

Abstract
Inspired by the ability of the sea cucumber to (reversibly) increase the stiffness of its dermis upon exposure to a stimulus, we herein report a stimuli-responsive nanocomposite that can reversibly increase its stiffness upon exposure to warm water. Nanocomposites composed of cellulose nanocrystals (CNCs) that are grafted with a lower critical solution temperature (LCST) polymer embedded within a poly(vinyl acetate) (PVAc) matrix show a dramatic increase in modulus, for example, from 1 to 350 MPa upon exposure to warm water, the hypothesis being that grafting the polymers from the CNCs disrupts the interactions between the nanofibers and minimizes the mechanical reinforcement of the film. However, exposure to water above the LCST leads to the collapse of the polymer chains and subsequent stiffening of the nanocomposite as a result of the enhanced CNC interactions. Backing up this hypothesis are energy conserving dissipative particle dynamics (EDPD) simulations which show that the attractive interactions between CNCs are switched on upon the temperature-induced collapse of the grafted polymer chains, resulting in the formation of a percolating reinforcing network.
Short abstract
Sea cucumber dermis inspired thermally stiffening polymer nanocomposites have been developed, using CNCs grafted with a lower critical solution temperature (LCST) polymer as the nanofiller.
Introduction
“Smart” materials with the ability to dramatically alter their mechanical properties have attracted attention in recent years, as they allow access to a range of responsive/adaptive materials that exhibit properties such as shape memory, actuation, and self-healing.1−7 Temperature is a commonly utilized stimulus that usually results in a reduction in the material’s mechanical strength upon the application of heat. Inspired by the mechanical adaptability of the sea cucumber dermis,8 we have previously reported chemosoftening CNC composites that exhibit dramatic softening upon exposure to an aqueous environment and body temperature.8−10 For example, PVAc/CNC composites exhibit mechanical switching between the dry state (∼5 GPA, room temperature) and the wet state (∼10 MPa, 37 °C) caused by water plasticization of the PVAc matrix combined with “switching off” the interactions between CNCs (and CNCs and matrix) through water solvation of the CNC surface.8 Here we show that this switching behavior can be reprogrammed to access a new class of stimuli-responsive materials that start soft (<1 MPa) and significantly stiffen (>300 MPa) upon exposure to water and heat. This is achieved by grafting lower critical solution temperature (LCST) polymers to the CNCs, which are then embedded in a poly(vinyl acetate) (PVAc) matrix. CNCs are rigid rodlike single crystals of native cellulose and have been incorporated into numerous polymer matrices on account of their inherent properties such as high stiffness, high strength, and low density. In addition, CNCs are also sustainable, biocompatible, and biodegradable and have been investigated in a number of application areas, including coatings, cosmetics, medical implants, drug delivery, and pharmaceuticals, to name a few.11−15 Using a combination of experimental results and computational modeling, the data shows that the stiffening is related to the heat-induced collapse of the grafted polymers, which results in the formation of a percolating reinforcing network. This process is fully reversible, and the switching temperature can be controlled by the nature of the grafted polymer, allowing access to composites that are soft in 25 °C water but stiffen in a biologically relevant environment at 37 °C. This unprecedented behavior opens the door to these materials being used as reinforcing medical implants, e.g., to aid in guided bone regeneration16 or in craniofacial implants.17
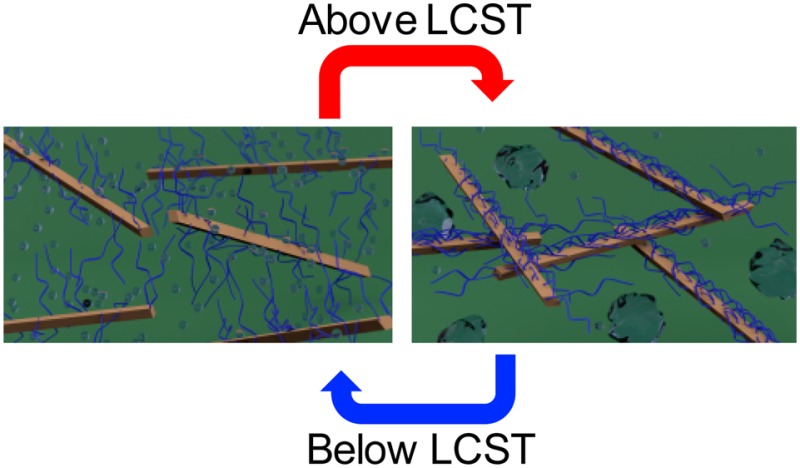
To our knowledge there are no materials that have the ability to switch reversibly between ca. 1 MPa at room temperature and >100 MPa when exposed to an increase in temperature and water (such as insertion in the body). However, it is known that the mechanical properties of nanocomposites are enhanced when there are strong interactions between the filler and the filler and matrix.18 Thus, an approach to materials that transition from “soft” to “stiff” upon insertion into a biologically relevant environment is the use of nanocomposites where the interactions between the nanofiller (and/or nanofiller and matrix) are “switched on” upon exposure to heat and water. One class of polymers that respond to both heat and water are LCST polymers,19−21 which are soluble in aqueous environments at lower temperatures but aggregate/precipitate upon heating above the LCST. Thin films of gels of such polymers have been shown to increase their elastic modulus from kPa’s below the LCST (chains swollen) to a few MPa above the LCST (chains collapsed).22,23 In order to access reversible heat-induced stiffening materials with a much larger mechanical contrast, nanocomposites were targeted in which CNCs grafted with LCST polymers were embedded in a PVAc matrix (Figure 1). Conceptually, below the LCST the water swollen grafted polymer would inhibit CNC–CNC interactions through steric considerations, resulting in relatively soft materials. However, upon heating above the LCST, the polymer chains collapse, allowing the engagement of a CNC network and subsequent stiffening of the material. To further examine this hypothesis, energy conserving dissipative particle dynamics (EDPD)24 was used as a mesoscale simulation technique to model/capture the thermoresponsive behavior of the nanocomposite. Unlike the standard dissipative particle dynamics (DPD),25 which only captures isothermal systems, EDPD can model nonisothermal systems due to the conservation of energy. For example, phase transitions in LCST microgels have recently been successfully captured using EDPD.26 It is worth noting that coarse-grained simulations have been used to investigate the temperature responsive dispersion/aggregation of polymer grafted nanospheres embedded in a polymer matrix, which showed that aggregation of these nanoparticles could be controlled with temperature.27,28
Figure 1.
Schematic showing the concept of a reversible, thermally stiffening water swollen composite below the LCST and a stiff reinforced composite above the LCST. The brown rods represent the CNCs, blue attachments represent the LCST polymers, and clear bubbles represent water. Below the LCST, polymer chains prevent CNC interactions; however, above LCST, polymer chains collapse, allowing interactions between the CNC.
CNCs grafted with LCST polymers,29−32 such as poly(N-isopropylacrylamide)31,32 and poly(N,N-dimethylaminoethyl methacrylate),33 are known. In this study the focus was on a different class of LCST polymer, namely, poly(oligo(ethylene glycol)monomethyl ether (meth)acrylates) (POEG(M)A), whose LCST transition can be tailored between 26 and 90 °C by altering the size of the oligo(ethylene glycol) chain.34 Furthermore, these thermoresponsive polymers are promising candidates for medical applications on account of their biocompatibility.34,35
Results and Discussion
To graft the polymer onto the surface of the CNCs (Scheme 1) an amine-end-capped polymer was synthesized via atom transfer radical polymerization (ATRP).36−38 2-(2-(2-Methoxyethoxy)ethoxy)ethyl acrylate39 was polymerized using a modified ATRP initiator with a terminal amine functionality, protected with a phthalimidyl group that can subsequently be converted to a primary amine by reacting with hydrazine. The amine-end-capped poly[2-(2-(2-methoxyethoxy)ethoxy)ethyl acrylate] (POEG3A) had a molecular weight of Mn = 12700 g/mol and dispersity (D) = 1.15 (Figure S1a). Carboxylic acid functionalized tunicate CNCs (t-CNC–COOH)40 with a carboxylic acid content of 950 mmol/kg were functionalized with the amine terminated polymer using standard carbodiimide-mediated amine coupling conditions. After functionalization, the polymer grafted t-CNCs (t-CNC-g-POEG3A) were thoroughly purified by extensive washing, sonication, and centrifuging to remove unreacted polymer. The weight of the t-CNC-g-POEG3A increased by 325% (200 mg of t-CNC–COOH is converted into 650 mg of t-CNC-g-POEG3A), consistent with the attachment of polymer chains on the t-CNC–COOH surface. Polymer attachment to the t-CNC–COOH was confirmed via a number of techniques. FT-IR (Figure S1b), AFM (Figures S1c and S1d), and wide-angle X-ray diffraction (Figure S2a) are consistent with attachment of the polymer onto the CNC and that little-to-no degradation of the CNC occurs during functionalization.
Scheme 1. Synthesis of the t-CNC-g-POEGy(M)A.

Furthermore, AFM height profiles (Figures 2a and 2b) show that the average profile area of the t-CNCs increases from 228 ± 47 nm2 for the t-CNC–COOH to 641 ± 49 nm2 for t-CNC-g-POEG3A, consistent with the attachment of the POEG3A to the t-CNC–COOH surface. Gratifyingly, the t-CNC-g-POEG3A exhibited an LCST of ca. 60 °C (Figure 2c), which is slightly larger than that of “free” POEG3A,39 as would be expected given the polar nature of the t-CNC. Neutron scattering results at different temperatures showed no changes in the shape or intensity of the scattering profiles of t-CNC–COOH as a function of temperature (Figure S2b), demonstrating that both the structure and solubility of the t-CNC–COOHs are not affected by such changes. However, t-CNC-g-POEG3A showed an increase in the scattering intensity for q < 0.04 Å–1 which is characteristic of an LCST behavior (Figure 2d). The increase in the slope of the scattering profiles for q > 0.02 Å–1 with an increase in temperature demonstrates that the grafted polymers are adopting more compact conformations as interactions with the solvent become more unfavorable. Fitting the scattering data to the Guinier–Porod model finds that the grafted chains are roughly ideal at 20 °C with a size that scales with the degree of polymerization (N) as R ∼ N0.47. At 60 °C, the chains are noticeably more compact with a size R ∼ N0.43. To better visualize the neutron scattering data, a Kratky plot41 (q2.14I(q) vs q) (Figure 2e) was generated in order to emphasize the change in slope of the scattering profiles. As expected, the Kratky plot showed a negative slope for the scattering profile above the LCST, indicative of a more compact conformation. Unfortunately, on account of the aggregation of t-CNC-g-POEG3A at 70 °C (above the LCST), it was not possible to obtain the polymer conformation information at this temperature.
Figure 2.

Characterization of t-CNC-g-POEG3A. (a) AFM height images (line in AFM height images corresponds to cross sections used to calculate the area and height of the t-CNCs). (b) Averaged height profiles of t-CNC–COOH and t-CNC-g-POEG3A. (c) Cloud point test as a function of temperature showing the LCST behavior of an aqueous dispersion of t-CNC-POEG3A. (d) Small angle neutron scattering profile of t-CNC-g-POEG3A as a function of temperature. (e) Kratky plot showing the difference in the slope of the scattering profile at different temperatures (linear fit is inserted to help guide the eye). (f) Simulation of the mean square radius of gyration of grafted polymer on nanorods with respect to temperature (kBT set at 1 for LCST); blue dots are the simulated radius of gyration, red dotted line is a guide for the reader’s eye, and errors bars represent the standard deviations of the simulated data.
In order to model the t-CNC-g-POEG3A temperature-sensitive behavior, polymer chains that exhibit LCST behavior are grafted on rigid nanorods. The LCST behavior is applied to the grafted chains through the mathematical implementation of an excess potential (equivalent to the Flory–Huggins χ-parameter) acting between the solvent molecules and the grafted chains. This potential is attractive below the LCST transition point and becomes highly repulsive as the temperature increases above the LCST (Figure S2c). Additionally, rods are considered to be interacting via a Morse potential which can mimic noncovalent bonding effects. This potential works similarly to a harmonic oscillator that generates bonds between the molecules; however, the bonds are dissociable when the distance between the interacting molecules reaches above a certain cutoff value (ca. 0.6 nm). In other words, the Morse potential between the rods is screened below the LCST transition point when the grafted chains are in an extended conformation. As the system is exposed to temperatures above the LCST, the grafted polymers collapse, resulting in a decrease in the distance between the rods and a subsequent activation of the Morse potential as the rods approach each other closely, which in this case mimics the interactions between the rods via hydrophobic effects or hydrogen bonding. EDPD simulations were performed to monitor the grafted polymers’ response to temperature. As a consequence of the presence of a heat flux between the system’s components, increasing the internal energy of the solvent molecules heats the whole simulated system. Gratifyingly, the EDPD simulations showed a dramatic decrease in the mean square radius of gyration of the grafted polymer chains as the system’s temperature is increased above the LCST transition point (Figure 2f), mimicking the real behavior of the grafted CNCs. Ensemble average of the radius of gyration of the polymer brushes in Figure 2f is calculated and averaged over the period of time required for the system to equilibrate at each temperature (error bars represent the standard deviation).
t-CNC-g-POEG3A/PVAc nanocomposites (composite set 1) were prepared via solution casting from DMF followed by drying. A range of compositions (2.5 to 40 wt % filler) were prepared and their mechanical properties studied by dynamic mechanical analysis (DMA) under different conditions (Figure 3a, Figures S3a–d, and Table S2). The dry, as-processed composites (1DAP) are flexible films whose modulus slightly increases with an increase in nanofiller (Figure 3a, black squares, and Figure S3a). Notably, while there does appear to be a broadening of the glass transition temperature (Tg) of the composites from the DMA data, the Tg obtained from DSC (by taking the midpoint of the step change, Figure S4a and Table S2b) ranged from 38 to 40 °C (slightly lower than neat PVAc Tg ca. 42 °C) no matter how much of the t-CNC-g-POEG3A are present, suggesting that the grafted polymer is partially (although not fully) phase separated (dewetted) from the PVAc matrix. DMA of the 40 wt % PVAc/t-CNC-g-POEG3A composite (Figure S4b) shows the presence of two Tg’s (similar to the Tg’s of the grafted polymer (ca. −40 °C) and PVAc (ca. 42 °C)), which is also consistent with phase separation in these films’ DMA. The modulus increase at room temperature is presumably a combination of a broadening of the Tg and an increase in the amount of reinforcing filler into the matrix. When placed in room temperature water, the films swell (Figure 3b) and show an increase in water swelling with the hydrophilic t-CNC content in PVAc, consistent with previous results.8−10 These wet films below the LCST (1WBL) are soft and flexible (Figure 3a, red circles), with the biggest decrease in modulus occurring for the higher weight fraction t-CNC films, attributed to water plasticization of the PVAc matrix (as observed by the drop in the wet composite Tg, Table S2b). Interestingly, when placed in 60 °C water (above the LCST), all the films (1WAL) show a significant increase in modulus (Figure 3a, blue triangles), e.g., from ca. 0.8 to 320 MPa for the higher volume fraction t-CNC composite, with a relatively low decrease (<50%) in the degree of swelling (Figure 3b), consistent with the proposed LCST collapse of the polymer chains. When the composites are redried above the LCST (1RDAL) (Figure 3a green triangles), the modulus at room temperature is at least as good as, if not slightly better than, the as-processed composites (1DAP), consistent with the recovery of the matrix Tg. It is worth noting that all the redried films have Tg’s closer to that of PVAc itself (Tg ca. 42 °C) than the as-processed films irrespective of t-CNC-g-POEG3A content (Figure S4c and Table S2b). This indicates that the POEG3A and PVAc are now more completely phase separated (dewetted), consistent with the collapse of the grafted POEG3A chains. This idea of phase separation is further proven by the dramatic reduction in the solubility parameter above the LCST transition point measured from the simulations (Figure 3c). Interestingly, the modulus above Tg of the films redried above the LCST (1RDAL) (Figure 3d) increases significantly with an increase in the filler content, suggesting that the collapse of the grafted polymer chains results in better mechanical reinforcement of the film.
Figure 3.

Thermomechanical properties and simulation results of t-CNC-g-POEG3A/PVAc nanocomposite above the LCST. (a) Tensile storage modulus (E′) of t-CNC-g-POEG3A/PVAc composites dry as-processed (1DAP, black squares), soaked in water (25 °C) below the LCST for 3 days (1WBL, red circles), placed in water (60 °C) above LCST for 1 h (1WAL, blue triangles), and redried above 60 °C (1RDAL, green triangles). (b) Percent of water uptake of the t-CNC-g-POEG3A/PVAc composites below and above LCST. (c) Solubility parameter of the simulated system as a function of temperature (kBT set at 1 for LCST). (d) Storage modulus of 1DAP and 1RDAL above glass transition temperature of the t-CNC-g-POEG3A/PVAc nanocomposites (75 °C).
The modulus values of the different filler volume 1RDAL composites above Tg were fitted to the percolation model42,43(see Methods in the Supporting Information), which calculates the effect that a percolating nanofiller phase has on the mechanical properties of the nanocomposite. As shown in Figure 4a, the above Tg modulus of the 1RDAL films fit this model, suggesting that the collapse of grafted polymer chains results in the formation of a percolating, reinforcing nanofiller network. In order to better understand these experimental results, EDPD simulations were also carried out. To access a percolating network each rod will require to have at least three contacts (Figure 4b inset). The EDPD simulations show that the average number of contacts for each rod increases to three as the system is heated up beyond the LCST transition point (Figure 4b). Snapshots taken at different time points (Figure 4c) further prove the formation of a network as the system’s temperature is heated above the LCST. In the figure the color of each rod changes based on the measured number of contacts; rods with three contacts are colored yellow while those with no contacts are blue.
Figure 4.

Network formation (experimental and simulation) of t-CNC-g-POEG3A/PVAc nanocomposite above the LCST. (a) Dry tensile storage modulus (E′) at 75 °C of the t-CNC-g-POEG3A/PVAc films with the films exposed to 60 °C water and dried (1RDAL) versus different volume fractions of t-CNC component (not including the grafted polymer); the dotted line represents the modulus predicted by the percolation model. (b) Simulated average number of contacts on each rod with respect to the system’s temperature. (c) Simulation snapshots at various time points. A color coding algorithm is used to identify the number of contacts between neighboring rods. Blue color represents rods without any contact, and as the number of contacts increases, the rod turns yellow.
The dramatic change in film modulus is visually demonstrated in Figure 5a (see also the Supporting Information video), where a soft 1DAL film is placed in 25 °C water (1WBL). The soft wet film is unable to visibly penetrate premade gelatin. However, when the film is placed in 60 °C water (1WAL), it stiffens to a degree that it can now easily penetrate the gelatin. It is important to note that, if left at room temperature, the wet film will soften as it cools, highlighting its thermally induced reversibility when wet. It is also worth noting that the soft wet film will retain a temporary shape if dried above the LCST (ca. 70 °C) as demonstrated in Figure S5a. The reversibility of switching can be seen over at least four cycles (Figure 5b), where the composite was kept wet but was cycled between 25 and 60 °C water. EDPD simulations show a similar reversibility effect in a heating and cooling cycle. The structural evolution of the system is monitored through the calculation of the ratio between the fraction of dispersed rods (zero number of bonds P(0)) and rods that form the network (three number of contacts P(3)). Figure 5c, represents this ratio as a function of the system’s temperature in both heating and cooling cycles. A transition is observed for the P(0)/P(3) ratio as the system is heated above the LCST. Reduction of this value upon heating is indicative of the network formation. Consequently, during the process of cooling, this ratio almost remains constant prior to dropping below the LCST of the system, which suggests a kinetic trapping of the engaged rods. Below the LCST, P(0)/P(3) increases, suggesting the disengagement of the rods in the network.
Figure 5.

Stiffness demonstration, kinetics, and reversibility (experimental and simulated) properties of t-CNC-g-POEG3A/PVAc nanocomposites. (a) Images of 30 wt % t-CNC-g-POEG3A/PVAc composite placed in 25 °C water, removed from water but unable to penetrate gelatin due to low mechanical strength, placed in 60 °C water for 2 min, and then removed from water but now strong enough to penetrate gelatin. (b) Tensile storage modulus (E′) of the 30 wt % t-CNC-g-POEG3A/PVAc composite cycled 4 times between 25 and 60 °C water. (c) EDPD simulation demonstrating the reversibility of the temperature responsive system. (d) Stiffness versus time of 30 and 15 wt % t-CNC-g-POEG3A/PVAc composites examined using DMA. Experiments start with the wet swollen films (1WBL), which are then exposed to 60 °C water (1WAL). (e) EDPD kinetic simulation of network formation as a function of time for different filler content.
Figure 5d shows the kinetics of the stiffening transition upon adding 60 °C water to wet, room temperature films. Increasing the amount of the hydrophilic filler reduces the stiffening time (from 4 min for 15 wt % filler to 2 min for 30 wt % filler), as would be expected given that this is a water-diffusion controlled process. Hypothetically, the stiffening behavior is also a consequence of the polymer collapse and filler network formation, and therefore EDPD simulation was used to study the kinetics of the network formation. Figure 5e shows the ratio of the fraction of dispersed rods and rods that form a network (P(0)/P(3)) as a function of time for two different filler contents, and as expected, systems with higher filler contents (higher concentration of rods) show faster kinetics in the formation of a perfect network above the LCST transition point.
One goal of this work was to access films that would switch in biologically relevant environments. Thus, it was important to show that these nanocomposites would stiffen in media that more closely mimic the body’s salt condition.19 Thus, a 40 wt % t-CNC-g-POEG3A/PVAc film was soaked in Earle’s balanced salt solution (HyClone), and these composites also reversibly stiffen in this biomimetic medium (Figure S5b), with only a slight difference in mechanical properties compared to the water exposed samples (9 MPa vs 0.8 MPa for 1WBL, and 303 MPa vs 320 MPa for 1WAL, in HyClone and water, respectively). Furthermore, for biomedical applications such materials ideally would be soft at room temperature but stiff in an aqueous environment at body temperature, 37 °C, which this first generation of the composites do not do. However, if the heat-induced collapse of the grafted polymer chains is critical to the stiffening mechanism, then it should be possible to change the switching temperature by simply changing the grafted polymer. Poly[2-(2-methoxyethoxy)ethyl methacrylate] (POEG2MA) has a lower LCST (26 °C)34 than POEG3A, and so POEG2MA (11 500 g/mol) was grafted onto the t-CNCs. It is worth noting that deprotecting the phthalate group on POEG2MA with hydrazine also resulted in the formation of a small percentage of polymer species that have the bromine chain end substituted by hydrazine (Figure S6). This effect was only observed in POEG2MA (and not the POEG3A studies above). Either way the amine end functionalized POEG2MA (with a bromine or hydrazine at the other chain end) could still be grafted onto the CNCs and purified in a similar manner. Kaiser tests of these dried t-CNC-g-POEG2MA did result in a purple color change, which is consistent with presence of free amines on the chain end of the grafted POEG2MA (Figure S6). PVAc composites with t-CNC-g-POEG2MA were prepared (composite 2, Figure S7, Table S3), and these films exhibit a thermal stiffening behavior similar to that of the t-CNC-g-POEG3A/PVAc composites but at much lower temperatures. This is highlighted in Figure 6a, which compares the tensile modulus of the two different PVAc composites (1 and 2) with 30 wt % filler upon exposure to water at different temperatures. Both are soft at 20 °C and stiff at 60 °C; however, only the t-CNC-g-POEG2MA is stiff at 40 °C, providing strong evidence for the LCST induced collapse of the grafted polymer chains being the cause of the mechanical stiffening. Furthermore, to show that these materials stiffen upon exposure to body temperature, a 25 °C water-soaked film of 30 wt % t-CNC-g-POEG2MA/PVAc was molded onto a human nose template and held in a hand for ca. 5 min, at which point the material was stiff enough to retain the shape of a human nose (Figure 6b).
Figure 6.

Properties and demonstration of the mechanical stiffening of the nanocomposites under biologically relevant conditions. (a) Comparison of the tensile storage modulus (E′) of 30 wt % t-CNC-g-POEG3A/PVAc (1) and t-CNC-g-POEG2MA/PVAc (2) nanocomposite films in water at different temperatures. (b) Images of a wet 30 wt % t-CNC-g-POEG2MA/PVAc composite stiffened enough to retain the shape of a human nose upon exposure to a warm hand.
Conclusion
In conclusion, by grafting LCST polymers from the surface of t-CNCs and embedding them in a PVAc matrix, a new class of stimuli-responsive material that reversibly stiffens with heat and water has been accessed. EDPD modeling and the mechanical studies suggest that this process occurs as a result of the collapse of the LCST polymer resulting in the formation of a percolating network and a subsequent stiffening of the composite material in moderately hot water. Furthermore, by simply drying the material above the LCST it will remain in its processed state. The t-CNC grafted polymer can be altered to access films that stiffen when exposed to body temperature, opening the door to an exploration of these materials as reinforcing implants in biomedical applications that require them to be initially pliable for positioning and become stiff upon exposure to the body’s environment.
Acknowledgments
This material is based on work supported by the National Science Foundation (NSF) under Grant No. DMR-1204948, the Army Research Office under Grant No. W911NF-15-1-0190, and the Kent H. Smith Charitable Trust (S.J.R.). This work also utilized neutron scattering facilities supported in part by the NSF under Agreement No. DMR-1508249. We would like to thank Dr. Arman Boromand and Dr. Safa Jamali for the fruitful discussions regarding the computational part of this work and Dr. Katie M. Weigandt (NIST) for help with the neutron scattering studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00215.
Author Contributions
S.J.R. designed the concept. E.C. and S.K. contributed equally to this work and carried out the experimental and modeling studies, respectively. A.E.W. carried out initial studies on this work. S.J.R. and J.M. guided the work. M.J.A.H. carried out the neutron scattering studies. All authors worked on and commented on the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Lendlein A.; Kelch S. Shape-Memory Polymers. Angew. Chem., Int. Ed. 2002, 41, 2034.. [DOI] [PubMed] [Google Scholar]
- Ohm C.; Brehmer M.; Zentel R. Liquid Crystalline Elastomers as Actuators and Sensors. Adv. Mater. 2010, 22, 3366–3387. 10.1002/adma.200904059. [DOI] [PubMed] [Google Scholar]
- Blaiszik B. J.; Kramer S. L. B.; Olugebefola S. C.; Moore J. S.; Sottos N. R.; White S. R. Self-Healing Polymers and Composites. Annu. Rev. Mater. Res. 2010, 40, 179–211. 10.1146/annurev-matsci-070909-104532. [DOI] [Google Scholar]
- Michal B. T.; Jaye C. A.; Spencer E. J.; Rowan S. J. Inherently Photohealable and Thermal Shape-Memory Polydisulfide Networks. ACS Macro Lett. 2013, 2, 694–699. 10.1021/mz400318m. [DOI] [PubMed] [Google Scholar]
- Michal B. T.; McKenzie B. M.; Felder S. E.; Rowan S. J. Metallo-, Thermo-, and Photoresponsive Shape Memory and Actuating Liquid Crystalline Elastomers. Macromolecules 2015, 48, 3239–3246. 10.1021/acs.macromol.5b00646. [DOI] [Google Scholar]
- Theato P.; Sumerlin B. S.; O’Reilly R. K.; Epps T. H. III Stimuli Responsive Materials. Chem. Soc. Rev. 2013, 42, 7055. 10.1039/c3cs90057f. [DOI] [PubMed] [Google Scholar]
- Xia Z.Stimuli-Responsive Materials. In Biomimetic Principles and Design of Advanced Engineering Materials; John Wiley & Sons, Ltd: Chichester, U.K., 2016; pp 188–209. [Google Scholar]
- Capadona J. R.; Shanmuganathan K.; Tyler D. J.; Rowan S. J.; Weder C. Stimuli-Responsive Polymer Nanocomposites Inspired by the Sea Cucumber Dermis. Science (Washington, DC, U. S.) 2008, 319, 1370–1374. 10.1126/science.1153307. [DOI] [PubMed] [Google Scholar]
- Dagnon K. L.; Shanmuganathan K.; Weder C.; Rowan S. J. Water-Triggered Modulus Changes of Cellulose Nanofiber Nanocomposites with Hydrophobic Polymer Matrices. Macromolecules 2012, 45, 4707–4715. 10.1021/ma300463y. [DOI] [Google Scholar]
- Dagnon K. L.; Way A. E.; Carson S. O.; Silva J.; Maia J.; Rowan S. J. Controlling the Rate of Water-Induced Switching in Mechanically Dynamic Cellulose Nanocrystal Composites. Macromolecules 2013, 46, 8203–8212. 10.1021/ma4008187. [DOI] [Google Scholar]
- Lee W. J.; Clancy A. J.; Kontturi E.; Bismarck A.; Shaffer M. S. P. Strong and Stiff: High-Performance Cellulose Nanocrystal/Poly(vinyl Alcohol) Composite Fibers. ACS Appl. Mater. Interfaces 2016, 8, 31500–31504. 10.1021/acsami.6b11578. [DOI] [PubMed] [Google Scholar]
- Yang J.; Zhao J.-J.; Xu F.; Sun R.-C. Revealing Strong Nanocomposite Hydrogels Reinforced by Cellulose Nanocrystals: Insight into Morphologies and Interactions. ACS Appl. Mater. Interfaces 2013, 5, 12960–12967. 10.1021/am403669n. [DOI] [PubMed] [Google Scholar]
- Pei A.; Malho J.-M.; Ruokolainen J.; Zhou Q.; Berglund L. A. Strong Nanocomposite Reinforcement Effects in Polyurethane Elastomer with Low Volume Fraction of Cellulose Nanocrystals. Macromolecules 2011, 44, 4422–4427. 10.1021/ma200318k. [DOI] [Google Scholar]
- Yang J.; Han C.-R.; Zhang X.-M.; Xu F.; Sun R.-C. Cellulose Nanocrystals Mechanical Reinforcement in Composite Hydrogels with Multiple Cross-Links: Correlations between Dissipation Properties and Deformation Mechanisms. Macromolecules 2014, 47, 4077–4086. 10.1021/ma500729q. [DOI] [Google Scholar]
- Moon R. J.; Martini A.; Nairn J.; Simonsen J.; Youngblood J. Cellulose Nanomaterials Review: Structure, Properties and Nanocomposites. Chem. Soc. Rev. 2011, 40, 3941. 10.1039/c0cs00108b. [DOI] [PubMed] [Google Scholar]
- Giannoudis P. V.; Dinopoulos H.; Tsiridis E. Bone Substitutes: An Update. Injury 2005, 36, S20–S27. 10.1016/j.injury.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Mantripragada V. P.; Lecka-Czernik B.; Ebraheim N. A.; Jayasuriya A. C. An Overview of Recent Advances in Designing Orthopedic and Craniofacial Implants. J. Biomed. Mater. Res., Part A 2012, 101, 3349–3364. 10.1002/jbm.a.34605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu L.; Weder C.; Rowan S. J. Stimuli-Responsive, Mechanically-Adaptive Polymer Nanocomposites. J. Mater. Chem. 2011, 21, 2812–2822. 10.1039/C0JM02383C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward M. A.; Georgiou T. K. Thermoresponsive Polymers for Biomedical Applications. Polymers (Basel, Switz.) 2011, 3, 1215–1242. 10.3390/polym3031215. [DOI] [Google Scholar]
- Gibson M. I.; O’Reilly R. K. To Aggregate, or Not to Aggregate? Considerations in the Design and Application of Polymeric Thermally-Responsive Nanoparticles. Chem. Soc. Rev. 2013, 42, 7204–7213. 10.1039/C3CS60035A. [DOI] [PubMed] [Google Scholar]
- Roy D.; Brooks W. L. A.; Sumerlin B. S. New Directions in Thermoresponsive Polymers. Chem. Soc. Rev. 2013, 42, 7214. 10.1039/c3cs35499g. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Canavan H. E.; Stein M. J.; Hull J. R.; Kweskin S. J.; Wagner M. S.; Somorjai G. A.; Castner D. G.; Ratner B. D. Surface Chemical and Mechanical Properties of Plasma-Polymerized N-Isopropylacrylamide. Langmuir 2005, 21, 7833–7841. 10.1021/la050417o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzak K. A.; Mateescu A.; Toca-Herrera J. L.; Jonas U. Simultaneous Measurement of Mechanical and Surface Properties in Thermoresponsive, Anchored Hydrogel Films. Langmuir 2012, 28, 12871–12878. 10.1021/la3019666. [DOI] [PubMed] [Google Scholar]
- Li Z.; Tang Y.-H.; Lei H.; Caswell B.; Karniadakis G. E. Energy-Conserving Dissipative Particle Dynamics with Temperature-Dependent Properties. J. Comput. Phys. 2014, 265, 113–127. 10.1016/j.jcp.2014.02.003. [DOI] [Google Scholar]
- Hoogerbrugge P. J.; Koelman J. M. V. A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. Europhys. Lett. 1992, 19, 155–160. 10.1209/0295-5075/19/3/001. [DOI] [Google Scholar]
- Li Z.; Tang Y.-H.; Li X.; Karniadakis G. E. Mesoscale Modeling of Phase Transition Dynamics of Thermoresponsive Polymers. Chem. Commun. 2015, 51, 11038–11040. 10.1039/C5CC01684C. [DOI] [PubMed] [Google Scholar]
- Martin T. B.; Mongcopa K. I. S.; Ashkar R.; Butler P.; Krishnamoorti R.; Jayaraman A. Wetting–Dewetting and Dispersion–Aggregation Transitions Are Distinct for Polymer Grafted Nanoparticles in Chemically Dissimilar Polymer Matrix. J. Am. Chem. Soc. 2015, 137, 10624–10631. 10.1021/jacs.5b05291. [DOI] [PubMed] [Google Scholar]
- Martin T. B.; Jayaraman A. Tuning the Wetting–dewetting and Dispersion–aggregation Transitions in Polymer Nanocomposites Using Composition of Graft and Matrix Polymers. Mater. Res. Express 2016, 3, 34001. 10.1088/2053-1591/3/3/034001. [DOI] [Google Scholar]
- Roy D.; Semsarilar M.; Guthrie J. T.; Perrier S. Cellulose Modification by Polymer Grafting: A Review. Chem. Soc. Rev. 2009, 38, 2046. 10.1039/b808639g. [DOI] [PubMed] [Google Scholar]
- Azzam F.; Heux L.; Putaux J.-L.; Jean B. Preparation By Grafting Onto, Characterization, and Properties of Thermally Responsive Polymer-Decorated Cellulose Nanocrystals. Biomacromolecules 2010, 11, 3652–3659. 10.1021/bm101106c. [DOI] [PubMed] [Google Scholar]
- Zoppe J. O.; Österberg M.; Venditti R. A.; Laine J.; Rojas O. J. Surface Interaction Forces of Cellulose Nanocrystals Grafted with Thermoresponsive Polymer Brushes. Biomacromolecules 2011, 12, 2788–2796. 10.1021/bm200551p. [DOI] [PubMed] [Google Scholar]
- Zoppe J. O.; Venditti R. A.; Rojas O. J. Pickering Emulsions Stabilized by Cellulose Nanocrystals Grafted with Thermo-Responsive Polymer Brushes. J. Colloid Interface Sci. 2012, 369, 202–209. 10.1016/j.jcis.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Yi J.; Xu Q.; Zhang X.; Zhang H. Temperature-Induced Chiral Nematic Phase Changes of Suspensions of poly(N,N-Dimethylaminoethyl Methacrylate)-Grafted Cellulose Nanocrystals. Cellulose 2009, 16, 989–997. 10.1007/s10570-009-9350-9. [DOI] [Google Scholar]
- Lutz J.-F. Polymerization of Oligo(ethylene Glycol) (Meth)acrylates: Toward New Generations of Smart Biocompatible Materials. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 3459–3470. 10.1002/pola.22706. [DOI] [Google Scholar]
- Lutz J.-F.; Andrieu J.; Üzgün S.; Rudolph C.; Agarwal S. Biocompatible, Thermoresponsive, and Biodegradable: Simple Preparation of “All-in-One” Biorelevant Polymers. Macromolecules 2007, 40, 8540–8543. 10.1021/ma7021474. [DOI] [Google Scholar]
- Hansson S.; Östmark E.; Carlmark A.; Malmström E. ARGET ATRP for Versatile Grafting of Cellulose Using Various Monomers. ACS Appl. Mater. Interfaces 2009, 1 (11), 2651–2659. 10.1021/am900547g. [DOI] [PubMed] [Google Scholar]
- Morandi G.; Heath L.; Thielemans W. Cellulose Nanocrystals Grafted with Polystyrene Chains through Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). Langmuir 2009, 25, 8280–8286. 10.1021/la900452a. [DOI] [PubMed] [Google Scholar]
- Harrisson S.; Drisko G. L.; Malmström E.; Hult A.; Wooley K. L. Hybrid Rigid/Soft and Biologic/Synthetic Materials: Polymers Grafted onto Cellulose Microcrystals. Biomacromolecules 2011, 12, 1214–1223. 10.1021/bm101506j. [DOI] [PubMed] [Google Scholar]
- Han S.; Hagiwara M.; Ishizone T. Synthesis of Thermally Sensitive Water-Soluble Polymethacrylates by Living Anionic Polymerizations of Oligo(ethylene Glycol) Methyl Ether Methacrylates. Macromolecules 2003, 36, 8312–8319. 10.1021/ma0347971. [DOI] [Google Scholar]
- Way A. E.; Hsu L.; Shanmuganathan K.; Weder C.; Rowan S. J. pH-Responsive Cellulose Nanocrystal Gels and Nanocomposites. ACS Macro Lett. 2012, 1, 1001–1006. 10.1021/mz3003006. [DOI] [PubMed] [Google Scholar]
- Rambo R. P.; Tainer J. A. Characterizing Flexible and Intrinsically Unstructured Biological Macromolecules by SAS Using the Porod-Debye Law. Biopolymers 2011, 95, 559–571. 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi M.; Uemura S.; Minami S. Application of Equivalent Model Method to Dynamic Rheo-Optical Properties of Crystalline Polymer. J. Polym. Sci., Part C: Polym. Symp. 1964, 5, 113–122. 10.1002/polc.5070050111. [DOI] [Google Scholar]
- Favier V.; Chanzy H.; Cavaillé J. Y. Polymer Nanocomposites Reinforced by Cellulose Whiskers. Macromolecules 1995, 28, 6365–6367. 10.1021/ma00122a053. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.