

Abstract
Transition-metal-based molecular complexes are a class of catalyst materials for electrochemical CO2 reduction to CO that can be rationally designed to deliver high catalytic performance. One common mechanistic feature of these electrocatalysts developed thus far is an electrogenerated reduced metal center associated with catalytic CO2 reduction. Here we report a heterogenized zinc–porphyrin complex (zinc(II) 5,10,15,20-tetramesitylporphyrin) as an electrocatalyst that delivers a turnover frequency as high as 14.4 site–1 s–1 and a Faradaic efficiency as high as 95% for CO2 electroreduction to CO at −1.7 V vs the standard hydrogen electrode in an organic/water mixed electrolyte. While the Zn center is critical to the observed catalysis, in situ and operando X-ray absorption spectroscopic studies reveal that it is redox-innocent throughout the potential range. Cyclic voltammetry indicates that the porphyrin ligand may act as a redox mediator. Chemical reduction of the zinc–porphyrin complex further confirms that the reduction is ligand-based and the reduced species can react with CO2. This represents the first example of a transition-metal complex for CO2 electroreduction catalysis with its metal center being redox-innocent under working conditions.
Short abstract
A zinc−porphyrin complex, with the redox-innocent Zn ion binding reaction intermediates and the ligand mediating electron transfer, catalyzes CO2 electroreduction to CO in high Faradaic efficiency.
Introduction
Environmental problems associated with the increasing level of atmospheric CO2 concentration have generated interest in converting CO2 to value-added products. Electrocatalytic reduction of CO2 is one attractive option as it operates under ambient conditions and could utilize electricity generated from renewable energy sources.1,2 Selective CO2 reduction to CO is one promising process as CO is an important feedstock chemical for production of methanol and liquid hydrocarbons.3 A wide range of materials, including metals (e.g., Au, Ag, and Zn),4−6 metal oxides (e.g., SnO2),7 metal dichalcogenides (e.g., MoS2 and WSe2),8,9 and transition-metal-based molecular complexes (e.g., Mn-, Fe-, Co-, Ni-, Cu-, Ru-, and Re-based complexes),1,10−12 have been explored for catalyzing electroreduction of CO2 to CO.
Compared to catalysts based on extended structures, transition-metal complexes bear distinct merits of well-defined molecular structures and atomic-level structural tunability that allow for rational catalytic performance optimization and mechanistic study. Thus far, a number of metal-complex-based CO2 electroreduction catalysts with interesting properties have been reported. For example, Savéant et al. appended phenolic hydroxyl groups to the second coordination sphere of an Fe porphyrin and dramatically boosted the catalytic activity for CO2 reduction to CO.13 Chang et al. realized significantly increased turnover rate for CO2 reduction to CO by building a Co porphyrin structure into a metal organic framework and a covalent organic framework.14,15 We introduced cyano substituents to a Co phthalocyanine and anchored the molecules on carbon nanotubes to render a hybrid electrocatalyst material that can reduce CO2 to CO with high current density comparable to the best heterogeneous catalysts and high per site activity comparable to the best molecular catalysts.16 We also discovered a Cu–porphyrin-based catalyst that can electrochemically reduce CO2 to hydrocarbons with a significant yield.17 In all these cases, the metal center accepts electrons from the working electrode by going to a lower valence state and then transfers the electrons to a bound CO2 species to catalyze the reduction reaction.
Here, we report the first example of a transition-metal complex that catalyzes electrochemical CO2 reduction with its metal center being redox-innocent under working conditions. We prepare a heterogenized electrocatalyst using a Zn–porphyrin complex, zinc(II) 5,10,15,20-tetramesitylporphyrin (PorZn). At −1.7 V vs the standard hydrogen electrode (SHE) in a mixed N,N-dimethylformamide (DMF)/H2O electrolyte system, the catalyst exhibits high selectivity and activity for CO2 reduction to CO, achieving a turnover frequency (TOF) of 14.4 site–1 s–1 and a Faradaic efficiency as high as 95%. The high Faradaic efficiency can be retained throughout 4 h of continuous electrolysis. In situ and operando X-ray absorption spectroscopy (XAS) reveals that the Zn(II) center maintains its oxidation state throughout the examined potential range. Chemical reduction of the PorZn complex further confirms that the ligand is redox-active and the reduced PorZn species can react with CO2. To the best of our knowledge, this is also the first Zn-based molecular catalyst that is able to electrochemically reduce CO2 with high product selectivity.18,19
Results and Discussion
The Zn porphyrin complex PorZn and its precursor H2Por (Figure 1A) were synthesized following a reported procedure (see the Supporting Information for detailed synthetic and purification procedures).20,21 Purified PorZn was characterized by mass spectrometry (Figure S1A) and 1H nuclear magnetic resonance (NMR) spectroscopy (Figure S1B). Elemental analysis (Table S1) and inductively coupled plasma mass spectrometry (ICP-MS) measurements (Table S2) both reveal no detectable impurities in the PorZn material. The PorZn catalyst electrode was prepared following our previous study on a heterogenized Cu porphyrin catalyst.17 Specifically, the PorZn complex was dissolved in dichloromethane (CH2Cl2) and then deposited on a commercially available polytetrafluoroethylene (PTFE)-treated carbon fiber paper with a porous coating layer as a gas diffusion layer (see the Supporting Information for details). The porous coating layer consisted of carbon nanoparticles with an average diameter of ca. 50 nm, as shown by scanning electron microscopy (SEM) (Figure 1B). The deposited PorZn adopts a morphology of cubic crystals, as confirmed by SEM imaging and energy-dispersive X-ray spectroscopy (EDS) mapping (Figures 1C and 1D).
Figure 1.

(A) The chemical structures of the Zn–porphyrin complex PorZn and its metal-free precursor H2Por. (B) SEM image of the porous coating layer on the carbon fiber paper. (C) SEM image of PorZn deposited on carbon fiber paper. (D) EDS map of Zn for the area in panel C.
The PorZn electrodes were investigated for electrochemical CO2 reduction at various potentials in a CO2-saturated solvent system consisting of 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in DMF/H2O (VH2O:VDMF = 1:9) (see Figure S2 for the electrochemical setup). The electrolyte solution was analyzed by 1H NMR after electrolysis, and no liquid product was observed (Figure S3). The gaseous products were analyzed by gas chromatography–mass spectrometry (GC–MS) (Figure S4) during electrolysis, and only H2 and CO were detected. The partial current densities for CO production along with the corresponding Faradaic efficiencies at different potentials are plotted in Figure 2A. At potentials more positive than −1.4 V vs SHE, H2 is the only product. CO2-to-CO conversion starts to be observable at −1.4 V, with a CO Faradaic efficiency of 22%. Both the current density and CO Faradaic efficiency increase as more negative potentials are applied. The maximum CO Faradaic efficiency of 95% is achieved at −1.7 V vs SHE with a current density of ca. 2.1 mA/cm2. At −1.7 V, both the CO Faradaic efficiency and total current density can be retained for at least 4 h (Figure 2B). Electrolysis under the same conditions was performed using blank carbon fiber paper as the working electrode, and H2 was the only product, confirming that PorZn is responsible for the observed electrocatalytic conversion of CO2 to CO (Table S3).
Figure 2.
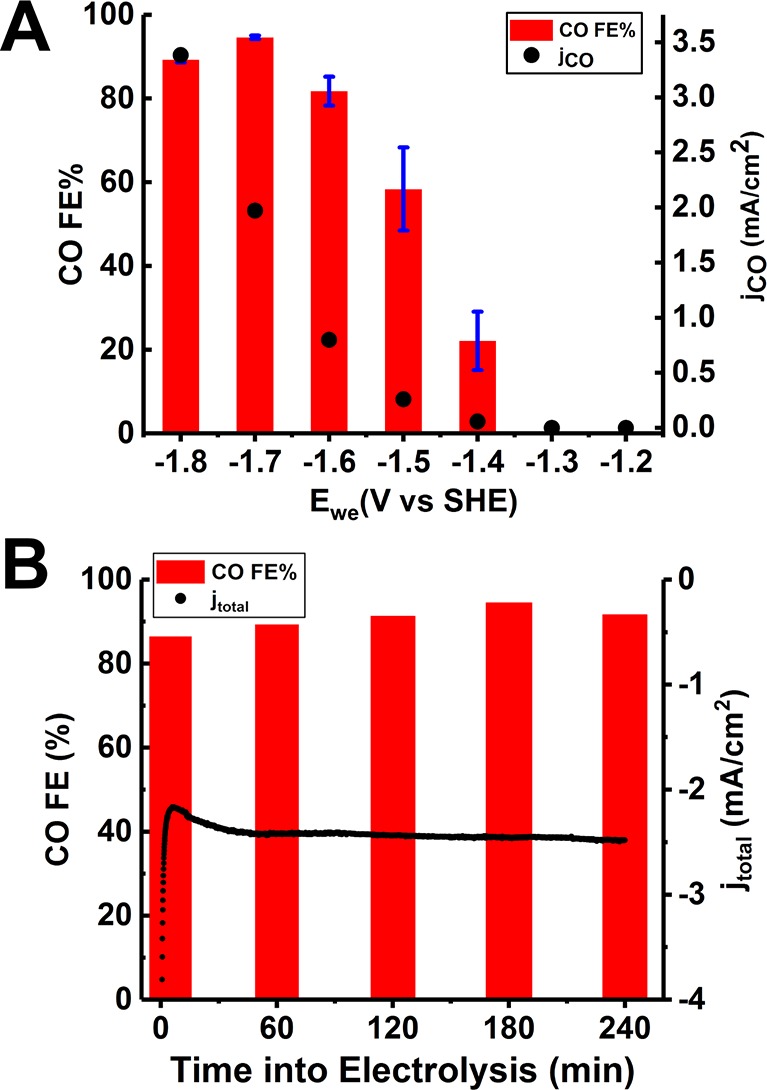
Electrochemical CO2 reduction catalyzed by the PorZn electrode in 0.1 M TBAPF6 DMF/H2O solution. (A) CO Faradaic efficiencies and CO partial current densities at different potentials averaged from three measurements. (B) CO Faradaic efficiencies and total current densities after 5, 60, 120, 180, and 240 min of electrolysis at −1.7 V vs SHE.
Control experiments were designed and performed to verify the molecular integrity of PorZn after electrocatalysis. After 1 h of continuous electrolysis at −1.7 V vs SHE, the PorZn electrode was removed from the electrochemical cell and thoroughly rinsed with CH2Cl2. The UV–vis spectrum of the obtained solution matches with that of pure PorZn dissolved in CH2Cl2 (Figure S5). No distinct absorption peaks of free-base porphyrin could be found. Subsequently, the electrolysis was continued with the used electrolyte using a blank carbon fiber paper as the working electrode. The total current density is ca. 20% of that of the original PorZn electrode (Figure S6), and little CO can be detected in the product (Table S3). Comparison of SEM images (Figure S7), EDS maps (Figure S8), and X-ray photoelectron spectroscopy (XPS) spectra (Figure S9) of the PorZn catalyst electrode before and after 1 h of electrolysis at −1.7 V shows that no morphological, structural, or compositional changes take place during electrocatalysis. These results are all consistent with our suggestion that the PorZn molecular structure is the active species that catalyzes the CO2 electroreduction to CO.
Given that Zn metal is known to be catalytically active for electrochemical CO2 reduction to CO,22−24 it is vital to exclude the formation of metallic Zn and its catalyzing the reaction. To monitor changes in the oxidation state and electronic structure of the Zn center under electrochemical CO2 reaction conditions, we performed in situ and operando XAS measurements using the reaction cell as described in our previous study.25 This technique has been successfully used to probe the oxidation state of Co in a solid-state cobalt–porphyrin-based catalyst material during electrocatalysis.14 No visible changes in the Zn K-edge X-ray absorption near edge structure (XANES) spectra are observed as the working electrode potential is adjusted from open circuit voltage (OCV) to −1.7 V vs SHE (Figure 3A) and then back to +0.2 V vs SHE, indicating that the oxidation state of the Zn center in the PorZn catalyst does not change in the examined potential range. A comparison to standard ZnO and Zn samples allows us to determine the oxidation state of Zn in the PorZn catalyst to be 2+, as the rising edges of the ZnO and PorZn XANES spectra overlap, which is also confirmed by the derivatives of the XANES spectra (Figure S10D). These results not only further exclude the formation of metallic zinc on the electrode during electrolysis but also suggest that the Zn center remains in its 2+ oxidation state throughout the electrolysis, which could be explained in part by the fact that Zn(II) already has a full 3d electron shell. To examine the local structure of the Zn ion in PorZn under reaction conditions, extended X-ray absorption fine structure (EXAFS) spectra were measured up to k = 13 Å–1 (Figures S10B and S10C). The high-quality data enable us to examine the local structures of PorZn. As shown in Figure 3B and Figure S10B, minimal changes in the Zn coordination number and bond distances are observed at the potentials examined and throughout the electrolysis. The minor changes in the Zn local structure could be due to reduction of the porphyrin ligand or binding of molecules on the Zn site. In another control experiment, we find that, when deposited on the carbon fiber paper electrode, the free-base porphyrin H2Por does not catalyze CO2 reduction to CO at the potentials we examined for PorZn electrodes (Table S4). This indicates that the Zn center is critical to the catalytic activity of PorZn even though it remains redox-innocent during electrocatalysis.
Figure 3.
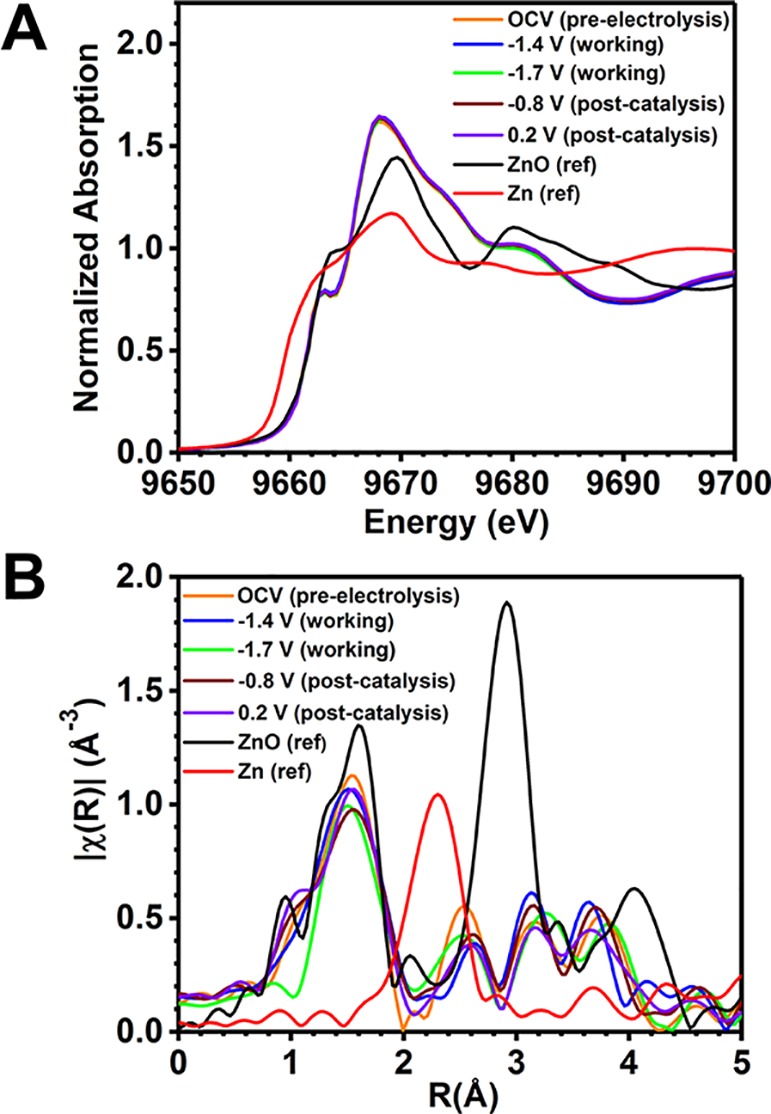
(A) Zn K-edge XANES spectra and (B) Fourier transforms of Zn K-edge EXAFS spectra of the PorZn catalyst electrode at different potentials (V vs SHE). ZnO and Zn are used as references.
We therefore conclude from the analyses that the Zn center does not experience redox reactions during the CO2 reduction process, but is still indispensable for the catalysis to take place. This feature is entirely distinct from all other transition-metal-based molecular electrocatalysts for CO2-to-CO conversion reported to date, where reduction of the metal center is correlated with catalysis of CO2 reduction (e.g., electrochemically generated Mn(0), Co(I), Fe(0), Ni(I), Ru(0), and Re(0)),26−32 although sometimes assignment of formal oxidation state to the metal center is less straightforward.33 To the best of our knowledge, this is the first report of a CO2 electroreduction molecular catalyst with a redox-innocent metal center that delivers high product selectivity. The Zn(II) center is likely to bind CO2 or CO2-derived reaction intermediates as a Lewis acid and thereby act in a way similar to the Mg(II) center in RuBisCo during biological CO2 fixation.34 Notably, a recent study highlighted an organometallic Zn complex that can activate CO2 for electroreduction through Lewis acid–base interactions.35 The exact role of the Zn center in our catalyst structure warrants further investigation.
The porphyrin ligand of PorZn is thus likely to be responsible for the two-electron reduction of CO2 to CO. Figure 4 shows the cyclic voltammograms (CVs) of PorZn in CO2- and Ar-saturated electrolyte. During the cathodic scan, the CV of PorZn in CO2-saturated electrolyte features a reduction wave that starts near −1.4 V vs SHE, matching with the observation of CO2 reduction to CO at the potential. The CV in Ar-saturated electrolyte exhibits a similar wave associated with reduction of PorZn, possibly coupled with protonation.36−39 The most striking difference between the two CVs is the returning wave pattern. The CV in CO2-saturated electrolyte shows a weak anodic wave around −1.45 V corresponding to the reduction wave around −1.6 V. In contrast, the CV in Ar-saturated electrolyte shows three anodic waves around −1.53 V, −1.17 V, and −0.43 V vs SHE. A similar three-wave anodic pattern was observed in a study on zinc(II) 5,10,15,20-tetraphenylporphyrin investigated at a similar scan rate.38 These anodic waves are proposed to correspond to sequential oxidation and deprotonation of the reduced porphyrin ring based on literature precedent.38 Note that the CVs of H2Por manifest similar redox features (Figure S11), suggesting that these redox events are indeed ligand-based. These data are consistent with the proposed mechanism of porphyrin-mediated charge transfer.
Figure 4.
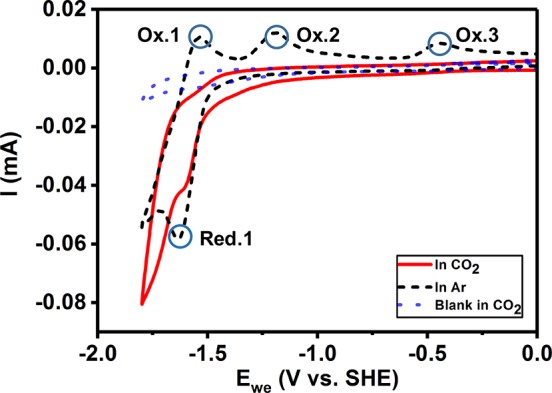
Cyclic voltammograms of PorZn coated on glassy carbon electrodes in Ar- (black) and CO2-saturated (red) electrolyte. The blue dotted trace shows the CV of blank glassy carbon electrode in CO2-saturated electrolyte solution.
To gain further mechanistic insights into PorZn-catalyzed CO2 reduction, we performed chemical reduction of PorZn in solution under inert conditions, using one or two equivalents of sodium naphthalenide in tetrahydrofuran (NaNap, ca. −2.4 V vs SHE in THF40) as the chemical reductant. The UV–vis spectra of the reduced PorZn species exhibit absorption bands at 710, 820, and 920 nm (Figure 5), which are characteristic features of transiently generated zinc–porphyrin compounds with reduced ligands.36,41 Upon exposure of the reduced PorZn species to air, rapid regeneration of PorZn was observed by UV–vis spectroscopy (Figure 5 and Figure S12). The instability of these reduced species has precluded full characterization thus far. However, when prepared in situ, the reduced species can react with CO2 to produce PorZn and unidentified species, according to the acquired UV–vis, NMR, and IR spectroscopy results (Figures S13–S15). These experiments support that PorZn undergoes ligand-centered reduction and that the reduced species can react with CO2 to form not only PorZn but also additional species as a result of chemical reactions that involve electron transfer from the ligand-reduced PorZn to CO2. The exact mechanism of this interesting reactivity warrants further investigation. For more detailed discussion and analysis of the IR, NMR, and UV–vis spectroscopy results, please refer to the Supporting Information.
Figure 5.
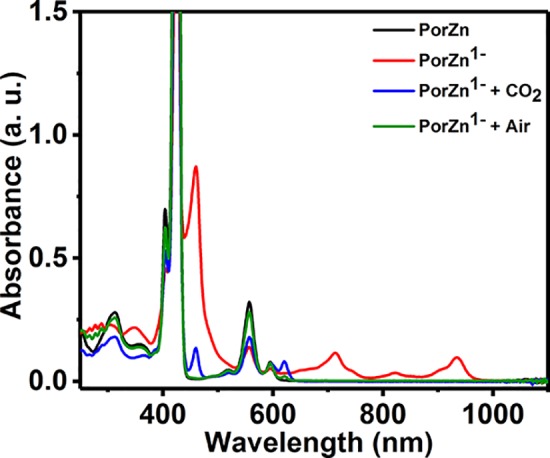
Absorption spectra of PorZn, its reduced species, and the reduced species after exposure to CO2 or air. The spectra were taken in THF.
Conclusion
In summary, we have discovered the first Zn-based molecularly structured catalyst for electrochemical reduction of CO2 to CO with high product selectivity. The Zn(II) center is shown to be redox-inactive yet still essential to the catalysis, which distinguishes our catalyst from other reported transition-metal-based molecular catalysts. The interesting observation that the ligand acts as the redox center during CO2 electroreduction calls for further mechanistic studies.
Acknowledgments
The work is supported by National Science Foundation (Grant CHE-1651717), American Chemical Society Petroleum Research Fund, and the Global Innovation Initiative from Institute of International Education. Funding was also provided by a generous donation from the TomKat Foundation. Additional support from the U.S. Department of Energy, Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science (DE-FG02-07ER15909), is gratefully acknowledged. The authors thank Interfacial Processes Group at Chemical Science and Engineering Division of Argonne National Lab for the onsite electrochemical instrument support. 5BM-D of DND-CAT was supported through E. I. duPont de Nemours & Co., Northwestern University, and The Dow Chemical Company. The use of Advanced Photon Source of Argonne National Laboratory was supported by Department of Energy under Contract No. DE-AC02-06CH11357. Z.F. thanks the Callahan Faculty Scholar Endowment Fund from Oregon State University. D.L.J. B. acknowledges the support from The Netherlands Organization for Scientific Research (Rubicon Postdoctoral Fellowship 680-50-1517). The authors also thank Jonas Karosas for assistance with ICP-MS measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00160.
Experimental details, characterization data, and discussion on the chemical reduction results (PDF)
Author Contributions
§ Y.W., J.J., and Z.W. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Qiao J.; Liu Y.; Hong F.; Zhang J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43 (2), 631–675. 10.1039/C3CS60323G. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Robert M.; Savéant J.-M. Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion. Acc. Chem. Res. 2015, 48 (12), 2996–3006. 10.1021/acs.accounts.5b00262. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Jacobs G.; Sparks D. E.; Dry M. E.; Davis B. H. CO and CO2 hydrogenation study on supported cobalt Fischer–Tropsch synthesis catalysts. Catal. Today 2002, 71 (3–4), 411–418. 10.1016/S0920-5861(01)00468-0. [DOI] [Google Scholar]
- Lu Q.; Rosen J.; Zhou Y.; Hutchings G. S.; Kimmel Y. C.; Chen J. G.; Jiao F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. 10.1038/ncomms4242. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Rosen J.; Jiao F. Nanostructured Metallic Electrocatalysts for Carbon Dioxide Reduction. ChemCatChem 2015, 7 (1), 38–47. 10.1002/cctc.201402669. [DOI] [Google Scholar]
- Hori Y.Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas C. G., White R. E., Gamboa-Aldeco M. E., Eds.; Springer New York: New York, NY, 2008; pp 89–189. [Google Scholar]
- Chen Y.; Kanan M. W. Tin Oxide Dependence of the CO2 Reduction Efficiency on Tin Electrodes and Enhanced Activity for Tin/Tin Oxide Thin-Film Catalysts. J. Am. Chem. Soc. 2012, 134 (4), 1986–1989. 10.1021/ja2108799. [DOI] [PubMed] [Google Scholar]
- Asadi M.; Kumar B.; Behranginia A.; Rosen B. A.; Baskin A.; Repnin N.; Pisasale D.; Phillips P.; Zhu W.; Haasch R.; Klie R. F.; Král P.; Abiade J.; Salehi-Khojin A. Robust carbon dioxide reduction on molybdenum disulphide edges. Nat. Commun. 2014, 5, 4470. 10.1038/ncomms5470. [DOI] [PubMed] [Google Scholar]
- Asadi M.; Kim K.; Liu C.; Addepalli A. V.; Abbasi P.; Yasaei P.; Phillips P.; Behranginia A.; Cerrato J. M.; Haasch R.; Zapol P.; Kumar B.; Klie R. F.; Abiade J.; Curtiss L. A.; Salehi-Khojin A. Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquid. Science 2016, 353 (6298), 467–470. 10.1126/science.aaf4767. [DOI] [PubMed] [Google Scholar]
- Collin J. P.; Sauvage J. P. Electrochemical reduction of carbon dioxide mediated by molecular catalysts. Coord. Chem. Rev. 1989, 93 (2), 245–268. 10.1016/0010-8545(89)80018-9. [DOI] [Google Scholar]
- Savéant J.-M. Molecular Catalysis of Electrochemical Reactions. Mechanistic Aspects. Chem. Rev. 2008, 108 (7), 2348–2378. 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- Takeda H.; Cometto C.; Ishitani O.; Robert M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7 (1), 70–88. 10.1021/acscatal.6b02181. [DOI] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Savéant J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338 (6103), 90–94. 10.1126/science.1224581. [DOI] [PubMed] [Google Scholar]
- Lin S.; Diercks C. S.; Zhang Y.-B.; Kornienko N.; Nichols E. M.; Zhao Y.; Paris A. R.; Kim D.; Yang P.; Yaghi O. M.; Chang C. J. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 2015, 349 (6253), 1208–1213. 10.1126/science.aac8343. [DOI] [PubMed] [Google Scholar]
- Kornienko N.; Zhao Y.; Kley C. S.; Zhu C.; Kim D.; Lin S.; Chang C. J.; Yaghi O. M.; Yang P. Metal–Organic Frameworks for Electrocatalytic Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2015, 137 (44), 14129–14135. 10.1021/jacs.5b08212. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Wu Z.; Zhang X.; Li L.; Li Y.; Xu H.; Li X.; Yu X.; Zhang Z.; Liang Y.; Wang H. Highly selective and active CO2 reduction electrocatalysts based on cobalt phthalocyanine/carbon nanotube hybrid structures. Nat. Commun. 2017, 8, 14675. 10.1038/ncomms14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Z.; Jiang J.; Wu Y.; Wu Z.; Guo X.; Materna K. L.; Liu W.; Batista V. S.; Brudvig G. W.; Wang H. Electrochemical CO2 Reduction to Hydrocarbons on a Heterogeneous Molecular Cu Catalyst in Aqueous Solution. J. Am. Chem. Soc. 2016, 138 (26), 8076–8079. 10.1021/jacs.6b04746. [DOI] [PubMed] [Google Scholar]
- Furuya N.; Matsui K. Electroreduction of carbon dioxide on gas-diffusion electrodes modified by metal phthalocyanines. J. Electroanal. Chem. Interfacial Electrochem. 1989, 271 (1), 181–191. 10.1016/0022-0728(89)80074-9. [DOI] [Google Scholar]
- Sonoyama N.; Kirii M.; Sakata T. Electrochemical reduction of CO2 at metal-porphyrin supported gas diffusion electrodes under high pressure CO2. Electrochem. Commun. 1999, 1 (6), 213–216. 10.1016/S1388-2481(99)00041-7. [DOI] [Google Scholar]
- Lindsey J. S.; Wagner R. W. Investigation of the synthesis of ortho-substituted tetraphenylporphyrins. J. Org. Chem. 1989, 54 (4), 828–836. 10.1021/jo00265a021. [DOI] [Google Scholar]
- Fang Y.; Koszelewski D.; Kadish K. M.; Gryko D. T. Facile electrosynthesis of [small pi]-extended porphyrins. Chem. Commun. 2014, 50 (64), 8864–8867. 10.1039/C4CC02759K. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Wakebe H.; Tsukamoto T.; Koga O. Electrocatalytic Process of Co Selectivity in Electrochemical Reduction of CO2 at Metal-Electrodes in Aqueous-Media. Electrochim. Acta 1994, 39 (11–12), 1833–1839. 10.1016/0013-4686(94)85172-7. [DOI] [Google Scholar]
- Hori Y.; Kikuchi K.; Suzuki S. Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 1985, 14 (11), 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Won D. H.; Shin H.; Koh J.; Chung J.; Lee H. S.; Kim H.; Woo S. I. Highly Efficient, Selective, and Stable CO2 Electroreduction on a Hexagonal Zn Catalyst. Angew. Chem., Int. Ed. 2016, 55 (32), 9297–9300. 10.1002/anie.201602888. [DOI] [PubMed] [Google Scholar]
- Wei C.; Feng Z.; Baisariyev M.; Yu L.; Zeng L.; Wu T.; Zhao H.; Huang Y.; Bedzyk M. J.; Sritharan T.; Xu Z. J. Valence Change Ability and Geometrical Occupation of Substitution Cations Determine the Pseudocapacitance of Spinel Ferrite XFe2O4 (X = Mn, Co, Ni, Fe). Chem. Mater. 2016, 28 (12), 4129–4133. 10.1021/acs.chemmater.6b00713. [DOI] [Google Scholar]
- Bourrez M.; Molton F.; Chardon-Noblat S.; Deronzier A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem., Int. Ed. 2011, 50 (42), 9903–9906. 10.1002/anie.201103616. [DOI] [PubMed] [Google Scholar]
- Hammouche M.; Lexa D.; Savéant J. M.; Momenteau M. Catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. J. Electroanal. Chem. Interfacial Electrochem. 1988, 249 (1), 347–351. 10.1016/0022-0728(88)80372-3. [DOI] [Google Scholar]
- Beley M.; Collin J.-P.; Ruppert R.; Sauvage J.-P. Nickel(II)-cyclam: an extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc., Chem. Commun. 1984, 19, 1315–1316. 10.1039/c39840001315. [DOI] [Google Scholar]
- Ishida H.; Tanaka K.; Tanaka T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the formation of CO and HCOO. Organometallics 1987, 6 (1), 181–186. 10.1021/om00144a033. [DOI] [Google Scholar]
- Sullivan B. P.; Bolinger C. M.; Conrad D.; Vining W. J.; Meyer T. J. One- and two-electron pathways in the electrocatalytic reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2[prime or minute]-bipyridine). J. Chem. Soc., Chem. Commun. 1985, 20, 1414–1416. 10.1039/C39850001414. [DOI] [Google Scholar]
- Isse A. A.; Gennaro A.; Vianello E.; Floriani C. Electrochemical reduction of carbon dioxide catalyzed by [CoI(salophen)Li]. J. Mol. Catal. 1991, 70 (2), 197–208. 10.1016/0304-5102(91)80161-U. [DOI] [Google Scholar]
- Thoi V. S.; Chang C. J. Nickel N-heterocyclic carbene-pyridine complexes that exhibit selectivity for electrocatalytic reduction of carbon dioxide over water. Chem. Commun. 2011, 47 (23), 6578–6580. 10.1039/c1cc10449g. [DOI] [PubMed] [Google Scholar]
- Römelt C.; Song J.; Tarrago M.; Rees J. A.; van Gastel M.; Weyhermüller T.; DeBeer S.; Bill E.; Neese F.; Ye S. Electronic Structure of a Formal Iron(0) Porphyrin Complex Relevant to CO2 Reduction. Inorg. Chem. 2017, 56 (8), 4745–4750. 10.1021/acs.inorgchem.7b00401. [DOI] [PubMed] [Google Scholar]
- Cleland W. W.; Andrews T. J.; Gutteridge S.; Hartman F. C.; Lorimer G. H. Mechanism of Rubisco: The Carbamate as General Base. Chem. Rev. 1998, 98 (2), 549–562. 10.1021/cr970010r. [DOI] [PubMed] [Google Scholar]
- Donovan E. S.; Barry B. M.; Larsen C. A.; Wirtz M. N.; Geiger W. E.; Kemp R. A. Facilitated carbon dioxide reduction using a Zn(ii) complex. Chem. Commun. 2016, 52 (8), 1685–1688. 10.1039/C5CC07318A. [DOI] [PubMed] [Google Scholar]
- Closs G. L.; Closs L. E. Negative Ions of Porphin Metal Complexes. J. Am. Chem. Soc. 1963, 85 (6), 818–819. 10.1021/ja00889a038. [DOI] [Google Scholar]
- Balducci G.; Chottard G.; Gueutin C.; Lexa D.; Saveant J.-M. Electrochemistry of iron(I) porphyrins in the presence of carbon monoxide. Comparison with zinc porphyrins. Inorg. Chem. 1994, 33 (9), 1972–1978. 10.1021/ic00087a038. [DOI] [Google Scholar]
- Lanese J. G.; Wilson G. S. Electrochemical Studies of Zinc Tetraphenylporphin. J. Electrochem. Soc. 1972, 119 (8), 1039–1043. 10.1149/1.2404391. [DOI] [Google Scholar]
- Lin C.-l.; Fang M.-Y.; Cheng S.-H. Substituent and axial ligand effects on the electrochemistry of zinc porphyrins. J. Electroanal. Chem. 2002, 531 (2), 155–162. 10.1016/S0022-0728(02)01056-2. [DOI] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96 (2), 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- Parkhots O. P.; Ivashin N. V. Study of the structure and spectral properties of radical anions of Zn complexes of porphyrins by the method of density functional theory. Opt. Spectrosc. 2009, 106 (2), 204–212. 10.1134/S0030400X0902009X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.