffer and were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using a Hoefer Minigel 250 apparatus (GE Healthcare, Little Chalfont, UK). Proteins were visualized by silver staining.
For mass spectrometry analysis, colloidal Coomassie staining was applied and selected protein bands were cut out with a clean scalpel, placed into Eppendorf tubes containing ethanol-distilled water (1:4), and stored at 4°C. In-gel digestion and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis were performed by the Mass Spectrometry and Proteomics Facility at the Department of Clinical Research of the University of Bern (Bern, Switzerland). The sequences obtained were compared with the sequences in the UniProt database (www.uniprot.org) by BLAST analysis.
ACKNOWLEDGMENTS
We acknowledge the financial support of the Swiss National Science Foundation (SNSF professorships PP00P2_133568 and PP00P2_157545 [to G.G.], SNSF grants 310030_165782 [to A.H.] and CRSII5_173718 [to J.F., A.H., and G.G.), the University of Bern (UniBe-ID; to J.F. and A.H.), the University of Zurich (to G.G.), the Stiftung für wissenschaftliche Forschung of the University of Zurich (to G.G.), the UBS Promedica Stiftung (to R.R. and G.G.), the Forschungskredit of the University of Zurich (to R.R.), and the Novartis Jubilee Foundation (to R.R. and G.G.). This work has received support under the program Investissements d'Avenir, launched by the French government and implemented by the ANR, with the reference ANR-10-IDEX-0001-02 PSL (to G.G.).
Many thanks are addressed to David Sibley (Washington University, St. Louis, MO, USA) for providing us with T. gondii beta-gal tachyzoites for screening purposes.
REFERENCES
- 1.Shaili E. 2014. Platinum anticancer drugs and photochemotherapeutic agents: recent advances and future developments. Sci Prog 97:20–40. doi: 10.3184/003685014X13904811808460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang CX, Lippard SJ. 2003. New metal complexes as potential therapeutics. Curr Opin Chem Biol 7:481–489. doi: 10.1016/S1367-5931(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 3.Ott I, Gust R. 2007. Preclinical and clinical studies on the use of platinum complexes for breast cancer treatment. Anti-Cancer Agents Med Chem 7:95–110. doi: 10.2174/187152007779314071. [DOI] [PubMed] [Google Scholar]
- 4.Ronconi L, Sadler PJ. 2007. Using coordination chemistry to design new medicines. Coord Chem Rev 251:1633–1648. doi: 10.1016/j.ccr.2006.11.017. [DOI] [Google Scholar]
- 5.Bruijnincx PCA, Sadler PJ. 2008. New trends for metal complexes with anticancer activity. Curr Opin Chem Biol 12:197–206. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meggers E. 2009. Targeting proteins with metal complexes. Chem Commun (Camb) 2009:1001–1010. [DOI] [PubMed] [Google Scholar]
- 7.Gasser G, Ott I, Metzler-Nolte N. 2011. Organometallic anticancer compounds. J Med Chem 54:3–25. doi: 10.1021/jm100020w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sava G, Bergamo A, Dyson PJ. 2011. Metal-based antitumour drugs in the post-genomic era: what comes next? Dalton Trans 40:9069–9075. doi: 10.1039/c1dt10522a. [DOI] [PubMed] [Google Scholar]
- 9.Hartinger CG, Metzler-Nolte N, Dyson PJ. 2012. Challenges and opportunities in the development of organometallic anticancer drugs. Organometallics 31:5677–5685. doi: 10.1021/om300373t. [DOI] [Google Scholar]
- 10.Komeda S, Casini A. 2012. Next-generation anticancer metallodrugs. Curr Top Med Chem 12:219–235. doi: 10.2174/156802612799078964. [DOI] [PubMed] [Google Scholar]
- 11.Casini A, Gabbiani C, Sorrentino F, Rigobello MP, Bindoli A, Geldbach TJ, Marrone A, Re N, Hartinger CG, Dyson PJ, Messori L. 2008. Emerging protein targets for anticancer metallodrugs: inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J Med Chem 51:6773–6781. doi: 10.1021/jm8006678. [DOI] [PubMed] [Google Scholar]
- 12.Oehninger L, Stefanopoulou M, Alborzinia H, Schur J, Ludewig S, Namikawa K, Munoz-Castro A, Koster RW, Baumann K, Wolfl S, Sheldrick WS, Ott I. 2013. Evaluation of arene ruthenium(II) N-heterocyclic carbene complexes as organometallics interacting with thiol and selenol containing biomolecules. Dalton Trans 42:1657–1666. doi: 10.1039/C2DT32319B. [DOI] [PubMed] [Google Scholar]
- 13.Clavel CM, Păunescu E, Nowak-Sliwinska P, Griffioen AW, Scopelliti R, Dyson PJ. 2014. Discovery of a highly tumor-selective organometallic ruthenium(II)-arene complex. J Med Chem 57:3546–3558. doi: 10.1021/jm5002748. [DOI] [PubMed] [Google Scholar]
- 14.Murray BS, Babak MV, Hartinger CG, Dyson PJ. 2016. The development of RAPTA compounds for the treatment of tumors. Coord Chem Rev 306(Pt 1):86–114. doi: 10.1016/j.ccr.2015.06.014. [DOI] [Google Scholar]
- 15.Gras M, Therrien B, Süss-Fink G, Zava O, Dyson PJ. 2010. Thiophenolato-bridged dinuclear arene ruthenium complexes: a new family of highly cytotoxic anticancer agents. Dalton Trans 39:10305–10313. doi: 10.1039/c0dt00887g. [DOI] [PubMed] [Google Scholar]
- 16.Giannini F, Süss-Fink G, Furrer J. 2011. Efficient oxidation of cysteine and glutathione catalyzed by a dinuclear areneruthenium trithiolato anticancer complex. Inorg Chem 50:10552–10554. doi: 10.1021/ic201941j. [DOI] [PubMed] [Google Scholar]
- 17.Giannini F, Furrer J, Ibao A-F, Süss-Fink G, Therrien B, Zava O, Baquie M, Dyson PJ, Stepnicka P. 2012. Highly cytotoxic trithiophenolatodiruthenium complexes of the type (eta(6)-p-MeC6H4Pr(i))(2)Ru-2(SC6H4-p-X)(3)(+): synthesis, molecular structure, electrochemistry, cytotoxicity, and glutathione oxidation potential. J Biol Inorg Chem 17:951–960. doi: 10.1007/s00775-012-0911-2. [DOI] [PubMed] [Google Scholar]
- 18.Giannini F, Paul LEH, Furrer J. 2012. Insights into the mechanism of action and cellular targets of ruthenium complexes from NMR spectroscopy. Chimia 66:775–780. doi: 10.2533/chimia.2012.775. [DOI] [PubMed] [Google Scholar]
- 19.Giannini F, Furrer J, Süss-Fink G, Clavel CM, Dyson PJ. 2013. Synthesis, characterization and in vitro anticancer activity of highly cytotoxic trithiolato diruthenium complexes of the type (eta(6)-p-(MeC6H4Pr)-Pr-i)(2)Ru-2(mu(2)-SR1)(2)(mu(2)-SR2)(+) containing different thiolato bridges. J Organomet Chem 744:41–48. doi: 10.1016/j.jorganchem.2013.04.049. [DOI] [Google Scholar]
- 20.Giannini F, Paul LEH, Furrer J, Therrien B, Süss-Fink G. 2013. Highly cytotoxic diruthenium trithiolato complexes of the type (eta(6)-p-MeC6H4Pri)(2)Ru-2(mu(2)-SR)(3)(+): synthesis, characterization, molecular structure and in vitro anticancer activity. New J Chem 37:3503–3511. doi: 10.1039/c3nj00476g. [DOI] [Google Scholar]
- 21.Furrer J, Süss-Fink G. 2016. Thiolato-bridged dinuclear arene ruthenium complexes and their potential as anticancer drugs. Coord Chem Rev 309:36–50. doi: 10.1016/j.ccr.2015.10.007. [DOI] [Google Scholar]
- 22.Tomsik P, Muthna D, Rezacova M, Micuda S, Cmielova J, Hroch M, Endlicher R, Cervinkova Z, Rudolf E, Hann S, Stibal D, Therrien B, Süss-Fink G. 2015. (p-MeC6H4Pri)(2)Ru-2(SC6H4-p-Bu-t)(3) Cl (diruthenium-1), a dinuclear arene ruthenium compound with very high anticancer activity: an in vitro and in vivo study. J Organomet Chem 782:42–51. doi: 10.1016/j.jorganchem.2014.10.050. [DOI] [Google Scholar]
- 23.Li F, Collins JG, Keene FR. 2015. Ruthenium complexes as antimicrobial agents. Chem Soc Rev 44:2529–2542. doi: 10.1039/C4CS00343H. [DOI] [PubMed] [Google Scholar]
- 24.Barna F, Debache K, Vock CA, Küster T, Hemphill A. 2013. In vitro effects of novel ruthenium complexes in Neospora caninum and Toxoplasma gondii tachyzoites. Antimicrob Agents Chemother 57:5747–5754. doi: 10.1128/AAC.02446-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess J, Keiser J, Gasser G. 2015. Toward organometallic antischistosomal drug candidates. Future Med Chem 7:821–830. doi: 10.4155/fmc.15.22. [DOI] [PubMed] [Google Scholar]
- 26.Kljun J, Scott AJ, Lanišnik Rižner T, Keiser J, Turel I. 2014. Synthesis and biological evaluation of organoruthenium complexes with azole antifungal agents. First crystal structure of a tioconazole metal complex. Organometallics 33:1594–1601. [Google Scholar]
- 27.Küster T, Lense N, Barna F, Hemphill A, Kindermann MK, Heinicke JW, Vock CA. 2012. A new promising application for highly cytotoxic metal compounds: η6-areneruthenium(II) phosphite complexes for the treatment of alveolar echinococcosis. J Med Chem 55:4178–4188. doi: 10.1021/jm300291a. [DOI] [PubMed] [Google Scholar]
- 28.Martínez A, Carreon T, Iniguez E, Anzellotti A, Sánchez A, Tyan M, Sattler A, Herrera L, Maldonado RA, Sánchez-Delgado RA. 2012. Searching for new chemotherapies for tropical diseases: ruthenium-clotrimazole complexes display high in vitro activity against Leishmania major and Trypanosoma cruzi and low toxicity toward normal mammalian cells. J Med Chem 55:3867–3877. doi: 10.1021/jm300070h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keiser J, Vargas M, Rubbiani R, Gasser G, Biot C. 2014. In vitro and in vivo antischistosomal activity of ferroquine derivatives. Parasit Vectors 7:424. doi: 10.1186/1756-3305-7-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biot C, Dive D. 2010. Bioorganometallic chemistry and malaria, p 155–193. In Jaouen G, Metzler-Nolte N (ed), Medicinal organometallic chemistry. Springer, Berlin, Germany. [Google Scholar]
- 31.Beagley P, Blackie MAL, Chibale K, Clarkson C, Moss JR, Smith PJ. 2002. Synthesis and antimalarial activity in vitro of new ruthenocene-chloroquine analogues. Dalton Trans 2002:4426–4433. [Google Scholar]
- 32.Smith GS, Therrien B. 2011. Targeted and multifunctional arene ruthenium chemotherapeutics. Dalton Trans 40:10793–10800. doi: 10.1039/c1dt11007a. [DOI] [PubMed] [Google Scholar]
- 33.Ali MI, Rauf MK, Badshah A, Kumar I, Forsyth CM, Junk PC, Kedzierski L, Andrews PC. 2013. Anti-leishmanial activity of heteroleptic organometallic Sb(V) compounds. Dalton Trans 42:16733–16741. doi: 10.1039/c3dt51382c. [DOI] [PubMed] [Google Scholar]
- 34.Simpson PV, Schmidt C, Ott I, Bruhn H, Schatzschneider U. 2013. Synthesis, cellular uptake and biological activity against pathogenic microorganisms and cancer cells of rhodium and iridium N-heterocyclic carbene complexes bearing charged substituents. Eur J Inorg Chem 2013:5547–5554. doi: 10.1002/ejic.201300820. [DOI] [Google Scholar]
- 35.Maia PIDS, Carneiro ZR, Lopes CD, Oliveira CG, Silva JS, Albuquerque S, Hagenbach A, Gust R, Deflon V, Abram U. 2017. Organometallic gold(III) complexes with hybrid SNS-donating thiosemicarbazone ligands: cytotoxicity and anti-Trypanosoma cruzi activity. Dalton Trans 46:2559–2571. doi: 10.1039/C6DT04307K. [DOI] [PubMed] [Google Scholar]
- 36.Clède S, Cowan N, Lambert F, Bertrand HC, Rubbiani R, Patra M, Hess J, Sandt C, Trcera N, Gasser G, Keiser J, Policar C. 2016. Bimodal X-ray and infrared imaging of an organometallic derivative of praziquantel in Schistosoma mansoni. Chembiochem 17:1004–1007. doi: 10.1002/cbic.201500688. [DOI] [PubMed] [Google Scholar]
- 37.Hess J, Patra M, Jabbar A, Pierroz V, Konatschnig S, Spingler B, Ferrari S, Gasser RB, Gasser G. 2016. Assessment of the nematocidal activity of metallocenyl analogues of monepantel. Dalton Trans 45:17662–17671. doi: 10.1039/C6DT03376H. [DOI] [PubMed] [Google Scholar]
- 38.Hess J, Patra M, Pierroz V, Spingler B, Jabbar A, Ferrari S, Gasser RB, Gasser G. 2016. Synthesis, characterization, and biological activity of ferrocenyl analogues of the anthelmintic drug monepantel. Organometallics 35:3369–3377. doi: 10.1021/acs.organomet.6b00577. [DOI] [Google Scholar]
- 39.Hess J, Patra M, Rangasamy L, Konatschnig S, Blacque O, Jabbar A, Mac P, Jorgensen EM, Gasser RB, Gasser G. 2016. Organometallic derivatization of the nematocidal drug monepantel leads to promising antiparasitic drug candidates. Chem Eur J 22:16602–16612. doi: 10.1002/chem.201602851. [DOI] [PubMed] [Google Scholar]
- 40.Patra M, Ingram K, Leonidova A, Pierroz V, Ferrari S, Robertson MN, Todd MH, Keiser J, Gasser G. 2013. In vitro metabolic profile and in vivo antischistosomal activity studies of (η6-praziquantel)Cr(CO)3 derivatives. J Med Chem 56:9192–9198. doi: 10.1021/jm401287m. [DOI] [PubMed] [Google Scholar]
- 41.Patra M, Ingram K, Pierroz V, Ferrari S, Spingler B, Gasser RB, Keiser J, Gasser G. 2013. [(η6-Praziquantel)Cr(CO)3] derivatives with remarkable in vitro anti-schistosomal activity. Chem Eur J 19:2232–2235. doi: 10.1002/chem.201204291. [DOI] [PubMed] [Google Scholar]
- 42.Nuralitha S, Siregar JE, Syafruddin D, Roelands J, Verhoef J, Hoepelman AIM, Marzuki S. 2015. Within-host selection of drug resistance in a mouse model of repeated incomplete malaria treatment: comparison between atovaquone and pyrimethamine. Antimicrob Agents Chemother 60:258–263. doi: 10.1128/AAC.00538-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson PV, Nagel C, Bruhn H, Schatzschneider U. 2015. Antibacterial and antiparasitic activity of manganese(I) tricarbonyl complexes with ketoconazole, miconazole, and clotrimazole ligands. Organometallics 34:3809–3815. doi: 10.1021/acs.organomet.5b00458. [DOI] [Google Scholar]
- 44.Halonen SK, Weiss LM. 2013. Toxoplasmosis. Handb Clin Neurol 114:125–145. doi: 10.1016/B978-0-444-53490-3.00008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaye A. 2011. Toxoplasmosis: diagnosis, treatment, and prevention in congenitally exposed infants. J Pediatr Health Care 25:355–364. doi: 10.1016/j.pedhc.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Fichera ME, Roos DS. 1997. A plastid organelle as a drug target in apicomplexan parasites. Nature 390:407–409. doi: 10.1038/37132. [DOI] [PubMed] [Google Scholar]
- 47.Stibal D, Therrien B, Giannini F, Paul LEH, Furrer J, Suss-Fink G. 2014. Monothiolato-bridged dinuclear arene ruthenium complexes: the missing link in the reaction of arene ruthenium dichloride dimers with thiols. Eur J Inorg Chem 2014:5925–5931. [Google Scholar]
- 48.Ibao A-F, Gras M, Therrien B, Süss-Fink G, Zava O, Dyson PJ. 2012. Thiolato-bridged arene-ruthenium complexes: synthesis, molecular structure, reactivity, and anticancer activity of the dinuclear complexes [(arene)2Ru2(SR)2Cl2]. Eur J Inorg Chem 2012:1531–1535. doi: 10.1002/ejic.201101057. [DOI] [Google Scholar]
- 49.Stíbal D, Therrien B, Süss-Fink G, Nowak-Sliwinska P, Dyson PJ, Čermáková E, Řezáčová M, Tomšík P. 2016. Chlorambucil conjugates of dinuclear p-cymene ruthenium trithiolato complexes: synthesis, characterization and cytotoxicity study in vitro and in vivo. J Biol Inorg Chem 21:443–452. doi: 10.1007/s00775-016-1353-z. [DOI] [PubMed] [Google Scholar]
- 50.Klinkert MQ, Heussler V. 2006. The use of anticancer drugs in antiparasitic chemotherapy. Mini-Rev Med Chem 6:131–143. doi: 10.2174/138955706775475939. [DOI] [PubMed] [Google Scholar]
- 51.McFadden DC, Seeber F, Boothroyd JC. 1997. Use of Toxoplasma gondii expressing beta-galactosidase for colorimetric assessment of drug activity in vitro. Antimicrob Agents Chemother 41:1849–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ojo KK, Reid MC, Kallur Siddaramaiah L, Müller J, Winzer P, Zhang Z, Keyloun KR, Vidadala RSR, Merritt EA, Hol WGJ, Maly DJ, Fan E, Van Voorhis WC, Hemphill A. 2014. Neospora caninum calcium-dependent protein kinase 1 is an effective drug target for neosporosis therapy. PLoS One 9:e92929. doi: 10.1371/journal.pone.0092929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kropf C, Debache K, Rampa C, Barna F, Schorer M, Stephens CE, Ismail MA, Boykin DW, Hemphill A. 2012. The adaptive potential of a survival artist: characterization of the in vitro interactions of Toxoplasma gondii tachyzoites with di-cationic compounds in human fibroblast cell cultures. Parasitology 139:208–220. doi: 10.1017/S0031182011001776. [DOI] [PubMed] [Google Scholar]
- 54.Müller J, Aguado-Martinez A, Manser V, Balmer V, Winzer P, Ritler D, Hostettler I, Solís D, Ortega-Mora LM, Hemphill A. 2015. Buparvaquone is active against Neospora caninum in vitro and in experimentally infected mice. Int J Parasitol Drugs Drug Resist 5:16–25. doi: 10.1016/j.ijpddr.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winzer P, Müller J, Aguado-Martínez A, Rahman M, Balmer V, Manser V, Ortega-Mora LM, Ojo KK, Fan E, Maly DJ, Van Voorhis WC, Hemphill A. 2015. In vitro and in vivo effects of the bumped kinase inhibitor 1294 in the related cyst-forming apicomplexans Toxoplasma gondii and Neospora caninum. Antimicrob Agents Chemother 59:6361–6374. doi: 10.1128/AAC.01236-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Syafruddin D, Siregar JE, Marzuki S. 1999. Mutations in the cytochrome b gene of Plasmodium berghei conferring resistance to atovaquone. Mol Biochem Parasitol 104:185–194. doi: 10.1016/S0166-6851(99)00148-6. [DOI] [PubMed] [Google Scholar]
- 57.McFadden DC, Tomavo S, Berry EA, Boothroyd JC. 2000. Characterization of cytochrome b from Toxoplasma gondii and Qo domain mutations as a mechanism of atovaquone-resistance. Mol Biochem Parasitol 108:1–12. doi: 10.1016/S0166-6851(00)00184-5. [DOI] [PubMed] [Google Scholar]
- 58.Sharifiyazdi H, Namazi F, Oryan A, Shahriari R, Razavi M. 2012. Point mutations in the Theileria annulata cytochrome b gene is associated with buparvaquone treatment failure. Vet Parasitol 187:431–435. doi: 10.1016/j.vetpar.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 59.Marsolier J, Perichon M, DeBarry JD, Villoutreix BO, Chluba J, Lopez T, Garrido C, Zhou XZ, Lu KP, Fritsch L, Ait-Si-Ali S, Mhadhbi M, Medjkane S, Weitzman JB. 2015. Theileria parasites secrete a prolyl isomerase to maintain host leukocyte transformation. Nature 520:378–382. doi: 10.1038/nature14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yousif LF, Stewart KM, Kelley SO. 2009. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem 10:1939–1950. doi: 10.1002/cbic.200900185. [DOI] [PubMed] [Google Scholar]
- 61.Chen LB. 1988. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- 62.Mulcahy SP, Grundler K, Frias C, Wagner L, Prokop A, Meggers E. 2010. Discovery of a strongly apoptotic ruthenium complex through combinatorial coordination chemistry. Dalton Trans 39:8177–8182. doi: 10.1039/c0dt00034e. [DOI] [PubMed] [Google Scholar]
- 63.Pierroz V, Joshi T, Leonidova A, Mari C, Schur J, Ott I, Spiccia L, Ferrari S, Gasser G. 2012. Molecular and cellular characterization of the biological effects of ruthenium(II) complexes incorporating 2-pyridyl-2-pyrimidine-4-carboxylic acid. J Am Chem Soc 134:20376–20387. doi: 10.1021/ja307288s. [DOI] [PubMed] [Google Scholar]
- 64.Li L, Wong Y-S, Chen T, Fan C, Zheng W. 2012. Ruthenium complexes containing bis-benzimidazole derivatives as a new class of apoptosis inducers. Dalton Trans 41:1138–1141. doi: 10.1039/C1DT11950H. [DOI] [PubMed] [Google Scholar]
- 65.Wang J-Q, Zhang P-Y, Qian C, Hou X-J, Ji L-N, Chao H. 2014. Mitochondria are the primary target in the induction of apoptosis by chiral ruthenium(II) polypyridyl complexes in cancer cells. J Biol Inorg Chem 19:335–348. doi: 10.1007/s00775-013-1069-2. [DOI] [PubMed] [Google Scholar]
- 66.Jacot D, Waller RF, Soldati-Favre D, MacPherson DA, MacRae JI. 2016. Apicomplexan energy metabolism: carbon source promiscuity and the quiescence hyperbole. Trends Parasitol 32:56–70. doi: 10.1016/j.pt.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 67.Kristensen R, Torp M, Kosiak B, Holst-Jensen A. 2005. Phylogeny and toxigenic potential is correlated in Fusarium species as revealed by partial translation elongation factor 1 alpha gene sequences. Mycol Res 109:173–186. doi: 10.1017/S0953756204002114. [DOI] [PubMed] [Google Scholar]
- 68.Ridgley EL, Xiong Z-H, Kaur KJ, Ruben L. 1996. Genomic organization and expression of elongation factor-1α genes in Trypanosoma brucei. Mol Biochem Parasitol 79:119–123. doi: 10.1016/0166-6851(96)02639-4. [DOI] [PubMed] [Google Scholar]
- 69.Toueille M, Saint-Jean B, Castroviejo M, Benedetto J-P. 2007. The elongation factor 1A: a novel regulator in the DNA replication/repair protein network in wheat cells? Plant Physiol Biochem 45:113–118. doi: 10.1016/j.plaphy.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 70.Lamberti A, Longo O, Marra M, Tagliaferri P, Bismuto E, Fiengo A, Viscomi C, Budillon A, Rapp UR, Wang E, Venuta S, Abbruzzese A, Arcari P, Caraglia M. 2007. C-Raf antagonizes apoptosis induced by IFN-[alpha] in human lung cancer cells by phosphorylation and increase of the intracellular content of elongation factor 1A. Cell Death Differ 14:952–962. [DOI] [PubMed] [Google Scholar]
- 71.Bouzaidi-Tiali N, Aeby E, Charrière F, Pusnik M, Schneider A. 2007. Elongation factor 1a mediates the specificity of mitochondrial tRNA import in T. brucei. EMBO J 26:4302–4312. doi: 10.1038/sj.emboj.7601857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Esseiva AC, Naguleswaran A, Hemphill A, Schneider A. 2004. Mitochondrial tRNA import in Toxoplasma gondii. J Biol Chem 279:42363–42368. doi: 10.1074/jbc.M404519200. [DOI] [PubMed] [Google Scholar]
- 73.Matsubayashi M, Teramoto-Kimata I, Uni S, Lillehoj HS, Matsuda H, Furuya M, Tani H, Sasai K. 2013. Elongation factor-1α is a novel protein associated with host cell invasion and a potential protective antigen of Cryptosporidium parvum. J Biol Chem 288:34111–34120. doi: 10.1074/jbc.M113.515544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang S, Zhang Z, Wang Y, Gadahi JA, Xu L, Yan R, Song X, Li X. 2017. Toxoplasma gondii elongation factor 1-alpha (TgEF-1α) is a novel vaccine candidate antigen against toxoplasmosis. Front Microbiol 8:168. doi: 10.3389/fmicb.2017.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang S, Wang Y, Sun X, Zhang Z, Liu T, Gadahi JA, Hassan IA, Xu L, Yan R, Song X, Li X. 2015. Protective immunity against acute toxoplasmosis in BALB/c mice induced by a DNA vaccine encoding Toxoplasma gondii elongation factor 1α. BMC Infect Dis 15:448. doi: 10.1186/s12879-015-1220-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Müller J, Hemphill A. 2013. New approaches for the identification of drug targets in protozoan parasites. Int Rev Cell Mol Biol 301:359–401. doi: 10.1016/B978-0-12-407704-1.00007-5. [DOI] [PubMed] [Google Scholar]