

Abstract
Hypoxia stimulates excessive growth of vascular smooth muscle cells (VSMCs) contributing to vascular remodelling. Recent studies have shown that histone deacetylase inhibitors (HDIs) suppress VSMC proliferation and activate eNOS expression. However, the effects of HDI on hypoxia‐induced VSMC growth and the role of activated eNOS in VSMCs are unclear. Using an EdU incorporation assay and flow cytometry analysis, we found that the HDIs, butyrate (Bur) and suberoylanilide hydroxamic acid (SAHA) significantly suppressed the proliferation of hypoxic VSMC lines and induced apoptosis. Remarkable induction of cleaved caspase 3, p21 expression and reduction of PCNA expression were also observed. Increased eNOS expression and enhanced NO secretion by hypoxic VSMC lines were detected using Bur or SAHA treatment. Knockdown of eNOS by siRNA transfection or exposure of hypoxic VSMCs to NO scavengers weakened the effects of Bur and SAHA on the growth of hypoxic VSMCs. In animal experiments, administration of Bur to Wistar rats exposed to hypobaric hypoxia for 28 days ameliorated the thickness and collagen deposition in pulmonary artery walls. Although the mean pulmonary arterial pressure (mPAP) was not obviously decreased with Bur in hypoxic rats, right ventricle hypertrophy index (RVHI) was decreased and the oxygen partial pressure of arterial blood was elevated. Furthermore, cell viability was decreased and eNOS and cleaved caspase 3 were induced in HDI‐treated rat pulmonary arterial SMCs. These findings imply that HDIs prevent hypoxia‐induced VSMC growth, in correlation with activated eNOS expression and activity in hypoxic VSMCs.
Keywords: endothelial nitric oxide synthase, vascular smooth muscle cell, vascular remodelling, histone deacetylase inhibitor, hypoxia
Introduction
Vascular remodelling is a common feature of cardiovascular diseases such as atherosclerosis and pulmonary arterial hypertension 1, 2, 3. A considerable change in the vasculature under these pathological conditions is thickening of medial layers of muscular vessels, occurring mainly due to the accumulation of vascular smooth muscle cells (VSMCs). Hypoxia, a common cause and result of cardiovascular disease, stimulates excessive growth of VSMCs and contributes significantly to vascular remodelling.
Histone deacetylase inhibitors (HDIs), which apparently inhibit histone deacetylase (HDAC) activity, suppress cell growth by modulating the expression of several genes related to the cell cycle and apoptosis 4, 5. Two HDIs, suberoylanilide hydroxamic acid (SAHA) and romidepsin (FK228), have been approved for treating cutaneous T cell lymphoma 6. Others, including butyrate, are under clinical trials. Recently, the suppressive effect of HDI on VSMC proliferation has been shown via the induction of p21 expression and subsequent cell cycle arrest with reduction in the phosphorylation of Rb protein at the G1–S phase 7. Either short interfering RNA‐mediated knockdown of HDAC or the pharmacological inhibition of HDAC prevented mitogen‐induced SMC proliferation 4, 8. However, the effects of HDI on hypoxia‐induced VSMC proliferation and vascular remodelling are unclear.
HDIs are a group of proteins that regulate histone acetylation in nucleosomes and mediate changes in chromatin conformation, leading to the regulation of gene expression 5, 6, 9, 10. Accumulating evidence shows that HDIs modulate histone acetylation states for the transcriptional control of proliferative genes such as p21 and p27 7, 11, 12, 13, 14. However, the epigenetic mechanism involved in the HDI‐mediated suppression of VSMC proliferation is not completely understood. Previous studies indicate that eNOS expression could be activated by the HDI, butyrate and trichostatin A (TSA) in non‐endothelial cells, including VSMCs 15, 16, 17. As previously known, nitric oxide (NO) is mainly synthesized and secreted by vascular endothelial cells via eNOS in physiological vasculature, which acts as an essential regulator of VSMC proliferation by inducing production of cleaved caspase 3 and p21 expression 18, 19, 20, 21, 22, 23. However, EC‐derived NO was suppressed in many pathological situations due to EC disorders and/or eNOS dysfunction 20, 24, 25. eNOS transfection or treatment with NO donors can inhibit VSMC proliferation 26, 27, 28. Furthermore, the degree of NO donor inhibition was significantly enhanced in the presence of hypoxia 28. Therefore, it is interesting to test whether HDI activates eNOS expression in hypoxic VSMCs and contributes to cell growth regulation.
In this study, we tested the effect of Bur and SAHA on eNOS gene expression in hypoxic VSMCs and determined whether eNOS gene activation in VSMCs was sufficient to suppress hypoxia‐induced VSMC proliferation. We observed that HDI treatment stimulated eNOS expression and NO secretion by hypoxic VSMCs. Their antiproliferative and pro‐apoptotic effects were attenuated by NO scavengers and siRNA‐mediated eNOS knockdown. Furthermore, induction of p21 expression and cleaved caspase 3 by HDI in hypoxic VSMCs was decreased by NO scavengers and siRNA‐mediated eNOS knockdown. Finally, we observed that Bur prevented the thickening and collagen deposition in the pulmonary artery (PA) wall in a rat model of hypobaric hypoxia‐induced vascular remodelling (simulating high altitude at 5000 m) and protected the function of the cardiovascular system with the elevation of PaO2 and the decreased right ventricle hypertrophy index (RVHI). Cell viability was decreased and the expression of eNOS and cleaved caspase 3 was induced in HDI‐treated rat pulmonary arterial SMCs (rPASMCs).
Material and methods
Cell culture and experimental treatment
The A10 SMC line was purchased from ATCC and cultured in DMEM/F12 (Hyclone) containing 10% foetal bovine serum (Gibco) and 100 μg/ml Pen/Strep (Gibco) at 37°C with 5% CO2 and 95% air.
Isolation and culture of pulmonary arterial smooth muscle cells (PASMCs) was performed as previously described 29. Eight male Wistar rats were used for each independent isolation. All protocols and surgical procedures were approved by the Institutional Animal Use Committee of the Third Military Medical University and were in accordance with the guidelines of the National Institutes of Health and the American Physiological Society. Briefly, rats were heparinized, anaesthetized (intraperitoneal injection of sodium pentobarbital, at 50 mg/kg) and killed by exsanguination. The thorax was immediately opened and the lungs were removed under sterile conditions. The intrapulmonary arteries, third to fourth generation, were dissected free of parenchyma and kept in ice‐cold Hanks’ buffer. Vascular segments were free from adventitia and were dissected open. The endothelium was then removed by gently scraping the luminal surface of the vessel under a dissecting microscope. After recovery for 30 min in cold (4°C) physiological salt solution (PSS) that contained 130 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 10 mM HEPES and 10 mM glucose followed by 20 min in reduced‐Ca2+ PSS (20 μM CaCl2) at room temperature, the tissues were digested at 37°C for 20 min in reduced‐Ca2+ PSS containing collagenase (type I, 1750 U/ml), papain (9.5 U/ml), bovine serum albumin (2 mg/ml) and dithiothreitol (1 mM). Cells were then collected and grown in DMEM supplemented with 10% FBS. Contaminating fibroblasts were separated from the PASMCs by exploiting their differential adhesive ability. After three passages at a ratio of 1:2 with trypsin, the purity of PASMCs was confirmed by the typical ‘hill and valley’ morphology and by α‐SMA immunofluorescence staining. Cells that had been passaged four to eight times were used in the following experiments. Each experiment was performed with the same batch of cells in triplicate, and three batches of cells from independent isolations were used in this study.
For the experiments, serum‐starved cell cultures were treated with Bur or SAHA in DMEM/F12 at the indicated concentration and then placed in the hypoxic chamber with 1% O2, 5% CO2 and 94% N2 for the indicated times. Cells incubated in the chamber with 5% CO2 and 95% air for the same duration were used as the normoxic controls.
DNA content and/or cell cycle analysis
The changes in DNA content associated with cell cycle progression were quantified by propidium iodide (PI) staining 30. In brief, following the indicated treatment, cells were collected and washed twice with ice‐cold PBS and then fixed overnight in 70% ethanol at 4°C. After washing twice with cold PBS, cells were treated with DNA staining buffer (PBS containing 1 mg/ml PI and 10 mg/ml RNase A) (BD Biosciences) and incubated at 37°C in the dark for 30 min. The cells were washed and transferred to flow cytometry tubes for cell cycle analysis with a flow cytometer. The percentage of cells at G0, S or G2 phase was analysed. The percentage of cells at S and G2 phases served as an index of cell proliferation activity 31.
EdU incorporation assay
To confirm the effects of HDI on the rate of DNA synthesis, cells were seeded at a density of 2 × 104 cells per well in Corning 24‐well plates and cultured overnight. The culture medium was then removed, and cells were incubated in culture medium with 20 μM EdU, followed by the indicated treatment. Following each exposure, the cells were washed twice with phosphate‐buffered saline (PBS) and fixed in ice‐cold 4% (wt/vol) buffered paraformaldehyde for 20 min. After permeabilization using 0.5% Triton X‐100, EdU was conjugated with Apollo 643‐azide using the Cell‐Light™ EdU In Vitro Imaging kit (Ribobio, C10310‐2; Beijing, China) as per the manufacturer's instructions. Nuclei were visualized by Hoechst 33342 staining. EdU‐positive cells were imaged under a fluorescence microscope.
Apoptosis analysis (annexin V–PI staining)
Apoptosis was analysed by the annexin V–PI staining assay 30. The Annexin V‐fluorescence (BD; NJ, USA) staining kit was used to detect the externalization of phosphatidylserine (an early event in apoptosis), and PI uptake was used as a marker of cell death. In brief, after treatment for the desired time, cells were collected and washed twice with ice‐cold PBS. The cells were then resuspended to achieve a density of 1 × 106 cells/ml. To these, 100 μl of binding buffer containing 2.5 μl of annexin V–FITC and 1 μl PI (100 μg/ml) was added and incubated for 30 min in the dark. Finally, the samples were washed twice with ice‐cold PBS and transferred to flow cytometry tubes for analysis of apoptosis with a flow cytometer. The number of cells that were positive for annexin V, PI or both was then calculated.
Cell viability assay
Cell viability was measured by the MTT assay. Briefly, PASMCs were plated in 96‐well plates at a density of 5000 cells per well. Serum‐starved PASMCs were treated with hypoxia or hypoxia along with 4 mM Bur. At the end of the desired duration, 10 μl MTT (0.5 mg/ml; Sigma‐Aldrich; St. Louis, MO, USA) was added and incubated for 4 hr until purple precipitate was visible. To this, 200 μl DMSO (Sigma‐Aldrich) was added and kept at room temperature in the dark for 10 min. Absorbance was then recorded at 570 nm.
NO measurement
Following the indicated treatment, cell culture supernatants were collected and NO levels were measured within 30 min using the Griess method as recommended by the manufacturer (Beyotime; Shanghai,China). The lysates were extracted using RIPA buffer (Beyotime) supplemented with a cocktail of protease inhibitors (Roche; Basel, Switzerland). The protein concentration of the lysates was measured with a Bradford protein assay (Bio‐Rad; Hercules, CA, USA). NO levels were normalized to the protein contents of each dish and calculated as the ratio of the sum of nitrite and nitrate concentration (nmol) to the relevant protein amount (μg) in the cell cultures.
Western blotting
Following the indicated treatment, cells were washed twice with ice‐cold PBS and cell lysates were extracted using urea lysis buffer (8 M urea, 0.001 M Tris buffer, pH 6.8, 1% SDS, 5 mM DTT, 1% Triton X‐100, 10% glycerol) supplemented with a protease inhibitor (Roche). The protein concentrations of lysates were measured using a Bradford protein assay (Bio‐Rad). Equal amounts of protein from each sample were separated by 10% SDS‐PAGE and then transferred to a PVDF membrane. After blocking with 5% milk for 1 hr, the membrane was incubated with primary antibody in blocking buffer overnight at 4°C. Following three washes with blocking buffer, the appropriate alkaline phosphatase‐conjugated secondary antibody (Jackson ImmunoResearch Laboratories) at a dilution of 1:2000 in blocking buffer was added and the membrane was then incubated for 1 h at room temperature. Antibody binding was visualized with an ECL detection kit (Thermo Fisher Scientific; MA, USA), and images were captured with a scanner using Quality One software (Bio‐Rad).
The primary antibodies and dilutions used in this study are summarized as follows: anti‐eNOS antibody (1:500, Santa Cruz Biotechnology, SC‐654; Santa Cruz, CA, USA), anti‐iNOS antibody (1:500, Santa Cruz Biotechnology, SC‐7271), anti‐β‐actin antibody (1:5000, Sigma‐Aldrich, A5316), anti‐Ac‐H4K12/8/5/1 antibody (1:500, Santa Cruz Biotechnology, SC‐34263), anti‐Ac‐H3K9/14 antibody (1:500, Santa Cruz Biotechnology, SC‐8655), anti‐caspase 3 antibody (1:500, Abcam (Cambridge, UK), ab4051), anti‐caspase 3 (cleaved) antibody (1:500, Abcam, ab2302), anti‐PCNA antibody (1:500, Santa Cruz Biotechnology, SC‐34263) and anti‐p21 antibody (1:500, Abcam, ab109520).
Total RNA isolation and RT‐PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) as per the manufacturer's instructions. RNA (1 μg) was reverse‐transcribed to cDNA using the Superscript III Third‐Strand Synthesis Kit (Invitrogen) in a reaction volume of 10 μl. Subsequently, 1 μl of cDNA product was used for amplification with the PCR amplification kit (Takara). The sequences of the specific primers for eNOS and β‐actin are listed in Table 1. The reaction was performed at 94°C for 30 sec., 56°C for 30 sec. and 72°C for 30 sec. for 36 cycles. β‐Actin served as the loading control. The amplified DNA was examined by electrophoresis and analysed using Quality One software. Relative mRNA levels were calculated as the density ratio of the target gene to β‐actin.
Table 1.
Sequence of specific primers
| Target | Primer sequence | Product size (bp) |
|---|---|---|
| eNOS cDNA |
5′‐TGGCAGCCCTAAGACCTATG‐3′; 5′‐AGTCCGAAAATGTCCTCGTG‐3′ |
243 |
| β‐Actin cDNA |
5′‐TCATGAAGTGTGACGTTGACATCCGT‐3′; 5′‐CCTAGAAGCATTTGCGGTGCAGGATG‐3′ |
285 |
| eNOS promoter |
5′‐GCTTCCTGCTCCTTTGTGTC‐3′; 5′‐TCACCCTTGCTCCTCTCCTA‐3′ |
164 |
| GAPDH promoter |
5′‐TACTAGCGGTTTTACGGGCG‐3′; 5′‐TCGAACAGGAGGAGCAGAGAGCGA‐3′ |
150 |
Knockdown with siRNA
To knock down eNOS expression, transfection was performed with siRNA targeted to eNOS. In brief, to formulate the lipid–siRNA complex, siRNA (Origene Technologies) and Lipofectamine 2000 (Invitrogen) were diluted separately in Opti‐MEM (Invitrogen). After incubation for 5 min., siRNA and Lipofectamine 2000 solutions were combined and then incubated for an additional 20 min. The siRNA/Lipofectamine 2000 complex was gently layered on to the cell cultures. After incubation for 4 hr, the cell medium was replaced with complete culture medium. After transfection for 16–24 hr, the cells were treated as indicated followed by total protein extraction. Western blotting was performed to evaluate the eNOS protein level as mentioned above.
Chromatin immunoprecipitation (ChIP) assay
Following the indicated treatments, the protein–DNA complexes in the cells were cross‐linked using 1% formaldehyde solution in PBS on a rocking or shaking device for 15 min. at room temperature. After adding 125 mM glycine to quench the formaldehyde and rocking for 5 min. at room temperature, the cells were collected and treated with lysis buffer in a volume of 500 μl per 5 × 106 cells on ice for 10 min. Sonication was performed to shear the chromatin to an average length of about 1 kb. Following refrigerated ultracentrifugation at 12,000× g for 10 min, the supernatants were collected and immunoprecipitation was performed using the ChIP Assay kit (Beyotime, P2078) as recommended by the manufacturer. The chromatin mixture was immunoprecipitated using anti‐Ac‐H4‐K12/8/5/1 (Santa Cruz Biotechnology, SC‐34263) antibody at 3 μg/ml, or an irrelevant antibody (Rabbit IgG, Beyotime, A‐7016). After DNA purification, the presence of the selected DNA sequence was assessed by PCR. The primers for the sequences of the eNOS promoter and GAPDH promoter are described in Table 1. GAPDH was set as the positive control and the sample loading control.
Experimental animals and hypobaric hypoxia exposure
Male adult Wistar rats, weighing 180–200 g, were provided by the Animal Center of the Third Military Medical University and divided randomly into four groups: hypoxia with saline (n = 13), hypoxia with Bur (n = 13), normoxia with saline (n = 13) and normoxia with Bur (n = 13). Hypoxic rats were exposed to a simulated high altitude of 5000 m in a hypobaric chamber for 28 days. Others served as the normoxia control and were inbred outside the chamber (the height of Chongqing above the sea level is 308 m) and were treated either with saline or with Bur by intragastric injection. Bur was administered at a dose of 500 mg kg−1 day−1. All the rats had free access to food and water and were subjected to a 12‐hr light–dark cycle. Hypoxic rats were kept free from hypobaric treatment for half an hour each day for cleaning and intragastric injection. All the animal study protocols were approved by the Institutional Animal Use Committee of the Third Military Medical University and were in accordance with the guidelines of the National Institutes of Health and the American Physiological Society.
Measurement of mean pulmonary arterial pressure (mPAP)
Following hypobaric hypoxia treatment, the rats were heparinized and anaesthetized (sodium pentobarbital, 50 mg/kg, intraperitoneal injection). The systolic pressure and diastolic pressure of the pulmonary artery were measured using a physiological recorder (PowerLab System, ADInstruments, Castle Hill, NSW, Australia), and mPAP was calculated. After the measurement, arterial blood was collected for PaO2 detection. The anaesthetized animals were killed by exsanguination, and the heart was harvested for subsequent experiments. All the procedures mentioned above were performed in a hypobaric chamber simulated as a 4000‐m plateau.
Oxygen partial pressure of arterial blood (PaO2) measurement
Following the measurement of mPAP, arterial blood was drawn from the heart and used for PaO2 detection by a blood‐gas analyser following the manufacturer's instructions.
Histology assay
Six hypoxic rats (three administered with saline and three with Bur) and three normoxic rats with saline were killed by perfusion with ice‐cold saline until they were clear of blood. After perfusion with 4% buffered paraformaldehyde for 30 min., the lungs and hearts were dissected and fixed in 4% buffered paraformaldehyde until the organs sank to the bottom. The areas of interest in the lungs were selected and embedded in paraffin, and 5‐μm‐thick slices were sectioned and mounted on glass carrier slides. The sections were stained with H&E to detect pathological changes or with Sirius Red solution (saturated picric acid containing 0.1% Direct Red 80 and 0.1% Fast Green FCF) to visualize collagen deposition.
Evaluation of right ventricle hypertrophy
The right and left ventricles with the septum were isolated and were weighed individually. The right ventricle hypertrophy index (RVHI, the ratio of the right ventricle weight to the left ventricle plus septum weight) was calculated.
Statistical analysis
The data are expressed as mean ± standard deviation (SD) values. Comparisons between two groups were made with two‐tailed t‐tests. Statistical analysis was performed using SPSS 10.0 for Windows. P < 0.05 was considered as statistically significant.
Results
HDI suppresses hypoxia‐stimulated VSMC proliferation and promotes apoptosis
The HDIs, Bur and SAHA, are reported to suppress PDGF‐induced VSMC proliferation 32, 33, 34. Their effect on hypoxia‐induced VSMC growth was unknown. Considering the cytotoxicity of HDI, we first tested the effects of HDI on cell growth and viability under normoxic conditions. As shown in Figure 1A and B, cell viability and cell proliferation were remarkably increased in the groups under hypoxia treatment for 24 hr. We then treated normoxia‐treated VSMCs with a series of Bur or SAHA concentrations for 24 hr. Cell cycle analysis, apoptosis by annexin V–PI staining, and MTT assay were performed. As shown in Figure 1C and D, with a Bur treatment of more than 8 mM or with SAHA treatment of more than 5 μM, apoptosis was promoted and cell cycle progress was inhibited. There were no significant changes when treated with less than 4 mM Bur or less than 5 μM SAHA, respectively, in normoxic VSMCs (Fig. 1C and D). Further studies showed that the EdU‐positive cell number in normoxic VSMCs showed no changes when treated with 4 mM Bur or 5 μM SAHA for 24 hr (Fig. 1E). However, viability of hypoxic cells was decreased by 4 mM Bur or 5 μM SAHA (Fig. 1E). Therefore, 4 mM Bur or 5 μM SAHA was used in the following experiments.
Figure 1.
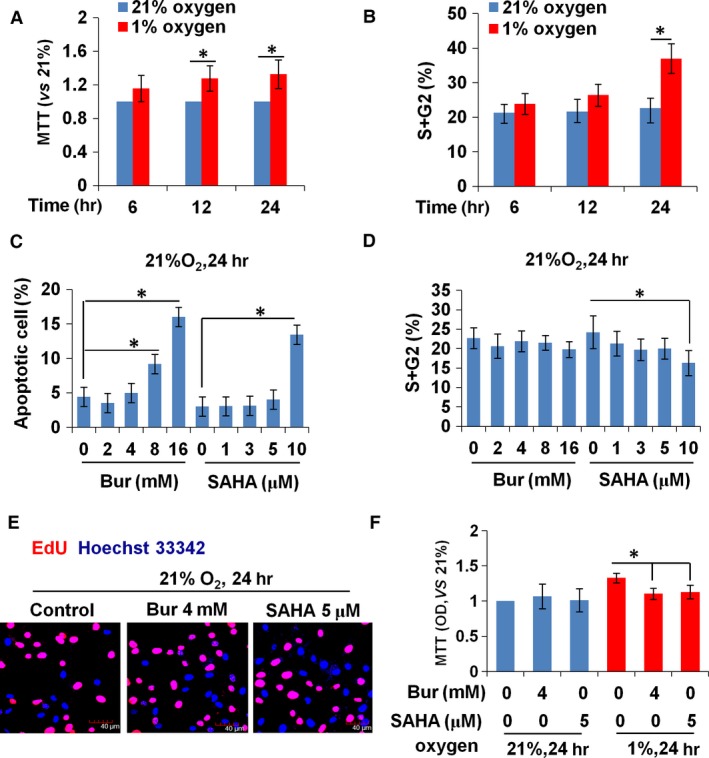
HDI at high concentrations inhibits the growth of VSMCs under normoxic conditions. (A) The proliferative activity of VSMCs, as detected by MTT assay, was increased significantly by hypoxia treatment for 12 and 24 hr. (B) Cell cycle analysis showed that the percentage of cells in the S + G2 phase was increased by 24‐hr hypoxic treatment. (C) The increased percentage of apoptotic cells was induced by more than 8 mM Bur or at 10 μM SAHA. (D) The percentage of cells in the S + G2 phase decreased in 10 μM SAHA‐treated normoxic VSMCs, but Bur had no obvious effect. (E) EdU incorporation was unchanged in normoxic VSMCs treated with 4 mM Bur or 5 μM SAHA. Scale bar: 40 μm. Red indicates the EdU‐positive signal and blue indicates the Hoechst 33342 staining. The experiment was performed in triplicate, and the representative images are shown. (F) MTT assay showed that 4 mM Bur or 5 μM SAHA suppressed the activity of hypoxic VSMCs, but had no effect on that of normoxic VSMCs.
We treated hypoxic VSMCs with 4 mM Bur or 5 μM SAHA and performed an EdU incorporation assay and cell cycle analysis to evaluate cell proliferation ability. The results in Figure 2A demonstrate that HDI remarkably decreased the EdU‐positive cell number in hypoxic VSMCs. The data from cell cycle analysis revealed that the percentage of cells at the S and G2 phases was significantly increased by hypoxia, but was suppressed by Bur and SAHA treatment (Fig. 2B and C). p21 is a potent cyclin‐dependent kinase inhibitor. Up‐regulation of p21 protein is a biomarker that reflects cell cycle arrest. The Western blot assay showed that HDI induced p21 expression under hypoxia (Fig. 2F). These data suggested that the proliferation ability of hypoxic VSMCs was significantly reduced when treated with HDI. To determine the effect of Bur and SAHA on apoptosis, we determined the rate of apoptotic cells by annexin V–PI staining. The data showed that 4 mM Bur or 5 μM SAHA treatment increased the rate of VSMCs that were positive for annexin V and/or PI staining (Fig. 2D and E). The levels of caspase 3 and cleaved caspase 3 are biomarkers for apoptosis activation. As shown in Figure 2F, cleaved caspase 3 was increased by HDIs in hypoxic VSMCs.
Figure 2.
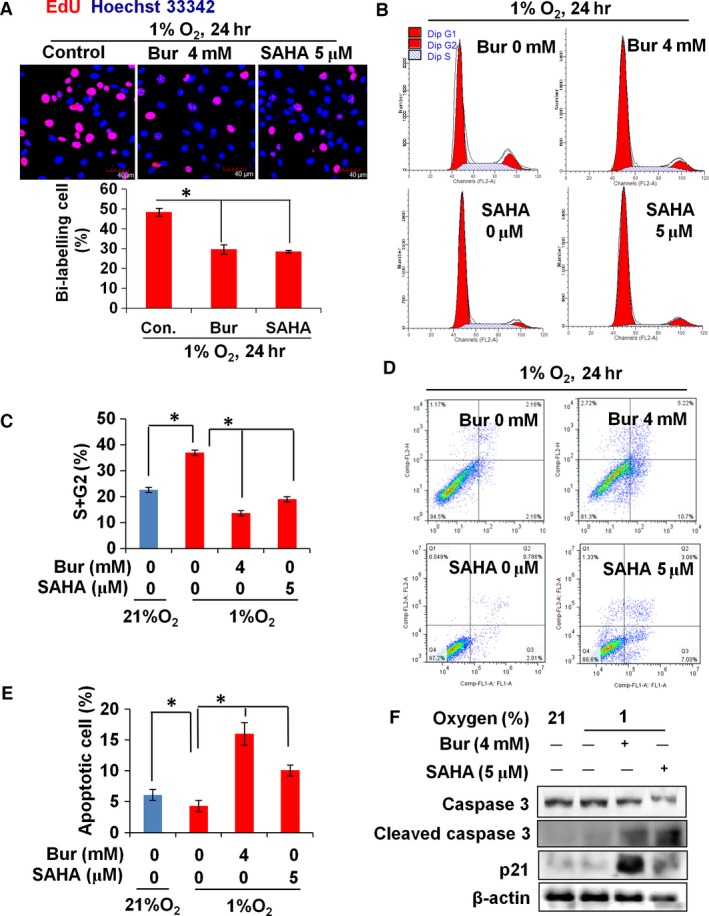
HDI suppresses proliferation and promotes apoptosis of hypoxic VSMCs. (A) EdU incorporation of hypoxic VSMCs was reduced significantly by the HDIs, Bur and SAHA. Scale bar: 40 μm. The experiment was performed in triplicate, and the representative images are shown. (B, C) The percentage of cells at the S and G2 phases was increased remarkably in Bur‐ or SAHA‐treated hypoxic VSMCs (*P < 0.05). The experiment was performed in triplicate, and the representative figure is shown. (D, E) The percentage of apoptotic VSMCs (indicated by positive annexin V staining and annexin V + PI double staining) was increased remarkably in Bur‐ or SAHA‐treated hypoxic VSMCs (*P < 0.05). (F) The protein level of cell growth regulators was detected by Western blot assay. β‐Actin served as the sample loading control. The experiment was performed in triplicate, and the representative images are shown.
Taken together, these results indicate that Bur and SAHA suppress VSMC proliferation and promote VSMC apoptosis under normoxic or hypoxic conditions. However, hypoxic cells are much sensitive to HDI treatment and are inhibited at lower concentrations of HDIs, in which the basal growth of normoxic VSMCs is not influenced. In our study, 4 mM Bur and 5 μM SAHA were used for the following experiments.
HDIs enhance eNOS expression and NO production in hypoxic VSMCs
To determine the effect of HDIs on eNOS expression, we cultured VSMCs in 1% oxygen with 4 mM Bur or 5 μM SAHA for 24 hr and examined eNOS levels by Western blotting. We found that eNOS protein was hardly detected in VSMCs cultured in either 21% or 1% oxygen, consistent with previous reports. In hypoxic VSMCs, the addition of Bur or SAHA obviously increased eNOS expression (Fig. 3A). To determine whether the HDI‐enhanced eNOS expression resulted in increased NO secretion by hypoxic VSMCs, we measured the NO concentrations in cell culture supernatants by the Griess method. Upon treatment with 4 mM Bur or 5 μM SAHA, the NO levels in the hypoxic VSMC culture media were increased significantly (Fig. 3B). Under physiological conditions, eNOS is not expressed in VSMCs. However, under pathological conditions such as hypoxia, inducible NOS (iNOS) is activated and acts as the main NO producer in VSMCs. We found that hypoxia‐induced iNOS expression was suppressed by Bur and SAHA treatment, while eNOS expression was promoted (Fig. 3A). This suggests that activated eNOS expression contributes to enhancement of NO secretion by HDI‐treated hypoxic VSMCs. In summary, HDI promotes eNOS expression and NO secretion in hypoxic VSMCs.
Figure 3.
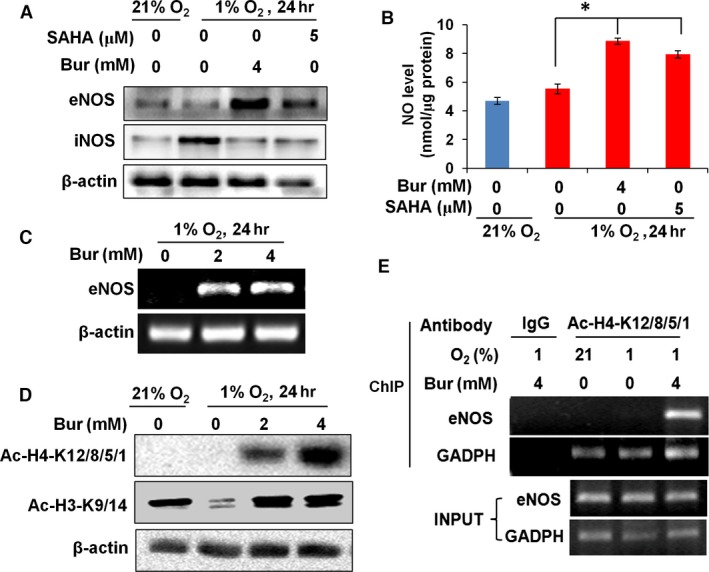
HDI enhances eNOS expression and NO secretion by hypoxic VSMCs. (A) Western blots showing that Bur and SAHA enhanced the protein levels of eNOS in hypoxic VSMCs. However, hypoxia‐induced iNOS expression was suppressed by HDI. β‐Actin was used as the sample loading control. The experiment was performed in triplicate, and the representative images are shown. (B) NO levels in the supernatant of hypoxic VSMCs were increased by Bur and SAHA treatment (*P < 0.05, n = 9). (C) Bur enhanced the eNOS mRNA levels in hypoxic VSMCs. β‐Actin mRNA levels were determined for internal normalization. The experiment was performed in triplicate, and the representative data are shown. (D) Total acetylation of histone H4‐K12/8/5/1 in VSMCs was barely detectable under either normoxic or hypoxic conditions, but was dramatically induced by Bur. Total acetylation of histone H3‐K9/14 was detected in VSMCs cultured under normoxic conditions, which was remarkably decreased after 24‐hr exposure to hypoxia. The addition of Bur increased the total acetylation of histone H3‐K9/14. β‐Actin was also used as the sample loading control. The experiment was performed in triplicate, and the representative data are shown. (E). ChIP assay was performed with anti‐Ac‐H4‐K8/12/5/1 antibody. A DNA fragment specific to the eNOS promoter was only detected in hypoxic VSMCs treated with 4 mM Bur. A DNA fragment of the GAPDH promoter was amplified as the positive control. The experiment was performed in triplicate, and the representative data are shown.
Concerning the promoter of active genes, one common feature is acetylation at lysine residues of histones H3 and H4, which leads to a less compact and transcriptionally active chromatin. HDAC removes the acetyl groups from lysine residues, leading to the formation of a condensed and transcriptionally silenced chromatin. HDAC inhibitors block this action and thereby affect gene expression. HDIs modulate the histone modification pattern and generate a novel genomic expression profile in VSMCs 10, 11, 12, 13, 33, 50, 51, 52, 53. To further confirm the induction of eNOS by HDIs and explore its mechanism, we examined the acetylation of histones H3 and H4 on the eNOS promoter region under Bur treatment. We first confirmed that eNOS mRNA expression was enhanced by Bur (Fig. 3C), further indicating that Bur induced transcriptional activation of eNOS in hypoxic VSMCs. Histone H4 acetylation at lysine 12 (K12) and histone H3 acetylation at lysine 9 (K9) are required for the specific transcription of eNOS in endothelial cells 15, 16, and HDI induces eNOS expression in non‐EC cells 16. To explore the effects of Bur on histone H4‐K12 and H3‐K9 acetylation in VSMCs, we treated hypoxic VSMCs with 2 or 4 mM Bur for 24 h, followed by examination of acetylation levels using respective specific antibodies. Acetylation of H4 at K12/8/5/1 was barely detectable and was not affected by hypoxia (Fig. 3D). Importantly, the addition of Bur remarkably increased the acetylation levels of H4 at K12/8/5/1 in hypoxic VSMCs (Fig. 3D). We detected high levels of basal acetylation of histone H3 at K9/14 under normoxic conditions. Hypoxia remarkably reduced acetylation levels of histone H3 at K9/14, which were restored by Bur treatment (Fig. 3D). We then explored whether Bur induced the acetylation of histone H4 on the eNOS promoter region. We treated hypoxic VSMCs with 4 mM Bur and performed chromatin immunoprecipitation (ChIP) assays using an antibody specific for Ac‐H4‐K12/8/5/1. The results shown in Figure 3E indicate that a specific fragment of the eNOS promoter was amplified and detected in the precipitated Ac‐H4‐K12/8/5/1 complexes from the Bur‐treated hypoxic VSMCs. We also examined histone H3 acetylation on the eNOS promoter by the ChIP assay with an anti‐Ac‐histone H3‐K9/14 antibody, but no specific DNA fragment of the eNOS promoter was found in the immune complexes (data not shown). Taken together, these data further indicate that the HDI Bur activates eNOS gene expression in hypoxic VSMCs by epigenetically increasing the acetylation levels of histone H4 at its promoter region.
Taken together, our results indicate that the eNOS–NO system is activated by HDI in hypoxic VSMCs.
eNOS expression in hypoxic VSMCs is required for HDI to suppress hypoxia‐induced cell growth
To confirm the role of HDI‐induced eNOS expression on the proliferation and apoptosis of VSMCs, we used siRNA to specifically knock down eNOS in VSMCs. The efficiency of eNOS knockdown in hypoxic VSMCs is shown in Figure 4A, which indicated that all three siRNAs effectively knocked down eNOS expression at the protein level and that siRNA #3 had the best effect. Next, we used siRNA #3 to knock down eNOS expression in hypoxic VSMCs. Apoptosis assays showed that knockdown of eNOS decreased the percentage of VSMCs with annexin V staining and annexin V–PI double staining (Fig. 4B). Cell cycle analysis showed that knockdown of eNOS increased the percentage of VSMCs at the S and G2 phases (Fig. 4C). EdU incorporation assay also demonstrated that the number of EdU‐positive cells was increased in hypoxic VSMCs with eNOS siRNA transfection (Fig. 4D). Consistently, up‐regulation of cleaved caspase 3 and p21 by Bur was abrogated in hypoxic cells by eNOS siRNA transfection (Fig. 4E). These data demonstrate that induced eNOS expression in VSMCs is an essential biological process for Bur and SAHA to suppress hypoxia‐induced cell proliferation and to promote apoptosis.
Figure 4.
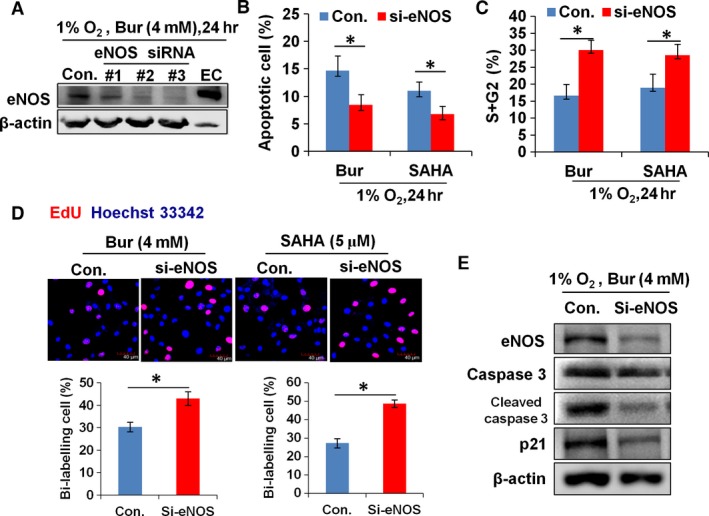
The antiproliferative and pro‐apoptotic effects of HDI on hypoxic VSMCs were weakened by eNOS siRNA transfection. (A) HDI‐induced expression of eNOS protein in hypoxic VSMCs was knocked down by eNOS siRNA transfection. β‐Actin protein was used as the sample loading control. Cell lysates from endothelial cell (EC) were used as the positive control. The experiment was performed in triplicate, and the representative data are shown. (B) The percentage of apoptotic VSMCs (indicated by positive annexin V staining and annexin V + PI double staining) was decreased remarkably in Bur‐ or SAHA‐treated hypoxic VSMCs upon eNOS siRNA transfection (*P < 0.05, n = 9). (C) Cell cycle analysis showed that the percentage of cells at the S and G2 phases was increased remarkably in both Bur‐ and SAHA‐treated hypoxic VSMCs upon eNOS siRNA transfection (*P < 0.05, n = 9). (D) The number of EdU+ cells was increased in both Bur‐ and SAHA‐treated hypoxic VSMCs upon eNOS siRNA transfection. Scale bar: 40 μm. The experiment was performed in triplicate, and the representative data are shown. (E) The protein levels of eNOS, caspase 3, cleaved caspase 3 and p21 were reduced in Bur‐treated hypoxic VSMCs that were transfected with eNOS siRNA. β‐Actin protein was used as the sample loading control.
Induced secretion of NO by hypoxic VSMCs is required for HDI to suppress hypoxia‐induced cell growth
To explore whether NO secretion by hypoxic VSMCs is required for HDI to suppress hypoxia‐induced VSMC proliferation, we examined the effects of the NO scavenger carboxy‐PTIO on the antiproliferative and pro‐apoptotic properties of Bur and SAHA. As shown in Figure 5A and B, the addition of carboxy‐PTIO increased the number of EdU‐positive cells and the percentage of VSMCs at the S and G2 phases. However, the percentage of cells with annexin V staining and annexin V–PI double staining was decreased (Fig. 5C). Meanwhile, the level of cleaved caspase 3 and p21 was decreased in SAHA‐ or Bur‐treated hypoxic VSMCs upon carboxy‐PTIO addition (Fig. 5D). These data indicated that elevated NO secretion is required for Bur or SAHA to achieve its antiproliferative and pro‐apoptotic effects on hypoxic VSMCs.
Figure 5.
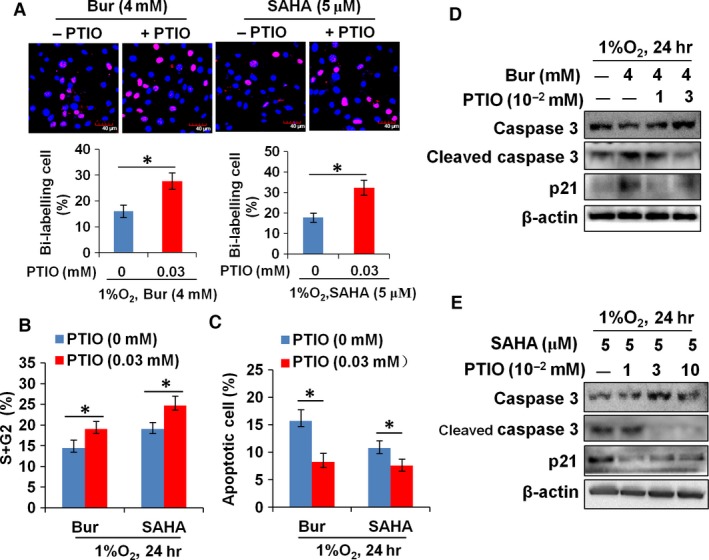
The NO scavenger carboxy‐PTIO restores the proliferation and survival of hypoxic VSMCs under HDI treatment. (A) The number of EdU+ cells was increased in both Bur‐ and SAHA‐treated hypoxic VSMCs upon the addition of the NO scavenger, carboxy‐PTIO. Scale bar: 40 μm. The experiment was performed in triplicate, and the representative image is shown. (B) Cell cycle analysis showed that the percentage of cells at the S and G2 phases was increased remarkably in both Bur‐ and SAHA‐treated hypoxic VSMCs upon the addition of the NO scavenger, carboxy‐PTIO (*P < 0.05, n = 5). (C) The percentage of apoptotic VSMCs (indicated by positive annexin V staining and annexin V + PI double staining) was decreased remarkably in Bur‐ or SAHA‐treated hypoxic VSMCs upon the addition of the NO scavenger, carboxy‐PTIO (*P < 0.05, n = 5). (D, E) HDI‐induced expression of cleaved caspase 3 and p21 was reduced in both Bur‐ and SAHA‐treated hypoxic VSMCs upon the addition of the NO scavenger, carboxy‐PTIO. β‐Actin protein was used as the sample loading control. The experiment was performed in triplicate, and the representative data are shown.
Bur attenuates pulmonary artery remodelling of rats exposed to chronic hypoxia
Hypoxia‐induced pulmonary arterial remodelling is characterized as thickening of medial layers of and collagen deposition at arterial walls. Both of them are related to VSMC activities 2, 3, 39. Hypoxia promotes proliferation and inhibits apoptosis of VSMCs, which contributes to excessive VSMC accumulation in the artery wall and to the thickening. Nevertheless, hypoxia induces a phenotype transformation of VSMCs from a contractile, differentiated to a synthetic, dedifferentiated state, which contributes to increased proliferative ability and collagen oversynthesis of VSMCs. To determine the effect of Bur on hypoxia‐induced VSMC accumulation in vivo, we established a rat model of pulmonary artery remodelling by exposing the rats to hypobaric hypoxia for 28 days. The thickness and collagen deposition of pulmonary arteries were then estimated. The greatest influence of pulmonary arterial wall thickness is the increase in pulmonary vascular resistance and right ventricular afterload, reflected by the elevation of mean pulmonary arterial pressure (mPAP) and the right ventricle hypertrophy index (RVHI, the ratio of right ventricle weight to left ventricle plus septum weight). To assess the function of pulmonary arteries, mPAP, RVHI and PaO2 were evaluated.
As shown in Figure 6A, the mPAP of hypoxic rats was much higher than that in normoxic rats. Consistently, the RVHI was increased with exposure to hypoxia (Fig. 6B). Similarly, rats exposed to hypobaric hypoxia showed increased thickness in the pulmonary artery wall when compared with that rats under normal conditions (308 m above sea level, Chongqing, China; Fig. 6C). Rats exposed to hypobaric hypoxia showed obvious collagen deposition at the pulmonary artery, which was barely observed in the control rat group (Fig. 6D). However, the thickness and collagen deposition of the pulmonary artery wall were less obvious in Bur‐treated hypoxic rats (Fig. 6C and D). Although mPAP showed no obvious changes in hypoxic rats with Bur administration, the RVHI was decreased significantly (Fig. 6A and B). Simultaneously, PaO2 was increased in rats exposed to long‐term hypobaric hypoxia and administrated Bur (Fig. 6E). To evaluate the effect of Bur on the viability of pulmonary arterial SMCs (PASMCs), we treated primarily cultured PASMCs with 4 mM Bur and cultured them in a hypoxic incubator for 24 hr. MTT assay showed that Bur treatment significantly reduced the viability of PASMCs (Fig. 6F). The protein levels of eNOS and cleaved caspase 3 in Bur‐treated hypoxic PASMCs were elevated (Fig. 6G). These results further suggest that Bur may effectively suppress hypoxia‐induced vascular remodelling in vivo.
Figure 6.
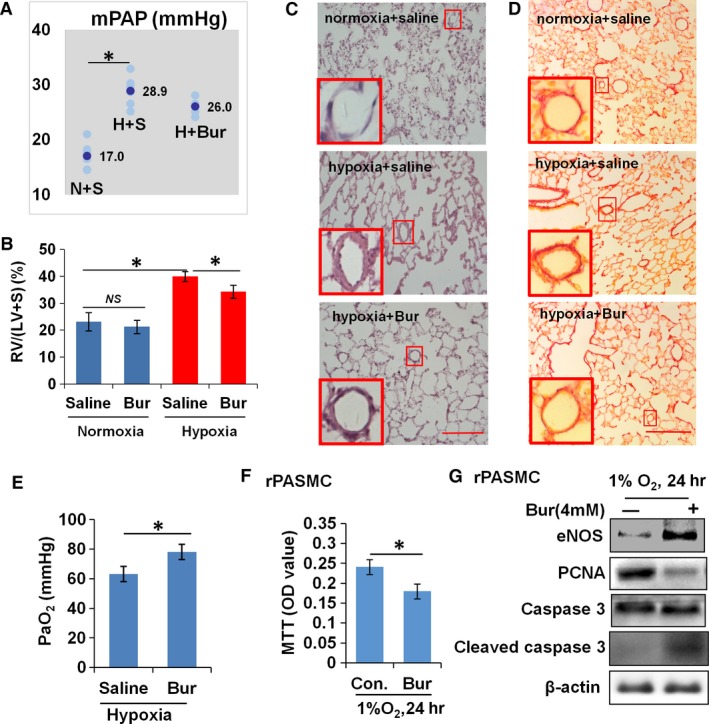
Bur abrogates hypoxia‐induced pulmonary artery remodelling. (A) Pulmonary arterial pressure was significantly elevated in rats under long‐term exposure (28 days) to hypobaric hypoxia (n = 6), but showed no obvious changes in hypoxic rats with Bur administration (n = 6). N + S, H + S and H + Bur indicate normoxia + saline, hypoxia + saline and hypoxia + Bur groups, respectively. The average value of mPAP in each group is shown. (B) Right ventricle hypertrophy index (RVHI) was significantly decreased in hypoxic rats upon Bur administration (n = 6). (C) H&E staining analysis showed that long‐term exposure (28 days) to hypobaric hypoxia increased the thickness of the pulmonary arterial wall in rats, which was partly prevented by Bur administration. Scale bar: 100 μm. Each experiment was performed in three cases, and the representative data are shown. (D) Sirius Red staining showing that exposure to hypobaric hypoxia promoted collagen deposition at the pulmonary arterial wall rats, which was prevented upon Bur administration. Scale bar: 100 μm. Each experiment was performed in three cases, and the representative is shown. (E) Bur administration protected the lung function in rats exposed to long‐term hypobaric hypoxia as indicated by the increase in PaO2 (*P < 0.05, n = 7). (F) Cell viability of hypoxic PASMCs was decreased upon Bur (4 mM) treatment (*P < 0.05, n = 9). (G) The protein level of PCNA was reduced in hypoxic PASMCs upon the addition of Bur (4 mM), while the levels of eNOS and cleaved caspase 3 were increased. β‐Actin protein was used as the sample loading control. The experiment was performed in triplicate, and the representative image is shown.
Discussion
HDIs have been widely used as an antiproliferation reagent in cancer research. Some of these such as Bur and SAHA have been tested in clinical trials. However, the effect of HDI on hypoxia‐induced VSMC proliferation is still unknown. In this study, we demonstrate that Bur and SAHA suppress the proliferation and promote apoptosis in hypoxic VSMCs in vitro. In vivo, the administration of Bur in rats exposed to hypobaric hypoxia for 28 days ameliorated the thickness and collagen deposition of pulmonary artery walls. These results indicate that Bur and SAHA are effective for suppressing hypoxia‐induced VSMC growth and prevent the development of vascular remodelling.
Previous studies reported that trichostatin A, another HDI, also suppresses VSMC proliferation 14, 40, 41. However, its definite cytotoxicity restricts its application potential 14, 40, 42. Alternatively, Bur is a naturally occurring short‐chain fatty acid and physiologically exists in the intestinal system and blood. It is a major metabolite of fibre‐enriched food 43, 44, 45, 46, 47, 48. Recent studies demonstrated the safety of butyrate in the protection of the cardiovascular system 32. SAHA is another HDI that is well tolerated intravenously 49. It has been approved by the FDA as an anticancer drug 50. In our study, we confirmed that Bur and SAHA are safe because they have no influence on the basal growth of normoxic VSMCs at low concentrations. Bur and SAHA are primarily proven to be anti‐inflammatory reagents and are effective at preventing the development of inflammatory bowel disease and lung fibrosis 8, 48, 50, 51, 52, 53. Our data demonstrate a novel application for these HDIs in maintaining cardiovascular homoeostasis.
The main clinical signs of PH include increased right ventricular systolic pressure, increased right ventricular weight and considerable pulmonary vascular remodelling 54. In this study, we found that vascular remodelling and right ventricular hypertrophy were partly suppressed by Bur administration in hypoxic rats. Unfortunately, a significant decrease in mPAP was not observed, although the average value appeared to have a declining trend in hypoxic rats upon Bur administration, which is unexpected but reasonable. The level of mPAP is dependent not only on the function of the pulmonary artery system but also on right ventricular function. Although elevated mPAP is an important contributor to right ventricular hypertrophy, direct responses of heart cells to hypoxia are also essential for cardiac remodelling. It has been revealed that hypoxia‐induced pulmonary vascular remodelling of rats was reversed after they were freed from the hypoxic environment for up to 6 weeks. However, significant recovery of PAP, right ventricular systolic pressure and right ventricular hypertrophy was not observed simultaneously 54, 55. In this regard, although HDIs show no obvious inhibition of hypoxia‐induced mPAP elevation, we believe that HPH development is suppressed by HDIs, which is reflected by the reversal of histological changes in the cardiovascular system. Further analysis of the benefit and feasibility of HDIs in the prevention of hypoxia‐induced vascular disorders needs to be properly defined in the future.
Hypoxia‐induced VSMC proliferation is associated with endogenous activity of VSMCs and the factors secreted by neighbouring cells or those transported from blood 30, 56, 57. Although growth factors, including PDGF, are contributors to hypoxia‐induced VSMC proliferation 58, 59, 60, 61, 62, they are mainly derived from either vascular endothelial cells or migrated inflammatory cells 56, 61, 63. Although HDIs suppressed PDGF‐induced VSMC proliferation, autosecretion of PDGF was not detected when VSMC was stimulated by hypoxia alone 63, 64. In this study, cultured VSMCs were free from the effects of other cell types or systemic responses to hypoxia. Nevertheless, we found that Bur and SAHA had no influence on the baseline of VSMC proliferation under normoxia at the concentration used in this study. Our data suggest for the first time that HDIs may inhibit hypoxia‐induced VSMC growth through a mechanism that is endogenous to VSMCs and independent of external sources of factors, such as PDGF.
As described in other reports, three functional classes of genes were dominantly changed by HDIs in VSMCs, which were associated with cell growth and differentiation, stress response or vascular function 10, 11. Examination of differentially expressed cell growth‐ and differentiation‐related genes indicated that butyrate‐inhibited VSMC proliferation appears to involve down‐regulation of genes that encode several positive regulators of cell growth and up‐regulation of some negative regulators of growth or differentiation inducers 10. Analysis of histone H3 modifications specific to butyrate‐arrested VSMC proliferation displayed induction of histone H3‐lysine 9 acetylation, inhibition of histone H3‐serine 10 phosphorylation, reduction of histone H3‐lysine 9 dimethylation and stimulation of histone H3‐lysine 4 dimethylation 13. The interplay of these site modifications might cause distinct chromatin alterations that allow distinct gene expression patterns 13. However, the epigenetic mechanism of HDI on cell growth regulation is still largely unknown.
In this study, we proved that endogenous eNOS expression was induced by Bur and SAHA treatment in hypoxic VSMCs. It has been reported that hyperacetylation of histone H4‐lysine 12 and H3‐lysine 9 is specifically related to eNOS expression in vascular endothelial cells. Increased acetylation levels at these sites are positively correlated with activation of the eNOS gene in non‐endothelial cells, such as cardiac cells 15, 16. We noticed that Bur treatment increased histone acetylation on the eNOS promoter in hypoxic VSMCs. However, only the hyperacetylation of histone H4‐lysine 12/8/5/1 was detected on the eNOS promoter in Bur‐treated hypoxic VSMCs, while acetylation of histone H3 at lysine 9 was undetected. Reduced global histone H4 acetylation and DNA methylation in the pulmonary vasculature of foetal lambs exposed to high‐altitude (3180 m) hypoxia was accompanied by the loss of p21 expression and contributed to SMC proliferation. These results suggest that acetylation of histone H4 at lysine 12/8/5/1 was remarkably induced by Bur in hypoxic VSMCs and was related to eNOS activation. It also implies that like other HDIs, Bur and SAHA may have biological effects on VSMC growth along with other mechanisms as well. Being the first such study, the role of HDI‐induced eNOS expression on cell growth was further analysed to facilitate a better understanding of the mechanism underlying HDI‐mediated protection of the cardiovascular system. Although the acetylation of histone H3 at lysine 9 was alleviated dramatically under hypoxia but was restored by Bur as declared by other reports, it did not participate in eNOS activation. However, it is widely believed that hypoacetylation of histone H3 at lysine 9 is closely related to gene repression. Primary studies had shown that p21 expression was suppressed by hypoxia and was restored by HDIs 10, 13, 14, 30, 42. Interestingly, we found that the alteration of p21 expression was positively correlated with the variation of histone H3 acetylation at lysine 9. Therefore, future studies on the modification of histone H3 at lysine 9 are necessary for a better understanding of the mechanism mediating hypoxia‐induced suppression of p21.
In physiological conditions, eNOS‐derived NO is secreted by vascular endothelial cells and this migrates into VSMCs. The effect of NO on VSMC growth is based on two main mechanisms. The first is to promote the cleavage of caspase 3, which contributes to apoptosis 65. The other mechanism is to promote p21 expression, which inhibits cell cycle progression 28, 65. We found that decreased proliferation and increased apoptosis induced by HDI treatment were alleviated by an NO scavenger or upon inhibition of eNOS expression in hypoxic VSMCs. Consistently, HDI‐induced expression of cleaved caspase 3 and p21 was also attenuated. These data suggest that induction of exogenous eNOS–NO activity in hypoxic VSMCs was directly involved in the antiproliferative and pro‐apoptotic effects of HDI. It has been mentioned above that HDI‐inhibited VSMC proliferation was based on the combined action of genes that serve as positive or negative regulators of cell growth 10, 11. Therefore, the positive integrators that combine the signals from all regulators are essential for understanding the protective role of HDI. A future study would be needed to investigate whether induced eNOS expression and bioactive NO secretion in VSMCs act as the combined mediator for HDI in cell growth regulation.
In many pathological conditions, eNOS‐derived NO production is attenuated 20, 24, 25. Due to endothelial cell injury and eNOS dysfunction, restoration of the endothelium‐based eNOS–NO system in pathological circumstances is limited and difficult 20, 23, 66. It has been demonstrated that eNOS transfection in adipose‐derived stem cells results in bioactive nitric oxide production 67. Another report has declared that eNOS gene transfection in VSMCs inhibits cell proliferation via up‐regulation of p27 and p21, resulting in delayed cell cycle progression, but no effect on apoptosis 26, 68, 69. Our study also proved that endogenous expression of eNOS in hypoxic VSMCs is achievable and effective for cell cycle suppression and apoptosis promotion. Interestingly, our study provides data to suggest an epigenetic strategy that activates endogenous eNOS–NO system in hypoxic VSMCs that can be easily realized by treatment with Bur and SAHA, two HDIs that have been approved clinically or are currently under clinical trials.
As schematically summarized in Figure 7, the HDIs, Bur and SAHA, activate the eNOS–NO system in hypoxic VSMCs. Increased NO production may mediate HDI‐induced cell cycle arrest and apoptosis. Our findings not only indicate a novel mechanism by which HDI suppresses hypoxia‐triggered VSMC growth, but also pave a possibility for the clinical utilization of HDIs, Bur and SAHA, to prevent vascular remodelling related to hypoxic stimulation.
Figure 7.
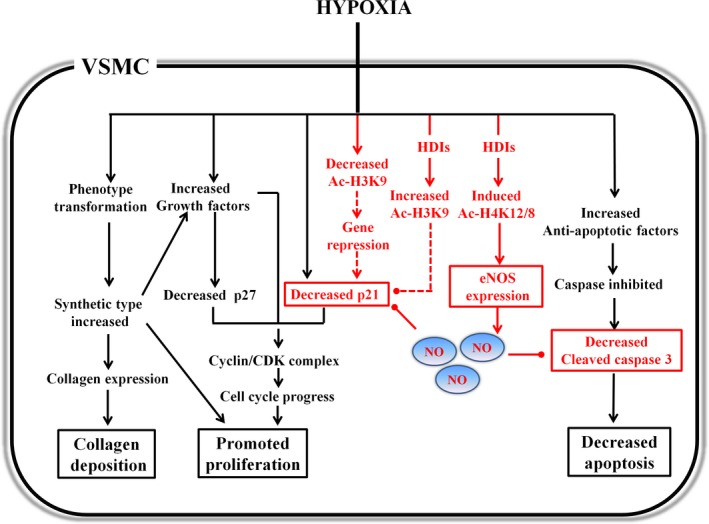
A schematic summary of the suppression of hypoxia‐induced VSMC growth by HDIs involving eNOS–NO activation. The point arrows are used to indicate promotion effects. The round‐headed arrows are used to indicate suppression. The parts with dotted lines are shown as speculative views. The abbreviations are described as follows: HDI, histone deacetylase inhibitor; Ac‐H3K9, acetylated histone H3 at lysine 9; Ac‐H4K12/8, acetylated histone H4 at lysine 12/lysine 8; eNOS, endothelial nitric oxide synthase.
Conflict of interest
The authors confirm that there are no conflict of interests.
Funding
The work was supported by the National Natural Science Foundation of China (grant numbers: 81270108; 30971198) and the National Basic Research Program of China (973 Program, grant number: 2012CB518201).
Acknowledgements
We thank Dewei Chen for A10 SMC, Wenjuan He and Gang Xu for their technical support, Prof. Nianli Sang for the preparation of the manuscript and the other members of our department for suggestions during the course of this work.
References
- 1. Hanze J, Weissmann N, Grimminger F, et al Cellular and molecular mechanisms of hypoxia‐inducible factor driven vascular remodeling. Thromb Haemost. 2007; 97: 774–87. [PubMed] [Google Scholar]
- 2. Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther. 2001; 92: 1–20. [DOI] [PubMed] [Google Scholar]
- 3. Stenmark KR, Fagan KA, Frid MG. Hypoxia‐induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006; 99: 675–91. [DOI] [PubMed] [Google Scholar]
- 4. Chen JS, Faller DV, Spanjaard RA. Short‐chain fatty acid inhibitors of histone deacetylases: promising anticancer therapeutics? Curr Cancer Drug Targets. 2003; 3: 219–36. [DOI] [PubMed] [Google Scholar]
- 5. Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor‐induced apoptosis. Adv Exp Med Biol. 2008; 615: 261–98. [DOI] [PubMed] [Google Scholar]
- 6. New M, Olzscha H, La Thangue NB. HDAC inhibitor‐based therapies: can we interpret the code? Mol Oncol. 2012; 6: 637–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cantoni S, Galletti M, Zambelli F, et al Sodium butyrate inhibits platelet‐derived growth factor‐induced proliferation and migration in pulmonary artery smooth muscle cells through Akt inhibition. FEBS J. 2013; 280: 2042–55. [DOI] [PubMed] [Google Scholar]
- 8. Findeisen HM, Gizard F, Zhao Y, et al Epigenetic regulation of vascular smooth muscle cell proliferation and neointima formation by histone deacetylase inhibition. Arterioscler Thromb Vasc Biol. 2011; 31: 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S, Sang N. Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia‐inducible factors. J Biomed Biotechnol. 2011; 2011: 197946–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ranganna K, Yousefipour Z, Yatsu FM, et al Gene expression profile of butyrate‐inhibited vascular smooth muscle cell proliferation. Mol Cell Biochem. 2003; 254: 21–36. [DOI] [PubMed] [Google Scholar]
- 11. Feng P, Ge L, Akyhani N, et al Sodium butyrate is a potent modulator of smooth muscle cell proliferation and gene expression. Cell Prolif. 1996; 29: 231–41. [DOI] [PubMed] [Google Scholar]
- 12. Mathew OP, Ranganna K, Milton SG. Involvement of the antioxidant effect and anti‐inflammatory response in butyrate‐inhibited vascular smooth muscle cell proliferation. Pharmaceuticals (Basel). 2014; 7: 1008–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mathew OP, Ranganna K, Yatsu FM. Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1‐specific cell cycle proteins in vascular smooth muscle cells. Biomed Pharmacother. 2010; 64: 733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Okamoto H, Fujioka Y, Takahashi A, et al Trichostatin A, an inhibitor of histone deacetylase, inhibits smooth muscle cell proliferation via induction of p21(WAF1). J Atheroscler Thromb. 2006; 13: 183–91. [DOI] [PubMed] [Google Scholar]
- 15. Fish JE, Matouk CC, Rachlis A, et al The expression of endothelial nitric‐oxide synthase is controlled by a cell‐specific histone code. J Biol Chem. 2005; 280: 24824–38. [DOI] [PubMed] [Google Scholar]
- 16. Gan Y, Shen YH, Wang J, et al Role of histone deacetylation in cell‐specific expression of endothelial nitric‐oxide synthase. J Biol Chem. 2005; 280: 16467–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rossig L, Li H, Fisslthaler B, et al Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002; 91: 837–44. [DOI] [PubMed] [Google Scholar]
- 18. Chen HI, Chang HR, Wu CY, et al Nitric oxide in the cardiovascular and pulmonary circulation–a brief review of literatures and historical landmarks. Chin J Physiol. 2007; 50: 43–50. [PubMed] [Google Scholar]
- 19. Crosswhite P, Sun Z. Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension. J Hypertens. 2010; 28: 201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ho JJ, Man HS, Marsden PA. Nitric oxide signaling in hypoxia. J Mol Med (Berl). 2012; 90: 217–31. [DOI] [PubMed] [Google Scholar]
- 21. Hubloue I, Biarent D Abdel Kafi S, et al Endogenous endothelins and nitric oxide in hypoxic pulmonary vasoconstriction. Eur Respir J. 2003; 21: 19–24. [DOI] [PubMed] [Google Scholar]
- 22. Le Cras TD, McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol. 2001; 280: L575–82. [DOI] [PubMed] [Google Scholar]
- 23. Napoli C, Loscalzo J. Nitric oxide and other novel therapies for pulmonary hypertension. J Cardiovasc Pharmacol Ther. 2004; 9: 1–8. [DOI] [PubMed] [Google Scholar]
- 24. De Pascali F, Hemann C, Samons K, et al Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S‐glutathionylation. Biochemistry. 2014; 53: 3679–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rochette L, Lorin J, Zeller M, et al Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: possible therapeutic targets? Pharmacol Ther. 2013; 140: 239–57. [DOI] [PubMed] [Google Scholar]
- 26. Sato J, Nair K, Hiddinga J, et al eNOS gene transfer to vascular smooth muscle cells inhibits cell proliferation via upregulation of p27 and p21 and not apoptosis. Cardiovasc Res. 2000; 47: 697–706. [DOI] [PubMed] [Google Scholar]
- 27. Sharma RV, Tan E, Fang S, et al NOS gene transfer inhibits expression of cell cycle regulatory molecules in vascular smooth muscle cells. Am J Physiol. 1999; 276: H1450–9. [DOI] [PubMed] [Google Scholar]
- 28. Stotz WH, Li D, Johns RA. Exogenous nitric oxide upregulates p21(waf1/cip1) in pulmonary microvascular smooth muscle cells. J Vasc Res. 2004; 41: 211–9. [DOI] [PubMed] [Google Scholar]
- 29. Hou X, Chen J, Luo Y, et al Silencing of STIM1 attenuates hypoxia‐induced PASMCs proliferation via inhibition of the SOC/Ca2+/NFAT pathway. Respir Res. 2013; 14: 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ray JB, Arab S, Deng Y, et al Oxygen regulation of arterial smooth muscle cell proliferation and survival. Am J Physiol Heart Circ Physiol. 2008; 294: H839–52. [DOI] [PubMed] [Google Scholar]
- 31. Lv G, Li Y, Wang Z, et al Hypoxia stimulates the proliferation of neonatal rat vascular smooth muscle cells through activation of hypoxia‐inducible factor‐1alpha. Int J Clin Exp Med. 2015; 8: 496–503. [PMC free article] [PubMed] [Google Scholar]
- 32. Ranganna K, Joshi T, Yatsu FM. Sodium butyrate inhibits platelet‐derived growth factor‐induced proliferation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1995; 15: 2273–83. [DOI] [PubMed] [Google Scholar]
- 33. Ranganna K, Yatsu FM. Inhibition of platelet‐derived growth factor BB‐induced expression of glyceraldehyde‐3‐phosphate dehydrogenase by sodium butyrate in rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997; 17: 3420–7. [DOI] [PubMed] [Google Scholar]
- 34. Ranganna K, Yatsu FM, Hayes BE, et al Butyrate inhibits proliferation‐induced proliferating cell nuclear antigen expression (PCNA) in rat vascular smooth muscle cells. Mol Cell Biochem. 2000; 205: 149–61. [DOI] [PubMed] [Google Scholar]
- 35. Patel P, Nankova BB, LaGamma EF. Butyrate, a gut‐derived environmental signal, regulates tyrosine hydroxylase gene expression via a novel promoter element. Brain Res Dev Brain Res. 2005; 160: 53–62. [DOI] [PubMed] [Google Scholar]
- 36. Ranganna K, Mathew OP, Yatsu FM, et al Involvement of glutathione/glutathione S‐transferase antioxidant system in butyrate‐inhibited vascular smooth muscle cell proliferation. FEBS J. 2007; 274: 5962–78. [DOI] [PubMed] [Google Scholar]
- 37. Safaya S, Klings ES, Odhiambo A, et al Effect of sodium butyrate on lung vascular TNFSF15 (TL1A) expression: differential expression patterns in pulmonary artery and microvascular endothelial cells. Cytokine. 2009; 46: 72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zapolska‐Downar D, Siennicka A, Kaczmarczyk M, et al Butyrate inhibits cytokine‐induced VCAM‐1 and ICAM‐1 expression in cultured endothelial cells: the role of NF‐kappaB and PPARalpha. J Nutr Biochem. 2004; 15: 220–8. [DOI] [PubMed] [Google Scholar]
- 39. Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002; 45: 173–202. [DOI] [PubMed] [Google Scholar]
- 40. Milton SG, Mathew OP, Yatsu FM, et al Differential cellular and molecular effects of butyrate and trichostatin a on vascular smooth muscle cells. Pharmaceuticals (Basel). 2012; 5: 925–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Song S, Kang SW, Choi C. Trichostatin A enhances proliferation and migration of vascular smooth muscle cells by downregulating thioredoxin 1. Cardiovasc Res. 2010; 85: 241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Q, Lu Z, Ramchandran R, et al Pulmonary artery smooth muscle cell proliferation and migration in fetal lambs acclimatized to high‐altitude long‐term hypoxia: role of histone acetylation. Am J Physiol Lung Cell Mol Physiol. 2012; 303: L1001–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dashwood RH, Ho E. Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol. 2007; 17: 363–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Licciardi PV, Ververis K, Karagiannis TC. Histone deacetylase inhibition and dietary short‐chain Fatty acids. ISRN Allergy. 2011; 2011: 869647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Myzak MC, Ho E, Dashwood RH. Dietary agents as histone deacetylase inhibitors. Mol Carcinog. 2006; 45: 443–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pham TX, Lee J. Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients. 2012; 4: 1868–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steliou K, Boosalis MS, Perrine SP, et al Butyrate histone deacetylase inhibitors. Biores Open Access. 2012; 1: 192–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vinolo MA, Rodrigues HG, Nachbar RT, et al Regulation of inflammation by short chain fatty acids. Nutrients. 2011; 3: 858–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kelly WK, Richon VM, O'Connor O, et al Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003; 9: 3578–88. [PubMed] [Google Scholar]
- 50. Wang Z, Chen C, Finger SN, et al Suberoylanilide hydroxamic acid: a potential epigenetic therapeutic agent for lung fibrosis? Eur Respir J. 2009; 34: 145–55. [DOI] [PubMed] [Google Scholar]
- 51. Felice C, Lewis A, Armuzzi A, et al Review article: selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment Pharmacol Ther. 2015; 41: 26–38. [DOI] [PubMed] [Google Scholar]
- 52. Khan S, Jena G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF‐beta1‐induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem Toxicol. 2014; 73: 127–39. [DOI] [PubMed] [Google Scholar]
- 53. Wang W, Yan M, Ji Q, et al Suberoylanilide hydroxamic acid suppresses hepatic stellate cells activation by HMGB1 dependent reduction of NF‐kappaB1. PeerJ. 2015; 3: e1362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sluiter I, van Heijst A, Haasdijk R, et al Reversal of pulmonary vascular remodeling in pulmonary hypertensive rats. Exp Mol Pathol. 2012; 93: 66–73. [DOI] [PubMed] [Google Scholar]
- 55. Church AC, Martin DH, Wadsworth R, et al The reversal of pulmonary vascular remodeling through inhibition of p38 MAPK‐alpha: a potential novel anti‐inflammatory strategy in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2015; 309: L333–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia‐inducible factor‐1alpha promote growth factor‐induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006; 290: H2528–34. [DOI] [PubMed] [Google Scholar]
- 57. Tajsic T, Morrell NW. Smooth muscle cell hypertrophy, proliferation, migration and apoptosis in pulmonary hypertension. Compr Physiol. 2011; 1: 295–317. [DOI] [PubMed] [Google Scholar]
- 58. Cai Y, Han M, Luo L, et al Increased expression of PDGF and c‐myc genes in lungs and pulmonary arteries of pulmonary hypertensive rats induced by hypoxia. Chin Med Sci J. 1996; 11: 152–6. [PubMed] [Google Scholar]
- 59. Chanakira A, Dutta R, Charboneau R, et al Hypoxia differentially regulates arterial and venous smooth muscle cell proliferation via PDGFR‐beta and VEGFR‐2 expression. Am J Physiol Heart Circ Physiol. 2012; 302: H1173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dahal BK, Heuchel R, Pullamsetti SS, et al Hypoxic pulmonary hypertension in mice with constitutively active platelet‐derived growth factor receptor‐beta. Pulm Circ. 2011; 1: 259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. ten Freyhaus H, Dagnell M, Leuchs M, et al Hypoxia enhances platelet‐derived growth factor signaling in the pulmonary vasculature by down‐regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med. 2011; 183: 1092–102. [DOI] [PubMed] [Google Scholar]
- 62. Zhang L, Ma J, Shen T, et al Platelet‐derived growth factor (PDGF) induces pulmonary vascular remodeling through 15‐LO/15‐HETE pathway under hypoxic condition. Cell Signal. 2012; 24: 1931–9. [DOI] [PubMed] [Google Scholar]
- 63. Ambalavanan N, Bulger A, Philips IJ. Hypoxia‐induced release of peptide growth factors from neonatal porcine pulmonary artery smooth muscle cells. Biol Neonate. 1999; 76: 311–9. [DOI] [PubMed] [Google Scholar]
- 64. Li F, Zhang Y, Che D. Platelet‐derived growth factor (PDGF) mRNA expression of porcine pulmonary artery smooth muscle cells induced by hypoxic endothelial cells conditioned medium. Zhonghua Bing Li Xue Za Zhi. 1998; 27: 425–8. [PubMed] [Google Scholar]
- 65. Postolow F, Fediuk J, Nolette N, et al Hypoxia and nitric oxide exposure promote apoptotic signaling in contractile pulmonary arterial smooth muscle but not in pulmonary epithelium. Pediatr Pulmonol. 2011; 46: 1194–208. [DOI] [PubMed] [Google Scholar]
- 66. Nathan SD, Hassoun PM. Pulmonary hypertension due to lung disease and/or hypoxia. Clin Chest Med. 2013; 34: 695–705. [DOI] [PubMed] [Google Scholar]
- 67. McIlhenny S, Zhang P, Tulenko T, et al eNOS transfection of adipose‐derived stem cells yields bioactive nitric oxide production and improved results in vascular tissue engineering. J Tissue Eng Regen Med. 2015; 9: 1277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. D'Souza FM, Sparks RL, Chen H, et al Mechanism of eNOS gene transfer inhibition of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2003; 284: C191–9. [DOI] [PubMed] [Google Scholar]
- 69. Kullo IJ, Schwartz RS, Pompili VJ, et al Expression and function of recombinant endothelial NO synthase in coronary artery smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997; 17: 2405–12. [DOI] [PubMed] [Google Scholar]