

Abstract
Electrocatalytic CO2 reduction to generate multicarbon products is of interest for applications in artificial photosynthetic schemes. This is a particularly attractive goal for CO2 reduction by copper electrodes, where a broad range of hydrocarbon products can be generated but where selectivity for C–C coupled products relative to CH4 and H2 remains an impediment. Herein we report a simple yet highly selective catalytic system for CO2 reduction to C≥2 hydrocarbons on a polycrystalline Cu electrode in bicarbonate aqueous solution that uses N-substituted pyridinium additives. Selectivities of 70–80% for C2 and C3 products with a hydrocarbon ratio of C≥2/CH4 significantly greater than 100 have been observed with several additives. 13C-labeling studies verify CO2 to be the sole carbon source in the C≥2 hydrocarbons produced. Upon electroreduction, the N-substituted pyridinium additives lead to film deposition on the Cu electrode, identified in one case as the reductive coupling product of N-arylpyridinium. Product selectivity can also be tuned from C≥2 species to H2 (∼90%) while suppressing methane with certain N-heterocyclic additives.
Short abstract
A simple catalytic system for the electrochemical reduction of CO2 that achieves high selectivity of 70-80% for C≥2 products on polycrystalline copper by addition of N-aryl substituted pyridinium compounds is reported.
The electrochemical reduction of CO2 to commodity chemicals or fuels driven by solar energy provides an appealing strategy to utilize an inexpensive carbon feedstock to close the anthropogenic carbon cycle via artificial photosynthesis.1−5 The use of electrochemical systems comprising earth abundant materials has certain advantages with respect to scalability and ultimate implementation. Toward accessing value added products and high energy density liquid fuels, the generation of multicarbon products is highly desirable. Cu is one of very few materials capable of converting CO2 to C≥2 products, including hydrocarbons, alcohols, and aldehydes, with significant efficiencies.6−9 Importantly, Cu-catalyzed CO2 reduction can be performed in aqueous bicarbonate.
Typically, a distribution of products is obtained with a copper electrode, with the highest Faradaic efficiency (FE) for C≥2 products being observed at ca. −1.1 V vs. RHE (VRHE) on polycrystalline Cu.7 The nature of the Cu electrocatalyst has been tuned to increase the selectivity for multicarbon products. For example, it has been shown that single crystal electrodes displaying Cu(100) facets promote C–C coupling and thus the formation of products such as ethylene and ethanol.10−12 Stepped (100) surfaces, such as Cu(911) and Cu(711), are able to convert CO2 into C≥2 products with efficiencies approaching 80%.10 The selectivity toward C≥2 products on less expensive polycrystalline copper can be increased by several means, including optimization of the process conditions such as the buffer strength of the electrolyte and CO2 pressure,13,14 nanostructuring the electrode,15−22 and plasma activation.23 A higher pH near the electrode surface and surfaces with increased density of bound CO generated under these conditions have been proposed to shift the selectivity from C1 to C≥2 products.13,14 The pH has also been shown to affect the CO adsorption to the electrode and product selectivity.24−27 CO can be an intermediate of Cu-catalyzed CO2 reduction,6,28 and oxide derived Cu shows increased conversion of CO to C≥2 products.29
Organic additives, such as ionic liquids and pyridines, have also been employed previously to affect product selectivity of metal electrodes.30−33 The use of pyridines is particularly notable in this context as they have been reported to facilitate methanol generation, a six electron reduction product of CO2, on Pd30 and Pt electrodes31 with FE ≈ 30%. Although the role of pyridine in such processes has been debated, proposed mechanisms have invoked mediation of electron and proton transfers via either singly or multiply reduced and protonated species, such as dihydropyridine (DHP).31,34−37 Redox mediators that can transfer both protons and electrons are appealing given the multiproton multielectron nature of CO2 reduction. Mediators with hydridic moieties or weak element–H bonds (e.g., N–H or C–H) may be particularly suitable for facilitating reactions of species generated in CO2 reduction.38,39 Here, we report the ability of N-substituted arylpyridinium additives to dramatically tune the selectivity of electrochemical CO2 reduction on polycrystalline copper for C≥2 products.
Electrochemical reduction of CO2 was performed on a polycrystalline copper electrode with CO2-saturated 0.1 M KHCO3 electrolyte at pH 6.8 using a recently reported cell design with high catalyst surface area to electrolyte volume ratio.40 Briefly, the cell design used in this study consists of a cathode compartment and an anode compartment with a volume of 2.0 mL each, separated by a Selemion AMV anion-exchange membrane. CO2 gas sparged into the cell was dispersed using a PEEK frit at the inlet of the cell. A copper foil was used as a working electrode and platinum foil as a counter electrode, both with a surface area of 1 cm2. Potentials were measured versus a leakless Ag/AgCl electrode and converted to the RHE scale. The gaseous products were analyzed by gas chromatography coupled with FID and TCD detectors. The performance of polycrystalline copper in this cell is similar to what has been previously reported in terms of product distribution at various potentials (Table S2, Figure S17).7,8,40 In our setup, ethylene and C≥2 products are observed at potentials negative of −0.9 VRHE, with the peak FE for ethylene (14%) at −1.14 VRHE, for ethanol (7%) at −1.06 VRHE, and for methane (32%) at −1.18 VRHE. Although substantial yields of C2 products are observed between −1.06 and −1.14 VRHE, they remain minor, with dihydrogen evolution reaction (HER, FE ≈ 40%) and methane production (FE = 20–30%) being the major processes at these potentials (Table 1).
Table 1. Faradaic Efficiency toward Different Products Produced during CO2 Reduction on a Polycrystalline Copper Electrode in a CO2 Saturated 0.1 M KHCO3 Electrolyte with 10 mM 1–14 at an applied potential of −1.1 VRHEa.
| Faradaic
efficiencies (%) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compound | CH4 | C2H4 | C2H5OH | C3H7OH | CO | H2 | HCOO– | C≥2 | total | C2/CH4b | j (mA/cm2) |
| none | 20.2 | 12.4 | 7.2 | 2.8 | 1.7 | 42.8 | 4.7 | 26.0 | 96.4 | ≥0.7 | –4.46 |
| 1 | 1.0 | 40.5 | 30.6 | 7.1 | l.8 | 15.5 | 6.5 | 78.2 | 103.1 | ≥36 | –1.02 |
| 3 | 3.1 | 29.3 | 29.6 | 0 | 2.5 | 21.8 | 10.1 | 58.9 | 96.4 | ≥15 | –0.70 |
| 4 | 0.3 | 37.7 | 22.3 | 8.7 | 2.1 | 16.6 | 10.6 | 68.6 | 98.3 | ≥130 | –1.46 |
| 5 | 0.1 | 40.8 | 26.7 | 8.6 | 2.1 | 12.4 | 8.8 | 76.1 | 99.5 | ≥450 | –1.34 |
| 6 | 2.1 | 18.2 | 16.0 | 0 | 3.7 | 52.1 | 6.9 | 34.2 | 99.0 | ≥9 | –1.40 |
| 7 | 0.07 | 33.6 | 27.1 | 11.8 | 3.1 | 10.0 | 13.0 | 72.4 | 98.7 | ≥830 | –1.10 |
| 8 | 5.1 | 1.7 | 0 | 5.3 | 2.6 | 50.5 | 10.6 | 7.0 | 75.8 | ≥0.5 | –3.32 |
| 9 | 0.4 | 0.1 | 0 | 2.1 | 0.4 | 88.5 | 5.6 | 2.2 | 97.1 | –6.28 | |
| 10 | 0.04 | 0 | 0 | 0 | 0.2 | 65.9 | 9.5 | 0 | 75.6 | –2.95 | |
| 11 | 0 | 3.2 | 0 | 0 | 0.7 | 28.6 | 9.0 | 3.2 | 41.5 | –0.97 | |
| 12 | 4.2 | 4.0 | 0 | 0 | 0.3 | 61.6 | 5.5 | 4.0 | 79.6 | –4.03 | |
| 13 | 0.01 | 0.07 | 0 | 0 | 0.5 | 76.6 | 1.3 | 0.07 | 78.5 | –6.44 | |
| 14 | 0.2 | 0.01 | 0 | 0 | 0.3 | 91.3 | 5.3 | 0.01 | 97.1 | –4.20 | |
All values represent an average of at least two runs. See Supporting Information for raw data.
Ratios shown are the lower of the independent values measured.
The effects of pyridine-based and other N-containing heterocycle additives on the electrocatalytic CO2 reduction with a Cu electrode were studied (see Figures 1 and 2 and Table 1). The electrochemical properties of water-soluble N-tolylpyridinium chloride (1) salt were studied by cyclic voltammetry (CV) using a Cu disk electrode in a 0.1 M KHCO3 electrolyte solution (see Figure S14).41 Under N2, a reduction wave at ∼−0.6 VRHE appears in the first CV scan and disappears completely after a few cycles. A white layer of material was observed on the electrode after multiple CV scans. Similar CV features and deposition behavior were observed when performing the CV under CO2. However, higher currents were observed in the presence of CO2, indicating that this system promotes electrocatalytic reduction. To quantify the products formed during CO2 reduction, chronoamperometry was performed. Addition of water-soluble 1, at a concentration of 10 mM, results in remarkable changes in the selectivity profile (Table 1 and Figure 2). Ethylene (FE ≈ 40%) and ethanol (FE ≈ 30%) become the major products at −1.1 VRHE with combined FE of ∼70%. 1-Propanol is also obtained in significant yield (FE ≈ 7%). Suppression of HER is observed at potentials more positive than −1.1 VRHE, illustrated by a decrease in FE to 15%. A sharp rise in hydrogen FE is observed at more negative potentials. Notably, methane is a very minor product (FE ≈ 1% or less) at potentials positive of −1.1 VRHE. At more negative potentials, methane production rises to FE between 5 and 10%, which is still significantly lower than in the absence of 1. The combined FE of C≥2 products at −1.1 VRHE is approximately 80%, to our knowledge unprecedented for polycrystalline copper electrodes. The increased selectivity for ethylene vs. methane at −1.1 VRHE remains stable over prolonged electrolysis for up to 10 h (Figure S18).
Figure 1.

Overview of the N-heterocyclic chloride salt additives studied herein.
Figure 2.
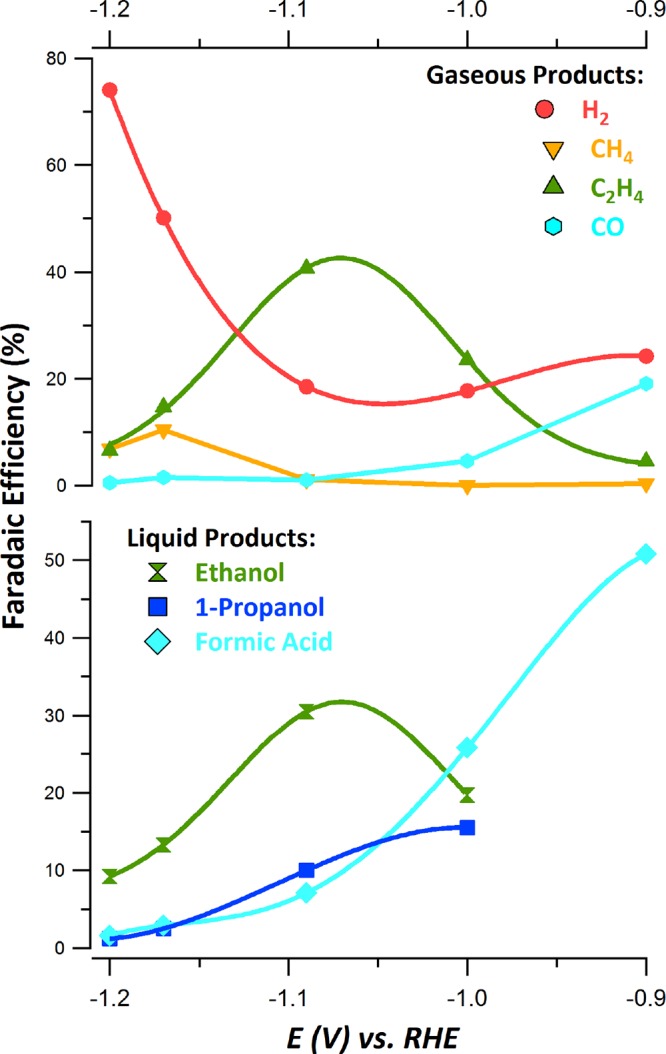
Faradaic efficiency toward products produced during CO2 reduction on a polycrystalline copper electrode in a CO2 saturated 0.1 M KHCO3 electrolyte with 10 mM N-tolylpyridinium chloride at different applied potentials, E (VRHE).
An examination of the partial current densities of products formed in the presence and the absence of 1 shows a decrease in the partial density toward hydrogen and methane production at −1.1 VRHE and more positive potentials (Figures S15). In contrast, the partial current densities toward ethylene, ethanol, and 1-propanol formation at these potentials are similar with and without 1 in the electrolyte. This suggests that the addition of 1 effectively suppresses the hydrogen evolution reaction and the methanation reaction, while not affecting the formation rates for C–C coupled products.
The effect of the concentration (1, 5, 10, 20 mM) of 1 on the product distribution for CO2 reduction at −1.1 VRHE was investigated (Table S6). With increasing concentration, the FE for ethylene and ethanol increases steadily up to 10 mM. In contrast, dihydrogen production decreases with increasing concentration of 1. Most dramatically, methane FE decreases from ∼20% in the absence of 1 to ∼1% at 10 and 20 mM 1. The dependence of product distribution on the concentration of 1 indicates that this additive plays a role in the overall reduction chemistry. Given that the highest yield of C≥2 was obtained with 10 mM 1, all other experiments were performed at this concentration. In all cases, the deposition of a colorless film on the electrode was observed.
Control experiments were performed to exclude the possibility of the detected products being decomposition products derived from 1. Electrolysis experiments using the same copper electrode with 10 mM 1 in a 0.1 M K2HPO4/0.1 M KH2PO4 buffer (pH 6.8) and a N2 flow showed no carbon-containing products, indicating that CO2 is required for hydrocarbon production. To confirm that CO2/bicarbonate is the source of carbon in the hydrocarbon products, isotopic labeling experiments were performed. The electrolysis experiments were carried out under the same conditions described above, but using 13CO2 and KH13CO3(aq).42 The GC-MS analysis (Figure 3a) of the gaseous products obtained from 13C-labeling experiments shows patterns diagnostic of 13C-ethylene (H213C=13CH2, m/z = 30). For comparison, the same analysis performed on the bulk electrolysis with natural abundance CO2 displays 12C-ethylene (H212C=12CH2, m/z = 28, identical to that of the calibration standard). The formation of 13C-ethylene is further verified by 1H NMR spectroscopy, which reveals a doublet (1JC–H = 154 Hz) at 5.40 ppm (Figure 3b). The liquid products obtained from the 13C-labeling experiments have also been subjected to 1H and 13C NMR analyses. The 1H NMR spectrum exhibits resonances of 13C2-ethanol, 13C3-1-propanol, and 13C-formate as indicated by their characteristic 1H–13C splitting patterns (Figure 3c), and their 1JC–H values are consistent with that obtained from the carbon satellites in natural abundant standards (Figure S24). For ethanol, <1% of the 12C signal was observed, presumably due to the presence of a trace (<1%) 12C impurity in the 13CO2 source.43 Lastly, the 13C NMR spectrum (Figure S25) displays the expected peaks for 13C2-ethanol and 13C3-1-propanol. Notably, 1JC–C splitting is observed, indicating that the products are essentially fully 13C labeled. Taken collectively, the 1H NMR, 13C NMR, and GC-MS analyses performed with 13C-labeling conclusively confirm that CO2 is the carbon source of the Cu-catalyzed electrocatalysis described herein.
Figure 3.

(a) GC-MS analyses and (b) 1H NMR spectra (400 MHz, CDCl3) of ethylene, and (c) 1H NMR spectra (400 MHz, H2O:D2O = 9:1) of (primarily) ethanol and propanol after bulk electrolysis at −1.1 VRHE with 1 (10 mM) and natural abundance (red) and 13C-enriched (blue) CO2-saturated KHCO3 (0.1 M). Black: Calibration standard of natural abundance ethylene. Purple dot indicates a trace impurity of methanol (<10 μM).
Although the exact nature of the species derived from 1 during electrocatalytic CO2 reduction remains unclear, the nature of the N-heterocyclic organic compounds present at the end of the electrolysis has been investigated for the case of the N-tolylpyridinium chloride additive. After 1 h of electrolysis at −1.1 VRHE, 1H NMR analysis shows that virtually all of compound 1 (>98%) remains in solution. However, the colorless deposit formed on the electrode is soluble in organic solvents and, based on subsequent analysis, is shown to arise from 1. Accordingly, upon rinsing the postelectrolysis electrode with water and then quickly transferring it to an inert atmosphere, the deposited film was extracted into CD2Cl2 for NMR analysis. The 1H NMR spectrum shows the same number of peaks as 1, but two of the peaks are shifted significantly upfield (Figure S5). A species with the same NMR spectrum was prepared independently by reduction of 1 with cobaltocene in dichloromethane. A solid-state structure of this compound (2) was determined by single crystal X-ray diffraction (XRD) (Figure 4). The C–C distances within the nitrogen containing ring are indicative of localization of single and double bond character resulting from dearomatization. The structure corresponds to two equivalents of 1 being reductively coupled at the para position on the pyridine ring, a net one-electron reduction per pyridinium. The mass of the deposited film corresponds to only ∼1% of 1, accounting for a small amount (∼0.6%) of reducing equivalents used in the electrolysis. A related dimerization process has been discussed in a computational study of the effect of pyridine on CO2 reduction, but was not in this case considered to be on the productive CO2 reduction pathway.34
Figure 4.
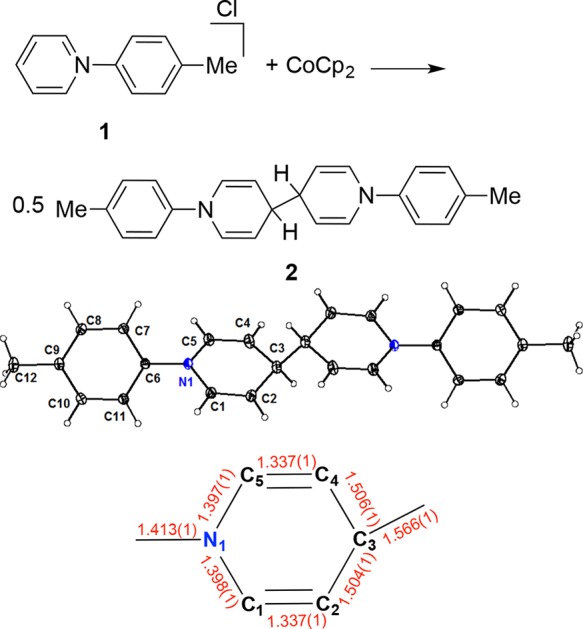
Synthesis, crystal structure, and selected structural parameters (Å) of 2. Thermal ellipsoids are shown at the 50% probability level.
To test whether this surface compound is indeed critical to the CO2 reduction chemistry observed, after 1 h electrolysis with 1, the solution was replaced by an electrolyte solution free of 1 and electrolysis was then performed for another hour. A similar product profile was observed for the second electrolysis run, strongly suggesting that the film rather than soluble 1 is required for the observed selectivity shift (Table S4).44 We dropcast independently prepared 2 from either dichloromethane (4.2 mg of 2) or benzene (0.5 mg of 2) solutions on the Cu electrodes but found that modified electrodes prepared as such did not result in the selectivity profile observed for electrodeposited 2. They instead afforded predominantly H2 (>75%). Additionally, disruption of the electrochemically deposited film, by manually removing it or by dissolution with organic solvent followed by redeposition by solvent evaporation, results in selectivity similar to polycrystalline Cu. These observations suggest that the film of 2 and factors such as its morphology and the nature of the contact between the Cu surface and the film as generated during electrodeposition may be instrumental to increased production of C≥2 species.
A structure–function study was performed by varying the nature of the N-substituent of the pyridinium additives (Figure 1). With additives 1, 3, 4, and 5, which are N-arylpyridinium compounds with substituents in the para position of the aryl group of varying electronic properties, C≥2 products were obtained with similarly high FE (>70%) while HER remained low (FE 15%). Notably, 6 displays a tert-butyl substituent and similar electronic properties to 1, but is more bulky, which may change its capacity to adhere to the Cu electrode. It gives very different results; 6 still enhances the selectivity of C≥2 over C1 products, but the efficiency toward hydrogen (FE ≈ 50%) is significantly higher than with compounds 1, 3, 4, and 5, indicating that the steric profile of the additives may be important for selectivity. Compound 7, with a phenyl substituent on the pyridine ring, is similar in catalytic behavior to 1. The most remarkable observation for compounds 1, 3, 4, 5, and 7 is the increase in the C≥2/CH4 ratio from 0.7 with bare Cu to >15 with 3, >36 with 1, >130 with 4, >450 with 5, and >830 with 7. Cu with N-butylpyridinium (8) as an additive shows significantly more HER (FE ≈ 50%) compared to N-arylsubstituted pyridinium. The low overall Faradaic efficiency (∼80%) observed for the alkyl substituted pyridinium suggests that nonproductive side reactions occur during electrolysis, possibly including degradation of N-butylpyridinium itself.
Inspired by previously reported changes in CO2 reduction selectivity on Pd and Pt, H-substituted pyridinium was also investigated as an additive to polycrystalline copper under analogous conditions.30,31 Including pyridinium chloride (9) as additive substantially enhanced the HER (FE > 88%). Other mono- and dicationic pyridinium and imidazolium chloride compounds, which could potentially deposit on the electrode as neutral species upon reduction, were also tested (see Table 1). However, all of these additives facilitate mostly HER activity and low overall Faradaic yields. Runs with methylene blue (13) result in coloration of the solution, indicating that reduction of heterocycle-derived species that are not precipitated on the electrode occurs, lowering the FE for productive reduction chemistry. 1-(4-Pyridyl)pyridinium (10) behaves more like H-substituted pyridinium rather than the phenyl-substituted pyridinium derivatives, which could be due to a similar binding effect of the pyridine to the Cu active sites. Although 1,1′-diphenyl-4,4′-bipyridinium (12) exhibits a structure related to 2 by the addition of two hydride equivalents, only small amounts of hydrocarbon products were observed when it was used as the additive. This compound (12) formed a black precipitate on the Cu electrode after electrolysis, which is in contrast with the white films deposited for the case of compounds 1–7, suggesting the possibility of a different reaction pathway. The high yield for HER, reaching FE of ∼90% with pyridinium (9) and imidazolium (14), and almost quantitative total FE, demonstrate that organic additives can be used to tune the selectivity of electrocatalysis on Cu electrodes for different products from C≥2 hydrocarbons to H2.
The mechanism of tuning selectivity with organic additives remains unclear. Materials displaying pyridine-like N-sites reminiscent of the simple pyridinium motifs reported here have been employed for CO2 reduction. For example, electrocatalysts based on N-doped carbon materials have shown good FE toward CO production at low overpotentials,45−47 and N-doped graphene quantum dots have been reported to produce hydrocarbon products, mostly ethylene, ethanol, and n-propanol.48 With Cu nanoparticles on an N-doped carbon nanospike film, highly selective production of ethanol (FE = 63%) was observed at 1.2 VRHE.49 The weak C–H bonds present in species like 2 may be involved in H-transfers to surface bound intermediates of CO2 reduction.34,37,39 To test this possibility, 1-d5 was prepared featuring a perdeuterated pyridine moiety. Electrocatalytic CO2 reduction experiments performed with 1-d5 as the additive in natural abundance buffer resulted in similar behavior as with 1. 1H NMR analysis of the resulting film (Figure S6) is consistent with formation of species 2-d10, without H/D exchange. This observation suggests that the C–D(H) bonds in 2-d10 (and 2) are not cleaved and regenerated during electrocatalysis. We cannot strictly exclude the possibility that such processes do occur, to a very limited extent and below the detection limit of 1H NMR spectroscopy, on the layers of film proximal to the electrode.
Alternatively, the film of 2 could selectively suppress hydrogen and methane formation. Analysis of partial current densities for each product shows that addition of 1 does not affect the C–C coupled products, but lowers the rates of formation of methane and hydrogen, consistent with this proposal. This could indicate that the film selectively poisons Cu sites that catalyze C1 or H2 product formation. Alternatively, the electrodeposited film might restrict proton diffusion, thereby increasing the local pH near the electrode surface. A related effect has been demonstrated for mesostructured Ag and Au catalysts.25,26 Pyridinium compounds with N-aryl substituents that are relatively flat show the highest increase in C≥2 selectivity, while tert-butyl substitution leads to increased H2 production. This behavior suggests that differences in binding to specific Cu sites, in packing of the film affected by the steric profile of the pyridinium moiety, or in electrode roughness factors may significantly affect overall selectivity.
In summary, a simple, inexpensive, and tunable system for the reduction of CO2 to multicarbon products with high selectivity has been reported herein. Upon reduction on polycrystalline Cu electrodes, N-tolylpyridinium chloride forms a deposited film consisting of its bimolecular reductive coupling product. This electrode electrocatalytically reduces CO2 to C≥2 species with higher than 75% FE. Formation of methane and hydrogen is significantly inhibited for this and several related N-arylpyridinium additives, with the ratio of FE for C≥2 species to CH4 being >800 for certain additives. An aryl-substituted imidazolium additive is shown to dramatically shift the product selectivity to H2. More generally, the combination of a metal electrode and a reducible organic additive with potential to precipitate and form a film allows for tuning of selectivity between hydrocarbons and hydrogen.
Acknowledgments
NMR and XPS spectra were collected at the NMR Facility (Division of CCE) and Molecular Materials Research Center (Beckman Institute) of the California Institute of Technology, respectively. This material is based upon work performed by the Joint Center for Artificial Photosynthesis, a DOE Energy Innovation Hub, supported through the Office of Science of the U.S. Department of Energy under Award Number DE-SC0004993.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00180.
Author Contributions
† Z.H. and R.K. contributed equally. All authors designed experiments, analyzed data, and prepared the manuscript. Z.H. performed the synthesis and characterization of additives and compound 2, and evaluated their effects on electrocatalysis at −1.1 VRHE. R.K. performed potential-dependent (with and without additives), concentration-dependent and long-term electrocatalytic measurements and compared Cu foils from different suppliers. H.-Y.C. performed experiments with 13CO2, XPS and the effect of 1 added in various order. Z.H. and H.-Y.C. characterized the deposited film.
The authors declare no competing financial interest.
Supplementary Material
References
- Appel A. M.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson E. E.; Kubiak C. P.; Sathrum A. J.; Smieja J. M. Electrocatalytic and Homogeneous Approaches to Conversion of CO2 to Liquid Fuels. Chem. Soc. Rev. 2009, 38, 89–99. 10.1039/B804323J. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Robert M.; Savéant J. M. Catalysis of the Electrochemical Reduction of Carbon Dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. 10.1039/C2CS35360A. [DOI] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Jhong H-R. M.; Ma S.; Kenis P. J. Electrochemical Conversion of CO2 to Useful Chemicals: Current Status, Remaining Challenges, and Future Opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. 10.1016/j.coche.2013.03.005. [DOI] [Google Scholar]
- Gattrell M.; Gupta N.; Co A. A Review of the Aqueous Electrochemical Reduction of CO2 to Hydrocarbons at Copper. J. Electroanal. Chem. 2006, 594, 1–19. 10.1016/j.jelechem.2006.05.013. [DOI] [Google Scholar]
- Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. 10.1039/c2ee21234j. [DOI] [Google Scholar]
- Hori Y.Electrochemical CO2 Reduction on Metal Electrodes; Springer: New York, 2008; Vol. 42. [Google Scholar]
- Hori Y.; Kikuchi K.; Murata A.; Suzuki S. Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide at Copper Electrode in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1986, 15, 897–898. 10.1246/cl.1986.897. [DOI] [Google Scholar]
- Hori Y.; Takahashi I.; Koga O.; Hoshi N. Selective Formation of C2 Compounds from Electrochemical Reduction of CO2 at a Series of Copper Single Crystal Electrodes. J. Phys. Chem. B 2002, 106, 15–17. 10.1021/jp013478d. [DOI] [Google Scholar]
- Schouten K. J. P.; Pérez Gallent E.; Koper M. T. M. Structure Sensitivity of the Electrochemical Reduction of Carbon Monoxide on Copper Single Crystals. ACS Catal. 2013, 3, 1292–1295. 10.1021/cs4002404. [DOI] [Google Scholar]
- Huang Y.; Handoko A. D.; Hirunsit P.; Yeo B. S. Electrochemical Reduction of CO2 Using Copper Single-Crystal Surfaces: Effects of CO* Coverage on the Selective Formation of Ethylene. ACS Catal. 2017, 7, 1749–1756. 10.1021/acscatal.6b03147. [DOI] [Google Scholar]
- Kas R.; Kortlever R.; Yılmaz H.; Koper M. T. M.; Mul G. Manipulating the Hydrocarbon Selectivity of Copper Nanoparticles in CO2 Electroreduction by Process Conditions. ChemElectroChem 2015, 2, 354–358. 10.1002/celc.201402373. [DOI] [Google Scholar]
- Varela A. S.; Kroschel M.; Reier T.; Strasser P. Controlling the Selectivity of CO2 Electroreduction on copper: The Effect of the Electrolyte Concentration and the Importance of the Local pH. Catal. Today 2016, 260, 8–13. 10.1016/j.cattod.2015.06.009. [DOI] [Google Scholar]
- Kas R.; Kortlever R.; Milbrat A.; Koper M. T. M.; Mul G.; Baltrusaitis J. Electrochemical CO2 Reduction on Cu2O-Derived Copper Nanoparticles: Controlling the Catalytic Selectivity of Hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. 10.1039/C4CP01520G. [DOI] [PubMed] [Google Scholar]
- Gonçalves M. R.; Gomes A.; Condeço J.; Fernandes T. R. C.; Pardal T.; Sequeira C. A. C.; Branco J. B. Electrochemical Conversion of CO2 to C2 Hydrocarbons Using Different Ex Situ Copper Electrodeposits. Electrochim. Acta 2013, 102, 388–392. 10.1016/j.electacta.2013.04.015. [DOI] [Google Scholar]
- Tang W.; Peterson A. A.; Varela A. S.; Jovanov Z. P.; Bech L.; Durand W. J.; Dahl S.; Nørskov J. K.; Chorkendorff I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Electroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. 10.1039/C1CP22700A. [DOI] [PubMed] [Google Scholar]
- Gonçalves M. R.; Gomes A.; Condeço J.; Fernandes R.; Pardal T.; Sequeira C. A. C.; Branco J. B. Selective Electrochemical Conversion of CO2 to C2 Hydrocarbons. Energy Convers. Manage. 2010, 51, 30–32. 10.1016/j.enconman.2009.08.002. [DOI] [Google Scholar]
- Ren D.; Deng Y.; Handoko A. D.; Chen C. S.; Malkhandi S.; Yeo B. S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. 10.1021/cs502128q. [DOI] [Google Scholar]
- Handoko A. D.; Ong C. W.; Huang Y.; Lee Z. G.; Lin L.; Panetti G. B.; Yeo B. S. Mechanistic Insights into the Selective Electroreduction of Carbon Dioxide to Ethylene on Cu2O-Derived Copper Catalysts. J. Phys. Chem. C 2016, 120, 20058–20067. 10.1021/acs.jpcc.6b07128. [DOI] [Google Scholar]
- Roberts F. S.; Kuhl K. P.; Nilsson A. High Selectivity for Ethylene from Carbon Dioxide Reduction over Copper Nanocube Electrocatalysts. Angew. Chem., Int. Ed. 2015, 54, 5179–5182. 10.1002/anie.201412214. [DOI] [PubMed] [Google Scholar]
- Ma M.; Djanashvili K.; Smith W. A. Controllable Hydrocarbon Formation from the Electrochemical Reduction of CO2 over Cu Nanowire Arrays. Angew. Chem., Int. Ed. 2016, 55, 6680–6684. 10.1002/anie.201601282. [DOI] [PubMed] [Google Scholar]
- Mistry H.; Varela A. S.; Bonifacio C. S.; Zegkinoglou I.; Sinev I.; Choi Y.-W.; Kisslinger K.; Stach E. A.; Yang J. C.; Strasser P.; Cuenya B. R. Highly Selective Plasma-Activated Copper Catalysts for Carbon Dioxide Reduction to Ethylene. Nat. Commun. 2016, 7, 12123. 10.1038/ncomms12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttig A.; Liu C.; Peng Q.; Yaguchi M.; Hendon C. H.; Motobayashi K.; Ye S.; Osawa M.; Surendranath Y. Tracking a Common Surface-Bound Intermediate during CO2-to-Fuels Catalysis. ACS Cent. Sci. 2016, 2, 522–528. 10.1021/acscentsci.6b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. S.; Yoon Y.; Wuttig A.; Surendranath Y. Mesostructure-Induced Selectivity in CO2 Reduction Catalysis. J. Am. Chem. Soc. 2015, 137, 14834–14837. 10.1021/jacs.5b08259. [DOI] [PubMed] [Google Scholar]
- Yoon Y.; Hall A. S.; Surendranath Y. Tuning of Silver Catalyst Mesostructure Promotes Selective Carbon Dioxide Conversion into Fuels. Angew. Chem., Int. Ed. 2016, 55, 15282–15286. 10.1002/anie.201607942. [DOI] [PubMed] [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited Proton Transfer Enhances Au-Catalyzed CO2-to-Fuels Selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4585–E4593. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y.; Murata A.; Yoshinami Y. Adsorption of CO, Intermediately Formed in Electrochemical Reduction of CO2, at a Copper Electrode. J. Chem. Soc., Faraday Trans. 1991, 87, 125–128. 10.1039/ft9918700125. [DOI] [Google Scholar]
- Li C. W.; Ciston J.; Kanan M. W. Electroreduction of Carbon Monoxide to Liquid Fuel on Oxide-Derived Nanocrystalline Copper. Nature 2014, 508, 504–507. 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Seshadri G.; Lin C.; Bocarsly A. B. A New Homogeneous Electrocatalyst for the Reduction of CO2 to Methanol at Low Overpotentials. J. Electroanal. Chem. 1994, 372, 145–150. 10.1016/0022-0728(94)03300-5. [DOI] [Google Scholar]
- Barton Cole E.; Lakkaraju P. S.; Rampulla D. M.; Morris A. J.; Abelev E.; Bocarsly A. B. Using a One-Electron Shuttle for the Multielectron Reduction of CO2 to Methanol: Kinetic, Mechanistic, and Structural Insights. J. Am. Chem. Soc. 2010, 132, 11539–11551. 10.1021/ja1023496. [DOI] [PubMed] [Google Scholar]
- Rosen B. A.; Salehi-Khojin A.; Thorson M. R.; Zhu W.; Whipple D. T.; Kenis P. J.; Masel R. I. Ionic Liquid-Mediated Selective Conversion of CO2 to CO at Low Overpotentials. Science 2011, 334, 643–644. 10.1126/science.1209786. [DOI] [PubMed] [Google Scholar]
- Huan T. N.; Simon P.; Rousse G.; Génois I.; Artero V.; Fontecave M. Porous Dendritic Copper: an Electrocatalyst for Highly Selective CO2 Reduction to Formate in Water/Ionic Liquid Electrolyte. Chem. Sci. 2017, 8, 742–747. 10.1039/C6SC03194C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C.-H.; Holder A. M.; Hynes J. T.; Musgrave C. B. Reduction of CO2 to Methanol Catalyzed by a Biomimetic Organo-Hydride Produced from Pyridine. J. Am. Chem. Soc. 2014, 136, 16081–16095. 10.1021/ja510131a. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Zeitler E. L.; Gu J.; Hu Y.; Bocarsly A. B. Electrochemistry of Aqueous Pyridinium: Exploration of a Key Aspect of Electrocatalytic Reduction of CO2 to Methanol. J. Am. Chem. Soc. 2013, 135, 14020–14023. 10.1021/ja4064052. [DOI] [PubMed] [Google Scholar]
- Ertem M. Z.; Konezny S. J.; Araujo C. M.; Batista V. S. Functional Role of Pyridinium During Aqueous Electrochemical Reduction of CO2 on Pt(111). J. Phys. Chem. Lett. 2013, 4, 745–748. 10.1021/jz400183z. [DOI] [PubMed] [Google Scholar]
- Keith J. A.; Carter E. A. Theoretical Insights into Pyridinium-Based Photoelectrocatalytic Reduction of CO2. J. Am. Chem. Soc. 2012, 134, 7580–7583. 10.1021/ja300128e. [DOI] [PubMed] [Google Scholar]
- Marjolin A.; Keith J. A. Thermodynamic Descriptors for Molecules That Catalyze Efficient CO2 Electroreductions. ACS Catal. 2015, 5, 1123–1130. 10.1021/cs501406j. [DOI] [Google Scholar]
- McSkimming A.; Colbran S. B. The Coordination Chemistry of Organo-Hydride Donors: New Prospects for Efficient Multi-Electron Reduction. Chem. Soc. Rev. 2013, 42, 5439–5488. 10.1039/c3cs35466k. [DOI] [PubMed] [Google Scholar]
- Lobaccaro P.; Singh M. R.; Clark E. L.; Kwon Y.; Bell A. T.; Ager J. W. Effects of Temperature and Gas-Liquid Mass Transfer on the Operation of Small Electrochemical Cells for the Quantitative Evaluation of CO2 Reduction Electrocatalysts. Phys. Chem. Chem. Phys. 2016, 18, 26777–26785. 10.1039/C6CP05287H. [DOI] [PubMed] [Google Scholar]
- All N-substituted pyridinium additives studied herein have been prepared as their chloride salts. See Supporting Information for details.
- KH13CO3 was prepared from KOH and 13CO2. The current densities and product FE obtained with electrolyte prepared from KOH and natural abundance CO2 are nearly identical to those obtained in runs using commercial K2CO3 (Table S5), thus indicating comparable electrocatalytic behavior.
- Methanol (3.35 ppm) and part of the acetate (1.90 ppm) are impurities in the starting electrolytes and therefore appear as 12C isotopologues, the same as observed in the natural abundance CO2 experiment.
- Note that, while in our view unlikely, it is in principle also possible that a soluble form of 1 or its corresponding reduction product is released reversibly from the film during electrocatalysis.
- Wu J.; Yadav R. M.; Liu M.; Sharma P. P.; Tiwary C. S.; Ma L.; Zou X.; Zhou X. D.; Yakobson B. I.; Lou J.; Ajayan P. M. Achieving Highly Efficient, Selective and Stable CO2 Reduction on Nitrogen-Doped Carbon Nanotubes. ACS Nano 2015, 9, 5364–5371. 10.1021/acsnano.5b01079. [DOI] [PubMed] [Google Scholar]
- Wu J.; Liu M.; Sharma P. P.; Yadav R. M.; Ma L.; Yang Y.; Zou X.; Zhou X. D.; Vajtai R.; Yakobson B. I.; Lou J.; Ajayan P. M. Incorporation of Nitrogen Defects for Efficient Reduction of CO2 via Two-Electron Pathway on Three-Dimensional Graphene Foam. Nano Lett. 2016, 16, 466–470. 10.1021/acs.nanolett.5b04123. [DOI] [PubMed] [Google Scholar]
- Varela A. S.; Ranjbar Sahraie N.; Steinberg J.; Ju W.; Oh H.-S.; Strasser P. Metal-Doped Nitrogenated Carbon as an Efficient Catalyst for Direct CO2 Electroreduction to CO and Hydrocarbons. Angew. Chem., Int. Ed. 2015, 54, 10758–10762. 10.1002/anie.201502099. [DOI] [PubMed] [Google Scholar]
- Wu J.; Ma S.; Sun J.; Gold J. I.; Tiwary C.; Kim B.; Zhu L.; Chopra N.; Odeh I. N.; Vajtai R.; Yu A. Z.; Luo R.; Lou J.; Ding G.; Kenis P. J. A.; Ajayan P. M. A Metal-Free Electrocatalyst for Carbon Dioxide Reduction to Multi-Carbon Hydrocarbons and Oxygenates. Nat. Commun. 2016, 7, 13869. 10.1038/ncomms13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Peng R.; Hensley D. K.; Bonnesen P. V.; Liang L.; Wu Z.; Meyer H. M.; Chi M.; Ma C.; Sumpter B. G.; Rondinone A. J. High-Selectivity Electrochemical Conversion of CO2 to Ethanol Using a Copper Nanoparticle/N-Doped Graphene Electrode. ChemistrySelect 2016, 1, 6055–6061. 10.1002/slct.201601169. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.