

Abstract
Using a combination of Isothermal Titration Calorimetry and quantum and MD calculations, we demonstrate that relatively soft anions have an affinity for hydrophobic concavity. The results are consistent with the anions remaining partially hydrated upon binding, and suggest a novel strategy for anion recognition.
Graphical abstract
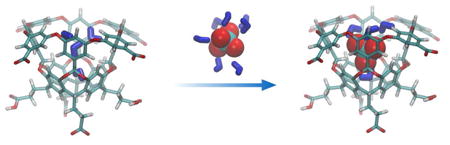
The challenges associated with anion recognition are well known.1 They have a range of geometries, may be pH sensitive, and are larger than the equivalent isoelectronic cations and have a lower charge to radius ratio. This diffuse nature means that across all the classes of functional groups utilized to bring about recognition,1-2 those involving Coulombic attraction and hydrogen bonding have proven to be the most popular. This is particularly true for anion recognition in aqueous solution, where these strategies have been utilized to counter the strong interactions between the anion and its hydration shell.3 Thus most of the reported hosts for anions that function in pure/buffered water are cationic.4 Moving away from this strategy is the idea of utilizing halogen bonds for recognition;5 an approach that takes advantage of the orthogonality between the requirements for forming halogen bonds, and those for forming hydrogen bonds; if the latter is not utilize, competition with water is less important.
An alternative strategy is not to compete with the waters of hydration but to bind the anion with its solvation shell. Although a wide range of ditopic receptors have been synthesized and studied,6 in general supramolecular chemistry has focused on the recognition of singular species. But why not recognize a hydrated anion rather than a “naked” one? Although this posses many challenges, it sidesteps the energetic requirements of ion desolvation, and has the potential to reveal subtleties about ion hydration of import to studies of the Hofmeister effect and how anions interact with biomacromolecules.7
One of the key requirements for the recognition of hydrated anions is undoubtedly a large, well-defined, binding pocket; the circumambient nature of which allows for multi-point recognition.8,9,10,11 But what are the specifics of such pockets? How many waters of hydration are easily removed from an anion, and is there a preferred hydration geometry for each anion type? Although much has been learned about isolated water clusters12 and the solvation requirements of ions,13 what we know of the structural requirements for recognizing hydrated ions is – to our knowledge – limited to the solid state.14
We recently showed that perchlorate (ClO4−) has an affinity for the hydrophobic pocket of cavitand 1, and that this association is dependent on the nature of other salts.15 Furthermore, ClO4− binding is able to induce Hofmeister effects in the binding of amphiphilic guests to 1.16 Here we demonstrate that anion binding to the concavity of 1 is general, use ITC to identify that these complexation events are strongly exothermic and entropically penalized, and show with a combination of quantum calculations and molecular dynamics simulations that partial solvation of the guest is key to binding.
We examined the affinity of twenty-six monovalent sodium salts for host 1. All associations were examined at pH = 11.5 to ensure sufficient solubility of the host and avoid protonation-state changes in the guest.17 Initial screening utilized 1H NMR to qualify, and where possible quantify, each association. This determined eighteen anions that bound. Those that did not bind were: cyanate (CNO−), formate (HCO2−) acetate (MeCO2−), ethane sulfonate (EtSO3−), borohydride (BH4−), chloride (Cl−), fluoride (F−), and trifluoroacetate (CF3CO2−).18 Of the binding anions, four bound too weakly to give reliable data: bromide (Br−), azide (N3−), bromate (BrO3−), and tetrafluoroborate (BF4−). Finally, of the remaining fourteen guests, two proved problematic: MnO4− and AuCl4−. The former gave a reliable Ka value of 1055 M−1 by NMR, but presumably because ITC also detected background reaction of this guest we could not obtain consistent calorimetric data. Similarly, AuCl4− was qualified to bind strongly to the host, but no reliable quantification of this association was possible with either NMR or ITC; even after solutions of the guest were allowed to stand for 48 hours to ensure that any decomposition of the guest to form the corresponding tetrahydroxo gold complex (Au(OH)4−) was complete.
Table 1 shows the ITC data for the remaining anions. Because the Wiseman parameter for each salt was low (0.01 ≤ c ≤ 1), each titration required modification as outlined by Turnbull19 and Tellinghuisen.20 Relatedly, the protocols from Turnbull19 were required because some of the titrations were limited to a maximal 80% complexation. Furthermore, the relatively high salt (titrant) concentrations necessitated reference titrations to account for salt dilution. These provisions gave reliable data, and using the data from non-associating NaCl we confirmed that the effects of Na+ binding21 to R–CO2− of the host were negligible.
Table 1.
Thermodynamic data from NMR and ITC for the binding of anions of sodium salts to host 1.a
| Anion | Ka (M−1) (NMR)b | Ka (M−1) (ITC)c | ΔG° cal/mol | ΔH° cal/mol | −TΔS° cal/mol |
|---|---|---|---|---|---|
| Cl3CCO2− | 5383 | 6337 | −5188 | −6499 | 1311 |
| PF6− | 575 | 790 | −3950 | −11610 | 7658 |
| MeSO2S− | 391 | 660 | −3848 | −11983 | 8136 |
| ReO4− | 322 | 371 | −3509 | −7393 | 3884 |
| TfO− | 67 | 314 | −3406 | −4529 | 1124 |
| IO4− | 216 | 235 | −3201 | −8029 | 4828 |
| ClO4− | 95 | 160 | −3010 | −9049 | 6039 |
| BH3CN− | 67 | 152 | −2975 | −7842 | 4867 |
| Cl2CHCO2− | 50 | 52 | −2345 | −4129 | 1787 |
| SCN− | 33 | 44 | −2240 | −8508 | 6268 |
| N(CN)2− | 10 | 37 | −2152 | −4219 | 2067 |
| I− | 11 | 17 | −1680 | −5276 | 3596 |
The average of 2-3 individual experiments.
25 °C, 10 mM sodium phosphate buffer, pH = 11.3.
25 ° C, 50 mM sodium phosphate buffer, pH = 11.5.
Overall the Ka values from ITC were higher than those determined by NMR. These differences were attributed to the higher ionic strength of the solutions used in ITC (50 mM verses 10 mM).15 In general, the binding anions are relatively hydrophobic. In other words they are soft, weakly hydrated, and “weakly coordinating”;22 a term that has a distinct Coulombic force, organic solvent-based etymology contradicting the associations observed here. Although the charge on the octa-anionic host is attenuated by cation condensation to the exterior coat,23 it is quite surprisingly that all complexations were enthalpically favored and entropically penalized.
The strongest binding guest was found to be trichloroacetate (Cl3CCO2−), which also had a relatively small entropy penalty of complexation; interestingly dichloroacetate (Cl2CHCO2−) bound >2 orders of magnitude more weakly, with a much smaller enthalpy and a slightly larger entropic penalty. Furthermore, as noted above, acetate demonstrated no affinity for the host.16a The strong association of Cl3CCO2− may in part be attributed to the formation of C– H…X hydrogen bonds between the inward pointing benzal protons of the host and the halogen of the guest;24 a conclusion supported by NMR signal shifts upon complexation (SI). However, the much lower affinity of Cl2CHCO2− is unusual. In this case negligible shifts in the benzal proton NMR signals suggest no C–H…X hydrogen bonds, but other shifts do suggest a gross “carboxylate up” binding motif similar to Cl3CCO2− (SI). ITC also revealed that, although it would be expected to have more space to move within the rigid pocket, Cl2CHCO2− actually had a larger entropic penalty for binding than Cl3CCO2−. Hexafluorophosphate (PF6−) was also found to bind relatively strongly. Interestingly, this guest bound with the second highest enthalpy, but also the second highest entropic penalty. Thus, although 30% less voluminous than Cl3CCO2−, and as an octahedral ion (Oh, symmetry number σ = 24) would be expected to more freely tumble within the pocket of 1, its entropic penalty of complexation was six times larger. Similarly, methanethiosulfate (MeSO2S−) was found to have the strongest enthalpy of complexation but also the highest entropic penalty. Its binding was in sharp contrast to EtSO3− which showed no affinity. Presumably, the larger and more polarizable sulfur atom of the thiosulfate is an important electronic factor behind the binding of this anion.
Several tetrahedral anions less reactive than MnO4− gave reproducible binding data, specifically: ReO4−, IO4−, and ClO4−. As these posses the same geometry they gave the opportunity to investigate the effects of percentage occupancy (space filling) of the pocket on affinity. However, there was no correlation between their volume (IO4− = 65 Å3, ReO4− = 58 Å3, ClO4− = 54 Å3) and Ka, ΔH° or −TΔS°.
The binding of TfO− was considerably weaker than MeSO2S−, suggesting that the ‘thiolate’ sulfur atom of the latter plays a stronger role in binding than the inductive properties of the F-atoms of TfO−. However, the fact that EtSO3− does not bind indicates that the F-atoms of TfO− do aid complexation. Triflate was noted to bind to host 1 with the lowest entropic penalty.
Three cyano containing anions were found to weakly associate: cyanoborohydride (BH3CN−), thiocyanate (SCN−), and dicyanamide (N(CN)2−. BH3CN− and SCN− bound with relatively strong enthalpies. The fact that SCN− bound whereas CNO− did not, confirms that charge diffusion is important. One possibility for the strong enthalpy of complexation seen with these anions is an anti-parallel dipole alignment of the cyano dipole to that of the host (∼5 D pointing out of the cavity portal, SI); a hypothesis that would also support the observation that acetonitrile is the best common organic solvent for denaturing capsular complexes formed by 1.25 Again however, the binding of these three anions came with an entropic penalty.
Finally, the weakest anion that reliably bound to host 1 was I−. In this case binding was enthalpically dominated and entropically costly; the latter far more so than the larger organic anions such as Cl3CO2−.
These associations are relatively weak, but that binding does occur is surprising considering that the host is ostensibly anionic (but see23), and is comprised of electron rich not electron deficient rings.1-2, 26 Moreover, anion complexation is likely in competition with traces of dihydrogen phosphate in the buffer. But why are these complexations entropically penalized?27 And why is there a poor, but defined trend that the smaller the guest the greater the entropic penalty to binding? It has been shown that the addition or removal of an anion from aqueous solution results in a near perfect cancellation of the ion-water and water reorganization contributions to measured hydration entropies.”28 Consequently, it is the solvation state of the bound anion that must be key to the data in Table 1. To test this idea we performed quantum and molecular dynamics (MD) simulations of complexes between 1 and I−, ClO4−, and Cl− (SI). The simulations revealed that the larger ions retain about half their solvation shells upon binding (Figure 1). For example, ClO4− has 6.4 solvation shell water molecules on average in the bulk and 3.1 when in the host, whilst much smaller I− has 7.3 in the bulk but only 3.0 in the host. When restrained within the cavity Cl− kept most of its solvation shell; it has on average 6.6 in the bulk and 5.3 waters inside the host. These results are consistent with the overall free energy of hydration of these anions.29
Figure 1.
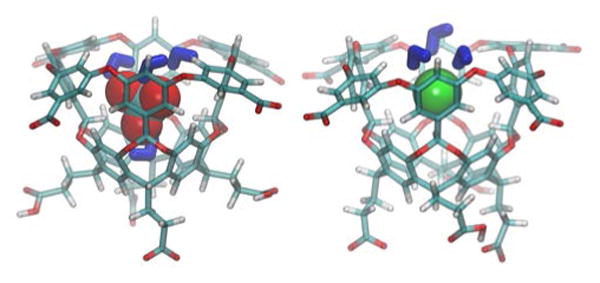
Representative calculated structures of the complexes between 1 and perchlorate (left) and iodide (right).
We subsequently carried out free energy calculations to qualify anion affinity. Specifically, we used thermodynamic integration calculations to find the free energy of binding to both the host and the bulk (SI). These calculations gave negative free energies of complexation for ClO4− and I− (ΔG° = −4.1 verses −8.2 kcal mol−1) and a positive value for Cl−. Considering these calculations involve differences that are a challenge for free energy calculations, and that they were determined under the standard state, it is perhaps not surprising that the empirical and calculated affinities of ClO4− and I− are reversed. Our interpretation of these quantum calculations is simply that: 1) they conform to which anions can bind and which cannot, and; 2) that binding ions are stabilized by stronger dispersion interactions with the host than they are with water, whereas non-binding ions are stabilized by stronger electrostatic interactions with their hydration shell than with the host (SI).
MD simulations have previously demonstrated that filling the cavity of 1 with water is an exergonic process (ΔGhyd ≈ −5 kcal mol−1) dominated by enthalpy (ΔHhyd ≈ −20 kcal mol−1, and with a sizable entropic penalty (−TΔShyd ≈ 15 kcal mol−1).30 These simulations also revealed that host 1 binds on average 4-5 waters within its hydrophobic cavity, but can bind up to 7.30 Considering these points and the data presented here, our working hypothesis is that large anions such as Cl3CCO2− bind with a minimum of (or no) co-complexing waters, but that smaller, “harder” anions are partially solvated by highly organized, entropically costly waters. However, there is no simple relationship between the size of the anion and the entropic cost of binding because the number and arrangement of co-bound waters is intimately tied to the ion-specific thermodynamics of desolvation, and how the corresponding, stable, partially solvated anions complement the shape of the binding pocket.
These results have manifold implications. The fact that anion binding in water can be affected without complete desolvation – even in the absence of traditionally strong supramolecular motifs for anion recognition – suggests an alternative approach to this difficult task. Moreover, these results dovetail with what is known about anions at the air-water interface31 and the macromolecule-water interface,32 and regarding the latter, point to other ways than cation-anion or hydrogen bonding that anions can interact with proteins.
Acknowledgments
Funding Sources: No competing financial interests have been declared. National Institutes of Health (GM 098141)
PS and BCG gratefully acknowledge the support of the National Institutes of Health (GM 098141). BCG acknowledges Dor Ben-Amotz for helpful discussions. SWR acknowledges the National Institutes of Health (GM 098141).
Footnotes
Supporting Information. NMR and ITC titration experiments and details of the quantum and MD simulations.
References
- 1.Sessler JL, Gale PA, Cho WS. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]
- 2.(a) Gale PA, Busschaert N, Haynes CJ, Karagiannidis LE, Kirby IL. Chem Soc Rev. 2013;43:205–241. doi: 10.1039/c3cs60316d. [DOI] [PubMed] [Google Scholar]; (b) Gale PA, Steed JW. Supramolecular Chemistry: From Molecules to Nanomaterials. John Wiley and Sons; 2012. [Google Scholar]; (c) Caballero A, Zapata F, Beer PD. Coordination Chemistry Reviews. 2013;257(17-18):2434–2455. [Google Scholar]; (d) Hirsch AKH, Fischer FR, Diederich F. Angew Chem Int Ed. 2007;46:338–352. doi: 10.1002/anie.200603420. [DOI] [PubMed] [Google Scholar]; (e) Berryman O, Johnson DW. Chem Commun. 2009:3143–3153. doi: 10.1039/b823236a. [DOI] [PubMed] [Google Scholar]
- 3.Kubik S. Chem Soc Rev. 2010;39(10):3648–63. doi: 10.1039/b926166b. [DOI] [PubMed] [Google Scholar]
- 4.(a) García-España E, Díaz P, Llinares JM, Bianchi A. Coordination Chemistry Reviews. 2006;250(23-24):2952–2986. [Google Scholar]; (b) Schmidtchen FP. Coordination Chemistry Reviews. 2006;250(23-24):2918–2928. [Google Scholar]; (c) O'Neil EJ, Smith BD. Coordination Chemistry Reviews. 2006;250(23-24):3068–3080. [Google Scholar]; (d) Schiessl P, Schmidtchen FP. The Journal of Organic Chemistry. 1994;59(3):509–511. [Google Scholar]; (e) Schmuck C, Schwegmann M. J Am Chem Soc. 2005;127(10):3373–9. doi: 10.1021/ja0433469. [DOI] [PubMed] [Google Scholar]; (f) Metzger A, Lynch VM, Anslyn EV. Angewandte Chemie International Edition in English. 1997;36(8):862–865. [Google Scholar]; (g) Schmuck C, Wich P. The development of artificial receptors for small pepticles using combinatorial approaches. In: Schrader T, editor. Creative Chemical Sensor Systems. Vol. 277. Springer-Verlag Berlin; Berlin: 2007. pp. 3–30. [Google Scholar]
- 5.(a) Lim JY, Beer PD. Chem Commun (Camb) 2015;51(17):3686–8. doi: 10.1039/c4cc10130h. [DOI] [PubMed] [Google Scholar]; (b) Langton MJ, Robinson SW, Marques I, Felix V, Beer PD. Nature Chemistry. 2014;6(12):1039–1043. doi: 10.1038/nchem.2111. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kim SK, Sessler JL. Acc Chem Res. 2014;47(8):2525–2536. doi: 10.1021/ar500157a. [DOI] [PubMed] [Google Scholar]; (b) Durola F, Heitz V, Reviriego F, Roche C, Sauvage JP, Sour A, Trolez Y. Acc Chem Res. 2014;47(2):633–645. doi: 10.1021/ar4002153. [DOI] [PubMed] [Google Scholar]; (c) Matthews SE, Beer PD. Supramol Chem. 2005;17(6):411–435. [Google Scholar]; (d) Smith BD. Ion-pair recognition by ditopic macrocyclic receptors. Springer; 2005. pp. 137–151. [Google Scholar]; (e) Park JS, Karnas E, Ohkubo K, Chen P, Kadish KM, Fukuzumi S, Bielawski CW, Hudnall TW, Lynch VM, Sessler JL. Science. 2010;329:1324–1327. doi: 10.1126/science.1192044. [DOI] [PubMed] [Google Scholar]; (f) Jordan JH, Gibb BC. Chem Soc Rev. 2015;44(2):547–85. doi: 10.1039/c4cs00191e. [DOI] [PubMed] [Google Scholar]; (g) Palmer LC, Rebek J., Jr Org Biomol Chem. 2004;2(21):3051–9. doi: 10.1039/B412510J. [DOI] [PubMed] [Google Scholar]; (h) Gil-Ramírez G, Chas M, Ballester P. J Am Chem Soc. 2010;132:2520–2521. doi: 10.1021/ja910436v. [DOI] [PubMed] [Google Scholar]
- 7.(a) Jungwirth P, Cremer PS. Nature Chemistry. 2014;6(4):261–3. doi: 10.1038/nchem.1899. [DOI] [PubMed] [Google Scholar]; (b) Zhang YJ, Cremer PS. Annu Rev Phys Chem. 2010;61:63–83. doi: 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]; (c) Tobias DJ, Hemminger JC. Science. 2008;319(5867):1197–8. doi: 10.1126/science.1152799. [DOI] [PubMed] [Google Scholar]; (d) Lo Nostro P, Ninham BW. Chem Rev. 2012;112(4):2286–322. doi: 10.1021/cr200271j. [DOI] [PubMed] [Google Scholar]; (e) Lund M, Vrbka L, Jungwirth P. J Am Chem Soc. 2008;130:11582–11583. doi: 10.1021/ja803274p. [DOI] [PubMed] [Google Scholar]
- 8.For water-soluble bambusurils as strong anion binders see: Yawer MA, Havel V, Sindelar V. Angew Chem Int Ed. 2015;54:276–279. doi: 10.1002/anie.201409895.
- 9.For the binding of large anions to cyclodextrins to cyclodextrins see: Assaf KI, Ural MS, Pan F, Georgiev T, Simova S, Rissanen K, Gabel D, Nau WM. Angew Chem Int Ed Engl. 2015;54(23):6852–6. doi: 10.1002/anie.201412485.
- 10.For anion binding to the active site of Human Carbonic Anhydrase II see: Fox JM, Kang K, Sherman W, Heroux A, Sastry GM, Baghbanzadeh M, Lockett MR, Whitesides GM. J Am Chem Soc. 2015;137(11):3859–66. doi: 10.1021/jacs.5b00187.
- 11.For early work on the weak binding of select anions to cyclodextrins see: Thoma JA, French D. J Am Chem Soc. 1960;82:4144–4147.Gelb RI, Schwartz LM, Radeos M, Laufer DA. J Phys Chem. 1983;87:3349–3354.Matsui Y, Ono M, Tokunaga S. Bull Chem Soc Jpn. 1997;70(3):535–541.Terekhova IV, Chibunova ES, Kumeev RS, Alper GA. Carbohydrate polymers. 2014;110:472–9. doi: 10.1016/j.carbpol.2014.04.057.Godínez LA, Schulze-fiehn BG, Patel S, Criss CM, Evanseck JD, Kaifer AE. Supramolecular Chemistry. 1996;8(1):17–22.
- 12.(a) Liu K, Brown MG, Carter C, Saykally RJ, Gregory JK, Clary DC. Nature. 1996;381:501–503. [Google Scholar]; (b) Gregory JK, Clary DC, Liu K, Brown MG, Saykally RJ. Science. 1997;275:814–817. doi: 10.1126/science.275.5301.814. [DOI] [PubMed] [Google Scholar]; (c) Perez C, Muckle MT, Zaleski DP, Seifert NA, Temelso B, Shields GC, Kisiel Z, Pate BH. Science. 2012;336(6083):897–901. doi: 10.1126/science.1220574. [DOI] [PubMed] [Google Scholar]
- 13.(a) Dalleska NF, Honma K, Sunderlin LS, Armentrout PB. J Am Chem Soc. 1994;116:3519–3528. [Google Scholar]; (b) Robertson WH, Johnson MA. Annu Rev Phys Chem. 2003;54:173–213. doi: 10.1146/annurev.physchem.54.011002.103801. [DOI] [PubMed] [Google Scholar]; (c) O'Brien JT, Prell JS, Bush MF, Williams ER. J Am Chem Soc. 2010;132(24):8248–8249. doi: 10.1021/ja1024113. [DOI] [PubMed] [Google Scholar]; (d) Garand E, Wende T, Goebbert DJ, Bergmann R, Meijer G, Neumark DM, Asmis KR. J Am Chem Soc. 2010;132(2):849–56. doi: 10.1021/ja9093132. [DOI] [PubMed] [Google Scholar]; (e) Cheng TC, Bandyopadhyay B, Mosley JD, Duncan MA. J Am Chem Soc. 2012;134(31):13046–55. doi: 10.1021/ja3038245. [DOI] [PubMed] [Google Scholar]; (f) Duncan MA. Rev Sci Instrum. 2012;83(4):041101. doi: 10.1063/1.3697599. [DOI] [PubMed] [Google Scholar]
- 14.(a) Barbour LJ, Orr GW, Atwood JL. Nature. 1998;393:671–673. [Google Scholar]; (b) Yoshizawa M, Kusukawa T, Kawano M, Ohhara T, Tanaka I, Kurihara K, Niimura N, Fujita M. J Am Chem Soc. 2005;127:2798–2799. doi: 10.1021/ja043953w. [DOI] [PubMed] [Google Scholar]
- 15.Carnagie R, Gibb CLD, Gibb BC. Angew Chem Int Ed. 2014;53(43):11498–11500. doi: 10.1002/anie.201405796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Gibb CLD, Gibb BC. J Am Chem Soc. 2011;133(19):7344–7347. doi: 10.1021/ja202308n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gibb CLD, Oertling EE, Velaga S, Gibb BC. Journal of Physical Chemistry B. 2015;119(17):5624–5638. doi: 10.1021/acs.jpcb.5b01708. [DOI] [PubMed] [Google Scholar]
- 17.In free solution the pKa values of the conjugate acids of the anions examined range form −20 to +4.67. However, we have observed that the pKa of carboxylic acids inside 1 can be shifted as much as 3-4 pKa units (Wang K, Sokkalingam P, Gibb BC. Supramolecular Chemistry. doi: 10.1080/10610278.2015.1082563. http://dx.doi.org/10.1080/10610278.2015.1082563.) If this is the case, then at pH 7 species such as acetate, formate, azide, dichloroacetate, and ethyl sulfate would bind primarily in their protonated form.
- 18.Of the six anions that did not bind to the host and have reliable thermodynamic data for hydration, all have ΔHhyd values greater than −373 kcal mol−1. In contrast, for the six binding anions with reliable hydration data, all have ΔHhyd values less than −338 kcal mol−1. There is however no correlation between the hydration and binding thermodynamics discussed here.
- 19.Turnbull WB, Daranas AH. J Am Chem Soc. 2003;125:14859–14866. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 20.Tellinghuisen J. Anal Biochem. 2008;373:395–397. doi: 10.1016/j.ab.2007.08.039. [DOI] [PubMed] [Google Scholar]
- 21.Kherb J, Flores SC, Cremer PS. J Phys Chem B. 2012;116(25):7389–97. doi: 10.1021/jp212243c. [DOI] [PubMed] [Google Scholar]
- 22.Krossing I, Raabe I. Angew Chem Int Ed Engl. 2004;43(16):2066–90. doi: 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- 23.It is interesting to note that because cation condensation to host 1 (see ref. 15) attenuates its net charge, reducing the complexation elextrostatic barrier for the approaching anion and stabilizing the resulting complex. Therefore, the idea of a charged host binding a like-charged ion should not be considered anathema when working in high dielectric solvents such as water.
- 24.(a) Gibb CLD, Stevens ED, Gibb BC. J Am Chem Soc. 2001;123:5849–5850. doi: 10.1021/ja005931p. [DOI] [PubMed] [Google Scholar]; (b) Laughrey ZR, Upton T, Gibb BC. Chem Commun. 2006:970–972. doi: 10.1039/b515187b. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Gibb BC. Chemical Communications. 2011;47(12):3574–6. doi: 10.1039/c1cc10122f. [DOI] [PubMed] [Google Scholar]
- 26.Meyer EA, Castellano RK, Diederich F. Angew Chem Int Ed. 2003;42(11):1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 27.Baron has investigated the consequences of charge states of host and guest pairings but not of those between negative hosts and negative guests: Baron R, Setny P, McCammon JA. J Am Chem Soc. 2010;132(34):12091–7. doi: 10.1021/ja1050082.
- 28.Ben-Amotz D, Underwood R. Acc Chem Res. 2008;41(8):957–67. doi: 10.1021/ar7001478. [DOI] [PubMed] [Google Scholar]
- 29.Marcus Y. Ion Properties. 1. Marcel Dekker; New York: 1997. [Google Scholar]
- 30.Ewell J, Gibb BC, Rick SW. J Phys Chem B. 2008;112:10272–10279. doi: 10.1021/jp804429n. [DOI] [PubMed] [Google Scholar]
- 31.(a) Jungwirth P, Tobias DJ. Journal of Physical Chemistry B. 2002;106:6361–6373. [Google Scholar]; (b) Petersen PB, Saykally RJ, Mucha M, Jungwirth P. Journal of Physical Chemistry B. 2005;109:10915–10921. doi: 10.1021/jp050864c. [DOI] [PubMed] [Google Scholar]; (c) Jungwirth P, Tobias JW. Chem Rev. 2006;106:1259–1281. doi: 10.1021/cr0403741. [DOI] [PubMed] [Google Scholar]
- 32.(a) Rembert KB, Paterova J, Heyda J, Hilty C, Jungwirth P, Cremer PS. J Am Chem Soc. 2012;134(24):10039–46. doi: 10.1021/ja301297g. [DOI] [PubMed] [Google Scholar]; (b) Paterová J, Rembert KB, Heyda J, Kurra Y, Okur HI, Liu WR, Hilty C, Cremer PS, Jungwirth P. J Phys Chem B. 2013;117(27):8150–8. doi: 10.1021/jp405683s. [DOI] [PubMed] [Google Scholar]