

ABSTRACT
1,2-Distigmasterylhemisuccinoyl-sn-glycero-3-phosphocholine (DSHemsPC) is a new lipid in which two molecules of stigmasterol (an inexpensive plant sterol) are covalently linked via a succinic acid to glycerophosphocholine. Our previous study revealed that liposome (Lip)-intercalated amphotericin B (AMB) prepared from DSHemsPC (DSHemsPC-AMB-Lip) possesses excellent colloidal properties and in vitro antifungal and antileishmanial activities similar to those of the liposomal AMB preparation AmBisome. The aim of this study was to determine the biodistribution and evaluate the antileishmanial effects of DSHemsPC-AMB-Lip in Leishmania major-infected BALB/c mice. The serum profile and tissue concentrations of AMB were similar in DSHemsPC-AMB-Lip- and AmBisome-treated mice after intravenous (i.v.) injection. Multiple i.v. doses of the micellar formulation of AMB (Fungizone; 1 mg/kg of body weight), DSHemsPC-AMB-Lip (5 mg/kg), and AmBisome (5 mg/kg) were used in L. major-infected BALB/c mouse models of early and established lesions. In a model of the early lesions of cutaneous leishmaniasis (CL), the results indicated that the level of footpad inflammation was significantly (P < 0.001) lower in mice treated with DSHemsPC-AMB-Lip and AmBisome than mice treated with empty liposomes or 5% dextrose. The splenic and footpad parasite load was also significantly (P < 0.001) lower in these groups of mice than in control mice that received 5% DW or free liposome. The in vivo activity of DSHemsPC-AMB-Lip was comparable to that of AmBisome, and both provided improved results compared to those achieved with Fungizone at the designated doses. The results suggest that systemic DSHemsPC-AMB-Lip administration may be useful for the treatment of leishmaniasis, and because it costs less to produce DSHemsPC-AMB-Lip than AmBisome, DSHemsPC-AMB-Lip merits further investigation.
KEYWORDS: amphotericin B, distigmasteryl-modified phospholipids, liposomes, leishmaniasis, biodistribution
INTRODUCTION
Leishmaniasis is a term for a variety of diseases caused by protozoan parasites of the genus Leishmania (1). Clinical manifestations depend on the infecting species and the immune status of the host and can vary from self-healing skin lesions to a lethal systemic form of disease. Visceral leishmaniasis (VL) is the most serious form of disease and can be fatal if it is left untreated.
The polyene antibiotic amphotericin B (AMB), a drug widely used for the treatment of systemic fungal infection (2), is currently recommended as an alternative treatment for mucocutaneous and visceral leishmaniasis, especially in patients who have failed treatment with pentavalent antimony (1, 3). The selective activity of AMB against fungi and leishmania instead of mammalian cells is because of its higher affinity for ergosterol and episterol, which are found in parasite membranes, than for cholesterol, which is the mammalian sterol (4, 5). However, treatment with AMB is restricted by its dose-dependent toxicity and low therapeutic index in humans (6).
Several lipid-based complexes for the delivery of AMB (AmBisome, Abelcet, and Amphocil) have been developed for the treatment of leishmaniasis (7). Liposomal delivery systems significantly alter the pharmacokinetics, distribution, and clearance of their drug payload in the body and modulate the toxicity compared to that achieved with the free drug (8). Additionally, lipid carriers are generally cleared from the circulatory system by phagocytosis, particularly by macrophages in the liver and spleen, and taken to the reticuloendothelial system (RES) (9). Since visceral leishmaniasis is a generalized infection of the RES, the AmBisome liposomal formulation is often used for the treatment of VL due to its specific distribution into the intracellular compartments where parasites usually reside in the human host.
The development of lipid delivery systems has greatly reduced the toxicity of AMB for the treatment of VL, but less attention has been paid to cutaneous leishmaniasis (CL). Though previous studies showed the activity of AmBisome against experimental CL, it was suggested that drug accumulation within the dermis is insufficient (10) and that higher doses are required to reach the curative level at the site of infection (11).
AMB is anchored tightly in the AmBisome bilayer through a favorable interaction of the macrolide with the surrounding lipid and cholesterol (7). In our previous work, we used 1,2-distigmasterylhemisuccinoyl-sn-glycero-3-phosphocholine (DSHemsPC) to prepare several AMB liposomal formulations (12). DSHemsPC, an amphiphilic cholesterol analogue, can form a bilayer membrane; however, unlike cholesterol, it is unable to rapidly transfer between membranes (Fig. 1). Thus, by confining cholesterol in the lipid membrane through sterol-modified phospholipids (SMLs), bilayer cohesion properties improve, and these improved properties subsequently stabilize the formulation and prevent premature drug transfer in the biological milieu of serum at 37°C (13). A major drawback of AmBisome is its considerable cost, which is because of the high cost of injectable good manufacturing practice (GMP)-grade cholesterol. The cost of injectable GMP-grade cholesterol is higher than that of plant-derived stigmasterol because of its animal origin and the special processing that is required to make the cholesterol free of the agents of bovine spongiform encephalopathy/transmissible spongiform encephalopathy (BSE/TSE) and the related analysis procedures, which make the final product expensive (EMA BSE/TSE guideline EMA/410/01, revision 3). Thus, the use of stigmasterol, a plant-derived sterol, as an alternative to cholesterol might decrease the overall cost of the liposomal AMB preparation and reduce the risk of transfer of prions from mammal-derived cholesterol that might occur (6).
FIG 1.

Structures of DSHemsPC, DMPC, DMPG, and amphotericin B.
We devised and characterized a number of DSHemsPC-AMB liposomal formulations composed of DSHemsPC, phosphatidylcholine, and phosphatidylglycerol at various molar ratios; DSHemsPC–1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC)–1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt) (DMPG)–AMB at a molar ratio of 1.25:5.0:1.5:1.0 (prepared at pH 5.5) showed favorable characteristics, including excellent colloidal properties, a high concentration for hemolysis of 50% of red blood cells (RBCs) (the 50% inhibitory concentration [IC50]) for potassium release, in vitro antifungal and antileishmanial activities similar to those of AmBisome, and a maximum tolerated dose (MTD) of 60 mg/kg of body weight in BALB/c mice when it was administered intravenously (i.v.) (12).
In this study, we compared the biodistribution of the DSHemsPC-AMB-liposome (Lip) to that of AmBisome and the micellar formulation of AMB (Fungizone) in healthy and Leishmania major-infected BALB/c mice. We also compared the in vitro hemolytic activity of DSHemsPC-AMB-Lip to that of AmBisome and Fungizone. Finally, we describe the antileishmanial activity of DSHemsPC-AMB-Lip (DSHemsPC-DMPC-DMPG-AMB at a molar ratio of 1.25:5.0:1.5:1.0) in L. major-infected BALB/c mice and compare it to that of AmBisome and Fungizone.
RESULTS
Liposome characterization.
The size of the DSHemsPC-AMB liposome was about 10 nm smaller than that of the AmBisome liposome; however, the homogeneity (polydispersity) of the two formulations was almost similar (Table 1). The net charge (zeta potential) of the DSHemsPC-AMB liposome was almost half that of the AmBisome liposome. A control empty liposome prepared using molar ratios of phospholipids that were the same as those in the DSHemsPC liposome had a diameter that was notably smaller (approximately 40 nm) than that of the DSHemsPC-AMB and AmBisome liposomes.
TABLE 1.
Physical and hemolytic properties of a liposomal AMB formulation prepared from DSHemsPC
| AMB formulation | Avg liposome size ± SD (nm) | Mean polydispersity ± SD | Mean zeta potential ± SD (mV) | Hemolysis IC50 (mg/ml)a |
|---|---|---|---|---|
| DSHemsPC-DMPC-DMPG-AMB | 111.6 ± 1.0 | 0.21 ± 0.02 | −25.3 ± 9.05 | 42.6 (16.7–278.4) |
| DSHemsPC-DMPC-DMPG | 76.9 ± 0.4 | 0.21 ± 0.06 | −42.7 ± 0.5 | Inactive |
| AmBisome | 120.3 ± 1.5 | 0.2 ± 0.2 | −54.5 ± 1.1 | 11.6 (0.14–971) |
| Fungizone | 0.077 (0.0018–9.32) |
The values in parentheses are the lower and upper 95% limits. The IC50 of the DSHemsPC-AMB liposomal formulation was significantly (P < 0.001) greater than that of AmBisome and Fungizone; the IC50 of AmBisome was also significantly (P < 0.01) greater than that of Fungizone.
Determination of RBC hemolysis.
In vitro red blood cell (RBC) hemolysis was used to assess the potential toxicity of the DSHemsPC-AMB formulation. Higher hemolysis IC50s indicate that the formulation has lower levels of toxicity for RBCs. Fungizone had the lowest IC50 (0.0765 mg/ml) (Table 1). This has been ascribed to the dissociation of AMB from the deoxycholate micelle and also the hemolytic activity of the detergent. Control liposomes showed no hemolytic activity. The hemolysis IC50 of the DSHemsPC-AMB liposome was significantly (P < 0.001) greater than that of AmBisome and Fungizone (Table 1).
Serum profile and tissue distribution of AMB in uninfected (healthy) BALB/c mice.
The concentrations of AMB in the serum and tissues of DSHemsPC-AMB-treated mice were determined following i.v. administration and compared to those of AmBisome (Table 2). The serum profiles of AMB administered as DSHemsPC-AMB and AmBisome were similar at different times postadministration. No significant difference in the tissue concentration of AMB when it was delivered to mice as DSHemsPC-AMB or as AmBisome was observed. The maximum concentration of drug in the livers and spleens of the mice was observed at 24 h following AmBisome or DSHemsPC-AMB administration. DSHemsPC-AMB and AmBisome treatment resulted in a lower level of drug accumulation in the kidneys of mice, a finding which further supports the reduced nephrotoxicity associated with AMB in the liposomal formulation (Table 2).
TABLE 2.
AMB concentration in serum and selected tissues following i.v. injection of DSHemsPC-AMB-Lip and AmBisome in uninfected (healthy) BALB/c mice
| Formulation | Mean concn ± SDa |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tissues |
Serum |
||||||||
| Heart | Liver | Kidney | Spleen | Lung | 1 h | 6 h | 12 h | 24 h | |
| DSHemsPC-AMB-Lip | 44.2 ± 9.2 | 0.4 ± 0.2 | 16.5 ± 4.1 | 2.1 ± 2.1 | 34.1 ± 0.2 | 6.3 ± 2.3 | 1.7 ± 0.03 | 1.7 ± 0.01 | |
| AmBisomeb | 0.4 ± 0.2 | 36.8 ± 9.9 | 1 ± 0.9 | 13.8 ± 2.8 | 1.5 ± 0.6 | 36 ± 0.15 | 9.8 ± 2.2 | 2.2 ± 0.5 | 1.9 ± 0.01 |
Each value is the mean concentration ± standard deviation for three animals. Concentrations are in micrograms per gram of tissue for heart, liver, kidney, spleen, and lung tissues and micrograms per milliliter for serum. No drug was detected in serum and tissues from experimental animals receiving 5% dextrose. The serum AMB concentrations at each time were not significantly different (P > 0.05) among DSHemsPC-AMB-Lip- and AmBisome-treated mice. The AMB concentrations in each tissue were not significantly different (P > 0.05) among DSHemsPC-AMB-Lip- and AmBisome-treated mice.
AmBisome was administered at 10 mg/kg.
Activity of DSHemsPC-AMB in a susceptible BALB/c mouse model of early CL.
The activity of DSHemsPC-AMB liposomes following i.v. administration to BALB/c mice was determined on days 1, 2, 4, 7, 14, 21, and 28 postinfection. The i.v. administration of DSHemsPC-AMB significantly reduced the footpad lesion size. This effect was notable from week 2 (P < 0.05) onward (P ≤ 0.01) compared to the findings for the negative-control groups. DSHemsPC-AMB controlled footpad swelling slightly better than AmBisome did; however, the difference was not statistically significant. The effect of DSHemsPC-AMB was also significantly greater than that of Fungizone (P < 0.01) (Fig. 2).
FIG 2.
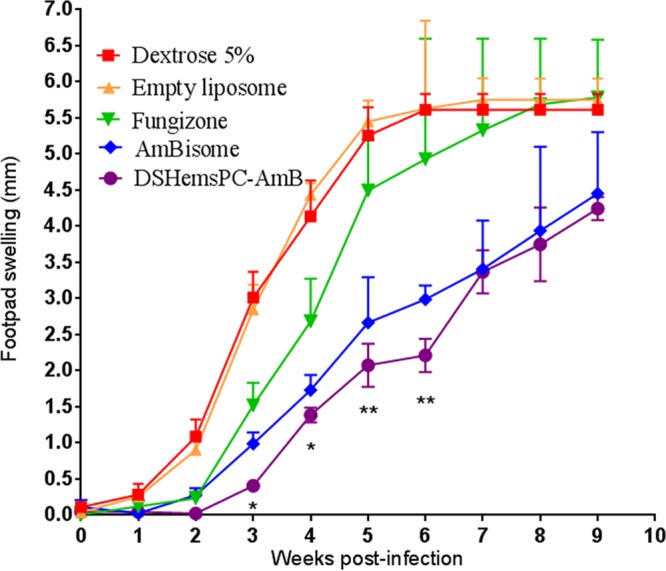
In vivo antileishmanial activity in model of early CL. The efficacies of DSHemsPC-AMB-Lip, AmBisome, and Fungizone in the susceptible L. major-infected BALB/c mouse model of early CL were determined. AMB formulations were i.v. administered on days 1, 2, 4, 7, 14, 21, and 28 after parasite injection. Values are the means ± standard errors of the means (weeks 0 to 3, n = 10; weeks 4 to 6, n = 7; weeks 7 to 9, n = 4). At weeks 2 and 3, there was a significant (*, P < 0.001) difference between the groups receiving DSHemsPC-AMB-Lip and AmBisome and the control groups (mice receiving 5% dextrose and empty liposome). At weeks 4 to 6, the difference between the DSHemsPC-AMB-Lip and control groups was significant (**, P ≤ 0.01). DSHemsPC-AMB showed greater activity than Fungizone (P ≤ 0.01).
Splenic parasite load in model of early CL.
The number of parasites in the spleens of the mice was determined at weeks 3, 6, and 9 postinfection. At week 3 postinfection, no parasite was detected in the spleens of mice treated with either DSHemsPC-AMB or AmBisome. Mice treated with Fungizone showed a significantly (P ≤ 0.01) lower parasite number than mice treated with 5% dextrose or empty liposomes (Fig. 3A); however, the parasite number after Fungizone treatment was not significantly different from that after DSHemsPC-AMB and AmBisome treatment. Interestingly, at week 6, no parasite was detected in the spleens of mice treated with either DSHemsPC-AMB or AmBisome. The number of parasites in Fungizone-treated mice was also significantly (P < 0.05) lower than that in the control groups (Fig. 3B). At week 9, as expected, parasites were undetectable in DSHemsPC-AMB- or AmBisome-treated mice; however, the splenic parasite load was significantly increased in the control group (Fig. 3C), in which the load was comparable to that in the Fungizone-treated group (P > 0.05).
FIG 3.
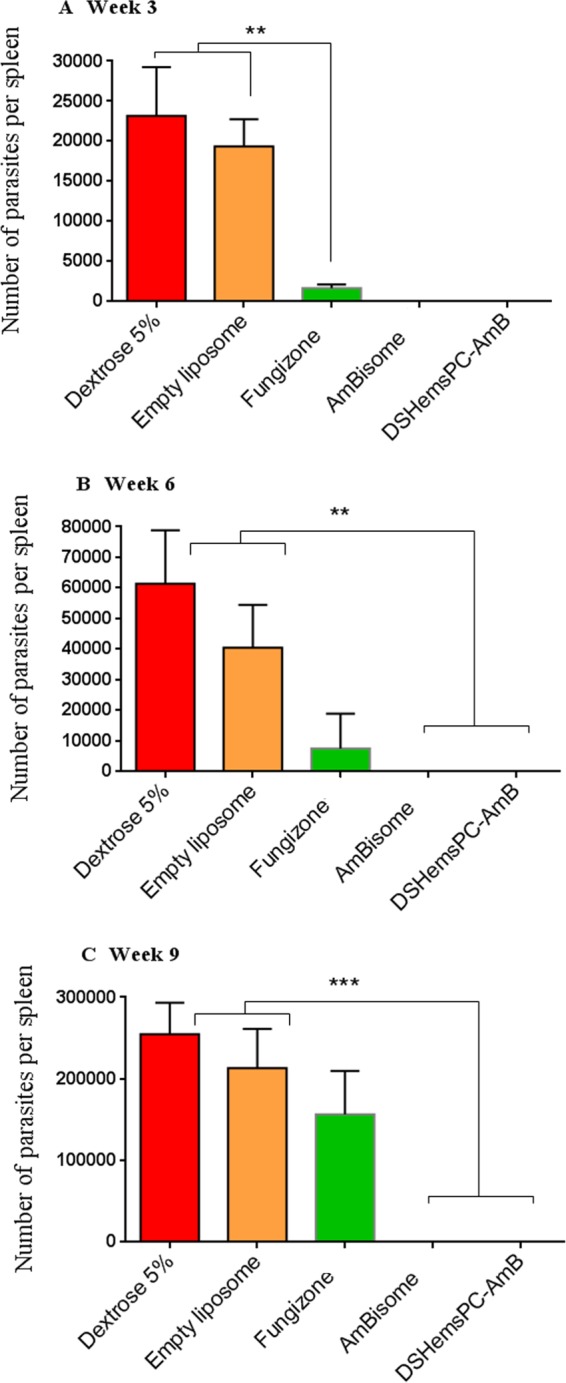
Splenic parasite burden in infected susceptible BALB/c mice in the model of early CL. The number of viable L. major parasites in the spleens of treated mice was quantified by limiting dilution assay at weeks 3 (A), 6 (B), and 9 (C) postinfection with L. major promastigotes. AMB formulations were administered i.v. on days 1, 2, 4, 7, 14, 21, and 28 after parasite injection. ** (P < 0.01) and *** (P < 0.001) indicate the significant differences when comparing splenic parasite loads for Fungizone (A) or DSHemsPC-AmB-Lip and AmBisome (B) to empty liposome and 5% dextrose.
Footpad parasite load in model of early CL.
At week 3 postinfection, no significant difference in the footpad parasite load between the DSHemsPC-AMB-treated mice and the AmBisome-treated mice was observed; however, both groups had significantly lower parasite loads than the Fungizone-treated and control groups (P < 0.001) (Fig. 4A). At week 6, the number of parasites in the footpads of mice in the DSHemsPC-AMB treatment group showed a significant decrease (P < 0.001) compared to that in the footpads of mice in the 5% dextrose or empty liposome (Fig. 4B) and Fungizone (P < 0.001) treatment groups. At week 9, the same trend could be seen, as DSHemsPC-AMB- and AmBisome-treated mice showed a significantly (P < 0.01) lower parasite load than 5% dextrose- or empty liposome-treated mice (Fig. 4C).
FIG 4.
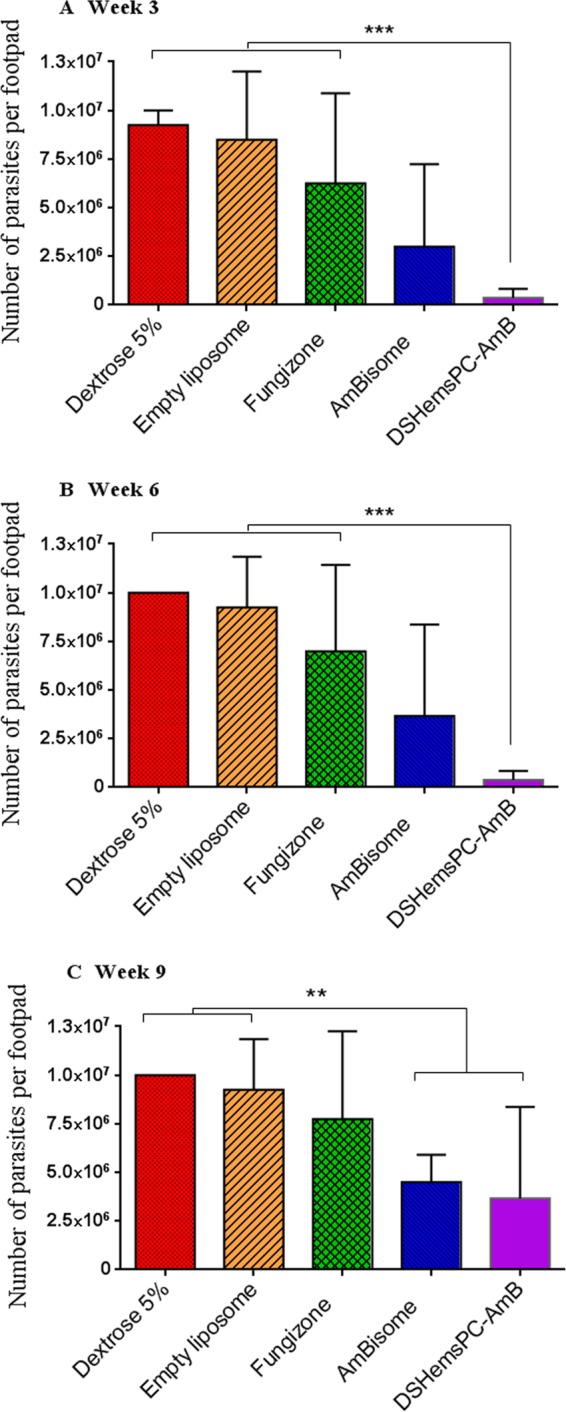
Footpad parasite load in infected susceptible BALB/c mice in the model of early CL. The number of viable L. major parasites in the spleens of treated mice was quantified by limiting dilution assay at weeks 3 (A), 6 (B), and 9 (C) postinfection with L. major promastigotes. *** (P < 0.001) indicates the significant difference when comparing the number of parasites per footpad for DSHemsPC-AmB-Lip to Fungizone, empty liposome, and 5% dextrose (A and B), and ** (P < 0.01) represents the significant difference when comparing DSHemsPC-AmB-Lip or AmBisome to empty liposome and 5% dextrose (C).
Serum and tissue distribution of AMB in model of early CL.
There was no significant difference in the concentration of AMB in the serum of DSHemsPC-AMB liposome- and AmBisome-treated mice following i.v. administration (Fig. 5). However, the serum AMB concentrations of Fungizone-treated mice were significantly lower than those of both DSHemsPC-AMB-Lip- and AmBisome-treated mice (at 1 h, P < 0.001; at 24 h, P < 0.01).
FIG 5.
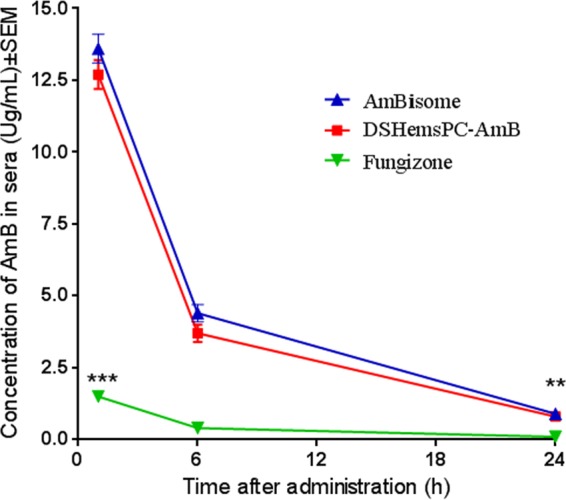
Pharmacokinetics of AMB in blood of L. major-infected mice treated with DSHemsPC-AMB-Lip, AmBisome, or Fungizone in the model of early CL lesions. The differences were significant when the serum AMB concentrations in DSHemsPC-AMB-Lip-treated mice (***, P < 0.001) and AmBisome-treated mice (**, P < 0.01) were compared to those in Fungizone-treated mice.
The tissue concentration of AMB was not significantly different between the DSHemsPC-AMB-Lip-treated group and the AmBisome-treated group (P > 0.05). The AMB levels in the livers of Fungizone-treated mice were significantly lower than those in the livers of DSHemsPC-AMB-Lip- and AmBisome-treated mice (P < 0.001) (Fig. 6).
FIG 6.
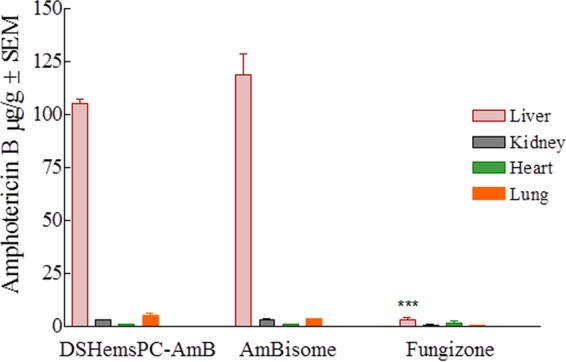
Tissue distribution of AMB in L. major-infected BALB/c mice treated with DSHemsPC-AMB-Lip, AmBisome, or Fungizone in the model of early CL. The liver AMB concentration in Fungizone-treated mice was significantly different from that in the DSHemsPC-AMB-Lip- and AmBisome-treated mice (***, P ≤ 0.001).
DSHemsPC-AMB in susceptible BALB/c mouse model of established CL.
In the model of established lesions, treatment was started at day 23, following the appearance of a lesion. As shown in Fig. 7, no significant differences (P > 0.05) in the footpad thicknesses of the different treatment groups were observed before the initiation of therapy (week 3 postinfection). From week 4 onward, i.v. administration of DSHemsPC-AMB and AmBisome significantly reduced the level of footpad swelling in the infected mice. In contrast, control mice receiving either 5% dextrose or empty liposomes exhibited a continuous increase in footpad thickness.
FIG 7.
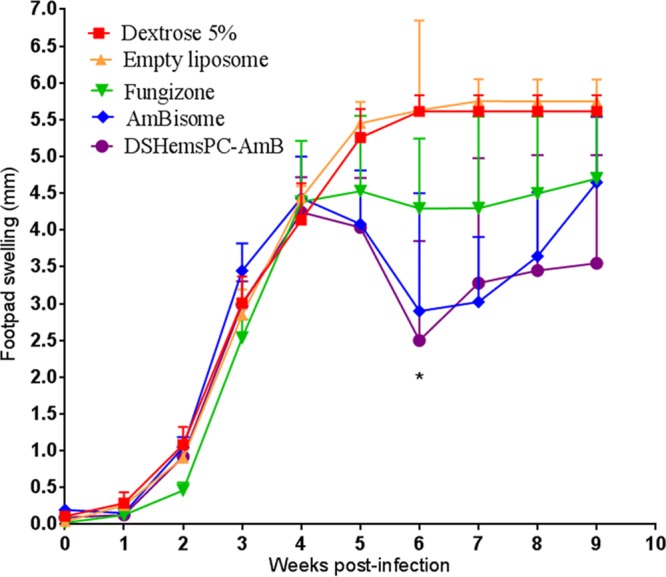
Antileishmania effect of DSHemsPC-AMB-Lip, AmBisome, and Fungizone in established CL lesions in susceptible BALB/c mice infected with L. major. AMB formulations were i.v. administered on days 23, 25, 27, 29, 31, 33, and 35 after parasite inoculation. Values represent means ± standard errors of the means (weeks 0 to 3, n = 10; weeks 4 to 6, n = 7; weeks 7 to 9, n = 4). The results for the DSHemsPC-AMB-Lip- and AmBisome-treated mice were significantly different from those for the control groups (mice receiving 5% dextrose and empty liposomes) (*, P < 0.05).
Splenic parasite load in model of established CL.
At week 3 postinfection and before the initiation of therapy, the splenic parasite load was not significantly different among the groups (P > 0.05) (Fig. 8A). As shown in Fig. 8B, at week 6, treatment with Fungizone, DSHemsPC-AMB, or AmBisome significantly (P < 0.01) reduced the parasite load in infected mice compared to that in the control groups. At week 9, the mice treated with the AMB formulations showed a significantly (P < 0.001) lower parasite burden than the mice in the control groups (Fig. 8C).
FIG 8.
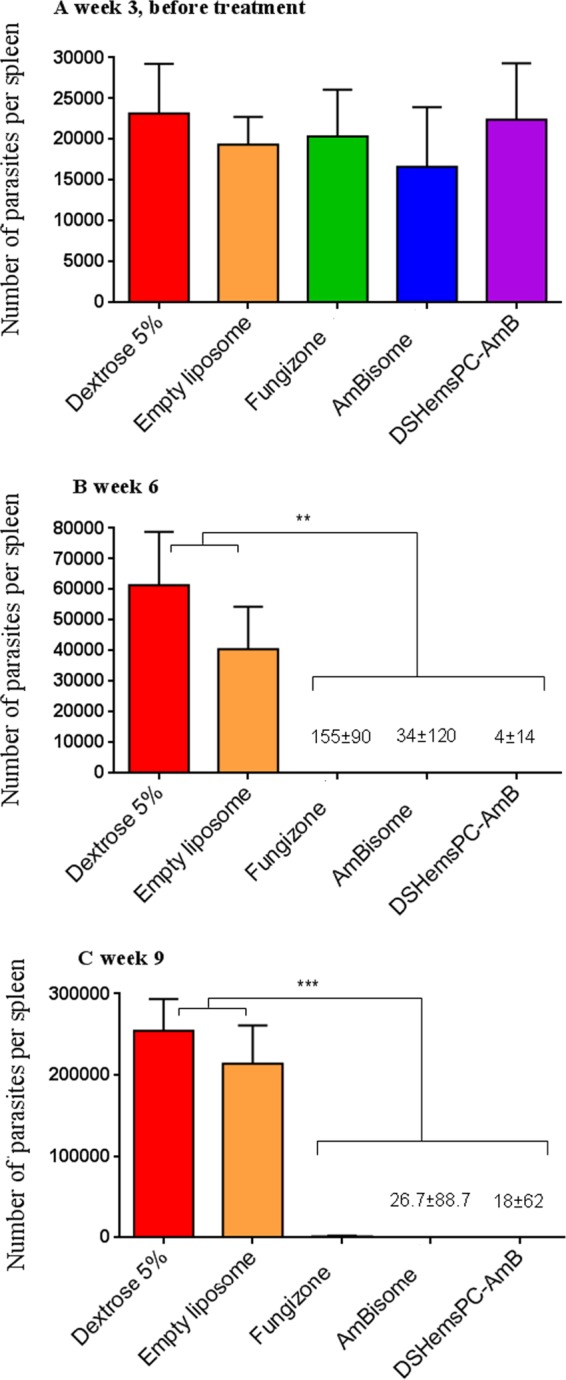
Splenic parasite burden in infected susceptible BALB/c mice in a model of established CL. The number of viable L. major parasites in the spleens of treated mice was quantified by limiting dilution assay at weeks 3 (A), 6 (B), and 9 (C) postinfection with L. major promastigotes. AMB formulations were administered i.v. on days 23, 25, 27, 29, 31, 33, and 35 after parasite inoculation. Values are the means ± standard errors of the means (n = 3). ** (P < 0.01) (B) and *** (P < 0.001) (C) indicate the significant difference when comparing splenic parasite loads for Fungizone, DSHemsPC-AmB-Lip, or AmBisome to empty liposome and 5% dextrose.
Footpad parasite load in model of established CL.
At week 3 and before the initiation of treatment, there was no significant difference (P > 0.05) in the parasite load in the footpads among the mice in the different groups (Fig. 9A). As shown in Fig. 9B, after treatment with DSHemsPC-AMB and AmBisome, the number of parasites per footpad in infected mice was significantly reduced (P ≤ 0.001) compared to that in the mice in the control groups. Significant differences (P < 0.05) in the footpad parasite load were also observed in the Fungizone- and 5% dextrose-treated groups (Fig. 9B). At week 9, the same trend continued and the parasite loads were lower in the DSHemsPC-AMB- or AmBisome-treated mice than the mice in the control groups (P ≤ 0.001) (Fig. 9C).
FIG 9.
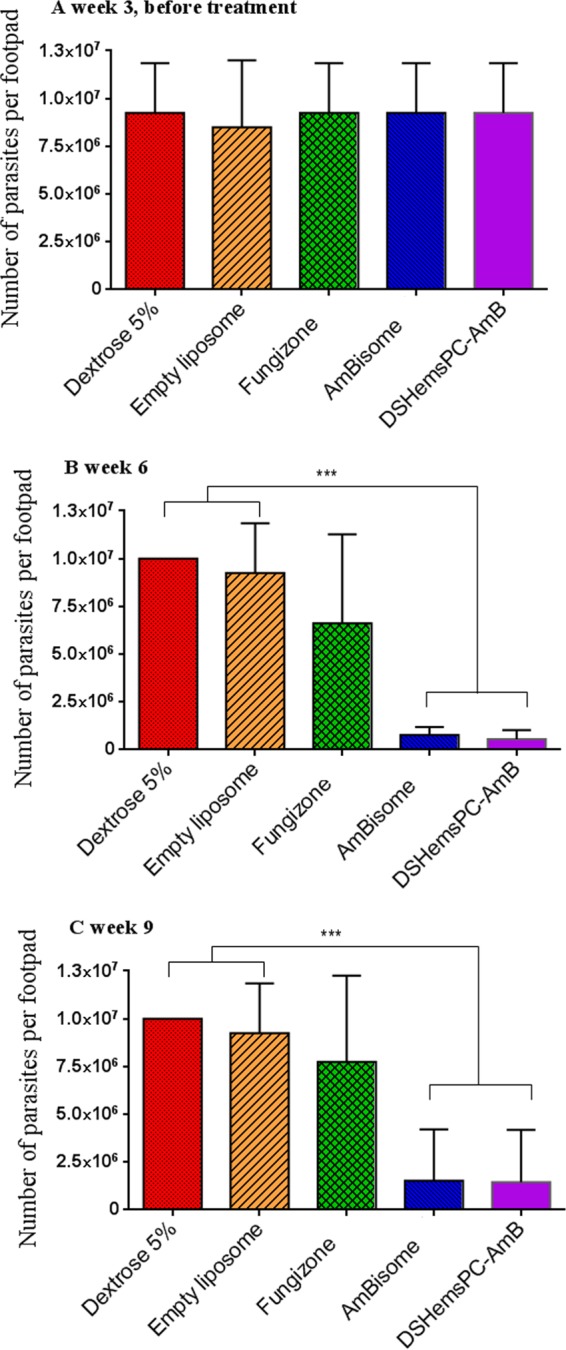
Footpad parasite burden in infected susceptible BALB/c mice in a model of established CL. The number of viable L. major parasites in the footpads of treated mice was quantified by limiting dilution assay at weeks 3 (A), 6 (B), and 9 (C) postinfection with L. major promastigotes. AMB formulations were administered i.v. on days 23, 25, 27, 29, 31, 33, and 35 after parasite inoculation. Values are means ± standard errors of the means (n = 3). *** (P < 0.001) indicates the significant difference when comparing the number of parasites per footpad for DSHemsPC-AmB-Lip or AmBisome to empty liposome and 5% dextrose (B and C).
Serum and tissue distribution of AMB in model of established CL.
There was no significant difference in the serum concentration of AMB following administration of DSHemsPC-AMB or AmBisome; however, both of these treatments resulted in serum levels of AMB significantly greater than those provided by treatment with Fungizone (P < 0.01) (Fig. 10). At 24 h after treatment with AmBisome or DSHemsPC-AMB, the highest concentration of AMB was observed in the liver. Treatment with all three formulations produced low levels of AMB in the kidneys, although the two liposome-based formulations were administered at five times the dose of Fungizone (Fig. 11).
FIG 10.
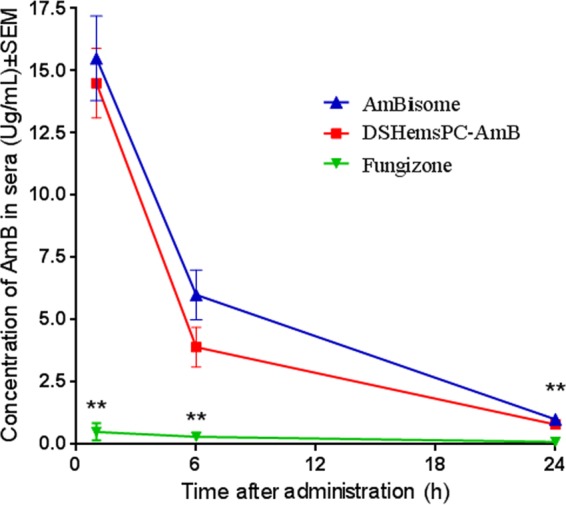
Pharmacokinetics of AMB in the blood of L. major-infected mice treated with DSHemsPC-AMB-Lip, AmBisome, or Fungizone in a model of established CL. BALB/c mice were infected in the footpad with 2 × 106 stationary-phase L. major promastigotes. The serum AMB concentrations were significantly different between Fungizone-treated mice and DSHemsPC-AMB-Lip- and AmBisome-treated mice (**, P ≤ 0.01).
FIG 11.
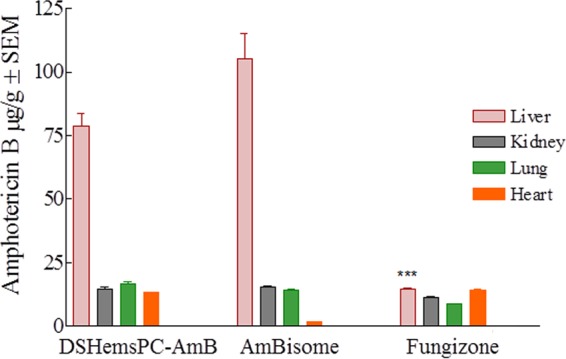
Tissue distribution of AMB in L. major-infected mice treated with DSHemsPC-AMB-Lip, AmBisome, or Fungizone in a model of established CL. The liver AMB concentrations were significantly different between Fungizone-treated mice and DSHemsPC-AMB-Lip- and AmBisome-treated mice (***, P ≤ 0.001).
DISCUSSION
Systemic (i.v.) administration of AMB is one of the most effective approaches to treat VL (1). Since Fungizone, the micellar formulation of AMB, quickly releases its payload following administration, it causes severe toxic adverse effects, including acute infusion-related toxicities and chronic nephrotoxicity, which limit its clinical use (14). Three different lipid-based nanoformulations of AMB (Amphotec, Abelcet, and AmBisome) have been developed and commercialized in the United States and Europe in order to increase tolerance and the therapeutic index of AMB and to reduce drug-related adverse effects (15). Liposomal AMB is widely used in the treatment of VL (11). In the visceral form of leishmaniasis, parasites live and propagate within the phagolysosome of infected macrophages of the spleen and liver (16, 17), which are the main cellular sites for the uptake of lipid-associated AMB preparations. Thus, the local release of AMB enhances the direct killing of the parasites in the phagolysosome of the infected macrophages (5).
Among these formulations, AmBisome has a significantly lower AMB toxicity profile (18–21). The AMB in AmBisome is firmly associated with the liposome bilayer and is not readily released while it remains in the circulation (22). Furthermore, due to its small diameter, negative surface charge, and rigid bilayer (phase transition temperature, approximately 55°C), AmBisome has a long circulation time in the bloodstream, which promotes its distribution to the site of inflammation (20, 22).
Liposomes containing cholesterol (up to 50%) are generally more stable than those not containing cholesterol. However, in biological milieus, free cholesterol could rapidly transfer from liposomes into biomembranes and serum lipoproteins (23, 24). This transfer decreases the stability of the liposome and results in the loss of the encapsulated contents.
In our previous work (12), we used DSHemsPC (Fig. 1) to formulate AMB in more stable AMB-lipid preparations and to compare their toxicity and efficacy with those of AmBisome. In the present study, on the basis of its colloidal and in vitro antileishmanial properties and MTD, the most promising formulation was DSHemsPC-AMB-Lip, which was examined for its hemolytic and in vivo antileishmanial activities in two different lesion models and also its biodistribution in healthy and infected mice.
The animal lethality test and in vitro incubation with red blood cells are the methods most commonly used for determining the toxicity of formulations (18, 25–27). In this study, the Nanotechnology Characterization Laboratory (NCL) hemolysis method was used. By this method, diluted rather than washed whole blood cells are used; thus, the presence of serum provides a rigorous challenge for the transfer of cholesterol from the formulation (28). Among the commercially available AMB formulations, AmBisome has the lowest hemolytic activity (25). Our hemolytic studies showed that DSHemsPC-AMB-Lip was slightly less hemolytic than AmBisome in this model (Table 1), which suggests the higher affinity of AMB for DSHemsPC-Lip than AmBisome. This may be due to the stabilizing effect of SML liposomes, which retain their contents more efficiently, since serum proteins play a major role in extracting cholesterol from the liposome bilayer, and action that results in content leakage (29).
On the other hand, the serum profile and biodistribution of AMB were similar in DSHemsPC-AMB-Lip- and AmBisome-treated mice. The highest AMB concentration was seen in the livers and spleens of healthy mice, which confirms a passive targeting of the drug to the macrophages of the spleen and liver. Additionally, DSHemsPC-AMB-Lip minimizes the concentration of AMB in the kidneys of mice as efficiently as AmBisome (30), suggesting that the level of nephrotoxicity of this formulation will be low.
Studies of the biodistribution of DSHemsPC-AMB-Lip, AmBisome, and Fungizone were also performed in infected mice to examine the effect of potential alterations of vascular permeability following infection on the distribution of AMB (Fig. 5, 6, 10, and 11). We found that the serum AMB concentrations were similar following administration of DSHemsPC-AMB-Lip and AmBisome and, again, were significantly higher than those achieved with Fungizone. This further confirms that AMB is tightly associated within the DSHemsPC-AMB-Lip and AmBisome formulations and does not transfer as readily as AMB from the deoxycholate in the Fungizone formulation.
For determination of in vivo antileishmanial activity, BALB/c mice were treated with 5 mg/kg of DSHemsPC-AMB-Lip or AmBisome, each of which was very well tolerated, while Fungizone was administered at 1 mg/kg, based on its MTD value. In mouse models of both acute and established lesions, treatment with DSHemsPC-AMB-Lip or AmBisome significantly reduced the parasite loads in the spleen. This effect was most likely due to the accumulation of DSHemsPC-AMB-Lip or AmBisome but not that of Fungizone in the spleens of infected mice (9).
The parasite numbers in the footpads of DSHemsPC-AMB-Lip- and AmBisome-treated mice did not decrease as quickly as those in the spleens, although the higher dose of the liposome formulations than the dose of Fungizone that can be administered provided a greater parasite reduction in the footpads (Fig. 4 and 9). This indicates that liposomal AMB completely prevents the distribution of the parasites to the liver and spleen, while the drug is less efficient in preventing the distribution of the parasites to the dermis. Hence, this formulation is more effective in the treatment of VL than the cutaneous form of the disease (9, 11). This provides an explanation for the lower levels of efficacy of the AMB formulations for the treatment of lesions according to the lesion size (footpad thickness) (Fig. 2 and 7).
When all of the in vivo antileishmanial results are considered, the activity of DSHemsPC-AMB-Lip was substantially better than that of Fungizone and comparable to that of AmBisome. Since both the DSHemsPC-AMB-Lip and AmBisome formulations have similar biodistribution patterns, this finding is perhaps to be expected. In the model of early footpad infection, DSHemsPC-AMB-Lip provided a greater reduction in the parasite load than AmBisome at 3 and 6 weeks, but this difference disappeared by week 9 (Fig. 4). In the model of established CL, the two liposome formulations provided equivalent levels of parasite reduction (Fig. 9).
In summary, DSHemsPC as a DSHemsPC-DMPC-DMPG-AMB formulation (molar ratio, 1.25:5.0:1.5:1.0, pH 5.5) provides a novel, stable matrix for solubilizing and delivering AMB in liposomes. The systemic liposomal AMB formulation is a successful treatment for VL, as the site of infection is an ideal target for passive drug delivery systems. However, in CL the main site of infection is within the dermis, which is a more difficult target for drug delivery systems after i.v. administration. Our studies demonstrate that i.v. administered DSHemsPC-AMB-Lip reduced the footpad thickness in mice with experimental CL infections and almost completely eradicated parasites from the spleen. The effect of this formulation in the treatment of leishmaniasis is comparable to that of AmBisome. Since it is possible to produce a liposomal AMB formulation in a more cost-effective manner using DSHemsPC, this formulation merits further investigation.
The breakeven cost of liposomal amphotericin B treatment in relationship to the cost of treatment of nephrotoxicity arising from amphotericin B deoxycholate therapy has been estimated to be between $72 and $87 (31). Thus, a liposomal formulation that could be produced for about 50% less than the current liposomal amphotericin B formulation could have a significant impact on which formulation is used for antifungal therapy. We do not know what the commercial production costs of the SML formulation would be; the cost of the stigmasterol component in a kilogram quantity has been quoted to us to be the same as that of cholesterol. If 10s of kilograms were ordered, the cost would drop. The manufacture of AmBisome requires precise control of the formation of the liposome, and no generic AmBisome formulation has ever appeared. Furthermore, amphotericin B formulations with physicochemical properties and lipid compositions very similar to those of AmBisome exhibit much higher toxicity than AmBisome (32). The SML preparation is reproducible and has been replicated in laboratories in two countries; these characteristics may make it a reasonable compound for further development.
MATERIALS AND METHODS
Animals and parasites.
Female BALB/c mice (age, 6 to 8 weeks) were obtained from the Pasteur Institute (Tehran, Iran). The mice were housed in a standard environment at a constant temperature of 25°C under a 12-h light and a 12-h dark cycle with free access to food and drinking water. All procedures involving animals and the proposal were approved by the Institutional Ethical Committee and the Research Advisory Committee of Mashhad University of Medical Sciences (Education Office, 26 February 2008, proposal code 87848), based on the Specific National Ethical Guidelines for Biomedical Research issued in 2005 by the Research and Technology Deputy of the Ministry of Health and Medicinal Education (MOHME) of Iran.
The virulence of Leishmania major strain MRHO/IR/75/ER was maintained by passage in BALB/c mice. The amastigotes were isolated from the spleen of an infected mouse, cultured on Novy-MacNeal-Nicolle (NNN) medium, and subcultured in RPMI 1640 (Sigma) containing 10% (vol/vol) heat-inactivated fetal calf serum (FCS), 2 mM glutamine, 100 U/ml of penicillin, and 100 μg/ml of streptomycin sulfate (RPMI-FCS) at 25 ± 1°C (33).
Chemicals.
DSHemsPC was synthesized and purified, and its structure was confirmed as described previously by Huang et al. (13). The phospholipids used in this study included 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dimyristoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt) (DMPG), which were purchased from Avanti Polar Lipids (Birmingham, AL, USA) (Fig. 1).
AMB and disodium succinate hexahydrate (DSSH) were purchased from Sigma (St. Louis, MO). Fungizone (Bristol-Myers Squibb Company, Princeton, NJ) and AmBisome (Gilead Sciences, Inc., Foster City, CA) were purchased and reconstituted according to the instructions in the manufacturers' package inserts. Chloroform and methanol were purchased from Merck (Germany).
Liposome preparation.
DSHemsPC liposome-intercalated AMB (DSHemsPC-AMB-Lip) was prepared by thin lipid film hydration followed by sonication (18, 34). Briefly, the lipid components (DSHemsPC, DMPC, and DMPG; molar ratio, 1.25:5.0:1.5) were weighed and dissolved in chloroform. AMB was dissolved in methanol at 0.2 mg/ml. The lipid and AMB solutions were mixed in a round-bottom flask. A thin lipid film was formed by removing the solvent on a rotary evaporator under reduced pressure. Liposomes were prepared by rehydrating the lipid film with succinate-buffered solution containing 9% sucrose (pH 5.5), followed by mixing on a vortex mixer for 10 to 15 min and sonication at 65°C for 60 min in a bath-type sonicator (Laboratory Supplies Company Inc., Hicksville, NY) under argon. The final total lipid concentration of all formulations was adjusted to 70 mM.
Liposome characterization.
The particle diameter of the samples was measured in triplicate using a dynamic light-scattering instrument (Nano-ZS; Malvern, United Kingdom). The zeta potential of the liposomes was determined on the same machine using the zeta potential mode and was the average of 20 measurements (12).
The amount of AMB intercalated into liposomes was determined after dissolving the liposomal formulation (1/1,000) in methanol and measuring the absorbance at 406 nm (12). The standard curve was linear up to 6 μg AMB per ml methanol. The intra- and interday variations in the amounts of AMB were determined, and there was no significant difference from day to day. Five concentrations were tested in triplicate to validate the reproducibility of the described principle (12, 34).
Analysis of hemolytic properties.
The hemolytic properties of DSHemsPC-AMB-Lip were determined according to the protocol (NCL method ITA-1, version 1.0) developed at the Nanotechnology Characterization Laboratory, National Cancer Institute at Frederick (SAIC-Frederick, Inc., Frederick, MD). Briefly, whole blood was collected from Sprague-Dawley rats, placed in a tube containing heparin as an anticoagulant, and stored at 2 to 8°C for up to 48 h. The total blood hemoglobin (TBH) concentration was determined using the Stanbio hemoglobin kit (Stanbio Laboratory, Boerne, TX). The whole blood was diluted with Ca2+- and Mg2+-free Dulbecco phosphate-buffered saline (DPBS; from the UCSF cell culture facility) to adjust the total hemoglobin concentration to 10 mg/ml (diluted TBH [TBHd]). In a 1.5-ml conical centrifuge tube, 100 μl of TBHd was mixed with 800 μl of Ca2+- and Mg2+-free DPBS. Then, blood was preincubated with various concentrations of DSHemsPC-AMB-Lip (0.0156 mg/ml to 4 mg/ml), a blank (5% dextrose), the positive control (1% Triton X-100 in water), and the negative control (40% polyethylene glycol 8000 in water) in triplicate. The tubes were gently rotated and incubated in a water bath set at 37°C for 3 h, and the samples were mixed every 60 min. After incubation, the tubes were centrifuged at 800 × g for 15 min. The supernatants were separated, and the hemoglobin concentration was measured using a Stanbio hemoglobin kit (Stanbio Laboratory, Boerne, TX). The hemoglobin (Hb) concentration in the supernatant of each DSHemsPC-AMB sample, AmBisome, and Fungizone was converted to a number ranging from 0 to 1 using the following formula: (Hb concentration of the sample − Hb concentration of the blank)/TBHd concentration. The concentration of AMB required to cause hemolysis of 50% of red blood cells (hemolysis IC50) by the DSHemsPC-AMB-Lip formulation (based on the AMB concentration), AmBisome, and Fungizone was calculated using CalcuSyn software, version 2.1 (Biosoft, Cambridge, UK).
Serum and tissue distribution of AMB in uninfected (healthy) mice.
Female BALB/c mice (five per group) were intravenously (i.v.) injected via the tail vein with DSHemsPC-AMB-Lip and AmBisome, each of which contained 10 mg/kg AMB, in 5% dextrose. Control mice received an equivalent volume of 5% dextrose. Blood samples were collected from the orbital sinus of the mice at 1, 6, 12, and 24 h postadministration. After 24 h, the mice were sacrificed, blood samples were taken by heart puncture, and then the kidneys, heart, liver, spleen, and lungs were removed and weighed. The blood was allowed to coagulate at 4°C and centrifuged for 10 min at 15,400 × g to remove the serum. Serum and tissues samples were stored frozen at −80°C until the day of assay for AMB by a high-performance liquid chromatography (HPLC) method (25, 35). The chromatographic separation was achieved in less than 12 min on a reverse-phase (C18) column using an acetonitrile-acetic acid-water (52:4.3:43.7, vol/vol/vol) mixture as the mobile phase. The flow rate was 1 ml/min, and the effluent was monitored at 406 nm (19, 20).
AMB extraction from tissue.
Twenty-four hours after the last injection, the animals were sacrificed; the organs were then removed and transferred into tubes containing 1 ml methanol and zirconia beads (18). The samples were completely homogenized by bead beating (Bead Beater; Biospec, Bartlesville, OK) at 5,000 rpm. The homogenates were incubated for 1 h at room temperature. The samples were then vortexed, transferred to Eppendorf polypropylene centrifuge tubes, and centrifuged for 10 min at 14,000 rpm. Twenty microliters of the supernatant was injected into the HPLC system to assay for AMB.
Activity of DSHemsPC-AMB-Lip in a susceptible BALB/c mouse model of early CL lesions.
For the model of early lesions, 40 female BALB/c mice were subcutaneously (s.c.) inoculated in the left footpad with stationary-phase L. major promastigotes (2 × 106/50 μl) (33). Mice (10 per group) were treated over a 4-week period on days 1, 2, 4, 7, 14, 21, and 28 postinfection with i.v. injections of DSHemsPC-AMB (5 mg/kg), AmBisome (5 mg/kg), or Fungizone (1 mg/kg). Control mice (5 per group) received i.v. injections of either 5% dextrose or empty liposomes.
Serum and tissue distribution of AMB in model of early CL lesions.
Blood was collected by orbital sinus puncture at 1, 6, and 24 h after the start of injection on day 21. After 24 h, three mice from each group were sacrificed, blood samples were removed via heart puncture, and then the kidneys, heart, liver, and lungs were removed and weighed. The AMB concentration was measured by HPLC as described previously (34).
DSHemsPC-AMB-Lip in susceptible BALB/c mouse model of established CL lesions.
The left footpad of female BALB/c mice was inoculated s.c. with L. major promastigotes (2 × 106/50 μl) that had been harvested at stationary phase. In the model of established lesions, treatment was initiated after the presence of an established infection was confirmed at 3 weeks after parasite inoculation and after the first splenic parasite load was determined. Thirty mice (10 per group) were treated with DSHemsPC-AMB (5 mg/kg), AmBisome (5 mg/kg), or Fungizone (1 mg/kg) on days 23, 25, 27, 29, 31, 33, and 35 postinfection. Two control groups of mice (5 per group) received either 5% dextrose or empty liposomes.
Serum and tissue distribution of AMB in model of established CL lesions.
Sampling of blood from the orbital sinus was performed at 1, 6, and 24 h postadministration on day 35 postinfection. After 24 h, three mice from each group were sacrificed, blood specimens were taken by heart puncture, and then the kidneys, heart, liver, and lungs were removed and weighed. The AMB concentration was assayed by an HPLC method (25, 35).
In vivo antileishmanial activity.
The in vivo activity of DSHemsPC-AMB-Lip in both early and established CL footpad lesions in infected BALB/c mice was determined using a metric caliper (Mitutoyo, Japan) (33). The lesion size was calculated by subtracting the thickness of the uninfected contralateral footpad from that of the infected one.
Quantitative parasite burden.
The number of viable L. major parasites in the spleens and infected feet of the mice was evaluated using a limiting dilution assay (36). Mice were sacrificed at 3, 6, and 9 weeks postinfection, and the spleens and infected feet were aseptically removed. The spleens were then homogenized in 1 ml RPMI-FCS with a sterile syringe piston. The infected feet were transferred into tubes containing 1 ml RPMI-FCS and zirconia beads and were completely homogenized with a bead beater for 20 s in one cycle. The homogenates were diluted with the same medium in 8 serial 10-fold dilutions in flat-bottom 96-well microtiter plates containing a solid layer of rabbit blood agar in triplicate and kept at 25 ± 1°C for 7 days. The positive wells (in which motile parasites were present) and the negative wells (in which motile parasites were absent) were detected using an invert microscope (CETI, UK). The data reported are the calculated mean and standard error of mean for the last positive well multiplied by the dilution factor (35, 37). The number of viable L. major parasites in the spleens or infected feet of the mice was enumerated by a limiting dilution assay.
Statistical analysis.
A one-way analysis of variance statistical test was used to assess the significance of the differences among the various groups. In the case of a significant F value, a multiple-comparison Tukey test was used to compare the means for the different treatment groups. Results with P values of <0.05 were considered statistically significant.
We are grateful to the Vice Chancellor for Research, Mashhad University of Medical Sciences, Mashhad, Iran, for the financial support of this project (grant number 900050). In addition, the authors would like to gratefully acknowledge partial financial support of this investigation from NIH grant GM061851.
ACKNOWLEDGMENT
We declare that there is no conflict of interest regarding the amphotericin B formulations described in this publication. We are grateful to the Vice Chancellor for Research, Mashhad University of Medical Sciences, Mashhad, Iran, for the financial support of this project (grant number 900050). In addition, the authors would like to gratefully acknowledge partial financial support of this investigation from NIH grant GM061851.
REFERENCES
- 1.Croft S, Coombs G. 2003. Leishmaniasis—current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol 19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Groll A, Walsh T. 2002. Antifungal chemotherapy: advances and perspectives. Swiss Med Wkly 132:303–311. [DOI] [PubMed] [Google Scholar]
- 3.Gradoni L, Bryceson A, Desjeux P. 1995. Treatment of Mediterranean visceral leishmaniasis. Bull World Health Organ 73:191–197. [PMC free article] [PubMed] [Google Scholar]
- 4.Ng A, Wasan K, Lopez-Berestein G. 2002. Development of liposomal polyene antibiotics: an historical perspective. J Pharm Pharm Sci 6:67–83. [PubMed] [Google Scholar]
- 5.Ahsan F, Rivas I, Khan M, Torres SA. 2002. Targeting to macrophages: role of physicochemical properties of particulate carriers—liposomes and microspheres—on the phagocytosis by macrophages. J Control Release 79:29–40. doi: 10.1016/S0168-3659(01)00549-1. [DOI] [PubMed] [Google Scholar]
- 6.Thornton S, Wasan K, Piecuch A, Lynd L, Wasan E. 2010. Barriers to treatment for visceral leishmaniasis in hyperendemic areas: India, Bangladesh, Nepal, Brazil and Sudan. Drug Dev Ind Pharm 36:1312–1319. doi: 10.3109/03639041003796648. [DOI] [PubMed] [Google Scholar]
- 7.Barratt G, Bretagne S. 2006. Optimizing efficacy of amphotericin B through nanomodification. Int J Nanomed 2:301–313. [PMC free article] [PubMed] [Google Scholar]
- 8.Bekersky I, Fielding R, Dressler D, Lee J, Buell D, Walsh T. 2002. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob Agents Chemother 46:828–833. doi: 10.1128/AAC.46.3.828-833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yardley V, Croft S. 2000. A comparison of the activities of three amphotericin B lipid formulations against experimental visceral and cutaneous leishmaniasis. Int J Antimicrob Agents 13:243–248. doi: 10.1016/S0924-8579(99)00133-8. [DOI] [PubMed] [Google Scholar]
- 10.Gregoriadis G. 1991. Overview of liposomes. J Antimicrob Chemother 28:39–48. [DOI] [PubMed] [Google Scholar]
- 11.Yardley V, Croft S. 1997. Activity of liposomal amphotericin B against experimental cutaneous leishmaniasis. Antimicrob Agents Chemother 41:752–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iman M, Huang Z, Szoka F Jr, Jaafari M. 2011. Characterization of the colloidal properties, in vitro antifungal activity, antileishmanial activity and toxicity in mice of a di-stigma-steryl-hemi-succinoyl-glycero-phosphocholine liposome-intercalated amphotericin B. Int J Pharm 408:163–172. doi: 10.1016/j.ijpharm.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Z, Jaafari M, Szoka F Jr. 2009. Disterolphospholipids: nonexchangeable lipids and their application to liposomal drug delivery. Angew Chem Int Ed Engl 48:4146–4149. doi: 10.1002/anie.200900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deray G. 2002. Amphotericin B nephrotoxicity. J Antimicrob Chemother 49:37–41. doi: 10.1093/jac/49.suppl_1.37. [DOI] [PubMed] [Google Scholar]
- 15.Stone N, Bicanic T, Salim R, Hope W. 2016. Liposomal amphotericin B (AmBisome (®)): a review of the pharmacokinetics, pharmacodynamics, clinical experience and future directions. Drugs 76:485–500. doi: 10.1007/s40265-016-0538-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croft S, Yardley V. 2002. Chemotherapy of leishmaniasis. Curr Pharm Des 8:319–342. doi: 10.2174/1381612023396258. [DOI] [PubMed] [Google Scholar]
- 17.Mauël J. 1990. Macrophage-parasite interactions in Leishmania infections. J Leukoc Biol 47:187–193. [DOI] [PubMed] [Google Scholar]
- 18.Szoka F Jr, Milholland D, Barza M. 1987. Effect of lipid composition and liposome size on toxicity and in vitro fungicidal activity of liposome-intercalated amphotericin B. Antimicrob Agents Chemother 31:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupont B. 2002. Overview of the lipid formulations of amphotericin B. J Antimicrob Chemother 49(Suppl 1):31–36. [DOI] [PubMed] [Google Scholar]
- 20.Herbrecht R, Natarajan-Amé S, Nivoix Y, Letscher-Bru V. 2003. The lipid formulations of amphotericin B. Expert Opin Pharmacother 4:1277–1287. doi: 10.1517/14656566.4.8.1277. [DOI] [PubMed] [Google Scholar]
- 21.Bellmann R, Cleary JD, Sterba J. 2009. Clinical roundtable monograph: safety and efficacy of lipid-based amphotericin B. Clin Adv Hematol Oncol 7:1–8. [PubMed] [Google Scholar]
- 22.Adler-Moore J, Proffitt R. 2008. Amphotericin B lipid preparations: what are the differences? Clin Microbiol Infect 14:25–36. doi: 10.1111/j.1469-0691.2008.01979.x. [DOI] [PubMed] [Google Scholar]
- 23.Kan C, Yan J, Bittman R. 1992. Rates of spontaneous exchange of synthetic radiolabeled sterols between lipid vesicles. Biochemistry 31:1866–1874. doi: 10.1021/bi00121a040. [DOI] [PubMed] [Google Scholar]
- 24.Fahr A, van Hoogevest P, May S, Bergstrand N. 2005. Transfer of lipophilic drugs between liposomal membranes and biological interfaces: consequences for drug delivery. Eur J Pharm Sci 26:251–265. doi: 10.1016/j.ejps.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Espada R, Valdespina S, Alfonso C, Rivas G, Ballesteros M, Torrado J. 2008. Effect of aggregation state on the toxicity of different amphotericin B preparations. Int J Pharm 361:64–69. doi: 10.1016/j.ijpharm.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 26.Larabi M, Pons F, Appel M, Gulik A, Schlatter J, Bouvet S, Barratt G. 2004. Study of the toxicity of a new lipid complex formulation of amphotericin B. J Antimicrob Chemother 53:81–88. [DOI] [PubMed] [Google Scholar]
- 27.Jensen G, Skenes C, Bunch T, Weissman C, Amirghahari N, Satorius A, Moynihan K, Eley C. 1999. Determination of the relative toxicity of amphotericin B formulations: a red blood cell potassium release assay. Drug Deliv 6:81–88. doi: 10.1080/107175499266995. [DOI] [Google Scholar]
- 28.Butler W, Cotlove E. 1971. Increased permeability of human erythrocytes induced by amphotericin B. J Infect Dis 123:341–350. doi: 10.1093/infdis/123.4.341. [DOI] [PubMed] [Google Scholar]
- 29.Huang Z, Szoka FC Jr. 2008. Sterol-modified phospholipids: cholesterol and phospholipid chimeras with improved biomembrane properties. J Am Chem Soc 130:15702–15712. doi: 10.1021/ja8065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCurdy D, Frederic M, Elkinton J. 1968. Renal tubular acidosis due to amphotericin B. N Engl J Med 278:124–130. doi: 10.1056/NEJM196801182780302. [DOI] [PubMed] [Google Scholar]
- 31.Cagnoni PJ, Walsh TJ, Prendergast MM, Bodensteiner D, Hiemenz S, Greenberg RN, Arndt CA, Schuster M, Seibel N, Yeldandi V, Tong KB. 2000. Pharmacoeconomic analysis of liposomal amphotericin B versus conventional amphotericin B in the empirical treatment of persistently febrile neutropenic patients. J Clin Oncol 18:2476–2483. doi: 10.1200/JCO.2000.18.12.2476. [DOI] [PubMed] [Google Scholar]
- 32.Olson JA, Adler-Moore JP, Jensen GM, Schwartz J, Dignani MC, Proffitt RT. 2008. Comparison of the physicochemical, antifungal, and toxic properties of two liposomal amphotericin B products. Antimicrob Agents Chemother 52:259–268. doi: 10.1128/AAC.00870-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaafari M, Bavarsad N, Bazzaz B, Samiei A, Soroush D, Ghorbani S, Heravi M, Khamesipour A. 2009. Effect of topical liposomes containing paromomycin sulfate in the course of Leishmania major infection in susceptible BALB/c mice. Antimicrob Agents Chemother 53:2259–2265. doi: 10.1128/AAC.01319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tremblay C, Barza M, Fiore C, Szoka F. 1984. Efficacy of liposome-intercalated amphotericin B in the treatment of systemic candidiasis in mice. Antimicrob Agents Chemother 26:170–173. doi: 10.1128/AAC.26.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith P, Olson J, Constable D, Schwartz J, Proffitt R, Adler-Moore J. 2007. Effects of dosing regimen on accumulation, retention and prophylactic efficacy of liposomal amphotericin B. J Antimicrob Chemother 59:941–951. doi: 10.1093/jac/dkm077. [DOI] [PubMed] [Google Scholar]
- 36.Titus R, Marchand M, Boon T, Louis J. 1985. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol 7:545–555. doi: 10.1111/j.1365-3024.1985.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 37.Jaafari M, Badiee A, Khamesipour A, Samiei A, Soroush D, Kheiri M, Barkhordari F, McMaster W, Mahboudi F. 2007. The role of CpG ODN in enhancement of immune response and protection in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63) encapsulated in cationic liposome. Vaccine 25:6107–6117. doi: 10.1016/j.vaccine.2007.05.009. [DOI] [PubMed] [Google Scholar]