

Abstract
Left ventricular (LV) remodelling after myocardial infarction (MI) is a crucial determinant of the clinical course of heart failure. Matrix metalloproteinase (MMP) activation is strongly associated with LV remodelling after MI. Elucidation of plasma membrane receptors related to the activation of specific MMPs is fundamental for treating adverse cardiac remodelling after MI. The aim of current investigation was to explore the potential association between the low‐density lipoprotein receptor‐related protein 1 (LRP1) and MMP‐9 and MMP‐2 spatiotemporal expression after MI. Real‐time PCR and Western blot analyses showed that LRP1 mRNA and protein expression levels, respectively, were significantly increased in peri‐infarct and infarct zones at 10 and 21 days after MI. Confocal microscopy demonstrated high colocalization between LRP1 and the fibroblast marker vimentin, indicating that LRP1 is mostly expressed by cardiac fibroblasts in peri‐infarct and infarct areas. LRP1 also colocalized with proline‐rich tyrosine kinase 2 (pPyk2) and MMP‐9 in cardiac fibroblasts in ischaemic areas at 10 and 21 days after MI. Cell culture experiments revealed that hypoxia increases LRP1, pPyk2 protein levels and MMP‐9 activity in fibroblasts, without significant changes in MMP‐2 activity. MMP‐9 activation by hypoxia requires LRP1 and Pyk2 phosphorylation in fibroblasts. Collectively, our in vivo and in vitro data support a major role of cardiac fibroblast LRP1 levels on MMP‐9 up‐regulation associated with ventricular remodelling after MI.
Keywords: LRP1, MMP‐9, cardiac remodelling, myocardial infarction, pPyk2
Introduction
Adverse myocardial remodelling is associated with poor patient outcomes in the setting of ischaemic heart disease and/or MI, cardiac hypertrophy and cardiomyopathy disease processes 1, 2, 3. In particular, adverse cardiac remodelling after MI causes ventricular functional impairment and heart failure (HF). The incidence of HF after MI is determined by infarct area size, infarct wound healing and chronic LV remodelling 4, 5, 6.
Cardiomyocyte apoptosis and necrosis involving infarcted regions and residual viable myocardium trigger a cascade of immune‐inflammatory pathways and cellular mechanisms that promote wound healing and a unique pattern of LV structural remodelling‐related changes 7, 8, 9. During its early phases, LV remodelling occurs secondary to thinning and dilation of the infarcted myocardial wall (infarct expansion). During its late phases, LV remodelling occurs secondary to architectural rearrangements of the surviving myocardium characterized by myocyte hypertrophy, interstitial fibrosis and LV dilation 4, 5. Perivascular fibrosis impairs myocyte oxygen availability, reduces coronary reserve and exacerbates myocardial ischaemia 10. LV remodelling thus plays a central role in clinical progression to HF. The identification of new receptors involved in LV remodelling has opened the door for finding new therapeutic targets for treating HF. Low‐density LRP1 is a lipoprotein receptor up‐regulated by hypoxia in human vascular smooth muscle cells (hVSMCs) 11, 12 and cardiomyocytes 13, 14. LRP1 was strongly up‐regulated in ischaemic myocardial tissue in a porcine model of acute MI 13, 14 and in ischaemic cardiomyopathy patients 13, 14. LRP1 plays a crucial role in regulating MMP‐9 and MMP‐2 expression 15, 16, 17 and mediates MMP‐9 cellular catabolism to regulate its extracellular levels 18, 19. Our group recently demonstrated that LRP1 modulates MMP‐9 expression and activation in hVSMCs exposed to hypoxia through Pyk2 phosphorylation 12. It is known that MMP‐9 plays a key role in MI‐related cardiac remodelling and adverse outcomes 20, 21; however, neither LRP1 spatiotemporal expression nor the relationship between LRP1 and MMP‐9 expression during the evolution of adverse myocardial remodelling has been previously investigated. The aim of current investigation was to explore the potential relationship between cardiac LRP1 spatiotemporal expression after MI and MMP‐9 and MMP‐2 expression and activation.
Materials and methods
Mouse model of MI
A total of 46 male C57/Bl6 mice 12–13 weeks of age (25–30 g; Charles River Laboratories, Inc.; Wilmington, MA) were used in this study. MI was induced as previously described 22. Briefly, each animal was intubated and anesthetized with a mixture of O2/isoflurane and was mechanically ventilated. The heart was exposed, and the left anterior descending coronary artery was permanently occluded. The animals were sacrificed at 1 day (n = 17), 10 days (n = 14) and 21 days (n = 15) after operation, and their hearts were excised and frozen in liquid nitrogen for molecular and lipid analyses. Six animals from each group were processed for histological analysis; their hearts were arrested during diastole using cardioplegic solution (68.4 mM NaCl, 59 mM KCl, 11.1 mM glucose, 1.9 mM NaHCO3, 29.7 mM 2,3‐butanedione monoxime, 1000 U heparin), excised, fixed, cryopreserved in 30% sucrose in phosphate‐buffered saline, embedded in Tissue‐Tek O.C.T. (Sakura) and snap‐frozen in liquid nitrogen‐cooled isopentane for histological evaluation. All animal handling procedures were approved by the Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Research (NIH Pub. No. 86‐23, Revised 1996).
Macrophage cell culture
Monocyte‐derived macrophages were isolated by standard protocols from buffy coats (35–40 ml) of healthy donors. This protocol complied with the Declaration of Helsinki and was approved by the local institutional committee on human research. Cells were applied on 15 ml of Ficoll–Hypaque and centrifuged at 400×g for 40 min. at 22°C, with no brake. Mononuclear cells were obtained from the central white band of the gradient, exhaustively washed in PBS and resuspended in RPMI medium supplemented with 10% human AB serum, 1% P/S and 1% HEPES. Cells were allowed to differentiate into macrophages by exposure to 10% human AB serum for 7 days, changing the medium every other day.
Fibroblast cell culture
Control (Lrp1 +/+, MEF, CRL‐2214) and LRP1‐deficient fibroblasts (Lrp1 −/−, PEA13, CRL‐2216) fibroblasts were obtained from American Type Culture Collection. Lrp1 +/+ and Lrp1 −/− were grown in DMEM supplemented with 10% foetal calf serum, 2 mM L‐glutamine, 100 U/ml penicillin G and 100 μg/ml streptomycin, as previously described 23. The cells were exposed to hypoxia (1% O2) in an H35 Hypoxic/Anoxic Workstation (Don Whitley Scientific Ltd.; West Yorkshire, UK) with 94% N2 and 5% CO2. The cells were then harvested via scraping in TriPure™ Isolation Reagent (Roche Molecular Diagnostics; Indianapolis, USA) for PCR and Western blot analysis, and the cell culture supernatants were collected for zymography studies.
Cell exposure to normoxic and hypoxic conditions
Macrophages and fibroblasts (Lrp1 +/+ and Lrp1 −/−) were serum‐deprived once they reached 80% confluence. Cells were exposed to normoxia (21% O2) in an incubator with gas mixtures consisting of 74% N2 and 5% CO2 or to hypoxia (1% O2) in a Hypoxic/Anoxic Workstation: H35 (Don Whitley Scientific Ltd.) with 94% N2 and 5% CO2. Cells were then harvested by scraping in TriPure™ Isolation Reagent (Roche Molecular Diagnostics) for Western blot analysis. Culture supernatants were finally collected for zymographic studies.
RNA extraction and cDNA synthesis
Frozen cardiac tissue samples were pulverized using a mortar and a pestle in liquid nitrogen to maintain RNA integrity. Cardiac tissue (40 mg) was weighed, and total RNA was isolated using TriPure™ Isolation Reagent (Roche Molecular Diagnostics, Indianapolis, USA), according to the manufacturer's instructions. RNA yield and quality were assessed by 1% agarose gel electrophoresis, and then the RNA was stored at −80°C until analysis. Reverse transcription was performed using 1.5 μg of total RNA and a High‐capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). The cDNA was stored at −20°C.
Gene expression analyses by RT‐PCR
Lrp1, Mmp‐9 and Mmp‐2 mRNA gene expression analyses were performed via semiquantitative real‐time reverse‐transcriptase polymerase chain reaction (RT‐PCR) using the assays‐on‐demand listed in Table 1. Endogenous eukaryotic 18S rRNA expression served as an internal gene amplification control. RT‐PCR was performed using 1 μl of reverse transcription products mixed with 10 μl of TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA), 1 μl of 20× assays and 8 μl of nuclease‐free water. After gentle mixing, the mixture was transferred to a real‐time PCR microplate.
Table 1.
Assays‐on‐demand used for real‐time PCR analysis
| Assays | Reference | Species | Company |
|---|---|---|---|
| Lrp1 | Mm00464608_m1 | Mouse | Applied Biosystems |
| Mmp‐9 | Rn00675898_m1 | Rat | Applied Biosystems |
| Mmp‐2 | Rn01538169_m1 | Rat | Applied Biosystems |
| 18s rRNA | 4319413E | Applied Biosystems |
PCR was performed in a PCR‐7600HT sequence detection system (Abiprism; Applied Biosystems) under the following conditions: 50°C for 2 min., 95°C for 10 min., 40 cycles at 95°C for 15 sec. and 60°C for 1 min. Relative gene expression levels were quantified and analysed using SDS 2.4 software, and the real‐time values of each sample were averaged and compared using the CT method, by which the target RNA expression level (2−ΔΔCT) was normalized to that of an endogenous control (ΔCT).
Western blotting
Frozen cardiac tissue samples were pulverized using a mortar and a pestle in liquid nitrogen to maintain protein integrity. Pulverized tissue aliquots (40 mg) were subsequently weighed, and protein was isolated using TriPure™ Isolation Reagent (Roche Molecular Diagnostics), according to the manufacturer's instructions. The protein was quantified using Pierce BCA Protein Assay (Thermo Scientific, Waltham, MA, USA). Equivalent amounts of total protein (25 or 30 μg) were loaded onto 10% (v/v) SDS‐polyacrylamide gels under reducing conditions. The samples were then electrotransferred to nitrocellulose membranes, which were saturated at room temperature for 1 hr in TTBS (20 mM Tris–HCl, pH 7.5, 500 mM NaCl, 0.01% Tween 20 and 5% non‐fat milk). Western blot analyses were performed using specific monoclonal antibodies (Table 2) and the corresponding secondary antibodies (1:10,000 dilution; Dako; Glostrup, Denmark).
Table 2.
Primary antibodies used for Western blot analysis
| Antibody | References | Company |
|---|---|---|
| LRP1 | Ab92544 | Abcam |
| pPyk2 | #3291 | Cell Signaling Technology |
| Total Pyk2 | #3480 | Cell Signaling Technology |
| pERK1,2 | #9101 | Cell Signaling Technology |
| Total ERK1,2 | KAP‐MA001 | Assay Design |
| Troponin T | #MS‐295‐P | Thermo Scientific |
Equal protein loading was verified via Ponceau staining and Western blotting for troponin T. Bands were detected using ECL Prime Western Blotting Detection Reagent (Amersham) and quantified via densitometry using a ChemiDoc system and Quantity One software (Bio‐Rad, Hercules, CA, USA). The results are expressed as arbitrary units of intensity.
Gelatin zymography
Relative MMP‐9 and MMP‐2 activity levels in infarcted myocardium were measured by zymography. Pulverized tissue aliquots (5 mg) were weighed, homogenized in 60 μl of lysis buffer [1 M Tris–HCl, pH 8, 1 M KCl supplemented with one tablet of complete protease inhibitor cocktail (Roche Molecular Diagnostics, IN, USA)], sonicated and centrifuged at 16, 000 × g for 15 min. The supernatants were subsequently quantified via Pierce BCA Protein Assay (Thermo Scientific, Waltham, MA, USA). Thirty micrograms of protein was then mixed with 6× non‐reducing loading buffer, after which the samples were loaded onto 10% (v/v) SDS‐polyacrylamide gels, with 1 mg/ml porcine skin A gelatin (Sigma‐Aldrich , St. Luis, MO, USA) serving as a substrate for MMP enzymatic activity, and run at 4°C for 4–6 hrs.
After electrophoresis, the gels were rinsed twice in 2.5% Triton X‐100 for 30 min. at room temperature and then incubated in substrate buffer (50 mM Tris–HCl, 10 mM CaCl2, 0.02% (w/v) N3Na, pH 8) for 18–20 hrs at 37°C. The gels were dyed with 10% acetic acid with one tablet of PhastGel™ Blue R. Gelatinolytic activity areas, in which the protease digested the substrate, appeared as clear bands on a blue background. The gels were ultimately scanned with a GS‐800 calibrated imaging densitometer, and quantitative densitometric analysis of the digested bands was performed using Quantity‐One software (Bio‐Rad, Hercules, CA, USA).
Immunofluorescence
Mouse heart cryosections were incubated with primary antibodies against LRP1 (2 μg/ml; Abcam, Cambridge, UK), cardiac troponin I (cTnI; 2 μg/ml; Abcam), vimentin (2 μg/ml; Abcam, Cambrigde, UK), pPky2 (4 μg/ml; Santa Cruz Biotechnology, Dallas, TX, USA), pERK1,2 (8 μg/ml; Santa Cruz Biotechnology, Dallas, TX, USA), MMP‐9 (20 μg/ml; AMS Biotechnology , Abingdon, UK) and MMP‐2 (20 μg/ml; AMS Biotechnology , Abingdon, UK) (Table 3). These samples were also stained with biotinylated GSLI B4 isolectin (Vectors Laboratories) for vessel detection. Cell nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI), and the results were analysed with an Axio Observer Z1 (Zeiss) laser confocal microscope.
Table 3.
Primary antibodies used for immunohistochemical analysis
| Antibody | References | Company |
|---|---|---|
| LRP1 | ab92544 | Abcam |
| LRP1 | 10R‐L107a | Fitzgerald |
| Vimentin | ab89996 | Abcam |
| pPyk2 | sc‐16824 | Santa Cruz Biotechnology |
| MMP‐9 | TP221 | AMS Biotechnology |
| pERK1,2 | sc‐16982 | Santa Cruz Biotechnology |
| MMP‐2 | TP220 | AMS Biotechnology |
| cTroponin I | Ab188877 | Abcam |
Statistical analysis
The statistical software package SPSS 15.0 for Windows (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses. Data normality was analysed using the Kolmogorov–Smirnov test. Continuous variables were compared between groups using one‐way anova. Comparisons between each subgroup were performed using Tukey post hoc test, and independent samples were analysed using Student's t‐test. Data are presented as the mean ± S.D. P < 0.05 was considered statistically significant.
Results
Lrp1 expression is up‐regulated in ischaemic myocardium at 10 and 21 days after MI
Lrp1 expression levels were analysed by RT‐PCR, and LRP1 protein expression levels in the non‐infarct or remote peri‐infarct and infarct regions were determined by Western blotting and confocal laser microscopy in all experimental groups (1, 10 and 21 days after MI). These analyses demonstrated significant increases in Lrp1 gene expression (Fig. 1A) and LRP1 protein levels (Fig. 1B) in the peri‐infarct and infarct regions compared to the non‐infarct remote zones at 10 and 21 days after MI. These results were confirmed by immunofluorescence. Confocal microscopy images demonstrated dramatic increases in LRP1 levels in the cells forming the scar at 10 and 21 days after MI (Fig. 1C). Slight LRP1 expression was also observed in the fibroblasts surrounding the remote cardiomyocytes. Precise colocalization of the fibroblast marker vimentin and LRP1 demonstrated that LRP1 is mostly expressed in cardiac fibroblasts (Fig. 2).
Figure 1.
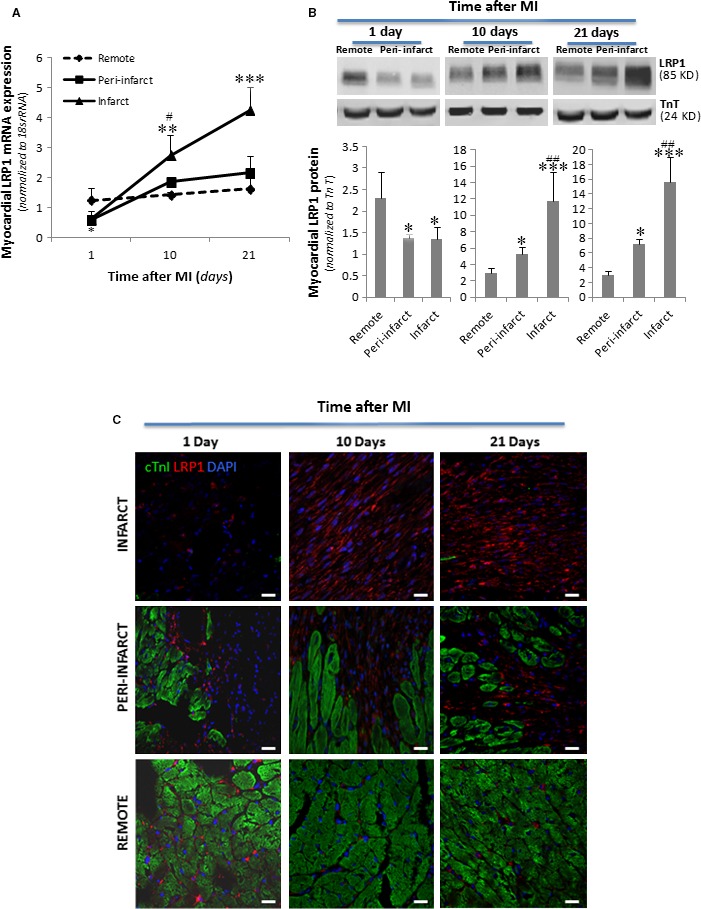
Changes in Lrp1 mRNA and LRP1 protein levels after MI. Frozen myocardial tissue samples (≈ 40 mg) from the remote, peri‐infarct and infarct zones were homogenized in TriPure™ Reagent. RNA and protein were isolated as explained in the Materials and Methods. (A) RT‐PCR showing Lrp1 mRNA expression levels. Data were processed using a specially designed software program based on the Ct values of each sample and normalized to 18s rRNA, which served as an endogenous control. (B) Representative Western blot analysis results showing LRP1 and troponin T protein expression and bar graphs showing the mean ± S.D. of LRP1 protein bands normalized to troponin T bands. n = 8. *P < 0.05 versus remote; # P < 0.05 versus peri‐infarct; **P < 0.01 versus remote; ## P < 0.01 versus peri‐infarct; ***P < 0.005 versus remote. (C) Representative immunostaining of heart cross sections showing the temporal evolution of LRP1 levels after MI. LRP1 is shown in red, and cTnI, in green. Cell nuclei were counterstained with DAPI (blue). Scale bars, 20 μm.
Figure 2.

Identification of LRP1‐expressing cells in infarcted heart cross sections. LRP1 is shown in red; vimentin, in green; cTnI, in grey; and Lrp1 and vimentin colocalization, in yellow. Cell nuclei were counterstained with DAPI (blue). Scale bars, 20 μm.
RT‐PCR and Western blot analysis showed that LRP1 mRNA and protein expression levels, respectively, were significantly reduced in the infarct zone at 1 day after MI, in contrast to the above results (Fig. 1A and B). Immunofluorescence imaging confirmed the reductions in LRP1 protein levels in the infarct areas compared to the peri‐infarct or remote areas (Fig. 1C).
pPyk2 and pERK1,2 are signal mediators that are differentially modulated at both the temporal level and the spatial level after MI
Western blot analysis showed that both, LRP1 and pPyk2 levels were significantly down‐regulated in the ischaemic zones at 1 day after MI (Fig. 3A and B). In contrast, pPyk2 expression was strongly up‐regulated in the peri‐infarct and infarct areas at 10 days (Fig. 3A and C) and 21 days after MI (Fig. 3A and D). In addition, pERK1,2 expression was strongly up‐regulated in the infarct areas at 1 (Fig. 3A and E), 10 (Fig. 3A and F) and 21 (Fig. 3A and G) days after MI.
Figure 3.

Temporal and spatial evolution of myocardial phosphorylated Pyk2 and ERK1,2 levels after MI. (A) Representative Western blot analysis showing pPyk2, total Pyk2, pERK1,2 and total ERK1,2 protein expression. Bar graphs showing the mean ± S.D. of the pPyk2/total Pyk2 ratio and the pERK1,2/total ERK1,2 ratio in the remote, peri‐infarct and infarct zones at 1 (B, E), 10 (C, F) and 21 (D, G) days after MI. n = 8. *P < 0.05 versus remote; # P < 0.05 versus peri‐infarct; **P < 0.01 versus remote; ## P < 0.01 versus peri‐infarct; ***P < 0.005 versus remote; ### P < 0.005 versus peri‐infarct.
MMP‐9 and MMP‐2 have a differential temporal and spatial modulation after MI
Zymography analysis showed that MMP‐9 activity was strongly increased in the infarct areas on day 1 after MI (Fig. 4A and B) and was moderately increased in the peri‐infarct and infarct areas on day 10 (Fig. 4A and C) and day 21 after MI (Fig. 4A and D) compared to the remote zone. Differently, MMP‐2 activity was extremely low in all tested zones on day 1 after MI (Fig. 4A and E). A strong increase in MMP‐2 activity was detected in all zones at 10 days after MI but especially in the infarct area (Fig. 4A and F). MMP‐2 overactivation was slightly maintained at 21 days after MI (Fig. 4A and G). RT‐PCR analysis yielded similar results than zymography with respect to temporal and spatial MMP‐9 (Fig. 6H, I and J) and MMP‐2 (Fig. 6K, L and M) mRNA expression patterns after MI.
Figure 4.
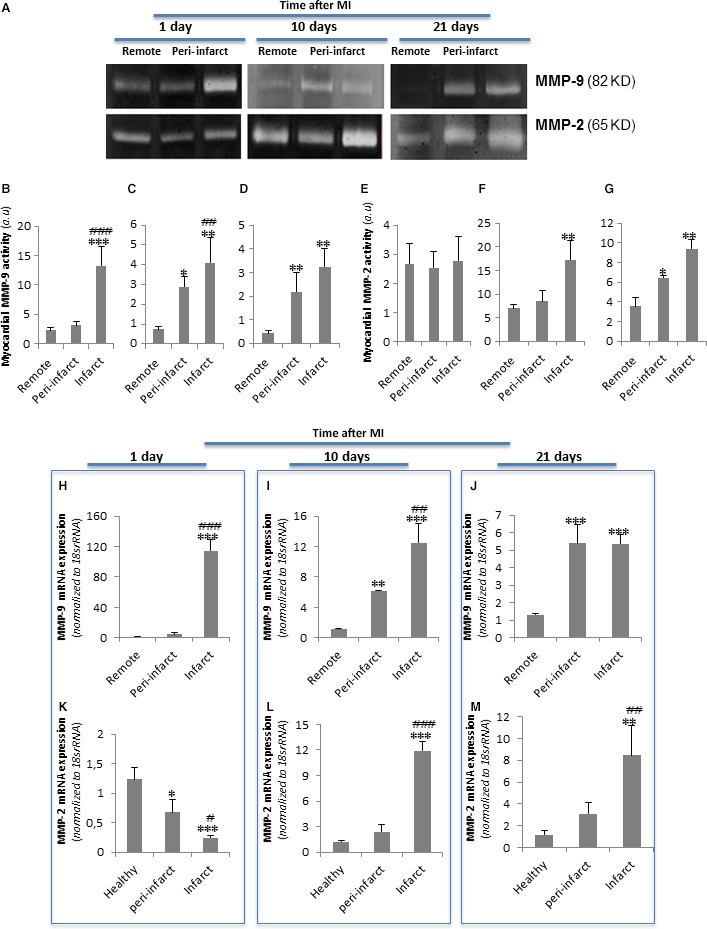
Temporal and spatial evolution of myocardial MMP‐9 and MMP‐2 activation and expression after MI. (A) Representative zymography analysis results showing myocardial MMP‐9 and MMP‐2 activity. Bar graphs showing the mean ± S.D. of MMP‐9 and MMP‐2 activity in the remote, peri‐infarct and infarct zones at 1 (B and E), 10 (C and F) and 21 (D and G) days after MI. (H–M) Real‐time PCR showing MMP‐9 and MMP‐2 mRNA expression levels. Data were processed using a specially designed software program based on the Ct values of each sample and normalized to 18s rRNA, which served as an endogenous control. The results are shown as the mean ± S.D. of MMP‐9 and MMP‐2 mRNA expression levels in the remote, peri‐infarct and infarct zones at 1 (H and K), 10 (I and L) and 21 (J and M) days after MI. n = 8. *P < 0.05 versus remote; **P < 0.01 versus remote; ## P < 0.01 versus peri‐infarct; ***P < 0.005 versus remote; ### P < 0.005 versus peri‐infarct.
Figure 6.

Representative confocal microscopy images showing LRP1, MMP‐9 and pERK1,2 expression levels in the remote, peri‐infarct and infarct zones after MI. (A) Immunostaining of heart cross sections showing the temporal evolution of pERK1,2 (grey), MMP‐9 (green) and LRP1 (red) expression levels after MI. (B) Immunostaining of heart cross sections showing the temporal evolution of pERK1,2 (grey), MMP‐2 (green) and LRP1 (red) expression levels after MI. Cell nuclei were counterstained with DAPI (blue). Scale bars, 50 μm.
MMP‐9 colocalized with pPyk2 and LRP1 in the cardiac fibroblasts of ischaemic myocardium at 10 and 21 days after MI
Confocal microscopy images demonstrated a strong colocalization among pPyk2, MMP‐9 and LRP1 in cardiac fibroblasts in the peri‐infarct and infarct zones at 10 and 21 days after MI (Fig. 5). In contrast, pERK1,2, which exhibited only slight expression, did not colocalize with MMP‐9 or LRP1 in any zones or stages after MI (Fig. 6A). Confocal microscopy also demonstrated no colocalization between MMP‐2 and pERK1,2, and only few cells expressed both LRP1 and MMP‐2 in all the areas analysed (Fig. 6B).
Figure 5.

Representative confocal microscopy images showing LRP1, MMP‐9 and pPyk2 levels in the remote, peri‐infarct and infarct zones after MI. Immunostaining of heart cross sections showing the temporal evolution of pPyk2, MMP‐9 and LRP1 expression levels after MI. LRP1 is shown in red; MMP‐9, in green; and pPyk2, in grey. A and B inserts are amplified in A’ and B’, respectively, to show colocalization of the three markers in light yellow. Cell nuclei were counterstained with DAPI (blue). Scale bars, 50 μm.
We next compared the role of hypoxia on LRP1 levels in cultured macrophages and fibroblasts. Western blot analysis showed that hypoxia significantly reduced Pyk2 phosphorylation (Fig. 7A and C) concomitantly with a decay on LRP1 protein levels (Fig. 7A and B) in macrophages. In contrast, both LRP1 protein (Fig. 7G and H) and Pyk2 phosphorylation (Fig. 7G and I) levels were significantly up‐regulated by hypoxia in fibroblasts. Zymography analysis showed that hypoxia reduces MMP‐9 activity in macrophages (Fig. 7D and E) but induces MMP‐9 activity in fibroblasts (Fig. 7J and K). MMP‐2 activity levels were similar in both macrophages (Fig. 7D and F) and fibroblasts (Fig. 7J and L). In addition, we compared the hypoxic effects on control (Lrp1 +/+) and LRP1‐deficient (Lrp1 −/−) fibroblasts. As shown in Figure 8, hypoxia failed to up‐regulate LRP1 (Fig. 8A and B) and Pyk2 phosphorylation (Fig. 8A and C) in Lrp1 −/− compared to Lrp1 +/+. Moreover, MMP‐9 activity (Fig. 8D and E) was significantly up‐regulated by hypoxia in Lrp1 +/+ but not in Lrp1 −/− fibroblasts, indicating that LRP1 is essential for Pyk2 phosphorylation and MMP‐9 activation in cardiac fibroblasts. MMP‐2 activity levels (Fig. 8D and F) were similar in all tested conditions.
Figure 7.
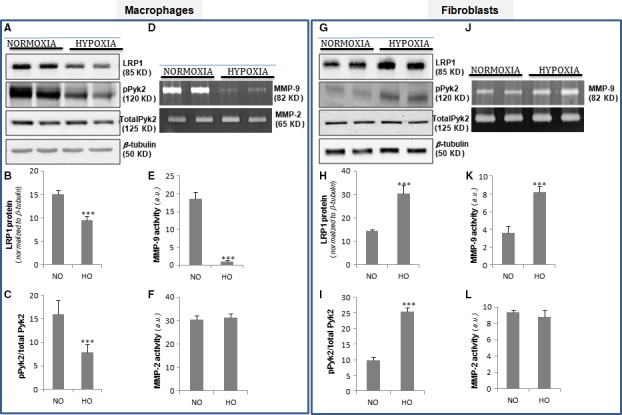
Comparison of the hypoxia impact on LRP1, pPyk2 levels and MMP‐9/MMP‐2 activation in macrophages and fibroblasts. Quiescent macrophages and fibroblasts were exposed to normoxic or hypoxic conditions for 18 hrs. Representative Western blot analysis showing LRP1, pPyk2, total Pyk2 and β‐tubulin bands (A&G). Bar graphs showing the mean ± S.D. of LRP1 normalized to β‐tubulin levels (B&H) and pPyk2/total Pyk2 ratio (C&I). (D&J) Representative zymography analysis showing MMP‐9 and MMP‐2 activity and bar graphs showing the mean ± S.D. of MMP‐9 (E&K) and MMP‐2 (F&L) activity levels. Results are shown as the mean ± S.E.M. of three independent experiments performed in triplicate. ***P < 0.001 versus normoxia. NO: normoxia; HO: hypoxia.
Figure 8.

Comparison of the hypoxia effect on pPyk2 levels and MMP‐9/MMP‐2 activation in control (Lrp1 +/+) and LRP1‐deficient fibroblasts (Lrp1 −/−). Quiescent Lrp1 +/+ and Lrp1 −/− fibroblasts were exposed to normoxic or hypoxic conditions for 18 hrs. (A) Representative Western blot analysis showing LRP1, pPyk2, total Pyk2 and β‐tubulin levels and bar graphs showing the mean ± S.D. of LRP1 levels normalized to β‐tubulin (B) and pPyk2/total Pyk2 ratio (C). (D) Representative zymography analysis showing MMP‐9 and MMP‐2 activity levels and bar graphs showing the mean ± S.D. of MMP‐9 (E) and MMP‐2 activity levels (F). Results are shown as the mean ± S.E.M. of three independent experiments performed in triplicate. ***P < 0.001 versus normoxia. (G) Representative scheme showing the crucial role of LRP1 up‐regulation in hypoxic cardiac fibroblasts in cardiac remodelling.
Discussion
LRP1 has been described as a key regulator of extracellular matrix remodelling 24, 25, 26, 27, 28, which is associated with morbidity and mortality after MI 29, 30, 31; however, no previous studies have analysed the spatial and temporal evolution of LRP1 after MI. Here, we report for the first time that the myocardial LRP1 strongly colocalizes with pPyk2 and MMP‐9 during the fibrotic stages of remodelling after MI. Although it is known that MMP‐9 activity plays a key role in remodelling after MI, these findings have not successfully facilitated improvements in human MI management. Our results indicate that LRP1 is a target for specifically modulating cardiac fibroblast MMP‐9 levels during the early stages of fibrosis after MI.
We demonstrated that LRP1 levels are extremely high in cardiac fibroblasts during the fibrotic states of remodelling after MI. In contrast, LRP1 was scarcely detected in the infarct areas during the inflammatory phase of remodelling after MI, whose main cellular components are neutrophils and macrophages. We have previously shown that inflammatory mediators reduce LRP1 levels in human macrophages through sterol regulatory element‐binding protein (SREBP)‐1 up‐regulation 32. The capacity of inflammatory mediators to up‐regulate SREBP‐1 levels has also been demonstrated in mouse macrophages 33, 34. These results suggest that the inflammatory state characterizing the infarct areas at 1 day after MI may be the cause of the reduced LRP1 levels in neutrophils and macrophages. In addition, macrophages with low LRP1 levels may contribute to the exacerbation of the inflammatory process, as it has been previously shown that macrophage LRP1 deficiency contributes to increased MMP‐9, MCP‐1 and TNF‐α secretion in atherosclerosis 35. Additional studies are required to establish whether the above‐mentioned low inflammatory cell LRP1 levels during the first stage of remodelling after MI are a cause of inflammation, a consequence of inflammation, or both. Consistent with the findings of previous studies 20, 36, 37, we observed high MMP‐9 levels in conjunction with neutrophil and macrophage infiltration during the inflammatory phase of remodelling after MI. Our confocal microscopy images demonstrated a lack of colocalization between MMP‐9 and LRP1 during the inflammatory phase after MI. Taken together, these results suggest that the robust MMP‐9 activation observed at 1 day after MI does not depend on LRP1. Remarkably, myocardial LRP1 was significantly up‐regulated in ischaemic areas during the fibrotic stages of remodelling after MI in conjunction with increases in cardiac fibroblast proliferation. It is well known that cardiac fibroblasts adopt a myofibroblast‐like phenotype in response to specific stimuli and that this acquired phenotype plays a critical role in remodelling after MI in the heart 38, 39. One potent stimulus that triggers the development of this myofibroblast‐like phenotype is hypoxia 40, which is also a potent inducer of LRP1 expression in hVSMCs 11, 12 and cardiomyocytes 13, 41. Therefore, hypoxia may contribute to significant LRP1 overexpression in cardiac fibroblasts located in ischaemic areas after MI. It has been previously reported that LRP1 depletion in fibroblasts inhibits transforming growth factor beta (TGFβ) expression 42, 43. LRP1 overexpression in cardiac fibroblasts may thus promote TGFβ signalling, a crucial inducer of the specialized phenotype that fibroblasts acquire in response to injury 44, 45. Therefore, LRP1 may act as an integrator of TGFβ and hypoxia signalling in cardiac fibroblasts.
Here, we observed increases in MMP‐9 mRNA expression and activation in the infarct areas at all time points after MI, consistent with the findings of previous studies 36, 46, 47, 48. Previous studies conducted by our group showed that the up‐regulatory effects exerted by hypoxia on LRP1 determine MMP‐9 activation and that this mechanism is related to Pyk2 phosphorylation in hVSMCs 12. Using confocal microscopy, we observed high level of colocalization among LRP1, Pyk2 and MMP‐9 in cardiac fibroblasts in the infarct zones at 10 and 21 days after MI. Western blot analysis demonstrated significant Pyk2 phosphorylation up‐regulation in the infarct zone at 10 days after MI, which persisted until 21 days after MI. Consistent with this finding, we observed high MMP‐9 expression and activity in the infarct zone at 10 and 21 days after MI. Factors other than hypoxia, including interleukin‐1α, have been reported to contribute to increases in MMP‐9 expression in cardiac fibroblasts 38, 49, 50, 51. Several factors can modulate Pyk2 and MMP‐9 levels in infarct areas independently of LRP1 and hypoxia.
The in vitro results comparing the effects of hypoxia on LRP1 and pPyk2 levels in macrophages and fibroblasts match with in vivo studies. On one side, LRP1 and pPyk2 down‐regulation in cultured macrophages exposed to hypoxia fits with down‐regulation of these molecules at early post‐infarct stages (1 day after MI) where the main component is inflammation and macrophages. On the other hand, LRP1 and pPyk2 up‐regulation in cultured fibroblasts fits with the high levels of these molecules at later post‐infarct stages (10 and 21 days after MI) where the main component is fibrosis and fibroblasts. MMP‐9 levels were extremely low in cultured hypoxic macrophages.
The high MMP‐9 activation that we have found in infarcted area at 1 day after MI suggests that inflammatory mediators present in vivo overcome the potential down‐regulatory effect of hypoxia on macrophage MMP‐9 levels. The activation of MMP‐9 levels found in infarcted areas at 10 and 21 days after MI is coherent with the up‐regulatory effect of hypoxia on fibroblast MMP‐9 activation. In agreement, our immunofluorescence imaging results show a high colocalization degree between LRP1, pPyk2 and MMP‐9 at 10 and 21 days after MI. Further, we showed that MMP‐9 activation by hypoxia does not take place in LRP1‐deficient fibroblasts. Taken together, our data suggest that by up‐regulating LRP1 in cardiac fibroblasts, hypoxia promotes pPyk2 phosphorylation and MMP‐9 activation in LV remodelling after MI (summarized in Fig. 8G).
We have previously shown that both LRP1 silencing and PP2, a Pyk2 phosphorylation inhibitor, abolished hypoxia‐induced MMP‐9 overexpression and activation in hVSMCs 12. We also showed that in contrast to LRP1 inhibition, PP2 expression did not alter vascular cell pro‐inflammatory phenotypes. PP2‐mediated Pyk2 phosphorylation inhibition has been shown to efficiently reverse fibrosis development in a load‐induced cardiac hypertrophy mouse model 52 and to attenuate fibrosis after MI 53. Here, we showed for the first time the temporal and spatial evolution of pPyk2 levels after MI. Our results suggest that pPyk2 could play a crucial role in up‐regulating MMP‐9 activity in cardiac fibroblasts, especially at 10 days after MI. We previously showed that hypoxia‐induced LRP1‐pPyk2‐NF‐kβ activation is important for MMP‐9 activation, but not MMP‐2 activation 12. The MMP‐9 and MMP‐2 promoter elements are very different 50, 54, suggesting that MMP‐9 and MMP‐2 are modulated by different pathways after MI.
In conclusion, our results suggest that LRP1 plays a major role in MMP‐9 up‐regulation in cardiac fibroblasts after MI and highlight the potential role of LRP1 modulation for treatment of cardiac remodelling.
Clinical implications
It has been consistently demonstrated that MMP activity modification is a key mechanism underlying remodelling after MI 55, 56. However, these results have not successfully facilitated improvements in human MI management. It has been suggested that temporal and cell‐specific MMP inhibition has therapeutic potential. LRP1 may enable clinicians to modulate cardiac fibroblast MMP‐9 levels during the early stage of fibrosis after MI.
Conflicts of interest
None.
Acknowledgements
The research leading to these results received funding from the Instituto de Salud Carlos III (FIS PI14/01729 and FIS PI14/01682) that was cofinanced by the European Fund for Regional Development, as well as from the Ministerio de Ciencia e Innovación (SAF2011‐30067‐C02‐01), the Fundació La Marató de TV3 (201521_10, 201502_30 and 080330), the Red de Terapia Celular‐TerCel (RD12/0019/0029), the Red Cardio‐vascular (RD12/0042/0027 and RD12/0042/0047), the Sociedad Española de Cardiología and the Fundació Privada Daniel Bravo Andreu. E.R.L. is a recipient of an FI‐DGR_B01008 grant from Generalitat de Catalunya, and D.G. is a recipient of CD14/0109 from Sara Borell of the Instituto de Salud Carlos III.
References
- 1. Drazner MH, Rame JE, Marino EK, et al Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: the Cardiovascular Health Study. J Am Coll Cardiol. 2004; 43: 2207–15. [DOI] [PubMed] [Google Scholar]
- 2. Lam CS, Grewal J, Borlaug BA, et al Size, shape, and stamina: the impact of left ventricular geometry on exercise capacity. Hypertension. 2010; 55: 1143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mann DL, Bristow MR. Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation. 2005; 111: 2837–49. [DOI] [PubMed] [Google Scholar]
- 4. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990; 81: 1161–72. [DOI] [PubMed] [Google Scholar]
- 5. Opie LH, Commerford PJ, Gersh BJ, et al Controversies in ventricular remodelling. Lancet. 2006; 367: 356–67. [DOI] [PubMed] [Google Scholar]
- 6. Dorn GW 2nd. Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nat Rev Cardiol. 2009; 6: 283–91. [DOI] [PubMed] [Google Scholar]
- 7. Frantz S, Bauersachs J, Ertl G. Post‐infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. 2009; 81: 474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002; 53: 31–47. [DOI] [PubMed] [Google Scholar]
- 9. Dobaczewski M, Gonzalez‐Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010; 48: 504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown RD, Ambler SK, Mitchell MD, et al The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005; 45: 657–87. [DOI] [PubMed] [Google Scholar]
- 11. Castellano J, Aledo R, Sendra J, et al Hypoxia stimulates low‐density lipoprotein receptor‐related protein‐1 expression through hypoxia‐inducible factor‐1alpha in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2011; 31: 1411–20. [DOI] [PubMed] [Google Scholar]
- 12. Revuelta‐Lopez E, Castellano J, Roura S, et al Hypoxia induces metalloproteinase‐9 activation and human vascular smooth muscle cell migration through low‐density lipoprotein receptor‐related protein 1‐mediated Pyk2 phosphorylation. Arterioscler Thromb Vasc Biol. 2013; 33: 2877–87. [DOI] [PubMed] [Google Scholar]
- 13. Cal R, Castellano J, Revuelta‐Lopez E, et al Low‐density lipoprotein receptor‐related protein 1 mediates hypoxia‐induced very low density lipoprotein‐cholesteryl ester uptake and accumulation in cardiomyocytes. Cardiovasc Res. 2012a; 94: 469–79. [DOI] [PubMed] [Google Scholar]
- 14. Cal R, Juan‐Babot O, Brossa V, et al Low density lipoprotein receptor‐related protein 1 expression correlates with cholesteryl ester accumulation in the myocardium of ischemic cardiomyopathy patients. J Transl Med. 2012b; 10: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jung HO, Uhm JS, Seo SM, et al Angiotensin II‐induced smooth muscle cell migration is mediated by LDL receptor‐related protein 1 via regulation of matrix metalloproteinase 2 expression. Biochem Biophys Res Commun. 2010; 402: 577–82. [DOI] [PubMed] [Google Scholar]
- 16. Montel V, Gaultier A, Lester RD, et al The low‐density lipoprotein receptor‐related protein regulates cancer cell survival and metastasis development. Cancer Res. 2007; 67: 9817–24. [DOI] [PubMed] [Google Scholar]
- 17. Song H, Li Y, Lee J, et al Low‐density lipoprotein receptor‐related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 2009; 69: 879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mantuano E, Jo M, Gonias SL, et al Low density lipoprotein receptor‐related protein (LRP1) regulates Rac1 and RhoA reciprocally to control Schwann cell adhesion and migration. J Biol Chem. 2010; 285: 14259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hahn‐Dantona E, Ruiz JF, Bornstein P, et al The low density lipoprotein receptor‐related protein modulates levels of matrix metalloproteinase 9 (MMP‐9) by mediating its cellular catabolism. J Biol Chem. 2001; 276: 15498–503. [DOI] [PubMed] [Google Scholar]
- 20. Lindsey ML, Zamilpa R. Temporal and spatial expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases following myocardial infarction. Cardiovasc Ther. 2012; 30: 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yabluchanskiy A, Li Y, Chilton RJ, et al Matrix metalloproteinases: drug targets for myocardial infarction. Curr Drug Targets. 2013; 14: 276–86. [PMC free article] [PubMed] [Google Scholar]
- 22. Bayes‐Genis A, Soler‐Botija C, Farre J, et al Human progenitor cells derived from cardiac adipose tissue ameliorate myocardial infarction in rodents. J Mol Cell Cardiol. 2010; 49: 771–80. [DOI] [PubMed] [Google Scholar]
- 23. Willnow TE, Herz J. Genetic deficiency in low density lipoprotein receptor‐related protein confers cellular resistance to Pseudomonas exotoxin A. Evidence that this protein is required for uptake and degradation of multiple ligands. J Cell Sci. 1994; 107: 719–26. [PubMed] [Google Scholar]
- 24. Etique N, Verzeaux L, Dedieu S, et al LRP‐1: a checkpoint for the extracellular matrix proteolysis. Biomed Res Int. 2013; 2013: 152163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamamoto K, Murphy G, Troeberg L. Extracellular regulation of metalloproteinases. Matrix Biol. 2015; 44–46: 255–63. [DOI] [PubMed] [Google Scholar]
- 26. Samouillan V, Revuelta‐Lopez E, Dandurand J, et al Cardiomyocyte intracellular cholesteryl ester accumulation promotes tropoelastin physical alteration and degradation: role of LRP1 and cathepsin S. Int J Biochem Cell Biol. 2014; 55: 209–19. [DOI] [PubMed] [Google Scholar]
- 27. Roura S, Cal R, Galvez‐Monton C, et al Inverse relationship between raft LRP1 localization and non‐raft ERK1,2/MMP9 activation in idiopathic dilated cardiomyopathy: potential impact in ventricular remodeling. Int J Cardiol. 2014; 176: 805–14. [DOI] [PubMed] [Google Scholar]
- 28. Gaultier A, Hollister M, Reynolds I, et al LRP1 regulates remodeling of the extracellular matrix by fibroblasts. Matrix Biol. 2010; 29: 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. White HD, Norris RM, Brown MA, et al Left ventricular end‐systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987; 76: 44–51. [DOI] [PubMed] [Google Scholar]
- 30. See F, Kompa A, Martin J, et al Fibrosis as a therapeutic target post‐myocardial infarction. Curr Pharm Des. 2005; 11: 477–87. [DOI] [PubMed] [Google Scholar]
- 31. Vanhoutte D, Schellings M, Pinto Y, et al Relevance of matrix metalloproteinases and their inhibitors after myocardial infarction: a temporal and spatial window. Cardiovasc Res. 2006; 69: 604–13. [DOI] [PubMed] [Google Scholar]
- 32. Costales P, Castellano J, Revuelta‐Lopez E, et al Lipopolysaccharide downregulates CD91/low‐density lipoprotein receptor‐related protein 1 expression through SREBP‐1 overexpression in human macrophages. Atherosclerosis. 2013; 227: 79–88. [DOI] [PubMed] [Google Scholar]
- 33. Im SS, Yousef L, Blaschitz C, et al Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein‐1a. Cell Metab. 2011; 13: 540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Im SS, Osborne TF. Protection from bacterial‐toxin‐induced apoptosis in macrophages requires the lipogenic transcription factor sterol regulatory element binding protein 1a. Mol Cell Biol. 2012; 32: 2196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Overton CD, Yancey PG, Major AS, et al Deletion of macrophage LDL receptor‐related protein increases atherogenesis in the mouse. Circ Res. 2007; 100: 670–7. [DOI] [PubMed] [Google Scholar]
- 36. Tao ZY, Cavasin MA, Yang F, et al Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004; 74: 1561–72. [DOI] [PubMed] [Google Scholar]
- 37. Lindsey ML, Gannon J, Aikawa M, et al Selective matrix metalloproteinase inhibition reduces left ventricular remodeling but does not inhibit angiogenesis after myocardial infarction. Circulation. 2002; 105: 753–8. [DOI] [PubMed] [Google Scholar]
- 38. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009; 123: 255–78. [DOI] [PubMed] [Google Scholar]
- 39. van den Borne SW, Diez J, Blankesteijn WM, et al Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010; 7: 30–7. [DOI] [PubMed] [Google Scholar]
- 40. Watson CJ, Collier P, Tea I, et al Hypoxia‐induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast‐like phenotype. Hum Mol Genet. 2014; 23: 2176–88. [DOI] [PubMed] [Google Scholar]
- 41. Revuelta‐Lopez E, Cal R, Herraiz‐Martinez A, et al Hypoxia‐driven sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) downregulation depends on low‐density lipoprotein receptor‐related protein 1 (LRP1)‐signalling in cardiomyocytes. J Mol Cell Cardiol. 2015; 85: 25–36. [DOI] [PubMed] [Google Scholar]
- 42. Cabello‐Verrugio C, Brandan E. A novel modulatory mechanism of transforming growth factor‐beta signaling through decorin and LRP‐1. J Biol Chem. 2007; 282: 18842–50. [DOI] [PubMed] [Google Scholar]
- 43. Llorente‐Cortes V, Barbarigo V, Badimon L. Low density lipoprotein receptor‐related protein 1 modulates the proliferation and migration of human hepatic stellate cells. J Cell Physiol. 2012; 227: 3528–33. [DOI] [PubMed] [Google Scholar]
- 44. Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)‐beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011; 51: 600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tomasek JJ, Gabbiani G, Hinz B, et al Myofibroblasts and mechano‐regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002; 3: 349–63. [DOI] [PubMed] [Google Scholar]
- 46. Chen J, Tung CH, Allport JR, et al Near‐infrared fluorescent imaging of matrix metalloproteinase activity after myocardial infarction. Circulation. 2005; 111: 1800–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cleutjens JP, Kandala JC, Guarda E, et al Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995; 27: 1281–92. [DOI] [PubMed] [Google Scholar]
- 48. Peterson JT, Li H, Dillon L, et al Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res. 2000; 46: 307–15. [DOI] [PubMed] [Google Scholar]
- 49. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007; 87: 1285–342. [DOI] [PubMed] [Google Scholar]
- 50. Mukherjee R, Mingoia JT, Bruce JA, et al Selective spatiotemporal induction of matrix metalloproteinase‐2 and matrix metalloproteinase‐9 transcription after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006; 291: H2216–28. [DOI] [PubMed] [Google Scholar]
- 51. Turner NA, Warburton P, O'Regan DJ, et al Modulatory effect of interleukin‐1alpha on expression of structural matrix proteins, MMPs and TIMPs in human cardiac myofibroblasts: role of p38 MAP kinase. Matrix Biol. 2010; 29: 613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clemente CF, Tornatore TF, Theizen TH, et al Targeting focal adhesion kinase with small interfering RNA prevents and reverses load‐induced cardiac hypertrophy in mice. Circ Res. 2007; 101: 1339–48. [DOI] [PubMed] [Google Scholar]
- 53. Fan GP, Wang W, Zhao H, et al Pharmacological inhibition of focal adhesion kinase attenuates cardiac fibrosis in mice cardiac fibroblast and post‐myocardial‐infarction models. Cell Physiol Biochem. 2015; 37: 515–26. [DOI] [PubMed] [Google Scholar]
- 54. Bergman MR, Cheng S, Honbo N, et al A functional activating protein 1 (AP‐1) site regulates matrix metalloproteinase 2 (MMP‐2) transcription by cardiac cells through interactions with JunB‐Fra1 and JunB‐FosB heterodimers. Biochem J. 2003; 369: 485–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mukherjee R, Brinsa TA, Dowdy KB, et al Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation. 2003; 107: 618–25. [DOI] [PubMed] [Google Scholar]
- 56. Creemers EE, Cleutjens JP, Smits JF, et al Matrix metalloproteinase inhibition after myocardial infarction: a new approach to prevent heart failure? Circ Res. 2001; 89: 201–10. [DOI] [PubMed] [Google Scholar]