

Abstract
This review focuses in papers published since 2000 on the topic of the properties of solutes in water. More specifically, it evaluates the state-of-the-art of our understanding of the complex relationship between the shape of a hydrophobe and the Hydrophobic effect. To highlight this a selection of references covering both empirical and molecular dynamics studies of small (molecular-scale) solutes are presented. These include empirical studies of small molecules, synthetic hosts, crystalline monolayers, and proteins, as well as in silico investigations of entities including idealized hard and soft spheres, small solutes, hydrophobic plates, artificial concavity, molecular hosts, carbon nanotubes and spheres, and proteins.
Keywords: Water, non-covalent interactions, supramolecular chemistry, Hydrophobic Effect, hydration
Exordium
Water is found in many parts of the universe, but planet Earth truly is a water-world. All four spheres consist to some extent of water: the hydrosphere, biosphere, atmosphere and lithosphere all contain (respectively decreasing) proportions of water. This review pertains to the hydrosphere and biosphere. More specifically, this review concerns the complex relationship between hydrophobic solutes and the solvent of life.(1) For understanding how solute shape and functionality control solvation, and how this solvation folds into measurable spectroscopic and thermodynamic changes, is key to understanding our world.
The ubiquity of water is reflected in a massive literature and consequently boundaries must be set for any review. First, although there is a continuum between a hydrophobe and a hard ion, and cases where the Hydrophobic effect (HE) and the Hofmeister effect(2-4) meet,(5; 6) where possible the latter is avoided. Second, this review focuses on the molecular-scale HE(7) and avoids the micro-scale.(8) Third, temperature and pressure can have a considerable effect on the HE,(9) but with only a few exceptions this review concerns ambient conditions. Fourth, bio-membranes are not discussed.(10) Finally, the bulk of this review concerns work performed since 2000.
The layout of this review follows the broad types of curvature as defined by mathematics: positive curvature (convex), zero curvature (flat surface) and negative curvature (concave). Historically it is positive curvature that has received the most attention, and as a result our picture of its solvation is relatively strong. Overall, there is less known about the solvation of flat surfaces or negative curvature, but the last decade or so has seen this gap close considerably.
A Word about Water
Water is small, highly polar (1.85 D, ε = 78), and a strong hydrogen bond (HB) donor (α = 1.17), and reasonable HB acceptor (β = 0.47).(11) Consequently, bulk liquid water is relatively structured. There is general consensus that bulk water molecule forms on average 3.5 HBs(12) with a lifetime on the order of 1-20 picoseconds. Although there is strong evidence that water forms (isolated) energy minimum clusters(13; 14) the extent of such clusters in the bulk is unclear. Indeed, probing water on the sub-femtosecond timescale indicates only two strong HBs between waters suggesting chains and rings are more dominant than cages.(15) This ambiguity means that the vague but descriptive term, “flickering clusters”(16) is often used to describe water.
A simple way to view water solvation is to consider the six different permutations of dangling HBs (those O–H bonds that point at a solute rather than HB to water) that can exist (Figure 1). Thus, there is a shift from no dangling HBs in the case of bulk water and small solutes, to four dangling HBs in the case of a totally isolated water. Undoubtedly, one of the keys to unraveling the HE is to be able to correlate these changes in the water HB pattern to corresponding spectroscopic and thermodynamic data.
Figure 1.
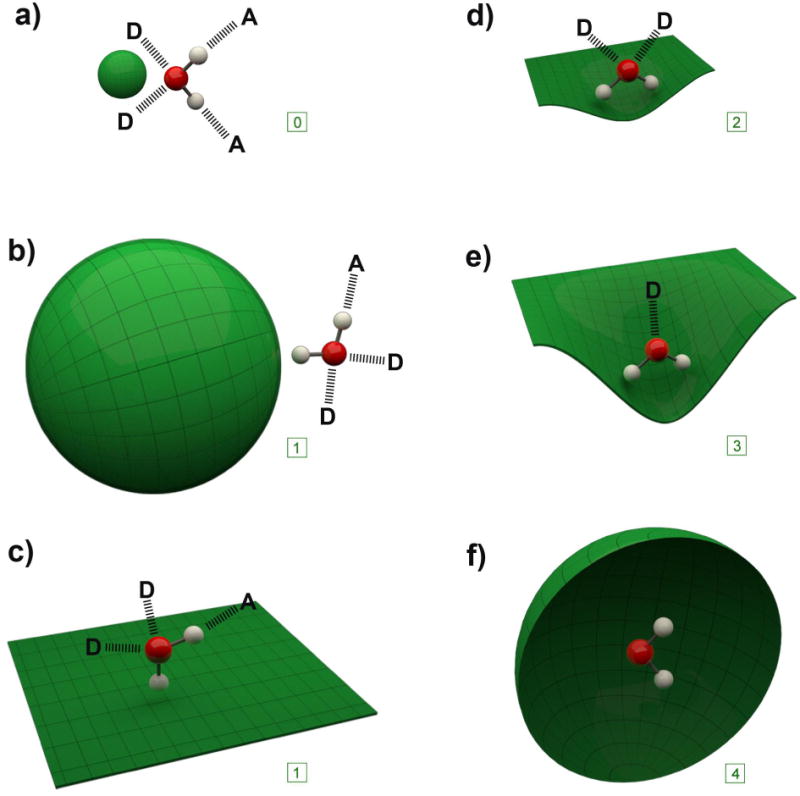
Idealized representations of the solvation of the surfaces discussed in this review. For each of the six cases one water molecule is shown along with the idealized number of HBs to donor (D) or acceptor (A) water molecules in the solvation shell. For each case, the corresponding number of dangling HBs is shown in the box: a) a small convex solute; b) a large convex molecule; c) a flat surface; d) a slightly concave surface; e) a highly concave surface; f) a fully encapsulating surface (front hemisphere removed for visualization).
Notes on The Hydrophobic Effect
As Ben-Amotz points out in a companion review,(17) our common notion that the HE is a powerful driver of binding and assembly in water is frequently misrepresented. Thus there is building evidence that interactions between nonpolar molecules in the gas phase are stronger than the corresponding interactions in water,(18) and like any solvent the solvation shell of water impedes other supramolecular interactions; it's just that water is not very good at this impeding. So it may be best to view the HE as pertaining to comparisons between water and other solvents. Additionally, there is evidence that the HE only becomes significant when ∼ 1 nm2 of water exposed surface area in buried in an assembly or complex.(18)
This noted, the hydration of small convex, hydrophobic solutes is generally enthalpically favorable.(19) Thus, the dissolution of methane in water (ΔG° = 25.5 kJ mol–1) is promoted by enthalpy (ΔH° =−13.8 kJ mol–1) but has a large entropic penalty (–TΔS° = 39.3 kJ mol–1). Such entropic penalties were observed by Frank and Evans,(20) and their interpretation of this phenomenon was that structured water, or “microscopic iceberg[s]” formed around hydrophobes. Some time later Kauzmann applied this concept to protein folding,(21) and in combination these two paper were largely responsible for the persisting textbook picture of the HE. This textbook description has proven to be too simplistic a viewpoint.(22; 23) Indeed, a review by Blokzijl and Engberts concluded that there is actually no good reason to suppose the iceberg model is true, and some evidence that it is not.(24) That stated, recent evidence highlighted here indicates that the jury is still out on the matter of icebergs around solutes.
Alternatives to this iceberg model have been proposed. For example, an entropic penalty might reflect the inability of the HB network of water to accommodate an apolar solute.(24) Alternatively, this entropy penalty may be due to the small molecular volume of water.(25-29) Consequently, the frequency of observing a cavity of sufficient volume to accommodate a large solute is low. In short, the most honest statement about the HE is that we far from fully understand it.
Positive Curvature
The key characteristic of positive curvature is that solvation is size dependent. As first suggested by Stillinger,(30) the surfaces of small solutes are wetted, i.e., the first solvation layer of water is mostly in direct contact with the solute, whereas large solutes are dewetted, i.e., their solvation shell is slightly remote from the surface. Although there is now good consensus that this is indeed the case, there is still disagreement about how this phenomenon is affected by the properties of the solute.
The only molecular-scale theory that successfully describes many of the structural and thermodynamic properties of infinitely dilute solutions of small, apolar solutes was devised by Pratt and Chandler (PC).(31; 32) This model gave accurate thermodynamic properties for aqueous solutions of apolar solutes, and successfully predicted the ΔG° of transfer of n-alkanes from a hydrocarbon solvent to water. However, this model could not account for observations from larger solutes systems. The first model that could successfully bridge the molecular and macro-scales and account for size dependent wetting/dewetting was described by Lum, Chandler, and Weeks (LCW).(33) To summarize, in the case of small solutes, HBing between waters is hindered yet persists near the surface. The result is a hydration layer denser than the bulk with roughly the same number but different patterns of HBs. In these cases, the ΔG° of solvation scales with solute volume. However, solutes > 10 Å diameter cannot be accommodated by even a distorted HB network, and consequently HBing is sterically depleted. The result is a reduction in the cohesive forces and a concomitant dewetting of the surface.(19; 34; 35) In such cases, ΔG° of solvation is dominated by interfacial free energetics and scales with surface area. Dewetting is reduced as Van der Waals forces between the solute and water are increased, however there is a negligible corresponding effect on the wetting of small solutes.
Because large solutes cannot be accommodated within the HB network of water, HBs can dangle over the solute (Figure 1). This accounts for the thermodynamics being scale dependent. As the breaking of HBs is dominated by enthalpy, but the spatial arranging of HBs is dominated by entropy, it is the latter that dominates in the solvation of a small solute. On the other hand, the entropy of solvating a large solute is favorable. Therefore there not only exists a wetting/ dewetting transition, but also an entropy crossover transition (or entropy-enthalpy crossover). These two types of solvation are fundamental to the assembly of hydrophobic solutes; the equilibrium is dominated by the difference between the entropically dominated solvation of the small solute and the enthalpically dominated solvation of the larger assembly.(19)
Small Solutes
A combination of neutron diffraction and H/D isotope substitution in the study of the formation and decomposition of methane clathrate suggest that the HE is not caused by the release of structured water;(36; 37) there is no significant differences in the water structure before, during, or upon clathrate formation, or after decomposition. Supporting this, ab initio molecular dynamic (MD) simulations of krypton in liquid water concurred with X-ray absorption fine-structure (XAFS) on krypton solution and were found to be significantly different from XAFS results for the radial hydration structure of Kr clathrate.(38)
Assembly of Small Solutes
The osmotic second virial coefficient (B2) offers a direct means to probe the HE, but there is a discrepancy between B2 measurements and the PC model. One explanation for this is that PC theory relies on hard-spheres rather than polarizable solutes. However, the former are intrinsically difficult to model in MD packages. As a step towards bridging this gap,(39) Pratt adapted simulation data to atomic-scale hard-core models of PC. The results show surprisingly strong attractive and endothermic, atomic-scale hydrophobic interactions between hard-sphere solutes, and constitute an initial step toward a molecular theory that account for attractive solute–water interactions.
Scheraga et al. has examined the potential of mean force for the assembly of dimers and trimers of methane.(40) By analyzing the packing and orientation of the solvation shell they determined that the ΔG° of association depended mostly on the difference in the number of waters in the first solvation shell of the cluster compared to the monomers. Additionally, they noted that during assembly unfavorable electrostatic interactions develop between waters in different regions of the solvation shell. These interactions make an anti-cooperative contribution to the potential of mean force. Thus, changes in the pattern of water interactions within the solvation shell make significant contributions to even straightforward assemblies.
Ashbaugh and Paulaitis have demonstrated that close-packed methane clusters and analogous hard-spheres have quite different hydration ΔG° values.(41) Clusters of 1 to 305 methanes showed no evidence of dewetting, but with hard-sphere analogues there was a decrease in water density as the radius increased from 3.25 to 16.45 Å. With small methane clusters a slight decrease in water density with increasing size was attributed to the greater surface roughness of the smaller assemblies.
The Crossover from Small to Large Solutes
Because of the low solubility of hydrocarbons in water, verification of a crossover is hard to confirm experimentally. However, relatively recent evidence of dewetting has been obtained by high-energy X-ray reflectivity measurements of the interface between water and octadecylsilane monolayers.(34; 35) Furthermore, evidence of thermodynamic crossover has been obtained from single molecule force spectroscopic studies of hydrophobic polymers.(42; 43)
Willard and Chandler have examined crossover in more detail using hard spheres.(44) Following the LCW model, the computed solvation ΔG° crossover occurred at ∼5-10 Å. Furthermore, when weak solute-solvent attractions were introduced, accurate predictions of the alkane-water interfacial tension were obtained.(45) In the absence of these attractions the water interface was found to be more than one solvent molecular diameter away from the surface, but in their presence wetting increased. For typical alkane-water interactions, there is still a dewetting (decrease in water density) adjacent to large hydrophobes, but it is in contact with the surface of the solute. However, within a temperature relevant to protein folding/unfolding these attractive interactions had little consequence to the temperature dependence of ΔG° of solvation. Parenthetically, MD simulations showed that increases in pressure and the addition of salt or ethanol resulted in a decrease in the crossover radius of the solute to sub-nanometer dimensions.(46)
The Garde group has examined the solvation of methane and spherical single and multisite C60 and C180 fullerenes. By examining water-solute interactions from purely repulsive to attractive, they determined that water structure around larger solutes is more sensitive to the strength of the solute-water attractions than smaller solutes are. Probing this further by separating the solute-water potential of mean force into solute-water and water-water interactions revealed that as the former was increased the latter became increasingly unfavorable. This “competitive expulsion” phenomenon captures the idea that the hydration shell opposes the introduction of a test water to replace one already present, and this was shown to be primarily enthalpic in origin. Mittal and Hummer have carried out a similar study.(47) An analysis of the radial distribution function of water around different solutes confirmed that the water density at contact gradually decreased with increasing solute size and that there was no evidence of surface wetting at a diameter of 20 Å or larger. These studies also examined the population and fluctuations in the water occupancy within the solute-water interface; for repulsive solutes, as the radius increased so the variance in the density fluctuation increased to four times the bulk. Moreover, probing the dynamics of the interfacial density fluctuations revealed a slow relaxation process attributed to transitions between locally wet and dry states, a process that slowed with increasing solute size.
There is an empirical correlation between the ΔG° of solvation of a molecule and the surface tension of its solution that allows the latter to be used to probe the thermodynamics of solvation. To bridge the difference between the molecular and macroscopic scales it has been necessary to introduce a curvature correction to surface tension measurements; a correction that must itself be temperature dependent to account for the fact that the entropies of hydrating small and large non-polar molecules are negative and positive respectively. Ashbaugh(48) has derived an expression predicting the solute size at which the solvation entropy is zero based on surface tension values, its first-order curvature correction, and their temperature dependencies. For hard-spheres at room temperature the estimated diameter was 16 Å.
Also examining crossover, the Berne group have determined the radial and orientational distribution functions of water around argon, methane, and neopentane.(49) Their results showed that only neopentane (diameter = 5.2 Å) displayed an orientational distribution of waters in the first hydration shell suggesting dewetting and the presence of dangling OH bonds. The authors also determined the potential of mean force between two neopentanes, ascertaining that there were two energy minima: the dimer, and the solvent separated dimer slightly higher in energy.
The Levy group have studied the solution thermodynamics of a series of linear, branched, and cyclic alkanes.(50) Their results support the idea that the classical HE has its roots in the small size of water rather than ‘icebergs’. They also found that their favorable hydration enthalpies arise from attractive solute-solvent dispersion interactions and by small water reorganization energies. Additionally, by separating solvation into cavitation and alkane solvation, they determined a complete thermodynamic description of the solvation of the corresponding cavity for each alkane. Their results suggest that the work of cavity formation is split roughly equally between unfavorable entropic and solvent reorganization energy effects.
The Ben-Amotz group has used Raman multivariate curve resolution (Raman-MCR) and simulations to quantify the extent of dangling HBs around hydrophobic groups.(51) For a series of alcohols they found a high-frequency OH band attributed to dangling OH bonds. Additionally, MD simulations of the vibrational spectra of waters in the hydration shell of neopentane and benzene revealed high-frequency OH features that closely resemble the experimentally observed dangling OH vibrational bands around (sufficiently soluble) neopentyl- and benzyl alcohol. In the latter, the red-shift was similar to that previously observed in benzene–water clusters. Expanding on this, the same group examined a series of different guests.(52) Their results were consistent with the idea that on average a hydrophobic moiety induces less than one dangling bond, and on average alcohols possessed more dangling HBs than alkyl ammoniums. Additionally, dangling HBs were stabilized (destabilized) by negatively (positively) charged groups. This study also revealed that both proximal and distal hydrocarbon group were important to the probability of forming a dangling HB, and that they were entropically stabilized.
Both the sign and the magnitude of ΔG° for the association of the alkyl groups of pairs of alcohol molecules remain unknown. To address this, Raman-MCR and polarization-resolved femtosecond IR experiments were combined with random mixing and molecular dynamics simulations.(53) The conclusion of these studies was that from methanol to tertiary butyl alcohol there is no association driven by the HE.
Raman-MCR has also been used to probe the hydration of linear alcohols,(54) carboxylic acids and tetraalkylammoniums.(55) These results showed that at low temperatures the hydration shells of each class has an enhanced water structure with greater tetrahedral order than the bulk, but for alcohols longer than 10 Å this structure disappeared as temperature increased. Additionally, at high pH onset of the hydration-shell structural transformation was suppressed by the carboxylate. Tetraalkylammonium cations were found to more strongly suppress this transformation.
Although short-chain alkanes are primarily in an extended conformation in water, there is still controversy about the extent of folding/hydrophobic collapse of longer chains. Simulations probing the properties of n-alkanes have found an approximately exponential decrease in solubility up to n-eicosane (C20H42), in excellent agreement with empirical data up to n-dodecane (C12H26).(56) Analysis of the ΔG° landscape of the alkanes revealed similarities between conformational preferences in the ideal gas and solution phases, suggesting that water does not heavily influence conformation; the authors found no evidence for hydrophobic collapse of n-alkane chains shorter than n-eicosane. However, water did increase the barriers between the minimal ΔG° conformations corresponding to compacted and extended chain conformations, and decreased the stability of the latter.
A recent paper examined the possibility of a hydrophobic to hydrophilic crossover for the hydration of sufficiently long n-alkane chains.(57) The authors combined previously obtained thermodynamic data, simulations, and fundamental thermodynamic relations to determine the intermolecular contributions to ΔG° and concluded that the hydrophobic to hydrophilic crossover would be in the region of hectane (C100H202).
A combination of femtosecond 2D-IR spectroscopy and femtosecond polarization-resolved vibrational pump-probe spectroscopy has revealed a correlated slowing down of both the vibrational frequency dynamics and orientational mobility of the waters around non-polar groups.(58) For different concentrations of four small solutes the fraction of slower water scaled with the number of methyl groups. Furthermore, the obtained results suggest that a common effect of hydrophobic moieties on solvating water is to restrict the formation of bifurcated HBs.
The Effects of Charge
Pascal et al. have examined the effects of introducing positive charge into spherical solutes.(59) The intrinsic solute was given helium-like parameters and ranged in diameter from 0-30 Å. As expected, as the neutral solute was increased in diameter so the solvation ΔG° became more repulsive. By examining the incremental addition of positive charge the authors observed a gradual shift in the solvation ΔG° to hydrophilic and attractive. The crossover was at 0.4e. Interestingly, with this charge the solvation ΔG° was approximately zero over all diameters. Breaking down the solvation ΔG° of these solutes revealed that in all cases solvation was dominated by enthalpy and entropically penalized, with the enthalpy becoming more negative as size increased being compensated by an increasing entropic penalty. Thus, unlike hydrophobic solutes the 0.4e charged species show no entropy-enthalpy crossover.
The Bakker group have studied the dynamics of solvating waters around tetra-n-alkylammonium bromides using polarization-resolved femtosecond IR spectroscopy(5) to reveal that both ions slow the orientational dynamics. In the case of the ammonium, they found that the number of retarded waters scaled with chain length. For the bromide, the solvation shell experienced a partial decay of its orientation via rapid wobbling and a subsequent slower decay. The former was found to be cation dependent. For the smallest salt Me4NBr the slow reorientation time was concentration independent, but with longer chains it increased dramatically with concentration suggesting aggregation. In complementary work dielectric relaxation spectroscopy was used to study the dynamics of waters around tetramethylurea.(58) They found that the reorientation dynamics of solvating waters around the methyls to be between three and ten times slower than the bulk; as the concentration increased, so the dynamics slowed. Their results also demonstrated that with increasing temperature the fraction of water contained in this hydrophobic hydration shell decreased.
The Ben-Amotz group has used Raman-MCR spectroscopy to quantify the interactions between the trimethyl domains of four solutes and the ions Na+, F−, and I−. Their results suggest that Na+ and F− are strongly expelled from the first hydration shells of the solute, while I− were observed to enter the hydrophobic hydration shell to an extent controlled by the partial charge on the methyl groups. However, the estimated association constants for the binding of I− suggested that its concentration in the first hydrophobic hydration shell was lower than the bulk. Contrasting this are recent studies utilizing more circumambient pockets of concave hosts. Thus both host-guest complexation studies(6; 60; 61) and extensive X-ray crystallography work on the binding site of carbonic anhydrase(62) have unequivocally demonstrated that relatively hydrophobic anions associate with hydrophobic pockets.
Zero Curvature
Because it is hard to control the dimerization of flat molecules, the study of these and the HE is dominated by computational work. Nevertheless, in addition to more established approaches such as X-ray reflectivity, newer techniques such as vibrational sum frequency generation spectroscopy and Raman-MCR have recently been applied to their study.
The Aqueous-Flat Surface Interface
Jensen et al. have experimentally confirmed the behavior of water at a hydrophobic surface.(63) Using X-ray reflectivity measurements and MD simulations they observed that there was significant dewetting of the surface of a crystalline monolayer of n-C36H74 that created a narrow “vacuum layer” 1.0 Å wide. This is smaller than predicted for hard spheres using the LCW model,(33) but larger than models which included attractive solute-water interactions.(64)
The Garde group have noted dewetting and dangling HBs in simulations of the water-octane interface.(65) Additionally, the water coordination number and extent of HBing between interfacial waters decreased moving from the bulk to the surface. Correspondingly, the models suggest an increasing probability of void formation closer to the surface. The same group have also shown that both the probability of cavity formation next to a surface and the ΔG° of binding of a hydrophobic solute correlate with macroscopic wetting.(66)
Malaspina et al. have probed the solvation of a rigid, planar graphene-like surface.(67) They observe that within 8-10 Å of the surface water dynamics slow relative to the bulk because of increased structure. This ordering was observed to be independent of temperature over the range of 240-320 K. Building on this, a comparison between flat and concave graphitic surfaces revealed no evidence of dangling HBs.(68)
Continuing with aromatic surfaces, the Ben-Amotz group have explored dangling HBs (OH–π bonds) between benzene and water using Raman-MCR.(69) In combination with computational studies, their results revealed the OH–π bond is ∼20% weaker and more flexible than bulk HBs. Furthermore, their results suggest that dangling HBs are more entropically favored but less enthalpically favored than water-water HBs.
The Chandler group have probed the mean water density and density fluctuations around plates to reveal the expected dewetting.(70) Additionally, they determine that near both repulsive and attractive hydrophobic surfaces, density fluctuations were larger than the bulk and akin to those at the water-vapor interface. Both of these factors contribute to a destabilization of water structure between surfaces and a driving force for dimerization.
Studies of hydrophilic and hydrophobic self-assembled monolayers have revealed that the hydration thermodynamics of hydrophobes at the former have a typical entropy-enthalpy crossover.(71) In contrast, near to hydrophobic surfaces effective surface tension ruled and hydration was enthalpic dominated and size independent. This work also revealed that small solutes bind entropically and increasingly more favorable with increasing temperature, whereas large solutes bind enthalpically in a temperature independent manner. Furthermore, the driving force for assembly of hydrophobes close to a hydrophobic surface was shown to be weaker than in the bulk and decreased with increasing temperature.
Vibrational generation spectroscopy demonstrated that D2O near a hydrophobic surface has enhanced orientation, structure, and stronger HB interactions relative to the D2O/air interface. The results were consistent with dangling HBs and water dipoles perpendicular to the surface;(72) an orientation preference that decreased with increased temperature.
Willard and Chandler have compared the air-water and hydrophobe-water interfaces.(73) Using a local instantaneous water interface as a dynamic frame of reference rather than a fixed Cartesian plane, the authors were able to define an “intrinsic interface”. The study revealed that the strength of attractive water-hydrophobic surface interactions had no significant effect on the intrinsic interface, and only a small effect on the mean density profile of water from the hydrophobic surface out to the bulk. In contrast however, attractive interactions did significantly affect the entropically driven capillary wave fluctuations of the instantaneous liquid interface.
Between Two Plates
The Berne group have modeled the solvation of two ellipsoid plates as a function of their size, separation, and the strength of the attraction between them and water.(74) They found that the critical distance for hydrophobic collapse was linearly proportional to interfacial area. Additionally, when the two plates were held at the critical distance an initially dry zone between them remained dry, but when initially wet the space remained wet. There was no hysteresis when the plates were held at a distance less that the critical distance; irrespective of the starting state, drying resulted. Hydrophobic collapse of the plates occurred only after an initial bubble nucleation and a subsequent cooperative large-scale drying fluctuation.
Electron density profiles and X-ray reflectivity data have revealed weak dewetting (wetting) between two hydrophobic (hydrophilic) surfaces.(75) In both cases characteristic water orientation was observed, with longer range ordering observed in the case of the hydrophilic surfaces. Additionally, the dynamic properties of water between the two pairs of surfaces were different from each other and the bulk. In particular, at the hydrophobic surface waters had characteristic diffusive behavior and orientational ordering due to the lack of interactions with the surface; observations suggesting that altered water dynamics, together with partial drying, are stronger signatures of the HE than structural ordering.
Phase transitions of water between two plates have been examined by Koga(76) and Giovambattista et al.(77) Koga observed two distinct phase transitions at separations of 9.4 Å and 8.2 Å: a liquid-to-bilayer amorphous solid phase transition and bilayer amorphous solid-to-liquid phase transition. The models predicted very few dangling HBs, with a bilayer structure comprised of 5-8 membered water-rings. Giovambattista et al. found water to take on one of two forms, monolayer ice and bilayer (either amorphous or crystalline) depending on the pressure and plate separation.(77) For hydrophobic surfaces, decreased pressure induced capillary evaporation, whilst increasing pressure pushed water closer to the surface and increased its order. However, the average orientation was not affected.
The Berne group have examined the properties of two hydrophobic plates as a function of salts of various charge density and inter-plate separation.(78) In pure water both enthalpy and entropy promote dimerization. Consistent with Hofmeister studies, high charge density ions formed strongly hydrated complexes independent from the hydrophobic plates that increased the hydrophobic interaction between the charged plates via an entropic effect. In contrast, low charge density ions exhibited an entropic or enthalpic-based weakening of the HE by adsorbing to the hydrophobic plates and causing a salting-in effect. The effect of 7 M urea upon a hydrophobic polymer, a particle-based hydrophobic surface, and graphene has also been examined.(79) Consistent among these simulations was the association of urea to all surfaces that caused a decrease in their hydrophobicity and a diminished potential of mean force. These effects were enthalpically dominated.
The Berne group have also studied plates composed of both hydrophobic and hydrophilic regions.(80) Their studies revealed the expected relationship between the degree of hydrophobicity and the presence or absence of a dewetting transition and the presence of attractive or repulsive interactions between plates. More interestingly, using these metrics an analysis of five plate pairs containing equal amounts of hydrophobic and hydrophilic groups arranged in different patterns revealed qualitative and quantitative differences between each. For example, if all the hydrophobic areas on the plate were group in the center dewetting between the plates readily occurred, whereas if the same number of hydrophobic groups were spaced out evenly on the surface no dewetting was observed. In pairing a purely hydrophilic and a purely hydrophobic plate the latter dominated wetting/dewetting. In related work, the affinity of a hydrophobic probe to a pair of hydrophobic plates was examined as a function of plate charge density.(81) Residency between the plates was observed to decrease linearly as the square of the magnitude of charge density of the plates, with the probe being attracted between the plates at q < 0.37; c.f. reference (59).
Sharma and Debenedetti found that paired 1 × 1 nm2 surfaces were too small to accommodate a stable vapor cavity except at distances of < 9 Å, whereas for all distances examined paired 3 × 3 nm2 surfaces exhibited gap-spanning tubular cavity formation.(82) The associated barriers corresponded to the formation of a critical-sized cavity for the latter, and to complete emptying of the gap region for the former. Calculated ΔG° barriers varied approximately linearly with gap size, with 1 Å increases leading to rate drops of ∼2 orders of magnitude. The authors also examined the evaporation rate of the confined water.(83) For the smaller plate examined the time necessary for nucleation of a surface-induced evaporation event showed a 10 order of magnitude increase from 9 to 14 Å separation, whilst the larger plates showed a 6 order of magnitude increase in this rate between 11 and 13 Å. The evaporation rate per unit area was also dependent on the plate size.
The Berne group have studied the kinetics of assembling two plates as a function of their hydrophobicity.(84) The author's models revealed that surfaces of high friction have large and slow water density fluctuations between the plates. Solvent confinement and drying played a critical role in the kinetics. The authors also found reasonable agreement between the calculated molecular- and Brownian-dynamics. Furthermore, they observed that molecular-scale hydrodynamic interactions were essential in describing the kinetics of assembly.
Ashbaugh has carried out 2D Lennard-Jones solvent Monte Carlo simulations between plates.(85) The results validated continuum thermodynamic theory and explored interfacially driven and pressure moderated evaporation. These linked the scale of simulation to the experimental realm. There was strong quantitative correlation between theory and simulation for inter-plate forces and critical evaporation lengths.
Using transmission electron microscopy, Algara-Siller et al. have imaged water between graphene sheets.(15) In contrast to tetrahedal bulk water, the water between the graphene had a “square-ice” structure, where the HBing angle within and between layers was 90°. This lattice had a high packing density. MD simulations supported this picture. Nearest neighbor measurements of oxygen-oxygen distances were in agreement for the plates that accommodated water monolayers, but for the bi- and trilayer cases the waters were found to have tetrahedral structure. This was attributed to the simulations not accounting for adhesion between the sheets imposing pressure on the water.
Aqueous-Protein Interfaces
Following pioneering work by Cheng and Rossky,(86) the Berne group have studied the hydrophobic collapse of two dimeric melittins to form the corresponding tetramer.(87) The fastest hydrophobic collapse was observed when the dimers were separated by a critical distance of 5.5-7.0 Å. Interestingly, when isoleucine-2 was mutated to valine, alanine or glycine no dewetting was observed. Similar results involving a different point mutation have also been observed.(88)
Work by Giovambattista et al. has also focused on the dimerization of melittin dimers.(89) In this instance, instead of using the natural “cupped” surface of a (truncated) melittin, a much smoother surface was obtained by flattening and also removing the surrounding charged groups. The ‘mutant’ surface was less hydrophobic than the wild-type, with desolvation only observed with a separation that sterically only permitted a single water layer. Furthermore, capillary dewetting occurred at lower pressures and separations than in the case of idealized hydrophobic flat surfaces, and was localized to a narrow central region between the dimers. When an amino acid residue in this central region was mutated to a polar residue cavitation was no longer observed.
The collapse of the two domains of BphC enzyme has been studied.(90) For separations > 4 Å both wetted or dewetted initial states ultimately led to a wet state with a water density 10-15% lower than in the bulk. At a separation of 4 Å only partial dewetting was seen unless the electrostatic, or the electrostatic and Van der Waals forces, were turned off. Switching off just Coulombic forces led to rapid and almost complete dewetting, whist switch off both forces led to a more rapid and complete dewetting. In related work with BphC, the Garde group also noted a permanently wetted inter-domain space even at 4 Å separations.(88) Similarly, turning off the charges on each protein resulted in dewetting of the inter-protein space.
The Berne group has also examined the water dynamics in the inter-domain space of the two BphC domains.(91) With the two surfaces held apart by 12 Å, the shortest transit time for a water was for those near the channel center, whilst the longest transit time was for those at a domain surface. Even the fastest waters were ∼30% slower than the bulk. A 4 Å gap showed slower water transitions, whilst a 20 Å gap exhibited bulk-like waters. In general, HB lifetime for waters near one of the domain surfaces was ∼3 times longer than those near the channel center. Interestingly, by this metric one surface alone only affected waters to a depth of 4-5 Å, but two surfaces held 20 Å apart had a similar effect to a depth of 8 Å.
The Bakker group has carried out vibrational sum frequency generation spectroscopic analysis of waters around antifreeze protein AFP-III at or above 0°C.(92) Even above freezing the investigators obtained strong evidence of ice-like waters at the flat, hydrophobic ice-binding site of the protein. A point mutation of threonine-18 residue to asparagine eliminated this structure and led to complete inactivity of the AFP. These results support a preordering-binding mechanism for the recognition of ice.
Negative Curvature
Negative curvature possesses circumambient properties; it can envelope, isolate, and simultaneously form multiple non-covalent interactions with whatever resides within. If water molecules lie there, then the cluster will possess multiple dangling HBs.
Complexation to negative curvature is commonly enthalpically driven. Ross and Subramanian first noted this for proteins,(93) and tabulation of thermodynamic data for guest binding to the cyclodextrins (Figure 2, 1) confirms that ∼99% are exothermic (and that 75% of these are entropically penalized).(94) In 1967 Bender suggested that the binding of guests to cyclodextrins was because, “water molecules in the cavity cannot form their full complement of HBs as a result of steric restrictions.”(95) Consequently, when a guest is bound and the waters liberated, there is a release of enthalpy. Recent evidence supports this idea of (high energy) waters with dangling HBs dominating the solvation and complexation properties of concavity.(96)
Figure 2.
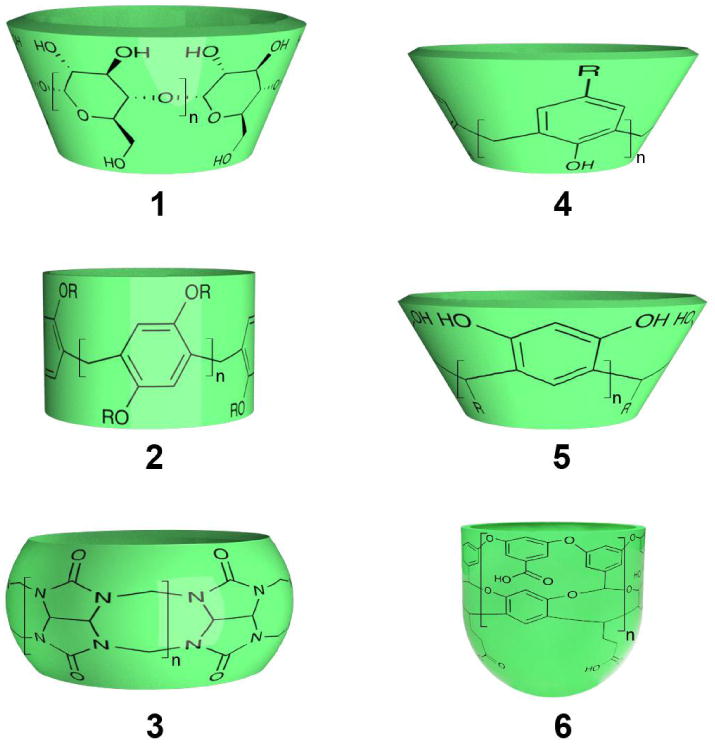
Structural representations of the host molecules 1-6 discussed in the text.
Synthetic Hosts
Figure 2 shows representative examples of water-soluble hosts of current interest in aqueous supramolecular chemistry.(96-102) These range from the very open to the truly concave. The diverse cyclophanes class of host,(103) including the recently synthesized pillar[n]arenes 2 (n = 5-10),(104; 105) are open in nature, possess bound waters with little in the way of dangling HBs and correspondingly bind guests relatively weakly. That stated, the growing class of artificial water channels(106) includes pillar[5]arenes that have proven to form water-wires and act as trans-membrane channels for protons,(107) and act as trans-membrane water channels.(108)
Cyclodextrins 1 (n = 6, 7 or 8) are toroidal hosts composed of either 6, 7 and 8 amylose units (α-, β-, and γ-cyclodextrin respectively).(98) These are more circumambient than cyclophanes, but the presence of hydroxy groups at both portals means that bound waters form relatively few dangling HBs and are not exceptionally high in energy.(96)
Cucurbiturils 3 (CBs, n = 5, 6, 7, 8 or 10) are bis-methylene linked oligomers of 5, 6, 7, 8 and 10 glycolurils. (109-111) Their pockets are comprised of a hydrophobic region with two portals rimmed by imide carbonyls. In general, guest complexations are strongly enthalpically promoted. By Biedermann's measure of high-energy water,(96) the host with the most hydrophobic binding site is CB7; it contains on average 7.9 waters each with an average 2.52 HBs. The resulting large number of dangling HBs is suspected to contribute to the general ability of CB7 to strongly bind guests.(112) Interestingly, CB8 can bind two guests. The inclusion of the first restricts HBing between any residual waters, and therefore the subsequent binding is strongly exothermic.(96)
Calix[4]arenes 4 (n = 4) and resorcinarenes 5 (n = 4) are truly concave but very shallow.(113) With open binding sites and proximal phenols, their hydrophobic surface is not effectively screened from the bulk. Unsurprisingly, only modest 1:1 complexations are observed. Resorcinarene-based, deep-cavity cavitands such as 6 (n = 4) possess truly hydrophobic pockets.(97) Host 6 has been shown to bind an average of 4.34 waters.(114) The base of the cavity is occupied by a singular water which accepts an average of 1.33 HBs, waters in a middle layer have on average 2.65 HBs, whilst those in the uppermost layer at the portal have on average 3.33 HBs. These bound waters have higher energy than the bulk, and the deeper a water resides the higher its energy and the more it derives stabilization from (dangling) host-water HBs rather than water-water HBs. Calculated translational diffusion constants revealed that bound waters possessed lower diffusion constants than the bulk, and the deeper the water was located the more it was constrained. The opposite was observed with rotational diffusion constants. Overall, the thermodynamics for hydration of the pocket were: ΔGhyd ≈−5 kcal mol–1, ΔHhyd ≈−20 kcal mol–1, and –TΔShyd ≈ 15 kcal mol–1.
In silico Carbon Nanotubes
The bore of carbon nanotubes (CNTs) of ∼8 Å diameter are solvated by a water-wire.(115; 116) These waters have only two relatively long-lived HBs and are less dense packed than the bulk. Possessing many dangling HBs only very small reductions of the Van der Waals attraction between water and tube are sufficient to induce emptying.(115) A combination of HBing and dipole–dipole interactions within the wire engenders rapid proton transfer via a Grotthuss relay mechanism.(116)
Water flows through CNTs in bursts and is limited only by the barriers of entry and egress; the tube itself is essentially frictionless and the flow rate nearly independent of its length.(117) Counterintuitively, as the diameter increases transport rates decrease because of increased HBing between waters.(118)
It has been shown that changing the diameter of the CNT induces phase transitions in water packing.(119; 120) Thus under ambient conditions, water within 8 Å CNTs is gas-like, whereas with 11 and 12 Å diameter CNTs the water is ice-like and stack into layers of pentagons or hexagons.(121; 122) For larger diameters still, e.g., 20-30 Å, bound waters form multiple nano-clusters.(120) It is not surprising therefore that there is a non-monotonic relationship between diameter and relative Helmholtz free energy of filling;(121) diameters of 8 and 12 Å represents energy minima. Overall entropy dominates slightly for tube diameters less than 10 Å or greater than 16 Å, and enthalpy dominates for tubes between 11 and 12 Å. With the exception of 11 and 12 Å nanotubes, confinement within the CNTs leads to an increase in the translational entropy of bound water. In passing it is worth noting that ice-structures are commonly observed in CNTs at lower temperatures(123) and elevated pressures.(124) Indeed, simulations of the thermodynamic states over the diameter-temperature plane reveal at least nine different ice phases(119) including stacked squares, pentagons, hexagons and heptagons. Pentagonal and hexagonal packed water is ferroelectric.(125) Interestingly, stacked octagonal water has only been observed in a coaxial-like system whereby small hydrophobes form a wire running down the central axis of stacked water-rings.(126)
In silico Hosts
In early work examining the properties of a hydrophobic pocket of 8 Å radius ‘cut’ into a paraffin slab, Setney observed that the average water density within the cavity was depleted relative to the bulk and that the pocket fluctuates between empty and filled states.(127) Overall the ΔG° values accompanying these fluctuations were small, suggesting that the selected radius is close to a critical size for promoting transitions between gas-like and liquid-like phases. In comparing 5 Å and 8 Å radii concavities to a corresponding flat surface,(128; 129) Setny observed vapor-like regions of significantly reduced water density in both pockets, with the smaller pocket possessing a substantial desolvation barrier to probe binding whereas entry into the 8 Å cavity was essentially barrierless. Contrary to the idea of high-energy waters dominating complexation, for both concavities dewetting was only a marginal contributor to the ΔG° of probe binding. Rather the observed ΔG° values were related to the hydration shell of the probe.
Building on this, Baron et al. estimated ΔG°, ΔH°, and –TΔS° along the concavity-guest binding trajectory as a function of the charge (neutral, +, or –) of host and guest.(130) They observed a broad range of thermodynamic signatures in which water enthalpic or entropic contributions drove cavity-guest binding or rejection. Thus, the binding of a neutral methane-like guest to a neutral host was driven by enthalpy and entropically penalized, with the roots of the former pinpointed to the formation of strong water-water interactions when bound waters moved into the bulk. Furthermore, their data implied that because of solvent fluctuations in the pocket, bound water was more entropically favored than the bulk. Overall, in this work it was high-energy bound waters with dangling HBs that drove complexation.(130; 131) Further simulations revealed that the reorientation kinetics of the waters around a methane-like guest increased as the host-guest distance was decreased, and that this was correlated with a decrease in the number of HBs between waters in the guest solvation shell.(132) Moreover, the authors calculated changes in the 1D and 2D IR spectra during complexation, suggesting a strategic route to correlating water structure changes and signature vibrational spectra differences. Most recently the role water plays in ligand binding kinetics has been investigated.(133) The empty pocket of the host exhibited solvated-desolvated oscillations whose magnitude and time-scale were modulated by the approaching guest. Furthermore, these fluctuations led to a friction-induced kinetic barrier to guest entry to the pocket.
Vaitheeswaran et al. have used Monte Carlo simulations to study the thermodynamic stability of water clusters inside smooth graphene-like spherical cavities and the fullerenes C140 and C180. Depending on the container size they observed thermodynamically stable water clusters composed of between three and nine waters. The smallest stable water cluster in a spherical cavity of 1.0 nm diameter was a trimer held together by three HBs. With a slightly smaller diameter of 0.9 nm a thermodynamically unstable dimer was observed. Overall the clusters observed were strikingly similar to gas-phase water clusters.(13; 14)
Proteins
The tubular binding site of Cox-2 was found to be unusual in that it possessed a drying transition.(134) To investigate this further, the Berne and Friesner groups sought to identify structural features important to drying. Screening the Protein Data Bank using two metrics of pocket hydrophobicity and one of narrowness identified twelve proteins.(135) Except for three of these where it was found that water could not exchange with the bulk, whether or not the remaining examples underwent a drying transition was found to be excellently correlated with a drying parameter; the product of the atom-based surface hydrophobicity and a measure of the narrowness of the cavity. Thus pockets that are relatively hydrophobic and narrow possessed drying transitions, whilst the others did not.
Water wires have been shown to form in 5.2 Å diameter hydrophobic channels formed by the assembly of penta-peptides into hexameric assemblies.(136) Interestingly, X-ray crystallographic analysis revealed two kinds of water wires characterized by different oxygen-oxygen distances. In both cases, the data suggested very weak interactions between the water and the tube. Furthermore, reinterpretation(136) of the solid-state structure of stacked, cyclic peptides composed of alternating D- and L-residues(137) also suggests that this family of peptides can template water wires.
Berne and Friesner examined three disparate binding sites: that of streptavidin, Cox-2, and the antibody DB3. The binding site of streptavidin has a hydrophobic floor and roof, yet possesses five co-planar HBing groups in its walls. In the case of Cox-2, its binding site is long, tubular and hydrophobic. Lastly, the pocket of DB3 has a less-pronounced hydrophobic enclosure. Simulations of streptavidin reveal hydration by five waters that form a persistent ring, an entropically costly water structure that in conjunction with little corresponding enthalpic gain, may contribute up to five orders of magnitude to ligand affinity. A less extreme example, the binding site of DB3 was found to be occupied by one water. Like those in streptavidin this water has very few energetically accessible configurations that contribute to an entropic penalty of hydration. In contrast, the binding site of Cox-2 was found to be devoid of water molecules 80% of the time; no energetically stable solvent configurations appeared to exist. Thus Cox-2 is an example of a protein solvated by high energy waters with dangling HBs.
The pocket in a protein need not be either entirely wet or dry. Using the WaterMap program (Schrödinger), Wang, Berne and Friesner examined the hydration properties of the binding sites of three proteins to identify wet and dry regions within their respective pockets.(138) The authors showed that calculated affinities were excellent in cases where a bound ligand only occupied space previously filled with water, but poor when a bound ligand displaced some or none of the waters. However, once the water free non-covalent contacts between ligand and protein in these dry regions were taken into account, the calculated affinities of congeneric ligands correlated with observed affinities.
Consideration of the enthalpic and entropic costs of transferring a single water molecule from the bulk to a binding site suggests that the ΔG° of transfer is relatively small.(139; 140) Olano and Rick calculated the ΔG° of transfer of a water from the bulk to an interior cavity of bovine pancreatic trypsin inhibitor (BPTI) and in the I76A mutant of barnase.(141) Previous X-ray crystallography revealed the BPTI binding site to be occupied by water, but gave ambiguous data concerning the barnase mutant. The former is polar and allows the formation of up to four water-protein HBs, whilst the latter is more hydrophobic and (potentially) allows only one HB. The calculations predicted that only the polar BPTI cavity would be hydrated, that the corresponding transfer entropies for BPTI are significantly entropically unfavorable, whilst the transfer to the cavity of the barnase mutant is entropically favorable.
The major domain of tetrabrachion consists of a right-handed four-helix bundle that defines a channel containing four cavities. Although the channel/cavities are lined with aliphatic residues X-ray crystallography revealed that all are filled with water: 1, 2, 5 and 9 waters from smallest to largest cavity respectively. Remarkably, recent MD simulations have revealed that at 25 and 92 °C the largest of these cavities was filled with clusters of seven to nine waters respectively.(142) Furthermore, filling of the cavity was found to be enthalpically driven and opposed by entropy. Only at 110 °C did the calculations suggest cluster instability and drying.
The Homans groups have studied ligand binding to mouse major urinary protein (MUP) using a variety of techniques.(143) Calculations revealed that the pockets of the wild type and mutant contained 3.74 and 4.07 waters respectively, and that in accord with X-ray crystallography water-density in the pocket was low. In the case of binding of 2-methoxy-3-isobutylpyrazine (IPMP), Isothermal Titration Calorimetry (ITC) revealed association was driven by favorable enthalpic contributions, even when the only ligand-protein HB was removed by mutagenesis. Furthermore, a combination of solvent isotopic substitution ITC and MD simulations suggested that neither solvation nor induced fit changes were major contributors to the overall binding enthalpy. Considering the poor solvation of the pocket, the binding thermodynamics were attributed to favorable dispersion interactions arising from the inequality of solvent-solute interactions before and after complexation. A related study of the binding of linear alcohols to MUP also supported this hypothesis.(144) The Homans group have also examined the contribution of the entropic costs of the desolvation of IPMP and found that the overall entropic penalty arose because the favorable entropic contribution from guest desolvation did not overcome the entropic penalty due to reduced degrees of freedom.(145) ITC studies of the MUP-n-alcohol complexes also revealed that although binding was exothermic and entropically penalized, the corresponding heat capacity change was typical of the HE, i.e., negative.(146)
Summary and Conclusions
The theoretical transition from a small convex solute, to a larger one, to a flat surface, through increasingly concave surfaces, and finally to full encapsulation (Figure 1), reveals the following trends. For positively curved hydrophobes there is an entropy-enthalpy crossover as a function of diameter. This crossover correlates with both a wetting/dewetting transition and a switch from the waters of solvation being fully HBed in the case of small solutes, to a solvation shell that geometrically allows dangling HBs to point towards the solute surface in the case of large solutes.
As may be expected, the case of a large solute and that of a flat hydrophobic surface are similar. However, when water between two flat surfaces is considered it is evident that they work in synergy to affect waters in the inter-surface zone. As a result, by the metrics of water molecule diffusion, number of dangling HBs, and phase transitions from wet to dry, there is a similarity between the two plates case and that of true concavity.
With respect to negative curvature, it is evident that the large number of dangling HBs of bound water plays a major role in regards to the properties of a tube or pocket. Quite simply, dangling HBs mean high energy waters that would rather not be encapsulated. And the more confining the hydrophobic binding, the more extreme the enthalpic domination; such that in the extreme case of total water confinement waters resemble gas-phase clusters.
En masse, although there is still some disagreement, the described results point to a switch from entropy dominated solvation to enthalpy domination and then to increasing enthalpy domination as a theoretical solute is morphed from a small convex entity, to large one, to a solute that is increasingly encapsulating. In addition though, it is evident that the HE is profoundly affected by all chemical and physical variables. It is therefore not surprising that we do not yet fully understand the HE. That means however, that there are rich pickings ahead for those immersed in water.
Summary Points.
The solvation of positive curvature is size-dependent. Small solutes are solvated without the breaking of hydrogen bonds (HBs) between waters. Large solutes (> 10 Å diameter) are not; it is geometrically impossible to maintain full HBing in the solvation shell. As a result, weaker dangling HBs to the solute are formed instead. The hydration layer of a small solute is denser (the solute is wetted) than the bulk, and the ΔG° of solvation scales with solute volume. In contrast large solutes undergo dewetting, with a ΔG° of solvation that scales with surface area.
Because the spatial arranging of HBs is dominated by entropy, and the breaking of HBs is dominated by enthalpy, entropy rules in the solvation of small positively curved solutes, whilst enthalpy rules in the solvation of large ones. Thermodynamically, this results in a size-dependent entropy crossover transition, or entropy-enthalpy crossover.
The solvation of negative curvature (concavity) means that bound waters cannot form their normal complement of HBs to other waters. The resulting dangling HBs increase in number as the curvature becomes more negative, and reaches an extreme of four dangling HBs in the case of a singular water molecule trapped inside a hydrophobic sphere. Thus, increasing concavity is associated with increasing enthalpic domination.
Between negative and positive curvature, zero curvature possesses intermediate properties. The solvation of a singular flat plate is reminiscent of that observed with large convex solutes, whilst the solvation of paired plates physicochemically resemble that of tubes and pockets.
Much work remains to be carried out to understand how subtle structural nuances such as functional group type and patterning, as well ‘exogenous’ variable such as temperature, pressure, and the presence of salts fold into the aforementioned solvation process to engender the Hydrophobic effect.
Acknowledgments
MBH would like to thank the Louisiana Board of Regents for a fellowship (LEQSF(2012-17)-GF-SREB-03). MBH and BCG gratefully acknowledge the National Institutes of Health (GM098141) and the National Science Foundation (CHE 1244024) for financial support. The authors would also like to thank Henry S. Ashbaugh for helpful discussions and Eric McCutcheon for help with the figures.
Literature Cited
- 1.Lynden-Bell RM, Morris SC, Barrow JD, Finney JL, Harper RLJ, editors. Water and Life. Boca Raton: CRC Press; 2010. [Google Scholar]
- 2.Lo Nostro P, Ninham BW. Hofmeister phenomena: an update on ion specificity in biology. Chem Rev. 2012;112:2286–322. doi: 10.1021/cr200271j. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Cremer PS. Chemistry of Hofmeister anions and osmolytes. Annu Rev Phys Chem. 2010;61:63–83. doi: 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]
- 4.Jungwirth P, Tobias DJ. Specific ion effects at the air/water interface. Chem Rev. 2006;106:1259–81. doi: 10.1021/cr0403741. [DOI] [PubMed] [Google Scholar]
- 5.van der Post ST, Scheidelaar S, Bakker HJ. Water dynamics in aqueous solutions of tetra-n-alkylammonium salts: hydrophobic and Coulomb interactions disentangled. J Phys Chem B. 2013;117:15101–10. doi: 10.1021/jp4085734. [DOI] [PubMed] [Google Scholar]
- 6.Assaf KI, Ural MS, Pan F, Georgiev T, Simova S, et al. Water structure recovery in chaotropic anion recognition: High-affinity binding of dodecaborate clusters to γ-cyclodextrin. Angew Chemie Int Ed. 2015;54:6852–6. doi: 10.1002/anie.201412485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hummer G, Garde S, García AE, Pratt LR. New perspectives on hydrophobic effects. Chem Phys. 2000;258:349–70. [Google Scholar]
- 8.Berne BJ, Weeks JD, Zhou R. Dewetting and hydrophobic interaction in physical and biological systems. Annu Rev Phys Chem. 2009;60:85–103. doi: 10.1146/annurev.physchem.58.032806.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koga K. Solvation of hydrophobes in water and simple liquids. Phys Chem Chem Phys. 2011;13:19749–58. doi: 10.1039/c1cp22344e. [DOI] [PubMed] [Google Scholar]
- 10.Pratt LR, Pohorille A. Hydrophobic effects and modeling of biophysical aqueous solution interfaces. Chem Rev. 2002;102:2671–92. doi: 10.1021/cr000692+. [DOI] [PubMed] [Google Scholar]
- 11.Sharp KA, Vanderkooi Water in the half shell: structure of water, focusing on angular structure and solvation. Acc Chem Res. 2010:231–9. doi: 10.1021/ar900154j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar R, Schmidt JR, Skinner JL. Hydrogen bonding definitions and dynamics in liquid water. J Chem Phys. 2007;126:204107. doi: 10.1063/1.2742385. [DOI] [PubMed] [Google Scholar]
- 13.Liu K, Brown MG, Carter C, Saykally RJ, Gregory JK, Clary DC. Characterization of a cage form of the water hexamer. Nature. 1996;381:501–3. [Google Scholar]
- 14.Perez C, Muckle MT, Zaleski DP, Seifert NA, Temelso B, et al. Structures of cage, prism, and book isomers of water hexamer from broadband rotational spectroscopy. Science. 2012;336:897–901. doi: 10.1126/science.1220574. [DOI] [PubMed] [Google Scholar]
- 15.Algara-Siller G, Lehtinen O, Wang FC, Nair RR, Kaiser U, et al. Square ice in graphene nanocapillaries. Nature. 2015;519:443–5. doi: 10.1038/nature14295. [DOI] [PubMed] [Google Scholar]
- 16.Frank HS, Wen WY. III. Ion-solvent interaction: Structural aspects of ion-solvent interaction in aqueous solutions: a suggested picture of water structure. Discussions of the Faraday Society. 1957;24:133–40. [Google Scholar]
- 17.Ben-Amotz D. Annu Rev Phys Chem Same special issue 2016 [Google Scholar]
- 18.Ben-Amotz D. Hydrophobic Ambivalence: Teetering on the edge of randomness. J Phys Chem Lett. 2015;6:1696–701. doi: 10.1021/acs.jpclett.5b00404. [DOI] [PubMed] [Google Scholar]
- 19.Chandler D. Interfaces and the driving force of hydrophobic assembly. Nature. 2005;437:640–7. doi: 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- 20.Frank HS, Evans MW. Free volume and entropy in condensed systems III. Entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J Chem Phys. 1945;13:507. [Google Scholar]
- 21.Kauzmann W. Some factors in the interpretation of proptein denaturation. Adv Protein Chem. 1959;14:1–63. doi: 10.1016/s0065-3233(08)60608-7. [DOI] [PubMed] [Google Scholar]
- 22.Snyder PW, Lockett MR, Moustakas DT, Whitesides GM. Is it the shape of the cavity, or the shape of the water in the cavity? Euro Phys J Special Topics. 2014;223:853–91. [Google Scholar]
- 23.Ball P. Water as an active constituent in cell biology. Chem Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 24.Blokzijl W, Engberts JBFN. Hydrophobic effects opinions and facts. Angew Chemie Int Ed. 1993;32:1545–79. [Google Scholar]
- 25.Lucas M. Size effect in transfer of nonpolar solutes from gas or solvent to another solvent with a view on hydrophobic behavior. J Phys Chem. 1976;80:359–62. [Google Scholar]
- 26.Lee B. Solvent reorganization contribution to the transfer thermodynamics of small nonpolar molecules. Biopolymers. 1991;31:993–1008. doi: 10.1002/bip.360310809. [DOI] [PubMed] [Google Scholar]
- 27.Lynden-Bell RM, Giovambattista N, Debenedetti PG, Head-Gordon T, Rossky PJ. Hydrogen bond strength and network structure effects on hydration of non-polar molecules. Phys Chem Chem Phys. 2011;13:2748–57. doi: 10.1039/c0cp01701a. [DOI] [PubMed] [Google Scholar]
- 28.Lazaridis T. Solvent size vs cohesive energy as the origin of hydrophobicity. Acc Chem Res. 2001;34:931–7. doi: 10.1021/ar010058y. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin RL. The new view of hydrophobic free energy. FEBS Lett. 2013;587:1062–6. doi: 10.1016/j.febslet.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Stillinger FH. Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled particle theory. J Soln Chem. 1973;2:141–58. [Google Scholar]
- 31.Pratt LR, Chandler D. Theory of the Hydrophobic effect. J Chem Phys. 1977;67:3683. [Google Scholar]
- 32.Pratt LR. Molecular theory of Hydrophobic effects: “She is too mean to have her name repeated”. Annu Rev Phys Chem. 2002;53:409–36. doi: 10.1146/annurev.physchem.53.090401.093500. [DOI] [PubMed] [Google Scholar]
- 33.Lum K, Chandler D, Weeks JD. Hydrophobicity at small and large length scales. J Phys Chem B. 1999;103:4570–7. [Google Scholar]
- 34.Poynor A, Hong L, Robinson IK, Granick S, Zhang Z, Fenter PA. How water meets a hydrophobic surface. Phys Rev Lett. 2006;97 doi: 10.1103/PhysRevLett.97.266101. [DOI] [PubMed] [Google Scholar]
- 35.Mezger M, Reichert H, Schoder S, Okasinski J, Schroder H, et al. High-resolution in situ X-ray study of the hydrophobic gap at the water-octadecyl-trichlorosilane interface. Proc Natl Acad Sci USA. 2006;103:18401–4. doi: 10.1073/pnas.0608827103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchanan P, Soper AK, Thompson H, Westacott RE, Creek JL, et al. Search for memory effects in methane hydrate: structure of water before hydrate formation and after hydrate decomposition. J Chem Phys. 2005;123:164507. doi: 10.1063/1.2074927. [DOI] [PubMed] [Google Scholar]
- 37.Buchanan P, Aldiwan N, Soper AK, Creek JL, Koh CA. Decreased structure on dissolving methane in water. Chem Phys Lett. 2005;415:89–93. [Google Scholar]
- 38.Ashbaugh H. Hydration of krypton and consideration of clathrate models of hydrophobic effects from the perspective of quasi-chemical theory. Biophysical Chemistry. 2003;105:323–38. doi: 10.1016/s0301-4622(03)00084-x. [DOI] [PubMed] [Google Scholar]
- 39.Chaudhari MI, Holleran SA, Ashbaugh HS, Pratt LR. Molecular-scale hydrophobic interactions between hard-sphere reference solutes are attractive and endothermic. Proc Natl Acad Sci USA. 2013;110:20557–62. doi: 10.1073/pnas.1312458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czaplewski C, Liwo A, Ripoll DR, Scheraga HA. Molecular origin of anticooperativity in hydrophobic association. J Phys Chem B. 2005;109:8108–19. doi: 10.1021/jp040691b. [DOI] [PubMed] [Google Scholar]
- 41.Ashbaugh HS, Paulaitis ME. Effect of solute size and solute–water attractive interactions on hydration water structure around hydrophobic solutes. J Am Chem Soc. 2001;123:10721–8. doi: 10.1021/ja016324k. [DOI] [PubMed] [Google Scholar]
- 42.Li ITS, Walker GC. Signature of hydrophobic hydration in a single polymer. Proc Natl Acad Sci USA. 2011;108:16527–32. doi: 10.1073/pnas.1105450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garde S, Patel AJ. Unraveling the hydrophobic effect, one molecule at a time. Proc Natl Acad Sci USA. 2011;108:16491–2. doi: 10.1073/pnas.1113256108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang DM, Geissler PL, Chandler D. Scaling of hydrophobic solvation free energies. J Phys Chem B. 2001;105:6704–9. [Google Scholar]
- 45.Huang DM, Chandler D. The Hydrophobic effect and the influence of solute–solvent attractions. J Phys Chem B. 2002;106:2047–53. [Google Scholar]
- 46.Rajamani S, Truskett TM, Garde S. Hydrophobic hydration from small to large lengthscales: Understanding and manipulating the crossover. Proc Natl Acad Sci USA. 2005;102:9475–80. doi: 10.1073/pnas.0504089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mittal J, Hummer G. Static and dynamic correlations in water at hydrophobic interfaces. Proc Natl Acad Sci USA. 2008;105:20130–5. doi: 10.1073/pnas.0809029105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ashbaugh HS. Entropy crossover from molecular to macroscopic cavity hydration. Chem Phys Lett. 2009;477:109–11. [Google Scholar]
- 49.Huang X, Margulis CJ, Berne BJ. Do molecules as small as neopentane induce a hydrophobic response similar to that of large hydrophobic surfaces? J Phys Chem B. 2003;107:11742–8. [Google Scholar]
- 50.Gallicchio E, Kubo MM, Levy RM. Enthalpy–entropy and cavity decomposition of alkane hydration free energies: numerical results and implications for theories of hydrophobic solvation. J Phys Chem B. 2000;104:6271–85. [Google Scholar]
- 51.Perera PN, Fega KR, Lawrence C, Sundstrom EJ, Tomlinson-Phillips J, Ben-Amotz D. Observation of water dangling OH bonds around dissolved nonpolar groups. Proc Natl Acad Sci USA. 2009;106:12230–4. doi: 10.1073/pnas.0903675106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis JG, Rankin BM, Gierszal KP, Ben-Amotz D. On the cooperative formation of non-hydrogen-bonded water at molecular hydrophobic interfaces. Nat Chem. 2013;5:796–802. doi: 10.1038/nchem.1716. [DOI] [PubMed] [Google Scholar]
- 53.Rankin BM, Ben-Amotz D, van der Post ST, Bakker HJ. Contacts between alcohols in water are random rather than hydrophobic. J Phys Chem Lett. 2015;6:688–92. doi: 10.1021/jz5027129. [DOI] [PubMed] [Google Scholar]
- 54.Davis JG, Gierszal KP, Wang P, Ben-Amotz D. Water structural transformation at molecular hydrophobic interfaces. Nature. 2012;491:582–5. doi: 10.1038/nature11570. [DOI] [PubMed] [Google Scholar]
- 55.Davis JG, Zukowski SR, Rankin BM, Ben-Amotz D. Influence of a neighboring charged group on hydrophobic hydration shell structure. J Phys Chem B. 2015;119:9417–22. doi: 10.1021/jp510641a. [DOI] [PubMed] [Google Scholar]
- 56.Ferguson AL, Debenedetti PG, Panagiotopoulos AZ. Solubility and molecular conformations of n-alkane chains in water. J Phys Chem B. 2009;113:6405–14. doi: 10.1021/jp811229q. [DOI] [PubMed] [Google Scholar]
- 57.Underwood R, Tomlinson-Phillips J, Ben-Amotz D. Are long-chain alkanes hydrophilic? J Phys Chem B. 2010;114:8646–51. doi: 10.1021/jp912089q. [DOI] [PubMed] [Google Scholar]
- 58.Bakulin AA, Pshenichnikov MS, Bakker HJ, Petersen C. Hydrophobic molecules slow down the hydrogen-bond dynamics of water. J Phys Chem A. 2011;115:1821–9. doi: 10.1021/jp107881j. [DOI] [PubMed] [Google Scholar]
- 59.Pascal TA, Lin ST, Goddard W, Jung Y. Stability of Positively Charged Solutes in Water: A Transition from Hydrophobic to Hydrophilic. J Phys Chem Lett. 2012:294–8. doi: 10.1021/jz201612y. [DOI] [PubMed] [Google Scholar]
- 60.Gibb CL, Gibb BC. Anion binding to hydrophobic concavity is central to the salting-in effects of Hofmeister chaotropes. J Am Chem Soc. 2011;133:7344–7. doi: 10.1021/ja202308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carnegie RS, Gibb CL, Gibb BC. Anion complexation and the Hofmeister effect. Angew Chemie Int Ed. 2014;53:11498–500. doi: 10.1002/anie.201405796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fox JM, Kang K, Sherman W, Heroux A, Sastry GM, et al. Interactions between Hofmeister Anions and the Binding Pocket of a Protein. J Am Chem Soc. 2015;137:3859–66. doi: 10.1021/jacs.5b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jensen T, Østergaard Jensen M, Reitzel N, Balashev K, Peters G, et al. Water in contact with extended hydrophobic surfaces: direct evidence of weak dewetting. Phys Rev Lett. 2003;90 doi: 10.1103/PhysRevLett.90.086101. [DOI] [PubMed] [Google Scholar]
- 64.Wallqvist A, Gallicchio E, Levy RM. A model for studying drying at hydrophobic interfaces: Structural and thermodynamic properties. J Phys Chem B. 2001;105:6745–53. [Google Scholar]
- 65.Patel HA, Nauman EB, Garde S. Molecular structure and hydrophobic solvation thermodynamics at an octane–water interface. J Chem Phys. 2003;119:9199. [Google Scholar]
- 66.Godawat R, Jamadagni SN, Garde S. Characterizing hydrophobicity of interfaces by using cavity formation, solute binding, and water correlations. Proc Natl Acad Sci USA. 2009;106:15119–24. doi: 10.1073/pnas.0902778106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malaspina DC, Schulz EP, Alarcon LM, Frechero MA, Appignanesi GA. Structural and dynamical aspects of water in contact with a hydrophobic surface. Euro Phys J E, Soft Matter. 2010;32:35–42. doi: 10.1140/epje/i2010-10594-2. [DOI] [PubMed] [Google Scholar]
- 68.Alarcón LM, Malaspina DC, Schulz EP, Frechero MA, Appignanesi GA. Structure and orientation of water molecules at model hydrophobic surfaces with curvature: From graphene sheets to carbon nanotubes and fullerenes. Chem Phys. 2011;388:47–56. [Google Scholar]
- 69.Gierszal KP, Davis JG, Hands MD, Wilcox DS, Slipchenko LV, Ben-Amotz D. π-Hydrogen bonding in liquid water. J Phys Chem Lett. 2011;2:2930–3. [Google Scholar]
- 70.Patel AJ, Varilly P, Chandler D. Fluctuations of water near extended hydrophobic and hydrophilic surfaces. J Phys Chem B. 2010;114:1632–7. doi: 10.1021/jp909048f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patel AJ, Varilly P, Jamadagni SN, Acharya H, Garde S, Chandler D. Extended surfaces modulate hydrophobic interactions of neighboring solutes. Proc Natl Acad Sci USA. 2011;108:17678–83. doi: 10.1073/pnas.1110703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Strazdaite S, Versluis J, Backus EH, Bakker HJ. Enhanced ordering of water at hydrophobic surfaces. J Chem Phys. 2014;140:054711. doi: 10.1063/1.4863558. [DOI] [PubMed] [Google Scholar]
- 73.Willard AP, Chandler D. The molecular structure of the interface between water and a hydrophobic substrate is liquid-vapor like. J Chem Phys. 2014;141:18C519. doi: 10.1063/1.4897249. [DOI] [PubMed] [Google Scholar]
- 74.Huang X, Margulis CJ, Berne BJ. Dewetting-induced collapse of hydrophobic particles. Proc Natl Acad Sci USA. 2003;100:11953–8. doi: 10.1073/pnas.1934837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jensen MO, Mouritsen OG, Peters GH. The hydrophobic effect: molecular dynamics simulations of water confined between extended hydrophobic and hydrophilic surfaces. J Chem Phys. 2004;120:9729–44. doi: 10.1063/1.1697379. [DOI] [PubMed] [Google Scholar]
- 76.Koga K. Solvation forces and liquid–solid phase equilibria for water confined between hydrophobic surfaces. J Chem Phys. 2002;116:10882. [Google Scholar]
- 77.Giovambattista N, Rossky P, Debenedetti P. Effect of pressure on the phase behavior and structure of water confined between nanoscale hydrophobic and hydrophilic plates. Phys Rev E. 2006;73 doi: 10.1103/PhysRevE.73.041604. [DOI] [PubMed] [Google Scholar]
- 78.Zangi R, Hagen M, Berne BJ. Effect of ions on the hydrophobic interaction between two plates. J Am Chem Soc. 2007;129:4678–86. doi: 10.1021/ja068305m. [DOI] [PubMed] [Google Scholar]
- 79.Zangi R, Zhou R, Berne BJ. Urea's action on hydrophobic interactions. J Am Chem Soc. 2009;131:1535–41. doi: 10.1021/ja807887g. [DOI] [PubMed] [Google Scholar]
- 80.Hua L, Zangi R, Berne BJ. Hydrophobic Interactions and Dewetting between Plates with Hydrophobic and Hydrophilic Domains. J Phys Chem C. 2009;113:5244–53. [Google Scholar]
- 81.Wang L, Friesner RA, Berne BJ. Competition of electrostatic and hydrophobic interactions between small hydrophobes and model enclosures. J Phys Chem B. 2010;114:7294–301. doi: 10.1021/jp100772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma S, Debenedetti PG. Free energy barriers to evaporation of water in hydrophobic confinement. J Phys Chem B. 2012;116:13282–9. doi: 10.1021/jp308362h. [DOI] [PubMed] [Google Scholar]
- 83.Sharma S, Debenedetti PG. Evaporation rate of water in hydrophobic confinement. Proc Natl Acad Sci USA. 2012;109:4365–70. doi: 10.1073/pnas.1116167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li J, Morrone JA, Berne BJ. Are hydrodynamic interactions important in the kinetics of hydrophobic collapse? J Phys Chem B. 2012;116:11537–44. doi: 10.1021/jp307466r. [DOI] [PubMed] [Google Scholar]
- 85.Ashbaugh HS. Solvent cavitation under solvophobic confinement. J Chem Phys. 2013;139:064702. doi: 10.1063/1.4817661. [DOI] [PubMed] [Google Scholar]
- 86.Cheng YK, Rossky PJ. Surface topography dependence of biomolecular hydrophobic hydration. Nature. 1998;392:696–9. doi: 10.1038/33653. [DOI] [PubMed] [Google Scholar]
- 87.Liu P, Huang X, Zhou R, Berne BJ. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature. 2005;437:159–62. doi: 10.1038/nature03926. [DOI] [PubMed] [Google Scholar]
- 88.Patel AJ, Varilly P, Jamadagni SN, Hagan MF, Chandler D, Garde S. Sitting at the edge: how biomolecules use hydrophobicity to tune their interactions and function. J Phys Chem B. 2012;116:2498–503. doi: 10.1021/jp2107523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Giovambattista N, Lopez CF, Rossky PJ, Debenedetti PG. Hydrophobicity of protein surfaces: Separating geometry from chemistry. Proc Natl Acad Sci USA. 2008;105:2274–9. doi: 10.1073/pnas.0708088105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou R, Huang X, Margulis CJ, Berne BJ. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–9. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 91.Hua L, Huang X, Zhou R, Berne BJ. Dynamics of water confined in the interdomain region of a multidomain protein. J Phys Chem B. 2006;110:3704–11. doi: 10.1021/jp055399y. [DOI] [PubMed] [Google Scholar]
- 92.Meister K, Strazdaite S, DeVries AL, Lotze S, Olijve LL, et al. Observation of ice-like water layers at an aqueous protein surface. Proc Natl Acad Sci USA. 2014;111:17732–6. doi: 10.1073/pnas.1414188111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ross PD, Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochem. 1981;20:3096–102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]
- 94.Rekharsky MV, Inoue Y. Complexation thermodynamics of cyclodextrins. Chem Rev. 1998;98:1875–917. doi: 10.1021/cr970015o. [DOI] [PubMed] [Google Scholar]
- 95.VanEtten RL, Sebastian JF, Clowes GA, Bender ML. Acceleration of phenyl ester cleavage by cycloamyloses. a model for enzymatic specificity J Am Chem Soc. 1967;89:3242. [Google Scholar]
- 96.Biedermann F, Nau WM, Schneider HJ. The hydrophobic effect revisited--studies with supramolecular complexes imply high-energy water as a noncovalent driving force. Angew Chemie Int Ed. 2014;53:11158–71. doi: 10.1002/anie.201310958. [DOI] [PubMed] [Google Scholar]
- 97.Jordan JH, Gibb BC. Molecular containers assembled through the hydrophobic effect. Chem Soc Rev. 2015;44:547–85. doi: 10.1039/c4cs00191e. [DOI] [PubMed] [Google Scholar]
- 98.Crini G. Review: A history of cyclodextrins. Chem Rev. 2014;114:10940–75. doi: 10.1021/cr500081p. [DOI] [PubMed] [Google Scholar]
- 99.Appel EA, del Barrio J, Loh XJ, Scherman OA. Supramolecular polymeric hydrogels. Chem Soc Rev. 2012;41:6195–214. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 100.Oshovsky GV, Reinhoudt DN, Verboom W. Supramolecular chemistry in water. Angew Chemie Int Ed. 2007;46:2366–93. doi: 10.1002/anie.200602815. [DOI] [PubMed] [Google Scholar]
- 101.Biros SM, Rebek J., Jr Structure and binding properties of water-soluble cavitands and capsules. Chem Soc Rev. 2007;36:93–104. doi: 10.1039/b508530f. [DOI] [PubMed] [Google Scholar]
- 102.Corbellini F, Knegtel RM, Grootenhuis PD, Crego-Calama M, Reinhoudt DN. Water-soluble molecular capsules: self-assembly and binding properties. Chemistry. 2004;11:298–307. doi: 10.1002/chem.200400849. [DOI] [PubMed] [Google Scholar]
- 103.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed. 2003;42:1210–50. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 104.Xue M, Yang Y, Chi X, Zhang Z, Huang F. Pillararenes, a new class of macrocycles for supramolecular chemistry. Acc Chem Res. 2012;45:1294–308. doi: 10.1021/ar2003418. [DOI] [PubMed] [Google Scholar]
- 105.Strutt NL, Zhang H, Schneebeli ST, Stoddart JF. Functionalizing pillar[n]arenes. Acc Chem Res. 2014;47:2631–42. doi: 10.1021/ar500177d. [DOI] [PubMed] [Google Scholar]
- 106.Barboiu M, Gilles A. From natural to bioassisted and biomimetic artificial water channel systems. Acc Chem Res. 2013;46:2814–23. doi: 10.1021/ar400025e. [DOI] [PubMed] [Google Scholar]
- 107.Si W, Chen L, Hu XB, Tang G, Chen Z, et al. Selective artificial transmembrane channels for protons by formation of water wires. Angew Chemie Int Ed. 2011;50:12564–8. doi: 10.1002/anie.201106857. [DOI] [PubMed] [Google Scholar]
- 108.Hu XB, Chen Z, Tang G, Hou JL, Li ZT. Single-molecular artificial transmembrane water channels. J Am Chem Soc. 2012;134:8384–7. doi: 10.1021/ja302292c. [DOI] [PubMed] [Google Scholar]
- 109.Freeman WA, Mock WL, Shih NY. Cucurbituril. J Am Chem Soc. 1981;103:7367–8. [Google Scholar]
- 110.Lagona J, Mukhopadhyay P, Chakrabarti S, Isaacs L. The Cucurbit[n]uril Family. Angew Chem Int Ed. 2005;44:4844–70. doi: 10.1002/anie.200460675. [DOI] [PubMed] [Google Scholar]
- 111.Moghaddam S, Yang C, Rekharsky M, Ko YH, Kim K, et al. New ultrahigh affinity host-guest complexes of cucurbit[7]uril with bicyclo[2.2.2]octane and adamantane guests: thermodynamic analysis and evaluation of M2 affinity calculations. J Am Chem Soc. 2011;133:3570–81. doi: 10.1021/ja109904u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cao L, Sekutor M, Zavalij PY, Mlinaric-Majerski K, Glaser R, Isaacs L. Cucurbit[7]uril-guest pair with an attomolar dissociation constant. Angew Chemie Int Ed. 2014;53:988–93. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]
- 113.Zouhair A, Böhmer V, Harrowfield J, Vicens J, Saadioui M, editors. Calixarenes 2001. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- 114.Ewell J, Gibb BC, Rick SW. Water inside a hydrophobic cavitand molecule. J Phys Chem B. 2008;112:10272–9. doi: 10.1021/jp804429n. [DOI] [PubMed] [Google Scholar]
- 115.Hummer G, Rasaiah JC, Noworyta JP. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–90. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 116.Kofinger J, Hummer G, Dellago C. Single-file water in nanopores. Phys Chem Chem Phys : PCCP. 2011;13:15403–17. doi: 10.1039/c1cp21086f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kalra A, Garde S, Hummer G. Osmotic water transport through carbon nanotube membranes. Proc Natl Acad Sci USA. 2003;100:10175–80. doi: 10.1073/pnas.1633354100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ohba T, Kaneko K, Endo M, Hata K, Kanoh H. Rapid water transportation through narrow one-dimensional channels by restricted hydrogen bonds. Langmuir. 2013;29:1077–82. doi: 10.1021/la303570u. [DOI] [PubMed] [Google Scholar]
- 119.Takaiwa D, Hatano I, Koga K, Tanaka H. Phase diagram of water in carbon nanotubes. Proc Natl Acad Sci USA. 2008;105:39–43. doi: 10.1073/pnas.0707917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ohba T. Size-dependent water structures in carbon nanotubes. Angew Chemie Int Ed. 2014;53:8032–6. doi: 10.1002/anie.201403839. [DOI] [PubMed] [Google Scholar]
- 121.Pascal TA, Goddard WA, Jung Y. Entropy and the driving force for the filling of carbon nanotubes with water. Proc Natl Acad Sci USA. 2011;108:11794–8. doi: 10.1073/pnas.1108073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mashl RJ, Joseph S, Aluru NR, Jakobsson E. Anomalously immobilized water: a new water phase induced by confinement in nanotubes. Nano Letters. 2003;3:589–92. [Google Scholar]
- 123.Kyakuno H, Matsuda K, Yahiro H, Inami Y, Fukuoka T, et al. Confined water inside single-walled carbon nanotubes: global phase diagram and effect of finite length. J Chem Phys. 2011;134:244501. doi: 10.1063/1.3593064. [DOI] [PubMed] [Google Scholar]
- 124.Koga K, Gao GT, Tanaka H, Zeng XC. Formation of ordered ice nanotubes inside carbon nanotubes. Nature. 2001;412:802–5. doi: 10.1038/35090532. [DOI] [PubMed] [Google Scholar]
- 125.Mikami F, Matsuda K, Kataura H, Maniwa Y. Dielectric properties of water inside single-walled carbon nanotubes. Nano. 2009;3:1279–87. doi: 10.1021/nn900221t. [DOI] [PubMed] [Google Scholar]
- 126.Tanaka H, Koga K. Formation of ice nanotube with hydrophobic guests inside carbon nanotube. J Chem Phys. 2005;123:94706. doi: 10.1063/1.2031127. [DOI] [PubMed] [Google Scholar]
- 127.Setny P, Geller M. Water properties inside nanoscopic hydrophobic pocket studied by computer simulations. J Chem Phys. 2006;125:144717. doi: 10.1063/1.2355487. [DOI] [PubMed] [Google Scholar]
- 128.Setny P. Water properties and potential of mean force for hydrophobic interactions of methane and nanoscopic pockets studied by computer simulations. J Chem Phys. 2007;127:054505. doi: 10.1063/1.2749250. [DOI] [PubMed] [Google Scholar]
- 129.Setny P. Hydrophobic interactions between methane and a nanoscopic pocket: three dimensional distribution of potential of mean force revealed by computer simulations. J Chem Phys. 2008;128:125105. doi: 10.1063/1.2839885. [DOI] [PubMed] [Google Scholar]
- 130.Baron R, Setny P, McCammon JA. Water in cavity-ligand recognition. J Am Chem Soc. 2010;132:12091–7. doi: 10.1021/ja1050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Setny P, Baron R, McCammon JA. How can hydrophobic association be enthalpy driven? J Chem Theory Comput. 2010;6:2866–71. doi: 10.1021/ct1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Baron R, Setny P, Paesani F. Water structure, dynamics, and spectral signatures: changes upon model cavity-ligand recognition. J Phys Chem B. 2012;116:13774–80. doi: 10.1021/jp309373q. [DOI] [PubMed] [Google Scholar]
- 133.Setny P, Baron R, Michael Kekenes-Huskey P, McCammon JA, Dzubiella J. Solvent fluctuations in hydrophobic cavity-ligand binding kinetics. Proc Natl Acad Sci USA. 2013;110:1197–202. doi: 10.1073/pnas.1221231110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Young T, Abel R, Kim B, Berne BJ, Friesner RA. Motifs for molecular recognition exploiting hydrophobic enclosure in protein-ligand binding. Proc Natl Acad Sci USA. 2007;104:808–13. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Young T, Hua L, Huang X, Abel R, Friesner R, Berne BJ. Dewetting transitions in protein cavities. Proteins. 2010;78:1856–69. doi: 10.1002/prot.22699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Raghavender US, Kantharaju, Aravinda S, Shamala N, Balaram P. Hydrophobic peptide channels and encapsulated water wires. J Am Chem Soc. 2010;132:1075–86. doi: 10.1021/ja9083978. [DOI] [PubMed] [Google Scholar]
- 137.Ghadiri MR, Kobayashi K, Granja JR, Chadha RK, McRee DE. The structural and thermodynamic basis for the formation of self-assembled peptide nanotubes. Angew Chemie Int Ed. 1995;34:93–5. [Google Scholar]
- 138.Wang L, Berne BJ, Friesner RA. Ligand binding to protein-binding pockets with wet and dry regions. Proc Natl Acad Sci USA. 2011;108:1326–30. doi: 10.1073/pnas.1016793108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dunitz JD. The entropic cost of bound water in crystals and biomolecules. Science. 1994;264:670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- 140.Ladbury JE. Just add water! The effect of water on the specificity of protein- ligand binding sites And its potential application to drug design. Chemistry and Biology. 1996;3:973–80. doi: 10.1016/s1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- 141.Olano LR, Rick SW. Hydration free energies and entropies for water in protein interiors. J Am Chem Soc. 2004;126:7991–8000. doi: 10.1021/ja049701c. [DOI] [PubMed] [Google Scholar]
- 142.Yin H, Hummer G, Rasaiah JC. Metastable water clusters in the nonpolar cavities of the thermostable protein tetrabrachion. J Am Chem Soc. 2007;129:7369–77. doi: 10.1021/ja070456h. [DOI] [PubMed] [Google Scholar]
- 143.Barratt E, Bingham RJ, Warner DJ, Laughton CA, Phillips SE, Homans SW. Van der Waals interactions dominate ligand-protein association in a protein binding site occluded from solvent water. J Am Chem Soc. 2005;127:11827–34. doi: 10.1021/ja0527525. [DOI] [PubMed] [Google Scholar]
- 144.Malham R, Johnstone S, Bingham RJ, Barratt E, Phillips SE, et al. Strong solute-solute dispersive interactions in a protein-ligand complex. J Am Chem Soc. 2005;127:17061–7. doi: 10.1021/ja055454g. [DOI] [PubMed] [Google Scholar]
- 145.Shimokhina N, Bronowska A, Homans SW. Contribution of ligand desolvation to binding thermodynamics in a ligand-protein interaction. Angew Chemie Int Ed. 2006;45:6374–6. doi: 10.1002/anie.200602227. [DOI] [PubMed] [Google Scholar]
- 146.Syme NR, Dennis C, Phillips SE, Homans SW. Origin of heat capacity changes in a “nonclassical” hydrophobic interaction. Chembiochem. 2007;8:1509–11. doi: 10.1002/cbic.200700281. [DOI] [PMC free article] [PubMed] [Google Scholar]