

Abstract
Epilepsy, characterized by spontaneous recurrent seizures (SRS), is a serious and common neurological disorder afflicting an estimated 1% of the population worldwide. Animal experiments, especially those utilizing small laboratory rodents, remain essential to understanding the fundamental mechanisms underlying epilepsy and to prevent, diagnose, and treat this disease. While much attention has been focused on epileptogenesis in animal models of epilepsy, there is little discussion on SRS, the hallmark of epilepsy. This is in part due to the technical difficulties of rigorous SRS detection. In this review, we comprehensively summarize both genetic and acquired models of SRS and discuss the methodology used to monitor and detect SRS in mice and rats.
Keywords: Spontaneous recurrent seizures, Animal model, Epilepsy
INTRODUCTION
Epilepsy, a chronic neurological disorder that is characterized by spontaneous recurrent seizures (SRS), is the fourth most common neurological disorder (Hirtz et al., 2007). Epilepsy was first described over 2 500 years ago, yet there is still relatively little known about the underlying cause and currently no disease-modifying therapies exist. Current treatment options include antiepileptic drugs (AEDs), ketogenic diet, neurosurgical resection, and electrical stimulation of the central nervous system (CNS), which work for some but not all afflicted individuals (Laxer et al., 2014). Thus, there is an urgent unmet clinical need to discover treatments for the entire epileptic population. Most currently available AEDs were first identified using simple acute seizure models (i.e., pentylenetetrazol induced seizure and maximal electroshock seizure models) (Löscher, 2011). These acute models fail to mirror the spontaneous nature of seizures seen in epilepsy. This issue is hypothesized to contribute to the large percentage of epileptic patients (∼30%) for whom AEDs fail to prevent or control SRS. Therefore, studying epilepsy using laboratory animals exhibiting SRS will provide an important tool to explore the underlying mechanism of epilepsy and develop novel therapeutic approaches.
Epilepsy has been studied in a wide range of species of laboratory animals from simple organisms (e.g., Drosophila melanogaster, Caenorhabditis elegans and Danio rerio) to non-human primates. Along this spectrum, Rattus norvegicus (rat) and Mus musculus (mouse) are the two most commonly used laboratory animals given their small size, docility, rapid breeding, and availability of advanced genetic tools. Importantly, rat and mouse models provide good construct, face, and predictive validities of epilepsy and demand relatively low cost and maintenance for chronic study of SRS. In this review, we discuss the methodology of SRS recording, and summarize both genetic and acquired models of SRS in rat and mouse, with particular emphasis on modeling and detection of SRS. Mechanism and treatment of epileptogenesis are addressed in other reviews (Goldberg & Coulter, 2013; Löscher et al., 2013; McNamara et al., 2006; Pitkänen & Lukasiuk, 2011; Varvel et al., 2015).
MONITORING AND DETECTION OF SRS IN RODENTS
Chronic recording and detection of SRS in rodents is fundamental for preclinical research of epilepsy. Rigorous monitoring of SRS requires continuous time-locked video-EEG 24/7 in freely moving rodents. To capture biopotentials of the brain, most studies utilize single or multiple unipolar or bipolar recording electrodes which are intracranially placed. Skull or intracerebral electrode arrays are also used to cover broader brain regions. EEGs are acquired via either tethered or telemetry (wireless) recording systems in free-roaming, conscious rodents (Figure 1A). If a telemeter is used, it is either directly mounted on the head or tunneled and secured subcutaneously on the back or abdomen of rodents, providing the advantage of eliminating a wired interface between the animal and instrumentation. This minimizes the electrical noise and movement artifacts inherent in a tethered system. An inductive charging technique enables the telemeter to work 24/7 without the interruption of recharging the batteries.
Figure 1.
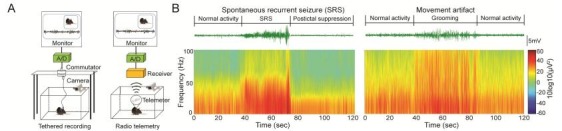
Schematic of video-EEG recording and EEG analyses
Given the rare, unpredictable nature and extremely diverse morphologies of SRS, identification of SRS is a technically challenging task. In most basic research settings, off-line visual inspection of EEG is performed by investigators to identify possible discrete epileptiform episodes, which are further confirmed by reviewing the time-locked video for behavioral correlates. Typical electrographic SRS features rhythmic neuronal firing characterized by increase of frequency and amplitude (especially in the gamma band) with clear initiation, propagation and termination (Figure 1B, left panel). In rodents, discrete epileptic discharges typically last seconds and are frequently followed by postictal suppression, which lasts minutes until normal electrographic activities resume. Electrographic SRS coincide with behavior phenotypes including rigid posture, facial automatisms, myoclonus, jumping and wild running, loss of postural control, tonic hindlimb extension, and death, which can be further semi-quantified using modified Racine’s scale (Ben-Ari, 1985; Racine, 1972). Spontaneous absence seizures characterized by spike-wave discharges (SWD) and behavioral arrest are also frequently observed in some models.
To achieve successful SRS monitoring and detection, the following factors also need to be considered: (1) depending on models, SRS are relatively rare and tend to cluster. The seizure-free latent or interictal period may last days or even weeks before first or subsequent SRS emerge. Therefore, long-term (weeks to months) recording is required to achieve meaningful interpretation; (2) in most studies, brain areas covered by electrodes are limited. Electrographic seizures may occur out of the recording site, and in the absence of overt behavior change; (3) rodents are commonly singly housed during monitoring to minimize damage of recording device and facilitate video analysis. How social isolation affects SRS needs to be evaluated; (4) to visualize animal behavior during dark cycles, in some studies, the recording area is illuminated, thereby disrupting the normal light/dark cycle of monitored animals. Infrared light and imaging devices are recommended for behavior monitoring during dark cycle if circadian rhythm is considered (Cho, 2012; Hofstra & De Weerd, 2009); (5) SRS automatic detection algorithm is available, but manual validation is strongly recommended.
SRS IN RODENT MODELS OF EPILEPSY
SRS in genetic models of epilepsy
Approximately 40% of epilepsies are idiopathic. Genetics play a significant role in the development, maintenance, and difficulty of treating epilepsy. A growing number of epilepsy-related single gene mutations have been identified. Animals possessing analogous genetic manipulations (engineered or spontaneous) have proven useful in the search for the possible treatment for idiopathic epilepsy (Table 1).
1.
SRS in transgenic models of epilepsy
| Gene | Modification | Latency | Frequency and features of SRS | References |
| *: model or strain dependent phenotype; ECS: editing site complementary sequence; OE: overexpression; SRS: spontaneous recurrent seizures; SUDEP: sudden unexpected death in epilepsy; SWD: spike-wave discharges. | ||||
| Scn1a | Scn1a-/- | P9 | Generalized convulsive SRS. SUDEP at P15 | Yu et al., 2006 |
| Scn1a+/- | P21–P27 | EEG and/or behavioral SRS lasted 20 s. Sporadic SUDEP from P21* | ||
| Scn1aFlox/+:: Zp3-Cre+/- | N/A | 12 out of 23 mice exhibited behavioral SRS (3 times/day, lasted 35 s). Lifespan of P33 | Dutton et al., 2013 | |
| Scn1aFlox/+:: Ppp1r2-Cre+/- | N/A | 2 out of 6 mice exhibited behavioral and/or EEG SRS | ||
| Scn1aR1407X/R1407X | P12–P16 | Multiple tonic-clonic SRS/day confirmed by EEG (lasted 1–3 min, interval: 1–4 hr). SUDEP by P21 | Ogiwara et al., 2007 | |
| Scn1aR1407X/+ | P18 | Sporadic SUDEP in 1–3 mo* | ||
| Scn1aR1648H/R1648H | P16 | Behavioral SRS lasted 30–90 s. SUDEP P16–P26 | Martin et al., 2010 | |
| Scn1aR1648H/+ | N/A | 2 out of 14 mice exhibited 21 SRS in total during 96 h EEG recording | ||
| Scn2a | Scn2aQ54 | 2 mo* | EEG and behavioral SRS. Frequency and duration of SRS increased with age* | Kearney et al., 2001 |
| Scn8a | Scn8a8J/+, Scn8amed/+ or Scn8amed-jo/+ | N/A | SWD with behavioral arrest* | Papale et al., 2009 |
| Scn8N1768D/N1768D | 3 wk | No SRS prior to day of SUDEP 3 wk. SRS lasted <1 min | Wagnon et al., 2015 | |
| Scn8N1768D/+ | 8–16 wk | 0–3 SRS/day. SRS lasted <1 min. SUDEP 14 wk | ||
| Scn8N1768D/- | 8 wk | As many as 25 SRS/day. SUDEP 9 wk | ||
| Scn1b | Scn1b-/- or Scn1bdel/del | P10 | EEG and behavioral SRS at random intervals with duration from seconds to minutes. SUDEP 3 wk | Chen et al., 2004; 2007 |
| Kcnq2 | Kcnq2A306T/A306T | P24 | Generalized EEG and behavioral SRS. SUDEP P16–P32* | Singh et al., 2008 |
| Kcnq3 | Kcnq3G311V/G311V | 2 wk | Generalized EEG and behavioral SRS. SUDEP P0–P73* | Singh et al., 2008 |
| Kcna1 | Kcna1-/- | 3 wk | Behavioral SRS lasted 20 s–2 min once or twice/hr throughout adult life. SUDEP 3–5 wk | Smart et al., 1998 |
| Kcna2 | Kcna2-/- | N/A | Tonic-clonic SRS. SUDEP at P17 | Brew et al., 2007; Douglas et al., 2007 |
| Kcnmb4 | Kcnmb4-/- | N/A | Generalized EEG seizures without overt behavioral manifestation | Brenner et al., 2005 |
| Cacna1a | Deletion (α1A−/−) | N/A | Absence seizures. SUDEP 3–4 wk | Jun et al., 1999 |
| Gria2 | Gria2+/ΔECS | P12 | Behavioral SRS (once/4 hr). SUDEP by P20 | Brusa et al., 1995 |
| Chrna4 | Chrna4S252F/S252F or + and Chrna4+L264/+L264 or + | N/A | SRS with high-amplitude, low-frequency cortical EEG activity, prominent theta and delta waves | Klaassen et al., 2006 |
| Gabrg2 | Gabrg2+/- or Gabrg2+/R43Q | P20 | Behavioral arrest and associated SWD*
(up to 50 times/hr and variable) |
Reid et al., 2013; Tan et al., 2007 |
| Tsc1/2 | Tsc1/2flox/flox::GFAP-Cre | 2–3 wk | Generalized tonic-clonic SRS. Few SRS at 3 wk, frequency increased over time. SUDEP 7–10 wk | Zeng et al., 2011 |
| Fgf13 | Fgf13+/- | P15 | Behavioral and EEG SRS. Frequency and duration varied by animal | Puranam et al., 2015 |
| Lgi1 | Lgi1-/- | P10 | Clonic SRS (1.6 seizures/hr at P14). SUDEP at P20 | Chabrol et al., 2010 |
| BACE1 | BACE1-/- | N/A | <40% rats exhibited generalized SRS and/or absence seizures | Hitt et al., 2010 |
| APP | APdE9 | N/A | 65% exhibited SRS, 10%–15% mortality at any age but peak around 3–4 mo | Minkeviciene et al., 2009 |
| hAPPFAD | N/A | Spontaneous nonconvulsive seizure activity. Occurrence of SUDEP | Palop et al., 2007 | |
| Ube3a | Ube3am+/p- | P18 | SWD accompanied by behavioral immobility or tonic-clonic SRS* | Miura et al., 2002, Jiang et al., 1998a; 2010 |
| Mecp2 | Viaat-Mecp2−/y Mecp2308/y | 5 wk N/A | Spontaneous rhythmic EEG activity including SWD* Spontaneous behavioral myoclonic jerks | Chao et al., 2010 D'Cruz et al., 2010 |
| Shank3 | Shank3 OE | N/A | Hyperexcitability discharges accompanied by EEG SRS | Han et al., 2013 |
| CNTNAP2 | CNTNAP2-/- | 6 mo | SRS with generalized interictal spike discharges | Peñagarikano et al., 2011 |
| Epm2A | Epm2A-/- | <9 mo | 80% exhibited myoclonic SRS, more frequent during dark cycle | Ganesh et al., 2002 |
| Celf4 | Celf4Ff/Ff or Celf4Ff/+ | 3 mo | Recurrent tonic-clonic seizures or absence seizures* | Yang et al., 2007 |
| Map2k1 | caMEK1flox/flox::Nestin-Cre | 6–8 wk | Lifetime behavioral arrest and forelimb myoclonus (6.2 SRS/7 hr) | Nateri et al., 2007 |
Ion channel genes
Ion channels control the electrical transduction of cells, thereby playing a pivotal role in regulating neuronal excitability. Most epilepsy-related genes encode proteins composing voltage- or ligand-gated ion channels. Below we summarize genetic models of epilepsy that result from mutations in various types of ion channels.
Of the many ion channels, a number of disruptions in genes encoding voltage-gated sodium channels have been described in multiple human epilepsies, including genetic epilepsy with febrile seizures plus (GEFS+) and Dravet syndrome. Disruptions of genes encoding either α (SCN1A, SCN2A and SCN8A) or β (SCN1B) subunits of voltage-gated sodium channels are sufficient to trigger SRS in rodents (Chen et al., 2004, 2007; Dutton et al., 2013; Kearney et al., 2001; Martin et al., 2010; Ogiwara et al., 2007; Papale et al., 2009; Wagnon et al., 2015; Yu et al., 2006). In addition, two modifier loci (Moe1 and Moe2) and multiple candidate modifier genes that influence the Scn2aQ54 epilepsy phenotype have also been identified and refined (Hawkins & Kearney, 2012).
Potassium channels also play an important role in action potentials by helping to return the neuron back to its resting membrane potential. Kcna1 and Kcna2 encode a pair of proteins (Kv1.1 and 1.2) which are members of the voltage-dependent potassium channel subfamily A. Kcna1 or Kcna2 knockout mice display frequent, severe SRS throughout their lives. In addition, SRS caused death in 50% of Kcna1 or Kcna2 knockout mice beginning from three weeks of age (Brew et al., 2007; Douglas et al., 2007; Smart et al., 1998). Mutations of Kcnq2 and Kcnq3, which encode subfamily Q of voltage-gated potassium channels have been found in patients with benign familial neonatal convulsions (BFNC). Kcnq2 or Kcnq3 mutant mice exhibit early onset generalized tonic-clonic SRS concurrent with M-current defects (Singh et al., 2008). Mice carrying Scn2aQ54 transgene together with Kcnq2 mutations (Szt1 or V182M) result in an exacerbated epileptic phenotype (Kearney et al., 2006). A gain-of-function mutation of gene Kcnmb4, which encodes calcium-activated potassium channel accessory β4 subunit also led to SRS (Brenner et al., 2005).
Calcium channels are important for neuronal excitability and intracellular signaling. Activation of T-type calcium channels evoke burst-firing in the thalamocortical circuitry that gives rise to SWD associated with absence epilepsy (Chen et al., 2014; Cheong & Shin, 2013). α1G T-type calcium currents play a critical role in the genesis of spontaneous absence seizures resulting from hypofunctioning P/Q-type channels (α1A−/−) (Jun et al., 1999; Song et al., 2004). These attacks have also been shown to reflect absence seizures in tottering (tg), leaner (tgla) and rocker (rkr) mice, which have spontaneously occurring mutant (Fletcher et al., 1996; Jun et al., 1999; Zwingman et al., 2001). In addition to pore-forming α1 subunit, loss of function mutations in ancillary subunits of calcium channels, including naturally occurring mutations in the β subunit gene Cchb4 in the lethargic (lh) mouse, loss of α2δ2 subunit protein in ducky mouse (du and du2j) and dysfunctional calcium channel γ2 subunits in stargazer (stg) and waggler (wgl) mice also result in SRS (Burgess et al., 1997; Zamponi et al., 2010).
In addition to voltage-gated ion channels, mutations of ligand-gated ion channel genes also result in SRS in mice. Heterozygous mice carrying an editing-deficient GRIA2 subunit allele express AMPA receptors with increased calcium permeability and develop SRS (Brusa et al., 1995). Fast ionotropic nicotinic acetylcholine receptor (nAChR) subunit genes, α2 (Chrna2), α4 (Chrna4) and β2 (Chrnb2), have been affiliated with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) when mutated. Mice with Chrna4 mutations (Chrna4S252F or Chrna4+L264) exhibited frequent SRS with diverse seizure semiology ranging from behavioral arrest to convulsive jerking (Klaassen et al., 2006). GABAA γ2-subunites have five known seizure associated mutations. Of these mutations, the R43Q mutation is of particular interest because it is related to childhood absence epilepsy and febrile seizures (Wallace et al., 2001). Both heterozygous Gabrg2 knock-out and R43Q knock-in mice exhibited spontaneous absence seizures accompanied by SWD (Reid et al., 2013; Tan et al., 2007).
Non-ion channel genes
SRS are also related to interruptions of non-ion channel genes that are involved in diverse neurological disorders including tuberous sclerosis complex (TSC), Alzheimer’s disease (AD) and autism. Notably, SRS can arise as a comorbid phenotype and/ or secondary consequence of gene modification from germline.
Epilepsy is the most common presenting symptom in TSC. Up to 80%–90% of individuals with TSC will develop epilepsy during their lifetime. Two genes, TSC1 and TSC2, encoding the proteins hamartin and tuberin, respectively, have been identified as causing TSC. Both genes, when conditionally inactivated in mice, have been shown to contribute to epileptic phenotype, among which Tsc2 led to more severe and frequent seizures (Zeng et al., 2011).
Prevalence of epilepsy in Alzheimer’s disease is significantly higher than in age-matched control populations. Manipulation of AD related genes (e.g., BACE1 and APP) can also cause SRS in mice. One study showed that BACE1 knockout mice were predisposed to both spontaneous and chemically induced seizures (Hitt et al., 2010). Autosomal-dominant mutations in amyloid precursor protein (APP) cause hereditary early-onset familial Alzheimer's disease (FAD). Transgenic mice overexpressing a mutant form of human APP (hAPPFAD) have spontaneous nonconvulsive seizure activity in cortical and hippocampal networks (Palop et al., 2007). It was shown that 65% of mice carrying human APP with Swedish double mutation (APPswe) cointegrated with human preselinin-1 with exon 9 deletion (PS1dE9) exhibited unprovoked seizures (Minkeviciene et al., 2009; Um et al., 2012).
Autism spectrum disorder (ASD) related genes are also extensively studied given the fact that epilepsy is common in individuals with autistic-like behavior resulting from particular genetic predisposition. A null mutation of maternal Ube3a gene (exon 1–2 or exon 15 and 16) results in core pathologies of Angelman syndrome including spontaneous EEG abnormality in mice (Jiang et al., 1998b; Miura et al., 2002). Spontaneous behavioral seizures were witnessed in mice with 1.6Mb large deletion (Ube3a to Babrb3) and loss of Ube3a selectively from the GABAergic neurons (Jiang et al., 2010; Judson et al., 2016). Global or conditional manipulation of Mecp2 gene in Rett syndrome model mice is also sufficient to elicit SRS, including spontaneous epileptiform discharges (Chao et al., 2010; D'Cruz et al., 2010; Shahbazian et al., 2002; Zhang et al., 2014). Mutations in the gene encoding SHANK3 and large duplications of the region spanning SHANK3 both cause ASD. Overexpression of SHANK3 in mice leads to SRS and maniac-like behavior (Han et al., 2013). The Cntnap2 gene which encodes a transmembrane protein that is essential in interactions between neurons and glia is strongly associated with ASD. Deletion of Cntnap2 leads to autistic-like behavior as well as SRS (Peñagarikano et al., 2011).
Along these lines, disruption of non-ion channel genes involved in many other disorders with epileptic manifestation also results in SRS in mice. Disruption of fibroblast growth factors 13 (FGF13) on the X chromosome is associated with GEFS+. Female mice in which one Fgf13 allele was deleted exhibited SRS (Puranam et al., 2015). Leucin-rich, glioma inactivated 1 (LGI1) is a secreted protein linked to human autosomal dominant epilepsy with auditory features (ADEAF). Lgi1 deletion in mice results in early onset SRS and seizure-related death. Selective deletion of Lgi1 in excitatory neurons, but not parvalbumin interneurons, contributes to the epileptic phenotype associated with LGI1 (Boillot et al., 2014; Chabrol et al., 2010). The gene Epm2a has been indicated in an autosomal recessive disorder known as Lafora Disease. Deletion of Epm2a can cause spontaneous myoclonic seizures with approximately 80% penetrance at the age of 9 months (Ganesh et al., 2002). Disruption of expression of doublecortin (Nosten-Bertrand et al., 2008), synapsin (Ketzef et al., 2011), CELF4 (Yang et al., 2007) or conditional expression of a constitutively active form of MAP/ERK kinases (Nateri et al., 2007) in the murine brain all led to SRS.
Besides genetically modified mice, SRS are also found in rats and mice with de novo mutations reported periodically in laboratories worldwide, like GAERS rat, WAG/Rij rat, lde/lde rat and tg, tgla, rkr, lh, du, stz, wgl mice (Noebels, 2006). Among these strains, GAERS rat and WAG/Rij rat are well validated genetic models of human absence epilepsy. Spontaneous absence seizures featuring SWD first appear at P30–P40 in GAERS rat, whereas they are observed at around P60–P80 in WAG/Rij rat. SWD in both strains are fully manifested with age and last throughout their lifetime (Coenen & van Luijtelaar, 2003; De Sarro et al., 2015; Marescaux et al., 1992). The progression of absence seizures with age in WAG/Rij and GAERS rats resembles genetically-determined epileptogenesis similar to post-brain insult epileptogenesis (Russo et al., 2016).
SRS in acquired models of epilepsy
It is estimated that up to 50% of all epilepsy cases are initiated by neurological insults also known as acquired epilepsy. To model acquired epilepsy in rodents, an episode of prolonged seizures, namely status epilepticus (SE), is commonly induced to trigger SRS (Table 2).
2.
SRS in acquired models of epilepsy
| Insult | Methods | Features | ||
| *: model or strain dependent phenotype; SE: status epilepticus; TBI: traumatic brain injury; KA: Kainic acid; DSP-4: N-(2-Chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride; TLE: temporal lobe epilepsy; SRS: spontaneous recurrent seizures; BLA: basolateral amygdala; AB: angular bundle: CCI: controlled cortical impact; LFP: lateral fluid percussion. | ||||
| SE | Pilocarpine (in the presence or absence of lithium) | Systemic or intracerebral injection | High mortality in general and wide spread brain damage* | |
| Kainic acid (KA) | Systemic or intracerebral injection | Hippocampal restricted damage. Short latent period (e.g., 3–5 days, KA amygdala infusion in mouse) | ||
| Bicuculine after a lesion induced by DSP-4 | Microinjection into anterior piriform cortex of rat | 30% developed SRS with mossy fiber sprouting | ||
| Tetanus toxin | Unilateral intrahippocampal injection in P10 rat | Early-life brain insult triggered diverse epileptiform response in adult rats | ||
| Febrile seizures | Hyperthermia in P10 rat | Mimic etiology of TLE. 35.2% rats developed SRS in adults | ||
| Sustained electrical stimulation | In BLA or AB of rat | Overall 80% (BLA) and 67% (AB) rats developed SRS | ||
| TBI | CCI or LFP | <50% developed SRS following TBI with long latent period* | ||
| Ischemia/hypoxia | Unilateral carotid ligation with hypoxia in P7 rat or global hypoxia in P10 rat | 100% rats developed SRS, which propagated along time | ||
| Methylazoxymethanol | In utero exposure | 2 out of 11 rats developed SRS | ||
| Virus infection | Intracerebral infection with Theiler’s murine encephalomyelitis virus | 75% mice developed seizures 3–10 days post infection* | ||
| Kindling | Over electrical kindling | Repeated daily electrical stimulus for weeks and months | Labor intensive, SRS have not been well characterized | |
| Flurothyl kindling | Repeated flurothyl induced convulsive seizures for 8 days (once/day) | SRS were observed within the first week following flurothyl kindling then remitted* | ||
Post-SE models
Kainic acid (KA, an ionotropic glutamate receptors agonist) and pilocarpine (a cholinergic muscarinic agonist) are two of the most commonly used chemicals to trigger SE (Ben-Ari, 1985; Ben-Ari et al., 1980; Turski et al., 1987, 1989). Systemic or intracerebral administration of KA causes SE followed by the emergence of SRS with remarkable histopathological correlation of hippocampal sclerosis in both rats and mice (Lévesque & Avoli, 2013). Compared to KA, pilocarpine-induced SE (in the presence or absence of lithium) results in higher mortality and wider spread brain damage in general along with SRS. The latency to onset of SRS and frequency of SRS varies depending on dose and administration route of chemicals as well as strains of animal. Convulsive SE can also be induced by microinjection of bicuculine into the anterior piriform cortex after a lesion of the locus coeruleus, which results in SRS in rat (Giorgi et al., 2006). In addition to chemically-induced convulsive SE, convulsive or non-convulsive SE can be induced by sustained electrical stimulation in the angular bundle or the basolateral amygdala of a rat, and can evoke SRS along with hippocampal sclerosis (Brandt et al., 2003; Gorter et al., 2001; Norwood et al., 2010). SE that occurred during early developmental stage can also cause SRS in adults. Unilateral injection of tetanus toxin into the hippocampus of P10 rats produces recurrent seizures for one week followed by epileptiform burst discharges (electrographic seizures on rare occasions) in adults (Jiang et al., 1998a; Lee et al., 1995). Both longitudinal and retrospective clinical studies reveal early life febrile SE causes temporal lobe epilepsy (TLE) in adults. Similarly, prolonged febrile seizures induced by hyperthermia in P10 rats render 35.2% of them epileptic in adulthood (Dubé et al., 2006).
Brain insults
SRS can also develop following direct brain insults such as traumatic brain injury (TBI), stroke and viral infection in both human and rodents in the absence of SE. TBI caused by controlled cortical impact (CCI) or lateral fluid-percussion injury (FPI) is able to elicit SRS in rats and mice (Bolkvadze & Pitkänen, 2012; D'ambrosio et al., 2004; Hunt et al., 2009; Kharatishvili et al., 2006). Rats that experienced global hypoxia at P10 or hypoxic-ischemic insult at P7 developed progressive SRS in adulthood (Kadam et al., 2010; Rakhade et al., 2011; Williams et al., 2004). Rats exposed to methylazoxymethanol in utero exhibited altered GluRs expression and developed sporadic SRS in adulthood (Harrington et al., 2007). Viral encephalitis of the CNS causes severe brain damage and epilepsy. Libbey et al. described the first mouse model of viral-induced epilepsy after intracerebral infection with Theiler's murine encephalomyelitis virus, where the seizures were transient and remitted after 10 days post infection (Libbey & Fujinami, 2011; Libbey et al., 2008).
Kindling models
Kindling is the process in which a train of repeated subconvulsive or subthreshold stimuli (electrical, audiogenic or chemical) renders a naïve animal more susceptible to subsequent stimuli. Kindling is a canonical model used for the study of epileptogenesis, yet it receives increasing criticism due to the lack of SRS. However, over-electrical kindling ultimately results in SRS (Kogure et al., 2000; McIntyre et al., 2002). Recent research revealed eight day consecutive flurothyl-kindling is sufficient to elicit SRS immediately after kindling, which remits weeks later (Kadiyala et al., 2016).
CONCLUDING REMARKS
Chronic rodent SRS recording is fundamental to preclinical study of epilepsy. A lack of standard methodology for SRS recording hampers the reproducibility of available models as well as the discovery of novel animal models of SRS. We recommend chronic 24/7 simultaneous video-EEG recording for rigorous study of SRS in rodents, and the recording period should vary from weeks to months depending on the model that is being used. Exclusive EEG recording often results in false positives because movement artifacts from grooming, drinking, and eating frequently generate epileptiform-like activity with rhythmic increases of frequency and amplitude (Figure 1B, right panel). Simultaneous analysis of behavior and EEG is necessary because exclusive video monitoring commonly fails to identify focal seizures or absence seizures since these lack overt behavioral manifestations.
While there are many ways to model SRS in rodents, the researcher first needs to decide what type of epilepsy they want to most closely recapitulate. Idiopathic or acquired epilepsy? TLE or absence seizures? Then the researcher needs to weigh the risks and benefits of each model that is chosen by studying the mortality and success rates and taking into consideration the developmental stage, length of latent period, frequency of SRS, electrographic and behavioral features of SRS, etc. Successful implication of rodent model of SRS will facilitate our knowledge of epilepsy and finally lead to discovery of potential biomarkers and therapies.
ACKNOWLEDGEMENTS
We thank Kamesh Krishnamurthy (Duke University, USA) for critical discussions and reading of the manuscript.
Funding Statement
This study was supported by the American Epilepsy Society Fellowship (2016)
REFERENCES
- 1. Ben-Ari Y, Tremblay E, Ottersen OP. 1980. Injections of kainic acid into the amygdaloid complex of the rat: an electrographic, clinical and histological study in relation to the pathology of epilepsy. Neuroscience, 5 (3): 515- 528. [DOI] [PubMed] [Google Scholar]
- 2. Ben-Ari Y. 1985. Limbic seizure and brain damage produced by kainic acid:mechanisms and relevance to human temporal lobe epilepsy. Neuroscience, 14 (2): 375- 403. [DOI] [PubMed] [Google Scholar]
- 3. Boillot M, Huneau C, Marsan E, Lehongre K, Navarro V, Ishida S, Dufresnois B, Ozkaynak E, Garrigue J, Miles R, Martin B, Leguern E, Anderson MP, Baulac S. 2014. Glutamatergic neuron-targeted loss of LGI1 epilepsy gene results in seizures. Brain, 137 2984- 2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bolkvadze T, Pitkänen A. 2012. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. Journal of Neurotrauma, 29 (5): 789- 812. [DOI] [PubMed] [Google Scholar]
- 5. Brandt C, Glien M, Potschka H, Volk H, Löscher W. 2003. Epileptogenesis and neuropathology after different types of status epilepticus induced by prolonged electrical stimulation of the basolateral amygdala in rats. Epilepsy Research, 55 (1-2): 83- 103. [DOI] [PubMed] [Google Scholar]
- 6. Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. 2005. BK channel β4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nature Neuroscience, 8 (12): 1752- 1759. [DOI] [PubMed] [Google Scholar]
- 7. Brew HM, Gittelman JX, Silverstein RS, Hanks TD, Demas VP, Robinson LC, Robbins CA, McKee-Johnson J, Chiu SY, Messing A, Tempel BL. 2007. Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. Journal of Neurophysiology, 98 (3): 1501- 1525. [DOI] [PubMed] [Google Scholar]
- 8. Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R. 1995. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science, 270 (5242): 1677- 1680. [DOI] [PubMed] [Google Scholar]
- 9. Burgess DL, Jones JM, Meisler MH, Noebels JL. 1997. Mutation of the Ca2+ channel β subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell, 88 (3): 385- 392. [DOI] [PubMed] [Google Scholar]
- 10. Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, RivaudPéchoux S, Fricker D, Baulac M, Miles R, LeGuern E, Baulac S. 2010. Electroclinical characterization of epileptic seizures in leucine-rich, gliomainactivated 1-deficient mice. Brain, 133 (9): 2749- 2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chao HT, Chen HM, Samaco RC, Xue MS, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JLR, Noebels JL, Rosenmund C, Zoghbi HY. 2010. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature, 468 (7321): 263- 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen CL, Westenbroek RE, Xu XR, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL. 2004. Mice lacking sodium channel β1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 24 (16): 4030- 4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen CL, Dickendesher TL, Oyama F, Miyazaki H, Nukina N, Isom LL. 2007. Floxed allele for conditional inactivation of the voltage-gated sodium channel β1 subunit Scn1b. Genesis, 45 (9): 547- 553. [DOI] [PubMed] [Google Scholar]
- 14. Chen YC, Parker WD, Wang KL. 2014. The role of T-type calcium channel genes in absence seizures. Frontiers in Neurology, 5 45- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheong E, Shin HS. 2013. T-type Ca2+ channels in absence epilepsy. Biochimica et Biophysica Acta (BBA)-Biomembranes, 1828 (7): 1560- 1571. [DOI] [PubMed] [Google Scholar]
- 16. Cho CH. 2012. Molecular mechanism of circadian rhythmicity of seizures in temporal lobe epilepsy. Frontiers in Cellular Neuroscience, 6 55- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coenen AM, van Luijtelaar ELJM. 2003. Genetic animal models for absence epilepsy: a review of the WAG/Rij strain of rats. Behavior Genetics, 33 (6): 635- 655. [DOI] [PubMed] [Google Scholar]
- 18. D'ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, Miller JW. 2004. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain, 127 304- 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. D'Cruz JA, Wu C, Zahid T, El-Hayek Y, Zhang L, Eubanks JH. 2010. Alterations of cortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiology of Disease, 38 (1): 8- 16. [DOI] [PubMed] [Google Scholar]
- 20. De Sarro G, Russo E, Citraro R, Meldrum BS. 2015. Genetically epilepsyprone rats (GEPRs) and DBA/2 mice: two animal models of audiogenic reflex epilepsy for the evaluation of new generation AEDs. Epilepsy & Behavior, 2015, 10.1016/j.yebeh.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 21. Douglas CL, Vyazovskiy V, Southard T, Chiu SY, Messing A, Tononi G, Cirelli C. 2007. Sleep in Kcna2 knockout mice. BMC Biology, 5 42- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dubé C, Richichi C, Bender RA, Chung G, Litt B, Baram TZ. 2006. Temporal lobe epilepsy after experimental prolonged febrile seizures:prospective analysis. Brain, 129 911- 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, Escayg A. 2013. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiology of Disease, 49 211- 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JD Jr, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. 1996. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell, 87 (4): 607- 617. [DOI] [PubMed] [Google Scholar]
- 25. Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, Akagi T, Gomi H, Suzuki T, Amano K, Agarwala KL, Hasegawa Y, Bai DS, Ishihara T, Hashikawa T, Itohara S, Cornford EM, Niki H, Yamakawa K. 2002. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Human Molecular Genetics, 11 (11): 1251- 1262. [DOI] [PubMed] [Google Scholar]
- 26. Giorgi FS, Mauceli G, Blandini F, Ruggieri S, Paparelli A, Murri L, Fornai F. 2006. Locus coeruleus and neuronal plasticity in a model of focal limbic epilepsy. Epilepsia, 47 (Suppl 5): 21- 25. [DOI] [PubMed] [Google Scholar]
- 27. Goldberg EM, Coulter DA. 2013. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nature Reviews Neuroscience, 14 (5): 337- 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gorter JA, Van Vliet EA, Aronica E, Da Silva FHL. 2001. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. European Journal of Neuroscience, 13 (4): 657- 669. [DOI] [PubMed] [Google Scholar]
- 29. Han K, Holder JL Jr, Schaaf CP, Lu H, Chen HM, Kang H, Tang JR, Wu ZY, Hao S, Cheung SW, Yu P, Sun H, Breman AM, Patel A, Lu HC, Zoghbi HY. 2013. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature, 503 (7474): 72- 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harrington EP, Möddel G, Najm IM, Baraban SC. 2007. Altered glutamate receptor-transporter expression and spontaneous seizures in rats exposed to methylazoxymethanol in utero. Epilepsia, 48 (1): 158- 168. [DOI] [PubMed] [Google Scholar]
- 31. Hawkins NA, Kearney JA. 2012. Confirmation of an epilepsy modifier locus on mouse chromosome 11 and candidate gene analysis by RNA-Seq. Genes, Brain and Behavavior, 11 (4): 452- 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, Zalutsky R. 2007. How common are the "common" neurologic disorders?. Neurology, 68 (5): 326- 337. [DOI] [PubMed] [Google Scholar]
- 33. Hitt BD, Jaramillo TC, Chetkovich DM, Vassar R. 2010. BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Molecular Neurodegener, 5 31- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hofstra WA, de Weerd AW. 2009. The circadian rhythm and its interaction with human epilepsy: a review of literature. Sleep Medicine Reviews, 13 (6): 413- 420. [DOI] [PubMed] [Google Scholar]
- 35. Hunt RF, Scheff SW, Smith BN. 2009. Posttraumatic epilepsy after controlled cortical impact injury in mice. Experimental Neurology, 215 (2): 243- 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang MH, Lee CL, Smith KL, Swann JW. 1998a. Spine loss and other persistent alterations of hippocampal pyramidal cell dendrites in a model of early-onset epilepsy. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 18 (20): 8356- 8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaud AL. 1998b. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron, 21 (4): 799- 811. [DOI] [PubMed] [Google Scholar]
- 38. Jiang YH, Pan YZ, Zhu L, Landa L, Yoo J, Spencer C, Lorenzo I, Brilliant M, Noebels J, Beaud AL. 2010. Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS One, 5 (8): e12278- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Judson MC, Wallace ML, Sidorov MS, Burette AC, Gu B, van Woerden GM, King IF, Han JE, Zylka MJ, Elgersma Y, Weinberg RJ, Philpot BD. 2016. GABAergic neuron-specific loss of Ube3a causes angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron, 90 (1): 56- 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jun K, Piedras-Rentería ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. 1999. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proceedings of the National Academy of Sciences of the United States of America, 96 (26): 15245- 15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kadam SD, White AM, Staley KJ, Dudek FE. 2010. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 30 (1): 404- 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kadiyala SB, Yannix JQ, Nalwalk JW, Papandrea D, Beyer BS, Herron BJ, Ferland RJ. 2016. Eight flurothyl-induced generalized seizures lead to the rapid evolution of spontaneous seizures in mice: a model of epileptogenesis with seizure remission. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 36 (28): 7485- 7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. 2001. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience, 102 (2): 307- 317. [DOI] [PubMed] [Google Scholar]
- 44. Kearney JA, Yang Y, Beyer B, Bergren SK, Claes L, DeJonghe P, Frankel WN. 2006. Severe epilepsy resulting from genetic interaction between Scn2a and Kcnq2. Human Molecular Genetics, 15 (6): 1043- 1048. [DOI] [PubMed] [Google Scholar]
- 45. Ketzef M, Kahn J, Weissberg I, Becker AJ, Friedman A, Gitler D. 2011. Compensatory network alterations upon onset of epilepsy in synapsin triple knock-out mice. Neuroscience, 189 108- 122. [DOI] [PubMed] [Google Scholar]
- 46. Kharatishvili I, Nissinen JP, Mcintosh TK, Pitkänen A. 2006. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience, 140 (2): 685- 697. [DOI] [PubMed] [Google Scholar]
- 47. Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. 2006. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 103 (50): 19152- 19157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kogure S, Kitayama M, Matsuda Y. 2000. Simultaneous kindling of the bilateral hippocampi: an advanced model for epilepsy research. Epilepsia, 41 (8): 929- 932. [DOI] [PubMed] [Google Scholar]
- 49. Laxer KD, Trinka E, Hirsch LJ, Cendes F, Langfitt J, Delanty N, Resnick T, Benbadis SR. 2014. The consequences of refractory epilepsy and its treatment. Epilepsy & Behavior, 37 59- 70. [DOI] [PubMed] [Google Scholar]
- 50. Lee CL, Hrachovy RA, Smith KL, Frost Jr JD, Swann JW. 1995. Tetanus toxin-induced seizures in infant rats and their effects on hippocampal excitability in adulthood. Brain Research, 677 (1): 97- 109. [DOI] [PubMed] [Google Scholar]
- 51. Lévesque M, Avoli M. 2013. The kainic acid model of temporal lobe epilepsy. Neuroscience & Biobehavioral Reviews, 37 (10): 2887- 2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Libbey JE, Kirkman NJ, Smith MCP, Tanaka T, Wilcox KS, White HS, Fujinami RS. 2008. Seizures following picornavirus infection. Epilepsia, 49 (6): 1066- 1074. [DOI] [PubMed] [Google Scholar]
- 53. Libbey JE, Fujinami RS. 2011. Neurotropic viral infections leading to epilepsy: focus on Theiler's murine encephalomyelitis virus. Future Virology, 6 (11): 1339- 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Löscher W. 2011. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure, 20 (5): 359- 368. [DOI] [PubMed] [Google Scholar]
- 55. Löscher W, Klitgaard H, Twyman RE, Schmidt D. 2013. New avenues for anti-epileptic drug discovery and development. Nature Reviews Drug discovery, 12 (10): 757- 776. [DOI] [PubMed] [Google Scholar]
- 56. Marescaux C, Vergnes M, Depaulis A. 1992. Genetic absence epilepsy in rats from strasbourg-a review. Journal of Neural Transmission. Supplementum, 35 37- 69. [DOI] [PubMed] [Google Scholar]
- 57. Martin MS, Dutt K, Papale LA, Dubé CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, Goldin AL, Escayg A. 2010. Altered function of the SCN1A voltage-gated sodium channel leads to γ-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. Journal of Biological Chemistry, 285 (13): 9823- 9834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McIntyre DC, Poulter MO, Gilby K. 2002. Kindling: some old and some new. Epilepsy Research, 50 (1-2): 79- 92. [DOI] [PubMed] [Google Scholar]
- 59. McNamara JO, Huang YZ, Leonard AS. 2006. Molecular signaling mechanisms underlying epileptogenesis. Science's STKE, 2006 (356): re12- [DOI] [PubMed] [Google Scholar]
- 60. Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. 2009. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 29 (11): 3453- 3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miura K, Kishino T, Li E, Webber H, Dikkes P, Holmes GL, Wagstaff J. 2002. Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiology of Disease, 9 (2): 149- 159. [DOI] [PubMed] [Google Scholar]
- 62. Nateri AS, Raivich G, Gebhardt C, Da Costa C, Naumann H, Vreugdenhil M, Makwana M, Brandner S, Adams RH, Jefferys JGR, Kann O, Behrens A. 2007. ERK activation causes epilepsy by stimulating NMDA receptor activity. The EMBO Journal, 26 (23): 4891- 4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Noebels JL. 2006. CHAPTER 17-spontaneous epileptic mutations in the mouse A2-Pitkänen, Asla. schwartzkroin PA and Moshé SL. In: Models of Seizures and Epilepsy. Burlington: Academic Press, 223- 232. [Google Scholar]
- 64. Norwood BA, Bumanglag AV, Osculati F, Sbarbati A, Marzola P, Nicolato E, Fabene PF, Sloviter RS. 2010. Classic hippocampal sclerosis and hippocampal-onset epilepsy produced by a single "cryptic" episode of focal hippocampal excitation in awake rats. The Journal of Comparative Neurology, 518 (16): 3381- 3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nosten-Bertrand M, Kappeler C, Dinocourt C, Denis C, Germain J, Tuy FPD, Verstraeten S, Alvarez C, Métin C, Chelly J, Giros B, Miles R, Depaulis A, Francis F. 2008. Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PLoS One, 3 (6): e2473- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. 2007. Nav1. 1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 27 (22): 5903- 5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. 2007. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron, 55 (5): 697- 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, Letts VA, Meisler MH, Frankel WN, Escayg A. 2009. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Human Molecular Genetics, 18 (9): 1633- 1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong HM, Sonnenblick LI, Gruver R, Almajano J, Bragin A, Golshani P, Trachtenberg JT, Peles E, Geschwind DH. 2011. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autismrelated deficits. Cell, 147 (1): 235- 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pitkänen A, Lukasiuk K. 2011. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol, 10 (2): 173- 186. [DOI] [PubMed] [Google Scholar]
- 71. Puranam RS, He XP, Yao LJ, Le T, Jang W, Rehder CW, Lewis DV, McNamara JO. 2015. Disruption of Fgf13 causes synaptic excitatoryinhibitory imbalance and genetic epilepsy and febrile seizures plus. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 35 (23): 8866- 8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Racine RJ. 1972. Modification of seizure activity by electrical stimulation: Ⅱ. Motor seizure. Electroencephalography and Clinical Neurophysiology, 32 (3): 281- 294. [DOI] [PubMed] [Google Scholar]
- 73. Rakhade SN, Klein PM, Huynh T, Hilario-Gomez C, Kosaras B, Rotenberg A, Jensen FE. 2011. Development of later life spontaneous seizures in a rodent model of hypoxia-induced neonatal seizures. Epilepsia, 52 (4): 753- 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Reid CA, Kim T, Phillips AM, Low J, Berkovic SF, Luscher B, Petrou S. 2013. Multiple molecular mechanisms for a single GABAA mutation in epilepsy. Neurology, 80 (11): 1003- 1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Russo E, Citraro R, Constanti A, Leo A, Lüttjohann A, van Luijtelaar G, De Sarro G. 2016. Upholding WAG/Rij rats as a model of absence epileptogenesis: hidden mechanisms and a new theory on seizure development. Neuroscience & Biobehavioral Reviews, 71 388- 408. [DOI] [PubMed] [Google Scholar]
- 76. Shahbazian M, Young JI, Yuva-Paylor LA, Spencer CM, Antalffy BA, Noebels JL, Armstrong DL, Paylor R, Zoghbi HY. 2002. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron, 35 (2): 243- 254. [DOI] [PubMed] [Google Scholar]
- 77. Singh NA, Otto JF, Dahle EJ, Pappas C, Leslie JD, Vilaythong A, Noebels JL, White HS, Wilcox KS, Leppert MF. 2008. Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. Journal of Physiology, 586 (14): 3405- 3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. 1998. Deletion of the KV1.1 potassium channel causes epilepsy in mice. Neuron, 20 (4): 809- 819. [DOI] [PubMed] [Google Scholar]
- 79. Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. 2004. Role of the α1G Ttype calcium channel in spontaneous absence seizures in mutant mice. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 24 (22): 5249- 5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tan HO, Reid CA, Single FN, Davies PJ, Chiu C, Murphy S, Clarke AL, Dibbens L, Krestel H, Mulley JC, Jones MV, Seeburg PH, Sakmann B, Berkovic SF, Sprengel R, Petrou S. 2007. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proceedings of the National Academy of Sciences of the United States of America, 104 (44): 17536- 17541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Turski L, Cavalheiro EA, Czuczwar SJ, Turski WA, Kleinrok Z. 1987. The seizures induced by pilocarpine: behavioral, electroencephalographic and neuropathological studies in rodents. Polish Journal of Pharmacology and Pharmacy, 39 (5): 545- 555. [PubMed] [Google Scholar]
- 82. Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. 1989. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy.Synapse, 3 (2): 154- 171. [DOI] [PubMed] [Google Scholar]
- 83. Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. 2012. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates fyn to impair neurons. Nature Neuroscience, 15 (9): 1227- 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Varvel NH, Jiang JX, Dingledine R. 2015. Candidate drug targets for prevention or modification of epilepsy. Annual Review of Pharmacology and Toxicology, 55 229- 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH. 2015. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Human Molecular Genetics, 24 (2): 506- 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. 2001. Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nature Genetics, 28 (1): 49- 52. [DOI] [PubMed] [Google Scholar]
- 87. Williams PA, Dou P, Dudek FE. 2004. Epilepsy and synaptic reorganization in a perinatal rat model of hypoxia-ischemia. Epilepsia, 45 (10): 1210- 1218. [DOI] [PubMed] [Google Scholar]
- 88. Yang Y, Mahaffey CL, Béerubé N, Maddatu TP, Cox GA, Frankel WN. 2007. Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genetics, 3 (7): e124- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. 2006. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature Neuroscience, 9 (9): 1142- 1149. [DOI] [PubMed] [Google Scholar]
- 90. Zamponi GW, Lory P, Perez-Reyes E. 2010. Role of voltage-gated calcium channels in epilepsy. Pflügers Archiv-European Journal of Physiology, 460 (2): 395- 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. 2011. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Human Molecular Genetics, 20 (3): 445- 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang W, Peterson M, Beyer B, Frankel WN, Zhang ZW. 2014. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 34 (7): 2754- 2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zwingman TA, Neumann PE, Noebels JL, Herrup K. 2001. Rocker is a new variant of the voltage-dependent calcium channel gene Cacna1a. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience, 21 (4): 1169- 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]