

Abstract
Mitochondrial function is central to many different processes in the cell, from oxidative phosphorylation to the synthesis of iron-sulfur clusters. Therefore, mitochondrial dysfunction underlies a diverse array of diseases, from neurodegenerative diseases to cancer. Stress can be communicated to the cytosol and nucleus from the mitochondria through many different signals, and in response the cell can effect everything from transcriptional to post-transcriptional responses to protect the mitochondrial network. How these responses are coordinated have only recently begun to be understood. In this review we explore how the cell maintains mitochondrial function, focusing on the UPRmt, a transcriptional response that can activate a wide array of programs to repair and restore mitochondrial function.
Graphical abstract
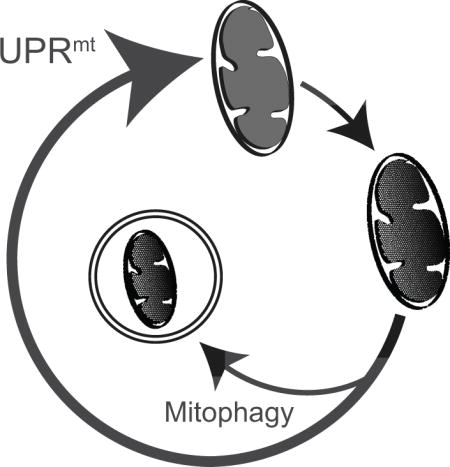
Mitochondria: More than energy production
Mitochondria are organelles descended from an endosymbioitic α-proteobacteria that was engulfed by pre-eukaryotic cells over a billion years ago (Allen, 2003, Lane and Martin, 2010, Gray, 2012). This engulfment event allowed energy production to be specialized to one area of the cell and contributed to the explosion of multicellular complexity found in metazoans. While mitochondria are associated with energy production, these organelles also serve important roles for production of essential metabolites and co-factors such as fatty acids (Osman et al., 2011), amino acids (Wellen and Thompson, 2012), and iron-sulfur clusters (Lill and Mühlenhoff, 2008). In addition to being a hub for the cell’s anabolic and catabolic reactions, mitochondria also serve as a signaling platform central for many processes in the cell, for example in the activation of apoptosis (Xiong et al., 2014). Because of these diverse functions, the preservation of mitochondrial homeostasis is vital to cellular and organismal health. Failure to maintain mitochondrial function results in a diverse array of diseases, from Parkinson’s disease (Vafai and Mootha, 2012) to cancer (Nunnari and Suomalainen, 2012).
Although the mitochondrial genome (mtDNA) is small in animals, about 16 kilobases in mammals, with a small number of genes encoded therein: 13 protein-coding, 22 tRNA, and 2 rRNA genes (Gray, 1999). In animal mtDNA there is a core of gene products always present that encodes components of the oxidative phosphorylation (OXPHOS) machinery that produce the majority of energy for the cell (Falkenberg et al., 2007). Transcription is polycistronic, producing two transcripts known as the heavy and light strands. While mitochondria harbor machinery to produce mtDNA gene products, the proteins encoded by mtDNA comprise only about 1% of the mitochondrial proteome. The other 99% of proteins located in mitochondria are encoded by the nuclear genome and synthesized in the cytosol. Because mitochondria are double-membrane bound organelles, proteins produced in the cytosol require a channel to be imported into mitochondria. This channel, a complex of proteins known as the translocase of the outer/inner membrane, transports unfolded proteins through the outer and inner membranes. Transport of a protein is dependent on the presence of a mitochondrial targeting sequence (MTS), the mitochondrial inner membrane potential (ΔΨ), ATP and molecular chaperones located within the mitochondrial matrix (Chacinska et al., 2009).
Mitochondrial defects affect a wide range of cellular processes and are associated with neurodegenerative disorders, cardiomyopathies, metabolic syndrome, cancer, and obesity. Mitochondrial disorders can manifest in any organ, ands at any age, depending on whether the mutations are autosomal, inherited from the X chromosome, or from the maternal line (Nunnari and Suomalainen, 2012). Furthermore, mtDNA mutations have pleiotropic effects due in part to heteroplasmy of mtDNA, the particular mix of wildtype and mutated mtDNA in a cell, and the severity of the mtDNA mutation (Wallace and Chalkia, 2013).
Cancer cells and mitochondrial dysfunction have long been linked, first in the description of the Warburg effect (Koppenol et al., 2011). The Warburg effect describes a phenomenon where glucose is fermented into lactic acid despite the ready availability of oxygen, often in cancer cells. While Warburg initially proposed that this phenomenon was due to mitochondrial dysfunction, the reality of how mitochondrial dysfunction interacts with cancer biology is now known to be much more complicated than previously hypothesized. Mitochondrial function is required for cancer cell viability, and depletion of functional mitochondria impairs tumor cell growth (Weinberg et al., 2010). MtDNA mutations are also associated with tumor growth, as many different types of cancers accumulate mtDNA mutations as they grow and some tumors contain homoplasmic mtDNA mutations (Polyak et al., 1998, Petros et al., 2005). These mutations can affect OXPHOS efficiency, and increase the production of reactive oxygen species (ROS) by mitochondria (Ralph et al., 2010, Ishikawa et al., 2008). Further evidence that mtDNA mutations drive cancer formation suggests that efficient mitochondrial function is tumor suppressive in some cases (Santidrian et al., 2013). While intriguing, it should be noted that the role of mtDNA mutations in cancer remains controversial as considerable data indicates that the mutations have little impact on cancer growth or survival (Ju et al., 2014).
Transcriptional Responses To Mitochondrial Dysfunction
As mitochondria are responsible for many essential cellular functions, maintenance of these organelles is critical. Mitochondrial activity is monitored by multiple mechanisms including mitochondria to nuclear communication, which occurs through several known pathways. Signals that convey mitochondrial status to the nucleus include calcium fluctuations (Biswas et al., 1999, Amuthan et al., 2001), ROS (Johnson et al., 2008), mitochondrial protein import efficiency (Nargund et al., 2012), metabolites (Mouchiroud et al., 2013), and energy production (Martinez-Reyes et al., 2012).
In yeast, mitochondrial dysfunction is signaled to the nucleus via the retrograde response (RTG). Mitochondrial stress caused by mtDNA depletion activates the transcription factors Rtg1 and Rtg3 (Liao and Butow, 1993, Jia et al., 1997). In the absence of mitochondrial stress, the Rtg1-Rtg3 complex is phosphorylated and sequestered in the cytosol. During stress, the phosphatase Rtg2 is activated resulting in dephosphorylation of Rtg1-Rtg3 and the transcription factors translocate to the nucleus and activate the RTG response. RTG activation causes induction of a number of metabolic genes including CIT2, a key enzyme in the glyoxylate cycle, which allows the cell to use alternate carbon sources for energy that can be processed independent of mitochondrial function (Butow and Avadhani, 2004, Liu and Butow, 1999). However, a pathway orthologous to the RTG pathway has not been found in metazoans. Rather, the RTG response seems to be fulfilled by multiple pathways. The transcription factors NRF1 and NRF2 control expression of OXPHOS genes, the voltage-dependent anion channel (VDAC), and mitochondrial transcription factors A and B (mtTFA and mtTFB) in mammals (Scarpulla, 2002). Peroxisome proliferator activated receptor (PPAR) α and γ control expression of genes involved in fatty acid metabolism and β-oxidation (Puigserver and Spiegelman, 2003) and PPARγ coactivator-1 (PGC-1) regulates mitochondrial biogenesis in tissues such as brown fat and skeletal muscle cells (Wu et al., 1999).
The UPRmt in Worms, Mice, and Humans
The mitochondrial unfolded protein response (UPRmt) is an adaptive transcriptional response that was initially described as a mechanism for cells to maintain mitochondrial protein homeostasis during mitochondrial dysfunction, as these organelles are constantly importing and processing proteins in an unfolded state. Hoogenraad and colleagues first described the pathway in rat hepatoma cells in which mtDNA had been depleted through ethidium bromide (EtBr) treatment (Martinus et al., 1996) and later found similar results when mitochondrial stress was caused by overexpression of a terminally misfolded mitochondrial protein (Zhao et al., 2002). However, many of the studies to determine how the UPRmt is regulated have been carried out in Caenorhabditis elegans (Figure 1). The UPRmt in C. elegans can be activated by conditions similar to those in mammalian cells that cause mitochondrial stress, such as depletion of mtDNA (Yoneda et al., 2004), OXPHOS components (Durieux et al., 2011), mitochondrial proteases (Nargund et al., 2012), perturbation of mitochondrial ribosomes (Moullan et al., 2015), or exposure to reagents that generate ROS (Yoneda et al., 2004). Genetic screens have identified multiple components required for UPRmt activation including the mitochondrial protease ClpP, the homeobox transcription factor DVE-1 (Haynes et al., 2007), the ubiquitin like protein UBL-5 (Benedetti et al., 2006), the mitochondrial peptide transporter HAF-1 (Haynes et al., 2010), multiple chromatin regulatory factors, (Merkwirth et al., 2016, Tian et al., 2016) and the transcription factor ATFS-1 (Nargund et al., 2015). Consistent with the UPRmt preserving mitochondrial function, worms lacking UPRmt components have impaired growth and survival during mitochondrial stress (Gatsi et al., 2014, Liu et al., 2014, Nargund et al., 2012).
Figure 1. Regulation of the UPRmt in C. elegans.
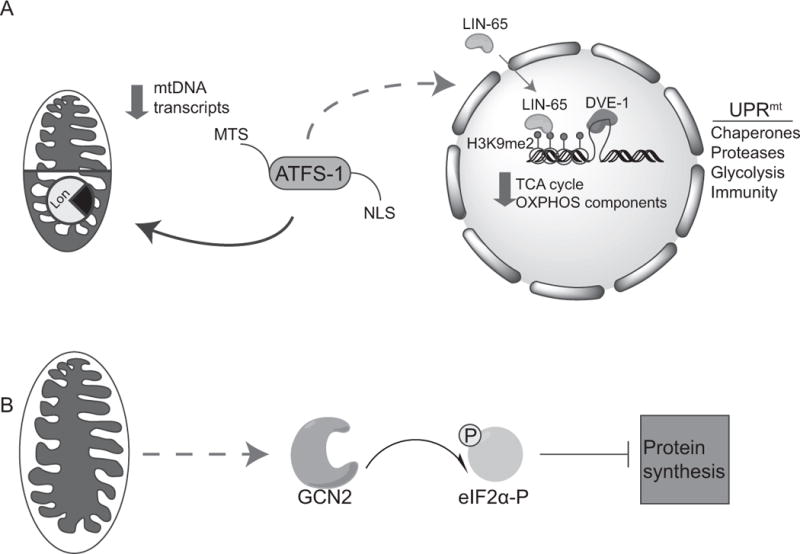
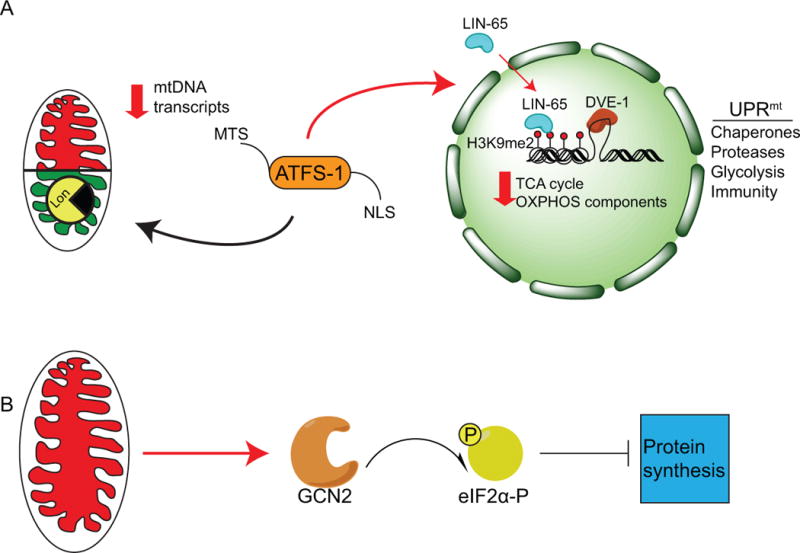
(A) The mitochondrial unfolded protein response (UPRmt) is regulated by the mitochondrial import efficiency of the transcription factor ATFS-1, which contains a mitochondrial targeting sequence (MTS) and a nuclear localization sequence (NLS). In the absence of stress, ATFS-1 is imported into mitochondria and subsequently degraded by the Lon protease. During stress import efficiency is reduced, and a percentage of ATFS-1 accumulates in the cytosol. ATFS-1 then translocates to the nucleus and activates a broad range of genes encoding mitochondrial chaperones and proteases as well as glycolysis and innate immune components, while limiting transcription of tricarboxylic acid (TCA) cycle genes, as well as oxidative phosphorylation (OXPHOS) genes encoded by both the nucleus and mitochondria. In addition to ATFS-1 signaling, the nuclear factor LIN-65 translocates to the nucleus during stress as well to modify chromatin, specifically the methylation of lysine 9 of histone H3 (H3K9me1/2) to allow the transcription factors DVE-1 and ATFS-1 to bind the promoters of UPRmt associated genes for transcription.
(B) In parallel, the eIF2α kinase GCN2 is activated during mitochondrial dysfunction and phosphorylates eIF2α reducing protein synthesis and the load of unfolded proteins in the stressed organelles. NB: Color version of this figure is available online.
Mitochondrial protein import efficiency, which is reliant on mitochondrial protein homeostasis and OXPHOS, of the bZip transcription factor ATFS-1 regulates the UPRmt. ATFS-1 is imported into the mitochondrial matrix via an N-terminal MTS where it is degraded by the Lon protease under normal conditions (Nargund et al., 2012). However during stress, import efficiency is reduced and a percentage of ATFS-1 accumulates in the cytosol. It then translocates to the nucleus via its nuclear localization sequence (NLS). This arrangement of two compartmental localization sequences within a single transcription factor couples activation of the UPRmt to mitochondrial import efficiency. Thus, mitochondrial import efficiency of ATFS-1 acts as a surrogate for the functional status of the entire mitochondrial network. Once activated, ATFS-1 upregulates over 400 genes involved in processes such as mitochondrial protein homeostasis, ROS detoxification, glycolysis, and interestingly, xenobiotic detoxification and innate immunity (Nargund et al., 2012). Perturbation of vital cellular functions activates an immune response, as multiple pathogens interfere with these vital functions (Melo and Ruvkun, 2012, Liu et al., 2014). Among those bacteria that activate the UPRmt are several that impair mitochondrial function. For example, exposure to the pathogen Pseudomonas aeruginosa activates the UPRmt as P. aeruginosa produces the OXPHOS inhibitor cyanide as a virulence factor resulting in activation of an innate immune response (Pellegrino et al., 2014, Liu et al., 2014).
Many of the genes upregulated by ATFS-1 contain a 14 base-pair sequence in the promoter region, termed the UPRmt element (UPRmtE), to which ATFS-1 directly binds. In addition to the genes that ATFS-1 upregulates, ATFS-1 limits transcription of genes encoding tricarboxylic acid (TCA) cycle enzymes, and OXPHOS components encoded by both the mitochondrial and nuclear genomes (Nargund et al., 2015). This potentially allows time for the recovery of mitochondrial protein homeostasis and regulates expression and efficient assembly of the highly expressed TCA cycle and OXPHOS complexes. In addition to these findings, during mitochondrial stress in mammals synthesis of mtDNA-encoded proteins is reduced (Munch and Harper, 2016).
In addition to transcriptional adaptions, mitochondrial stress during development affects long-term chromatin changes, and these epigenetic marks contribute to activation of the UPRmt in worms and mammals that maintains a “youthful” state (Tian et al., 2016, Merkwirth et al., 2016). Specifically, mitochondrial stress results in di-methylation of the histone H3K9, mediated by the methyltransferase met-2 and the nuclear factor lin-65. This change causes global silencing of chromatin, but opens up chromatin in regions associated with UPRmt activation and is required for DVE-1 and ATFS-1 to bind the appropriate promoters (Tian et al., 2016). Furthermore, two histone lysine demethylases, the Jumonji family proteins jmjd-1.2 and jmjd-3.1 are required for UPRmt activation and mitochondrial stress mediated longevity in worms (Merkwirth et al., 2016). The homologs of these proteins in mammals, PHF8 and JMJD3 respectively, positively correlate with methylation status, mRNA, and protein expression of UPRmt associated genes.
Interestingly, UPRmt activation can be communicated in a cell non-autonomous manner (Durieux et al., 2011, Berendzen et al., 2016). For example, mitochondrial dysfunction in neuronal cells activates a neuronal UPRmt, which in turn leads to activation of the UPRmt in intestinal cells. While the cell-to-cell communication is not completely understood, considerable data indicates a requirement for serotonin (Berendzen et al., 2016) and the secreted neuropeptide FLP-2. Furthermore, FLP-2 signaling involves a neural sub-circuit that includes two sensory neurons with environmental exposure and an interneuron. The sensory neurons communicate mitochondrial stress to the interneuron, which releases FLP-2 to signal to downstream neurons to activate the UPRmt in distal tissues (Shao et al., 2016), although how the intestinal cells receive the signal remains to be determined. Cell non-autonomous signaling potentially allows for metabolic coordination between cells (Berendzen et al., 2016) or pre-emptive UPRmt activation in distal cells in response to systemic mitochondrial stress, such as during bacterial infection.
ATF5 regulates a Mammalian Mitochondrial UPR
While C. elegans has proven useful to identify UPRmt signaling components, the initial observations were made in mammalian cells. Those studies used a mutated form of ornithine transcarbamylase (∆OTC), a protein that is terminally misfolded following import into mitochondria (Zhao et al., 2002) resulting in mitochondrial stress. ΔOTC expression results in upregulation of mitochondrial chaperones and proteases such as Cpn60 (HSP60) and ClpP. The transcription factor CHOP, which is also induced by ∆OTC expression, has been proposed to regulate the UPRmt but the mechanism by which CHOP is stimulated by mitochondrial stress and regulates UPRmt genes is unclear (Aldridge et al., 2007).
Using a combination of C. elegans and mammalian genetics, we found the mammalian bZIP transcription factor ATF5 to be required for UPRmt activation during a variety of mitochondrial stresses (Figure 2). ATF5, in addition to sharing homology with ATFS-1 and containing a putative MTS, is transcriptionally upregulated in several mitochondrial diseases such as the neurological disease spinocerebullar ataxia 28 (SCA28), which is caused by a mutation in the mitochondrial m-AAA protease AFG3L2 (Mancini et al., 2013). In addition, ATF5 is induced in a mouse model of mitochondrial myopathy caused by a dysfunctional mitochondrial helicase, Twinkle, which causes accumulation of mtDNA deletions (Tyynismaa et al., 2004, Tyynismaa et al., 2010). Similarly, ATF5 is induced in mice harboring a deletion in the mitochondrial aspartyl-tRNA synthetase DARS2, which results in mitochondrial stress (Tyynismaa et al., 2004, Dogan et al., 2014).
Figure 2. Regulation of a mammalian UPRmt.
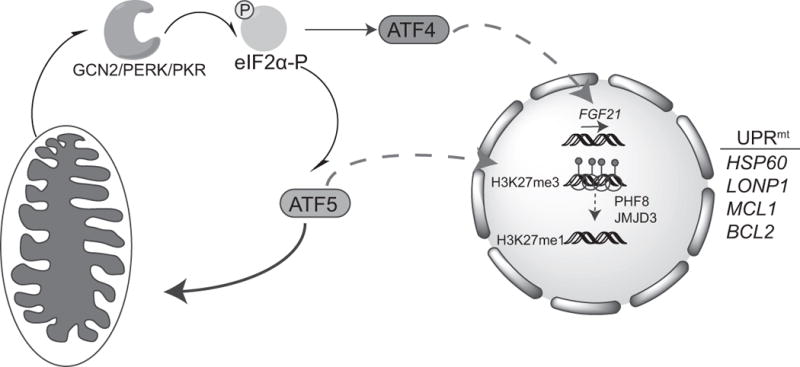
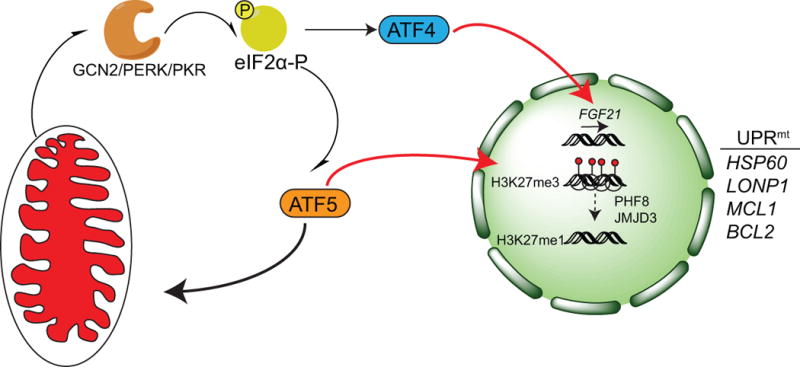
The bZIP transcription factor ATF5 has homology to ATFS-1 and localizes to mitochondria in the absence of stress. During mitochondrial stress, ATF5 translocates to the nucleus and upregulates chaperones (HSP60) and proteases (LONP1) associated with the UPRmt as well as anti-apoptotic genes like MCL1 and BCL2. ATF5 is also regulated by eIF2α–phosphorylation due to the presence of upstream open reading frames (uORFs) in one of the ATF5 mRNA splice variants, The eIF2α kinases GCN2, PERK, and PKR all phosphorylate eIF2α during mitochondrial stress but how they are activated is unclear. Thus, ATF5 activation is regulated by import efficiency and the Integrated Stress Response (ISR) to promote the recovery of dysfunctional mitochondria. The transcription factor ATF4 is also activated during mitochondrial stress via the ISR, which upregulates expression of the endocrine FGF21, which regulates metabolic changes in the organism. Like ATF5, the ATF4 transcript contains uORFs in the 5′ UTR of the transcript. Furthermore, mitochondrial stress results in chromatin reorganization by the histone demethylases PHF8 and JMJD3, which modify the methylation status of lysine 27 on histone H3 (H3K27me3/1) and mediate UPRmt gene expression. NB: Color version of this figure is available online.
Consistent with conservation of the UPRmt between worms and mammals, expression of ATF5 rescues UPRmt signaling in worms lacking atfs-1 (Fiorese et al., 2016). ATF5 is required for the increase of mitochondrial-protective transcripts in response to mitochondrial stress, such as the ROS-generating molecule paraquat, OXPHOS inhibition and transgenic expression of ΔOTC. And, transcriptional activation by ATF5 requires the same UPRmtE promoter element to which ATFS-1 binds in worms during mitochondrial stress (Nargund et al., 2015). And intriguingly, the promoters of those genes induced in the Twinkle mouse model of mitochondrial myopathy were enriched for the same UPRmtE (Tyynismaa et al., 2010).
In C. elegans, ATFS-1 responds to mitochondrial stress when mitochondrial protein import becomes impaired. It is well documented that ATF5 accumulates in the nucleus when activated (Monaco et al., 2007, Dalton et al., 2013), but in the absence of stress ATF5 accumulates in mitochondria in worm, mice, and human cells suggesting ATF5 is also regulated by mitochondrial import efficiency. Lastly, ATF5 is required for mitochondrial function and cellular recovery from a variety of mitochondrial perturbations including ∆OTC expression, mtDNA depletion and exposure to the ATP synthase inhibitor oligomycin (Fiorese et al., 2016). Thus, ATF5 is conceptually and experimentally similar to C. elegans ATFS-1.
It is important to note however, that differences between UPRmt signaling in worms and mammals are emerging. For example ClpP, a component required for UPRmt signaling in worms is likely dispensable for UPRmt signaling in mice and humans (Seiferling et al., 2016, Gispert et al., 2013). ClpP knockout in a DARS2 knockout model of cardiomyopathy has no effect on the induction of UPRmt genes. In fact, ClpP deletion seems to improve OXPHOS function and even extended the lifespan of DARS2 knockout mice, through a mechanism that results in decreased protein synthesis within mitochondria and increased protein turnover rates.
Additionally, there is the question of how ATF5 interacts with other proposed regulators of the UPRmt. For example ATF5 and CHOP have been show to regulate each other (Watatani et al., 2007, Teske et al., 2013), but how these two components interact during mitochondrial stress remains to be investigated. Additional transcription factors affected by mitochondrial stress include the estrogen receptor and FOXOA3, which upregulate expression of the mitochondrial protease Omi (Papa and Germain, 2011) and induce antioxidant genes (Papa and Germain, 2014) respectively. It will be interesting to determine how these pathways integrate with ATF5 and the UPRmt.
A Role for the Integrated Stress Response During Mitochondrial Stress
The integrated stress response (ISR) is characterized by the phosphorylation of eIF2α by one of four eIF2α kinases that are activated by different forms of stress. Endoplasmic reticulum stress activates the kinase PERK (Shi et al., 1998, Harding et al., 1999), PKR is activated by the accumulation of double stranded RNA in the cytosol (Meurs et al., 1990), general control nonderepressible 2 (GCN2) is activated by amino acid depletion and ROS (Harding et al., 2003, Shenton et al., 2006), and the heme regulated inhibitor kinase (HRI) is activated during heme depletion (Lu et al., 2001). Regardless of the upstream stressor, all four kinases phosphorylate eIF2α which represses global protein synthesis, but promotes translation of mRNAs harboring upstream open reading frames (uORFs) in the 5′ untranslated region (UTR) such as ATF4 and ATF5 (Lu et al., 2004, Zhou et al., 2008).
Interestingly, PKR (Rath et al., 2012), PERK (Hori et al., 2002) and GCN2 (Martinez-Reyes et al., 2012, Baker et al., 2012) can be activated during mitochondrial dysfunction although the respective modes of activation are unclear. Consistent with both mRNAs containing uORFs ATF4 and ATF5 are preferentially synthesized when eIF2α is phosphorylated (Harding et al., 2000, Zhou et al., 2008). As described above, ATF5 regulates expression of mitochondrial protective transcripts, as does ATF4. For example, ATF4 induces expression of LONP1, components of one carbon metabolism (Harding et al., 2003, Bao et al., 2016) as well as the endocrine hormone fibroblast growth factor 21 (FGF21) in the serum, which promotes metabolic coordination between tissues (Kim et al., 2013a, Kim et al., 2013b, Dogan et al., 2014).
Thus, considerable evidence in mammals supports a role for the ISR in the response to mitochondrial dysfunction, potentially through the UPRmt. In C. elegans, the ISR is not required for UPRmt activation as worms lacking all homologous eIF2α kinases are still able to activate the UPRmt. In fact, GCN2 promotes mitochondrial function during mitochondrial stress independent of ATFS-1, most likely by reducing protein synthesis and reducing the protein folding load on dysfunctional mitochondria (Baker et al., 2012). However, it should be noted that like ATF5, some atfs-1 mRNAs contain uORFs and some do not, suggesting ATFS-1 may be regulated by the ISR under specific stress conditions. Furthermore, most cultured mammalian cells only express the ATF5 splice variant harboring the uORF, thus requiring eIF2α phosphorylation to be translated. However, in mouse tissues such as liver, ATF5 mRNA and protein are expressed at relatively high levels likely due to relatively high steady state levels of eIF2α phosphorylation dependent of Gcn2 (Zhang et al., 2002, Fiorese et al., 2016, Pascual et al., 2008). Going forward, it will important to understand the contribution of the individual eIF2a kinases during mitochondrial stress, but also the role of each splice isoform of ATF5 and ATFS-1 both in tissue culture models, but more importantly in vivo.
Mitochondrial Quality Control
In addition to transcriptional adaptations to mitochondrial stress, cells also employ a number of quality control pathways that can degrade damaged mitochondrial proteins or whole organelles (Figure 3). The ubiquitin-proteasome system degrades outer membrane mitochondrial proteins as well as mitochondrial-targeted proteins that fail to be imported into mitochondria (Neutzner et al., 2008) while mitophagy sequesters mitochondria for degradation (Narendra et al., 2010).
Figure 3. Responses to mitochondrial dysfunction.
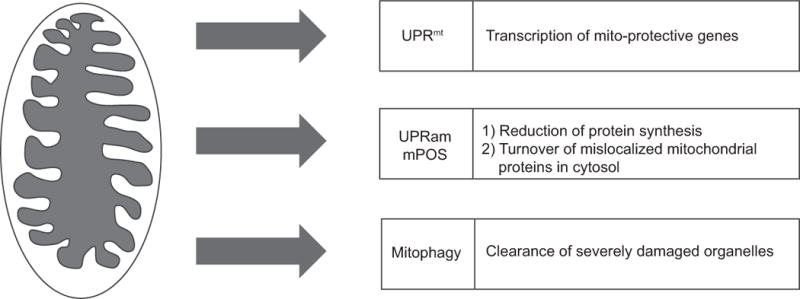
Mitochondrial stress activates quality control pathways such as the UPRmt, the UPRam/mPOS, and mitophagy. Once mitochondria are damaged, the UPRmt activates a transcriptional response to recover mitochondrial function, while the unfolded protein response activated by mistargeted proteins (UPRam) and mitochondrial precursor over-accumulation stress (mPOS) serve to reduce cytosolic protein synthesis and degrade mislocalized mitochondrial proteins that fail to be imported. The kinase PINK1 accumulates on the mitochondria outer membrane where it phosphorylates ubiquitin which recruits Parkin, leading to the clearance of severely damaged mitochondria via lysosome-dependent degradation. Coordination of these three pathways promotes the recovery of the mitochondrial network and cellular health. NB: Color version of this figure is available online.
During mitochondrial dysfunction, a reduction in protein import efficiency results in an accumulation of mislocalized mitochondria-targeted proteins in the cytosol. In response to this accumulation of precursor proteins, processes known as the unfolded protein response activated by mistargeted proteins (UPRam) (Wrobel et al., 2015), and mitochondrial precursor over-accumulation stress (mPOS) (Wang and Chen, 2015) are activated. Impressively, these programs increase cytosolic proteasome activity and reduce protein synthesis to rescue otherwise lethal mitochondrial dysfunction that impairs of mitochondrial protein import.
Mitophagy is a process to recognize and degrade mitochondria when the organelles have become severely damaged. The protein kinase PINK1 is a mitochondrial-localized protein that in the absence of stress is imported and degraded (Narendra et al., 2010). During stress, mitochondrial import is impaired causing PINK1 to accumulate on the outer membrane where it phosphorylates ubiquitin (Kane et al., 2014, Koyano et al., 2014, Ordureau et al., 2015). Decoration of mitochondrial proteins with ubiquitin serves to recruit the ubiquitin ligase Parkin, which in turn is phosphorylated and activated by PINK1 (Kazlauskaite et al., 2014). Parkin further ubiquitinates mitochondrial outer membrane proteins (Tanaka et al., 2010, Sarraf et al., 2013) leading to the recruitment of the autophagy receptors Optineurin (OPTN) and NDP52, which serves as a bridge between the cargo and the autophagosome. OPTN binds to polyubiquitin chains and recruits the kinase TBK1 to phosphorylate the autophagy receptors, which increases the affinity of the autophagy receptor for the autophagosome component LC3 and increases autophagic clearance (Lazarou et al., 2015, Richter et al., 2016, Heo et al., 2015, Ordureau et al., 2015). Thus, PINK1 initiates an amplification loop to increase mitophagy signaling ensuring the specific recognition and degradation of the least fit organelles.
Conclusion and Perspectives
Several strategies by which cells respond to mitochondrial stress have emerged in recent years including the UPRmt, mitophagy and pathways that protect the cytosol from mis-localized mitochondrial proteins. While the individual quality control pathways are still being elucidated, it will be interesting to understand how each pathway (Figure 3) integrates or interacts with one another to coordinate a successful defense against mitochondrial stress and maintenance of the mitochondrial network. The UPRmt, UPRam/mPOS, and mitophagy are all regulated by mitochondrial import efficiency. Do these pathways directly or indirectly antagonize each other, and what is the tipping point from recovery of mitochondrial function through the UPRmt and UPRam/mPOS to turnover of mitochondria via mitophagy?
The mammalian UPRmt appears to be regulated similarly to the C. elegans UPRmt, with ATF5 fulfilling many of the same roles as ATFS-1 in the worm. ATF5 localizes to mitochondria, and during mitochondrial stress translocates to the nucleus and upregulates mito-protective genes. However, much remains unknown about the UPRmt. For example, how does the UPRmt interact with the ISR? Perhaps it is through eIF2α kinases, many of which are activated by mitochondrial stress. Of particular interest are those known to signal downstream to ATF5 and ATF4, such as GCN2 (Zhou et al., 2008), PERK (Dalton et al., 2013), and PKR (Rath et al., 2012). Furthermore, additional mitochondrial stress activated transcriptional responses involving the estrogen receptor alpha and FOXOA3 have been identified (Papa and Germain, 2011), (Papa and Germain, 2014). It will be interesting to understand how these responses coordinate to promote cellular and organismal health.
The physiologic roles for the described pathways are expanding rapidly. Considerable data supports roles for the UPRmt during mitochondrial dysfunction associated with aging (Houtkooper et al., 2013), toxin exposure (Gatsi et al., 2014) and infection by pathogenic bacteria that perturb mitochondrial function (Pellegrino et al., 2014). However, recent work has demonstrated that prolonged UPRmt activation has potential negative consequences. In C. elegans, the UPRmt promotes the maintenance and propagation of deleterious mtDNAs in both aging somatic cells as well as between generations (Lin et al., 2016, Gitschlag et al., 2016). The UPRmt appears to both counter the mitophagy pathway in this respect, which serves to clear deleterious mtDNA, as well as promote deleterious mtDNA propagation directly. Furthermore, overexpression of Parkin eliminates mutant mtDNA in mammalian cells (Suen et al., 2010). It remains to be seen if the mammalian UPRmt fufills a similar role to the worm UPRmt in the maintenance of deleterious mtDNA.
ATF5 expression is required for survival of many transformed cells such as glioblastomas (Arias et al., 2012, Monaco et al., 2007), which are also affected by mitochondrial dysfunction (Deighton et al., 2014). How does ATF5, and by extension the UPRmt allow transformed cells to survive and grow in environments they normally would not? Cancer cells can gain advantage by aberrant activation of stress response pathways in other contexts (Paolicchi et al., 2016), and the UPRmt is an attractive candidate to target with inhibitors to prevent cancer cells from benefitting from this stress response pathway.
Acknowledgments
This work was supported by grants from the NIH (R01HL127891 and R01AG047182 to C.M.H.)
Footnotes
Declarations of Interest
The authors report no declarations of interest
References
- ALDRIDGE JE, HORIBE T, HOOGENRAAD NJ. Discovery of Genes Activated by the Mitochondrial Unfolded Protein Response (mtUPR) and Cognate Promoter Elements. PLoS ONE. 2007;2 doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLEN JF. The function of genomes in bioenergetic organelles. Philosophical Transactions of the Royal Society of London B: Biological Sciences. 2003;358:19–38. doi: 10.1098/rstb.2002.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AMUTHAN G, BISWAS G, ZHANG SY, KLEIN-SZANTO A, VIJAYASARATHY C, AVADHANI NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001;20:1910–20. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARIAS A, LAME MW, SANTARELLI L, HEN R, GREENE LA, ANGELASTRO JM. Regulated ATF5 loss-of-function in adult mice blocks formation and causes regression/eradication of gliomas. Oncogene. 2012;31:739–51. doi: 10.1038/onc.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKER BM, NARGUND AM, SUN T, HAYNES CM. Protective Coupling of Mitochondrial Function and Protein Synthesis via the eIF2? Kinase GCN-2. PLoS Genetics. 2012;8 doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAO XR, ONG SE, GOLDBERGER O, PENG J, SHARMA R, THOMPSON DA, VAFAI SB, COX AG, MARUTANI E, ICHINOSE F, GOESSLING W, REGEV A, CARR SA, CLISH CB, MOOTHA VK. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife. 2016;5 doi: 10.7554/eLife.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENEDETTI C, HAYNES CM, YANG Y, HARDING HP, RON D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174:229–39. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERENDZEN KM, DURIEUX J, SHAO LW, TIAN Y, KIM HE, WOLFF S, LIU Y, DILLIN A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell. 2016;166:1553–1563 e10. doi: 10.1016/j.cell.2016.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BISWAS G, ADEBANJO OA, FREEDMAN BD, ANANDATHEERTHAVARADA HK, VIJAYASARATHY C, ZAIDI M, KOTLIKOFF M, AVADHANI NG. Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 1999;18:522–33. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUTOW RA, AVADHANI NG. Mitochondrial signaling: the retrograde response. Molecular cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- CHACINSKA A, KOEHLER CM, MILENKOVIC D, LITHGOW T, PFANNER N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALTON RP, LYONS DB, LOMVARDAS S. Co-opting the unfolded protein response to elicit olfactory receptor feedback. Cell. 2013;155:321–32. doi: 10.1016/j.cell.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEIGHTON RF, LE BIHAN T, MARTIN SF, GERTH AM, MCCULLOCH M, EDGAR JM, KERR LE, WHITTLE IR, MCCULLOCH J. Interactions among mitochondrial proteins altered in glioblastoma. J Neurooncol. 2014;118:247–56. doi: 10.1007/s11060-014-1430-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOGAN SA, PUJOL C, MAITI P, KUKAT A, WANG S, HERMANS S, SENFT K, WIBOM R, RUGARLI EI, TRIFUNOVIC A. Tissue-specific loss of DARS2 activates stress responses independently of respiratory chain deficiency in the heart. Cell Metab. 2014;19:458–69. doi: 10.1016/j.cmet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- DURIEUX J, WOLFF S, DILLIN A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FALKENBERG M, LARSSON NG, GUSTAFSSON CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- FIORESE CJ, SCHULZ AM, LIN YF, ROSIN N, PELLEGRINO MW, HAYNES CM. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol. 2016;26:2037–43. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GATSI R, SCHULZE B, RODRIGUEZ-PALERO MJ, HERNANDO-RODRIGUEZ B, BAUMEISTER R, ARTAL-SANZ M. Prohibitin-mediated lifespan and mitochondrial stress implicate SGK-1, insulin/IGF and mTORC2 in C. elegans. PLoS One. 2014;9:e107671. doi: 10.1371/journal.pone.0107671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GISPERT S, PARGANLIJA D, KLINKENBERG M, DROSE S, WITTIG I, MITTELBRONN M, GRZMIL P, KOOB S, HAMANN A, WALTER M, BUCHEL F, ADLER T, HRABE DE ANGELIS M, BUSCH DH, ZELL A, REICHERT AS, BRANDT U, OSIEWACZ HD, JENDRACH M, AUBURGER G. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum Mol Genet. 2013;22:4871–87. doi: 10.1093/hmg/ddt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GITSCHLAG BL, KIRBY CS, SAMUELS DC, GANGULA RD, MALLAL SA, PATEL MR. Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C. elegans. Cell Metab. 2016;24:91–103. doi: 10.1016/j.cmet.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAY MW. Evolution of organellar genomes. Curr Opin Genet Dev. 1999;9:678–87. doi: 10.1016/s0959-437x(99)00030-1. [DOI] [PubMed] [Google Scholar]
- GRAY MW. Mitochondrial evolution. Cold Spring Harbor perspectives in biology. 2012;4:a011403. doi: 10.1101/cshperspect.a011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARDING HP, NOVOA I, ZHANG Y, ZENG H, WEK R, SCHAPIRA M, RON D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- HARDING HP, ZHANG Y, RON D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- HARDING HP, ZHANG Y, ZENG H, NOVOA I, LU PD, CALFON M, SADRI N, YUN C, POPKO B, PAULES R, STOJDL DF, BELL JC, HETTMANN T, LEIDEN JM, RON D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- HAYNES CM, PETROVA K, BENEDETTI C, YANG Y, RON D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–80. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- HAYNES CM, YANG Y, BLAIS SP, NEUBERT TA, RON D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–40. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEO JM, ORDUREAU A, PAULO JA, RINEHART J, HARPER JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015;60:7–20. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HORI O, ICHINODA F, TAMATANI T, YAMAGUCHI A, SATO N, OZAWA K, KITAO Y, MIYAZAKI M, HARDING HP, RON D, TOHYAMA M, STERN DM, OGAWA S. Transmission of cell stress from endoplasmic reticulum to mitochondria enhanced expression of Lon protease. The Journal of Cell Biology. 2002;157:1151–1160. doi: 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOUTKOOPER RH, MOUCHIROUD L, RYU D, MOULLAN N, KATSYUBA E, KNOTT G, WILLIAMS RW, AUWERX J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIKAWA K, TAKENAGA K, AKIMOTO M, KOSHIKAWA N, YAMAGUCHI A, IMANISHI H, NAKADA K, HONMA Y, HAYASHI J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–4. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- JIA Y, ROTHERMEL B, THORNTON J, BUTOW RA. A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol Cell Biol. 1997;17:1110–7. doi: 10.1128/mcb.17.3.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON JA, JOHNSON DA, KRAFT AD, CALKINS MJ, JAKEL RJ, VARGAS MR, CHEN PC. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann N Y Acad Sci. 2008;1147:61–9. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JU YS, ALEXANDROV LB, GERSTUNG M, MARTINCORENA I, NIK-ZAINAL S, RAMAKRISHNA M, DAVIES HR, PAPAEMMANUIL E, GUNDEM G, SHLIEN A, BOLLI N, BEHJATI S, TARPEY PS, NANGALIA J, MASSIE CE, BUTLER AP, TEAGUE JW, VASSILIOU GS, GREEN AR, DU MQ, UNNIKRISHNAN A, PIMANDA JE, TEH BT, MUNSHI N, GREAVES M, VYAS P, EL-NAGGAR AK, SANTARIUS T, COLLINS VP, GRUNDY R, TAYLOR JA, HAYES DN, MALKIN D, GROUP, I. B. C., GROUP, I. C. M. D., GROUP, I. P. C. FOSTER CS, WARREN AY, WHITAKER HC, BREWER D, EELES R, COOPER C, NEAL D, VISAKORPI T, ISAACS WB, BOVA GS, FLANAGAN AM, FUTREAL PA, LYNCH AG, CHINNERY PF, MCDERMOTT U, STRATTON MR, CAMPBELL PJ. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife. 2014;3 doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANE LA, LAZAROU M, FOGEL AI, LI Y, YAMANO K, SARRAF SA, BANERJEE S, YOULE RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205:143–53. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAZLAUSKAITE A, KONDAPALLI C, GOURLAY R, CAMPBELL DG, RITORTO MS, HOFMANN K, ALESSI DR, KNEBEL A, TROST M, MUQIT MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460:127–39. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM KH, JEONG YT, KIM SH, JUNG HS, PARK KS, LEE HY, LEE MS. Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem Biophys Res Commun. 2013a;440:76–81. doi: 10.1016/j.bbrc.2013.09.026. [DOI] [PubMed] [Google Scholar]
- KIM KH, JEONG YT, OH H, KIM SH, CHO JM, KIM YN, KIM SS, KIM DH, HUR KY, KIM HK, KO T, HAN J, KIM HL, KIM J, BACK SH, KOMATSU M, CHEN H, CHAN DC, KONISHI M, ITOH N, CHOI CS, LEE MS. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013b;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- KOPPENOL WH, BOUNDS PL, DANG CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- KOYANO F, OKATSU K, KOSAKO H, TAMURA Y, GO E, KIMURA M, KIMURA Y, TSUCHIYA H, YOSHIHARA H, HIROKAWA T, ENDO T, FON EA, TREMPE JF, SAEKI Y, TANAKA K, MATSUDA N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–6. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- LANE N, MARTIN W. The energetics of genome complexity. Nature. 2010;467:929–934. doi: 10.1038/nature09486. [DOI] [PubMed] [Google Scholar]
- LAZAROU M, SLITER DA, KANE LA, SARRAF SA, WANG C, BURMAN JL, SIDERIS DP, FOGEL AI, YOULE RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO X, BUTOW RA. RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993;72:61–71. doi: 10.1016/0092-8674(93)90050-z. [DOI] [PubMed] [Google Scholar]
- LILL R, MÜHLENHOFF U. Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- LIN YF, SCHULZ AM, PELLEGRINO MW, LU Y, SHAHAM S, HAYNES CM. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature. 2016;533:416–9. doi: 10.1038/nature17989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Y, SAMUEL BS, BREEN PC, RUVKUN G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature. 2014;508:406–10. doi: 10.1038/nature13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Z, BUTOW RA. A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol Cell Biol. 1999;19:6720–8. doi: 10.1128/mcb.19.10.6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU L, HAN AP, CHEN JJ. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol. 2001;21:7971–80. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU PD, HARDING HP, RON D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANCINI C, RONCAGLIA P, BRUSSINO A, STEVANIN G, LO BUONO N, KRMAC H, MALTECCA F, GAZZANO E, BARTOLETTI STELLA A, CALVARUSO MA, IOMMARINI L, CAGNOLI C, FORLANI S, LE BER I, DURR A, BRICE A, GHIGO D, CASARI G, PORCELLI AM, FUNARO A, GASPARRE G, GUSTINCICH S, BRUSCO A. Genome-wide expression profiling and functional characterization of SCA28 lymphoblastoid cell lines reveal impairment in cell growth and activation of apoptotic pathways. BMC Med Genomics. 2013;6:22. doi: 10.1186/1755-8794-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTINEZ-REYES I, SANCHEZ-ARAGO M, CUEZVA JM. AMPK and GCN2-ATF4 signal the repression of mitochondria in colon cancer cells. Biochem J. 2012;444:249–59. doi: 10.1042/BJ20111829. [DOI] [PubMed] [Google Scholar]
- MARTINUS RD, GARTH GP, WEBSTER TL, CARTWRIGHT P, NAYLOR DJ, HOJ PB, HOOGENRAAD NJ. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem. 1996;240:98–103. doi: 10.1111/j.1432-1033.1996.0098h.x. [DOI] [PubMed] [Google Scholar]
- MELO JA, RUVKUN G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149:452–66. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERKWIRTH C, JOVAISAITE V, DURIEUX J, MATILAINEN O, JORDAN SD, QUIROS PM, STEFFEN KK, WILLIAMS EG, MOUCHIROUD L, TRONNES SU, MURILLO V, WOLFF SC, SHAW RJ, AUWERX J, DILLIN A. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell. 2016;165:1209–23. doi: 10.1016/j.cell.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEURS E, CHONG K, GALABRU J, THOMAS NS, KERR IM, WILLIAMS BR, HOVANESSIAN AG. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–90. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- MONACO SE, ANGELASTRO JM, SZABOLCS M, GREENE LA. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int J Cancer. 2007;120:1883–90. doi: 10.1002/ijc.22469. [DOI] [PubMed] [Google Scholar]
- MOUCHIROUD L, HOUTKOOPER RH, MOULLAN N, KATSYUBA E, RYU D, CANTO C, MOTTIS A, JO YS, VISWANATHAN M, SCHOONJANS K, GUARENTE L, AUWERX J. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–41. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOULLAN N, MOUCHIROUD L, WANG X, RYU D, WILLIAMS EG, MOTTIS A, JOVAISAITE V, FROCHAUX MV, QUIROS PM, DEPLANCKE B, HOUTKOOPER RH, AUWERX J. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUNCH C, HARPER JW. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. 2016;534:710–3. doi: 10.1038/nature18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARENDRA DP, JIN SM, TANAKA A, SUEN DF, GAUTIER CA, SHEN J, COOKSON MR, YOULE RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARGUND AM, FIORESE CJ, PELLEGRINO MW, DENG P, HAYNES CM. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt) Mol Cell. 2015;58:123–33. doi: 10.1016/j.molcel.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARGUND AM, PELLEGRINO MW, FIORESE CJ, BAKER BM, HAYNES CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–90. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEUTZNER A, BENARD G, YOULE RJ, KARBOWSKI M. Role of the ubiquitin conjugation system in the maintenance of mitochondrial homeostasis. Ann N Y Acad Sci. 2008;1147:242–53. doi: 10.1196/annals.1427.012. [DOI] [PubMed] [Google Scholar]
- NUNNARI J, SUOMALAINEN A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ORDUREAU A, HEO JM, DUDA DM, PAULO JA, OLSZEWSKI JL, YANISHEVSKI D, RINEHART J, SCHULMAN BA, HARPER JW. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A. 2015;112:6637–42. doi: 10.1073/pnas.1506593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSMAN C, VOELKER DR, LANGER T. Making heads or tails of phospholipids in mitochondria. The Journal of Cell Biology. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAOLICCHI E, GEMIGNANI F, KRSTIC-DEMONACOS M, DEDHAR S, MUTTI L, LANDI S. Targeting hypoxic response for cancer therapy. Oncotarget. 2016;7:13464–78. doi: 10.18632/oncotarget.7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPA L, GERMAIN D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci. 2011;124:1396–402. doi: 10.1242/jcs.078220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPA L, GERMAIN D. SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol. 2014;34:699–710. doi: 10.1128/MCB.01337-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASCUAL M, GOMEZ-LECHON MJ, CASTELL JV, JOVER R. ATF5 is a highly abundant liver-enriched transcription factor that cooperates with constitutive androstane receptor in the transactivation of CYP2B6: implications in hepatic stress responses. Drug Metab Dispos. 2008;36:1063–72. doi: 10.1124/dmd.107.019380. [DOI] [PubMed] [Google Scholar]
- PELLEGRINO MW, NARGUND AM, KIRIENKO NV, GILLIS R, FIORESE CJ, HAYNES CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014;516:414–7. doi: 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETROS JA, BAUMANN AK, RUIZ-PESINI E, AMIN MB, SUN CQ, HALL J, LIM S, ISSA MM, FLANDERS WD, HOSSEINI SH, MARSHALL FF, WALLACE DC. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A. 2005;102:719–24. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLYAK K, LI Y, ZHU H, LENGAUER C, WILLSON JK, MARKOWITZ SD, TRUSH MA, KINZLER KW, VOGELSTEIN B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat Genet. 1998;20:291–3. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- PUIGSERVER P, SPIEGELMAN BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- RALPH SJ, RODRIGUEZ-ENRIQUEZ S, NEUZIL J, SAAVEDRA E, MORENO-SANCHEZ R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation - why mitochondria are targets for cancer therapy. Mol Aspects Med. 2010;31:145–70. doi: 10.1016/j.mam.2010.02.008. [DOI] [PubMed] [Google Scholar]
- RATH E, BERGER E, MESSLIK A, NUNES T, LIU B, KIM SC, HOOGENRAAD N, SANS M, SARTOR RB, HALLER D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61:1269–78. doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICHTER B, SLITER DA, HERHAUS L, STOLZ A, WANG C, BELI P, ZAFFAGNINI G, WILD P, MARTENS S, WAGNER SA, YOULE RJ, DIKIC I. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–44. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SANTIDRIAN AF, MATSUNO-YAGI A, RITLAND M, SEO BB, LEBOEUF SE, GAY LJ, YAGI T, FELDING-HABERMANN B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068–81. doi: 10.1172/JCI64264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARRAF SA, RAMAN M, GUARANI-PEREIRA V, SOWA ME, HUTTLIN EL, GYGI SP, HARPER JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–6. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCARPULLA RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- SEIFERLING D, SZCZEPANOWSKA K, BECKER C, SENFT K, HERMANS S, MAITI P, KONIG T, KUKAT A, TRIFUNOVIC A. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 2016;17:953–64. doi: 10.15252/embr.201642077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHAO LW, NIU R, LIU Y. Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res. 2016;26:1182–1196. doi: 10.1038/cr.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHENTON D, SMIRNOVA JB, SELLEY JN, CARROLL K, HUBBARD SJ, PAVITT GD, ASHE MP, GRANT CM. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem. 2006;281:29011–21. doi: 10.1074/jbc.M601545200. [DOI] [PubMed] [Google Scholar]
- SHI Y, VATTEM KM, SOOD R, AN J, LIANG J, STRAMM L, WEK RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUEN DF, NARENDRA DP, TANAKA A, MANFREDI G, YOULE RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107:11835–40. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANAKA A, CLELAND MM, XU S, NARENDRA DP, SUEN DF, KARBOWSKI M, YOULE RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TESKE BF, FUSAKIO ME, ZHOU D, SHAN J, MCCLINTICK JN, KILBERG MS, WEK RC. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. 2013 doi: 10.1091/mbc.E13-01-0067. [Online]. Available: http://www.molbiolcell.org files/53/F6.expansion.html. [DOI] [PMC free article] [PubMed]
- TIAN Y, GARCIA G, BIAN Q, STEFFEN KK, JOE L, WOLFF S, MEYER BJ, DILLIN A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt) Cell. 2016;165:1197–208. doi: 10.1016/j.cell.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TYYNISMAA H, CARROLL CJ, RAIMUNDO N, AHOLA-ERKKILA S, WENZ T, RUHANEN H, GUSE K, HEMMINKI A, PELTOLA-MJOSUND KE, TULKKI V, ORESIC M, MORAES CT, PIETILAINEN K, HOVATTA I, SUOMALAINEN A. Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet. 2010;19:3948–58. doi: 10.1093/hmg/ddq310. [DOI] [PubMed] [Google Scholar]
- TYYNISMAA H, SEMBONGI H, BOKORI-BROWN M, GRANYCOME C, ASHLEY N, POULTON J, JALANKO A, SPELBRINK JN, HOLT IJ, SUOMALAINEN A. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet. 2004;13:3219–27. doi: 10.1093/hmg/ddh342. [DOI] [PubMed] [Google Scholar]
- VAFAI SB, MOOTHA VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- WALLACE DC, CHALKIA D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X, CHEN XJ. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature. 2015;524:481–4. doi: 10.1038/nature14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATATANI Y, KIMURA N, SHIMIZU YI, AKIYAMA I, TONAKI D, HIROSE H, TAKAHASHI S, TAKAHASHI Y. Amino acid limitation induces expression of ATF5 mRNA at the post-transcriptional level. Life Sci. 2007;80:879–85. doi: 10.1016/j.lfs.2006.11.013. [DOI] [PubMed] [Google Scholar]
- WEINBERG F, HAMANAKA R, WHEATON WW, WEINBERG S, JOSEPH J, LOPEZ M, KALYANARAMAN B, MUTLU GM, BUDINGER GR, CHANDEL NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WELLEN KE, THOMPSON CB. A two-way street: reciprocal regulation of metabolism and signalling. Nature reviews Molecular cell biology. 2012;13:270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- WROBEL L, TOPF U, BRAGOSZEWSKI P, WIESE S, SZTOLSZTENER ME, OELJEKLAUS S, VARABYOVA A, LIRSKI M, CHROSCICKI P, MROCZEK S, JANUSZEWICZ E, DZIEMBOWSKI A, KOBLOWSKA M, WARSCHEID B, CHACINSKA A. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. 2015;524:485–8. doi: 10.1038/nature14951. [DOI] [PubMed] [Google Scholar]
- WU Z, PUIGSERVER P, ANDERSSON U, ZHANG C, ADELMANT G, MOOTHA V, TROY A, CINTI S, LOWELL B, SCARPULLA RC, SPIEGELMAN BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- XIONG S, MU T, WANG G, JIANG X. Mitochondria-mediated apoptosis in mammals. Protein & cell. 2014;5:737–749. doi: 10.1007/s13238-014-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YONEDA T, BENEDETTI C, URANO F, CLARK SG, HARDING HP, RON D. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci. 2004;117:4055–66. doi: 10.1242/jcs.01275. [DOI] [PubMed] [Google Scholar]
- ZHANG X, GRAND RJ, MCCABE CJ, FRANKLYN JA, GALLIMORE PH, TURNELL AS. Transcriptional regulation of the human glycoprotein hormone common alpha subunit gene by cAMP-response-element-binding protein (CREB)-binding protein (CBP)/p300 and p53. Biochem J. 2002;368:191–201. doi: 10.1042/BJ20020634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Q, WANG J, LEVICHKIN IV, STASINOPOULOS S, RYAN MT, HOOGENRAAD NJ. A mitochondrial specific stress response in mammalian cells. The EMBO journal. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU D, PALAM LR, JIANG L, NARASIMHAN J, STASCHKE KA, WEK RC. Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem. 2008;283:7064–73. doi: 10.1074/jbc.M708530200. [DOI] [PubMed] [Google Scholar]