

Supplemental Digital Content is available in the text
Keywords: dilated cardiomyopathy, familial, gene, KCNJ12 channel
Abstract
Dilated cardiomyopathy (DCM) is characterized by left ventricular dilation, and is associated with systolic dysfunction and increased action potential duration. Approximately 50% of DCM cases are caused by inherited gene mutations with genetic and phenotypic heterogeneity. Next generation sequencing may be useful in screening unknown mutations in such cases.
A family was identified with DCM, in which the affected family members developed heart failure, arrhythmia, and sudden death. Probands and 4 affected family members underwent whole exome sequencing (WES), bioinformatics methods, and gene annotation to identify potentially causative variants. The Sanger sequencing method was used to verify the candidate mutation.
WES yielded 2,238,831 variations. KCNJ12 (p.Glu334del) was identified as a candidate mutation, and the heterozygous mutation was verified by Sanger sequencing.
Our study emphasizes the application of WES in identifying causative mutations in DCM. This report is the first to describe the KCNJ12 gene as a cause of DCM in patients.
1. Introduction
Dilated cardiomyopathy (DCM) is a myocardial disorder characterized by left ventricular chamber enlargement and systolic dysfunction, which often leads to heart failure, arrhythmia, and sudden death. Approximately 50% of DCM cases have a genetic and heritable etiology.[1] Furthermore, approximately 90% of familial DCM cases are autosomal dominant, and <10% of these cases are X-linked recessive and autosomal recessive.[2] Based on the results of family studies, over 30 genes have been linked to familial DCM,[3] and there is considerable genetic heterogeneity with implicated genes that encode cytoskeletal, nuclear membrane, mitochondrial, and calcium-handing proteins.[4] In a previous study, we have proven that CHRM2 is a gene associated with FDCM.[5]
KCNJ12 encodes ATP-sensitive inward rectifier potassium channel 12, and contributes to the cardiac inwardly rectifying potassium current (IK1). IK1 stabilizes resting membrane potential, and is responsible for shaping the initial depolarization and final repolarization of the action potential.[6] Compared with ischemic cardiomyopathy (ICM), the IK1 channel characteristics in ventricular myocytes obtained from patients with DCM exhibited an action potential that had a longer duration and a slower repolarization phase, a lower resting membrane potential, and a smaller whole cell current slope conductance.[7]KCNJ12 mutations have been published in the COSMIC (v64) database, but were filtered out from head and neck squamous cell carcinoma[8] and colon and rectal cancer.[9] By using whole exome sequencing (WES) technology and gene annotation, we have shown that KCNJ12 is another candidate gene linked to FDCM; and these results were validated using the Sanger sequencing method.
2. Materials and methods
2.1. Sample collection and exome sequencing
Genomic DNA was purified from blood leukocytes obtained from 5 family members (patients) of this DCM pedigree. Exome capture was performed on an Illumina Nextera 62 M Rapid Capture array (Illumina Inc., San Diego, CA). Then, exon-enriched DNA libraries were sequenced by the Illumina Hiseq 2500 platform (Biomarker Technologies, South Faxinfu, Shunyi District).
2.2. Data mapping, SNP calling, and filtering
BWA software[10] was used to align the sequencing reads to the human genome reference assembly (hg 19). The mapping efficiency of these 5 exome sequencing data to the human genome was 95% to 96%, and the coverage of the target region was from 97.71% to 99.15%.
Next, SNP calling and indel calling were taken using GATK software tools.[11] With the mapping result file (SAM file or BAM file) of clean reads on the genome reference, the SAM tools software (a part of the GATK software) was used to first mark duplicates. Then, local realignment, base recalibration, SNP calling, and indel calling were performed.
Highly confident SNPs and indels, which were not deposited in the dbSNP database (v138) and 1000 Genomes database (MAF>0.05), were extracted for subsequent analysis.
2.3. Family-based linkage analysis
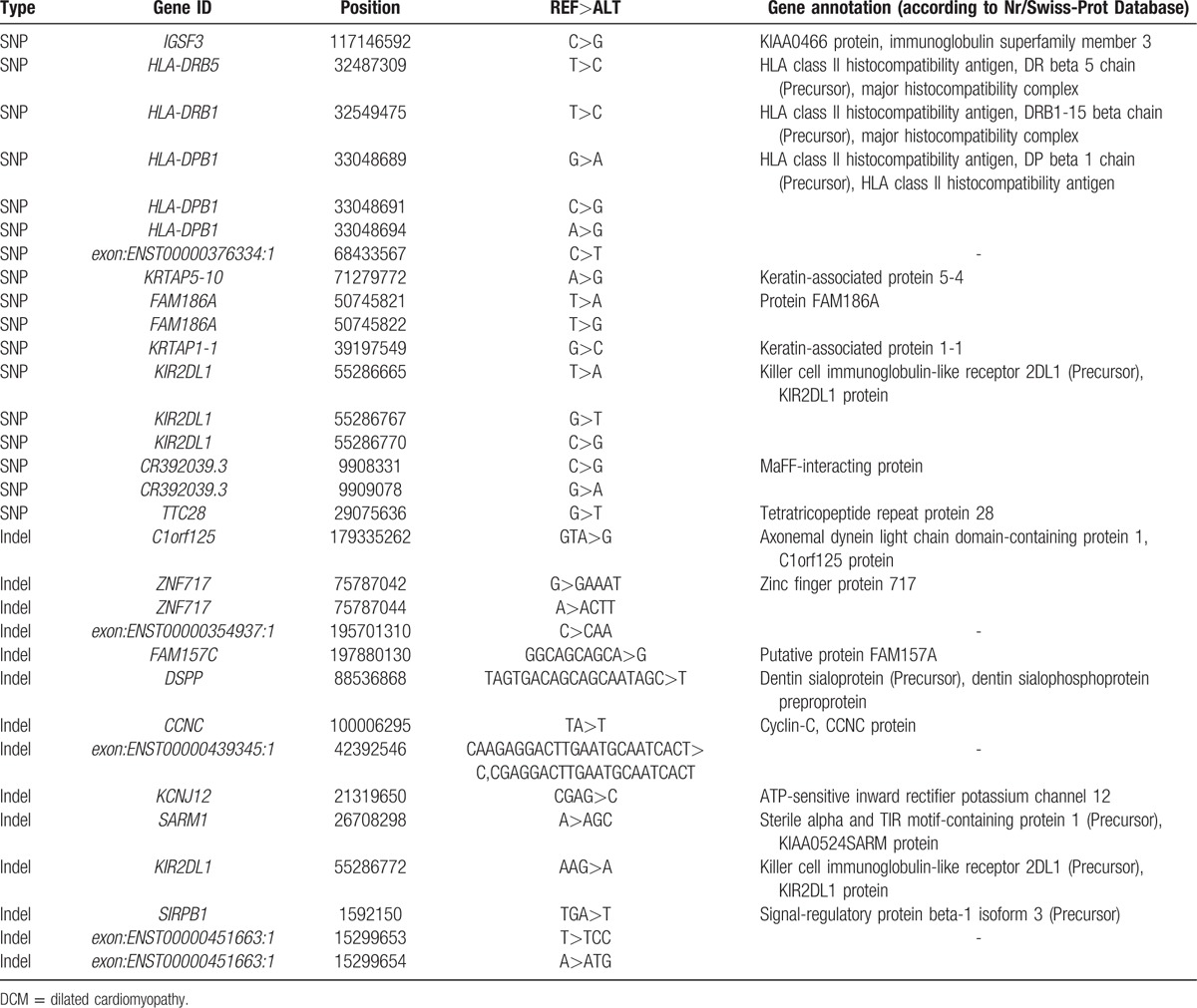
By using a family-based linkage analysis method, 11 genes with nonsynonymous SNPs and 12 genes with indel that were common to all 5 family members (patients) were found.
2.4. Sanger sequencing of KCNJ12 within the DCM pedigree
The mutation and flanking regions of the KCNJ12 gene for all 5 family members with DCM was sequenced using the Sanger sequencing method. The KCNJ12 gene has multiple similar sequences in the inwardly rectifying potassium channel subfamily members. To avoid nonspecial amplification, the PCR primer pairs were designed to mismatch similar sequences at the 3′ end, and were verified using primer BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome). The primer sequences used were 5′-TCGCCCATCACCATCTTGCATGAG-3′ for the sense strand and 5′-CCTGGGGGCTGAGGCCG-3′ for the antisense strand. Sequencing reactions were performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies; Thermo Fisher Scientific, Shanghai, China), and products were resolved on ABI Prism 3730 Analyzers (Life Technologies).
3. Results
3.1. Clinical characteristics
A Chinese family with autosomal dominant, familial DCM was studied (Fig. 1A). This family experienced progressive DCM that resulted in arrhythmias, heart failure, and sudden death. Individuals I:1 and II:1 died of sudden deaths at ages 61 and 47, respectively. Five members (II:3, II:5, III:1, III:3, and III:5) of the descendants had DCM and arrhythmia. IV:1 refused to attend the detection. The diagnosis of DCM was made in accordance with the criteria established by the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Classification of Cardiomyopathy. Familial DCM patients were evaluated by collecting their detailed clinical history, physical examination, 12-lead ECG, 24-hour ambulatory ECG, chest radiography, and transthoracic echocardiography (M-mode, 3D, and Doppler). The diagnostic criteria for DCM were defined as an ejection fraction <50% and/or left ventricular shortening fraction of 28% by echocardiography analysis, regional fractional shortening on M-mode analysis, or both in the presence of a left ventricular internal diastolic diameter ≥2.7 cm/m2 of the body surface area. The study protocol was approved by the Ethics Committee of Beijing Chao-Yang Hospital, and conforms to the Declaration of Helsinki. All participants provided an informed written consent.
Figure 1.
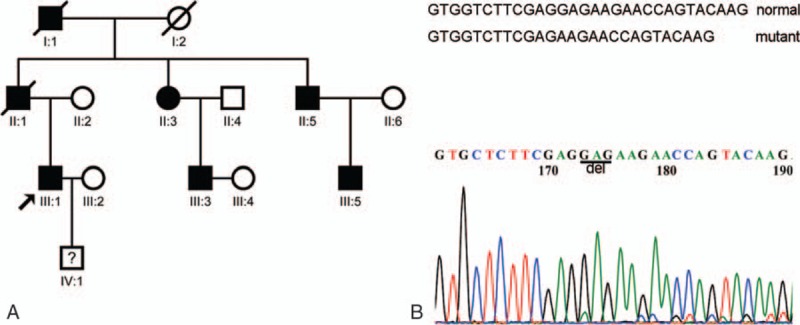
Mutation of the FDCM pedigree. (A) The pedigree shows an autosomal dominant inheritance pattern. Five affected members were selected to give samples for WES, the results of which were verified by the Sanger sequencing method. (B) Sanger sequencing confirmed the GAG base deletion in the KCNJ12 gene sequence, which produces a Glu deletion in the 333 amino acid sequence of the Kir2.2 protein. WES = whole exome sequencing.
3.2. Whole exome sequencing and variant calling
The whole exomes of II:3, II:5, III:1, III:3, and III:5 were sequenced using an NGS strategy to screen for the causal genes of the familial DCM pedigree. IV:1 did not have a DCM phenotype, but refused to provide a blood sample.
The read depth achieved by the exome sequencing ranged from 66.56- to 111.3-fold (Supplementary Table 1), and detected 1,961,054 SNPs (Supplementary Tables 2 and 3) and 277,777 small indels (Supplementary Tables 4 and 5). After removing variants with global MAF >0.05 in the database of the dbSNP138 or 1000 Genomes Project, highly confident SNPs and indels were extracted for subsequent analysis. Eleven genes with nonsynonymous SNPs and 12 genes with indels that appeared together in all 5 family members were found (Table 1). To narrow the list of candidates further, gene annotation was used to search the Swiss-Prot database and identify the KCNJ12 gene that codes for a protein that participates in maintaining the resting membrane potential and establishes the action potential in electrically excitable cells, allowing potassium to flow into rather than out of the cells.
Table 1.
Statistics of 11 genes with nonsynonymous SNP and 12 genes with indel in the DCM pedigree.
3.3. Sanger sequencing of KCNJ12 within the familial DCM pedigree
Using the Sanger sequencing method, the mutation of the KCNJ12 gene (p.Glu334del) in all 5 affected family members was detected. This indel mutation (CGAG>C) was fully heterozygous within the familial DCM phenotype. The mutation was searched in an exome sequencing database (http://evs.gs.washington.edu/), and the variant in either the 4300 individuals of European ancestry or 2202 individuals of African ancestry was not found. This result validates that the KCNJ12 gene (p.Glu334del) variant was the causal variation in this pedigree (Fig. 1B).
4. Discussion
Inwardly rectifying K+ (Kir) channels allow K+ ions to move more easily into rather than out of the cell. There are 7 Kir channel subfamilies that can be classified into 4 functional groups, depending on their type and location. Cardiac KATP channels are heteromultimers composed of Kir6.2 and the regulatory SUR2A subunit, which is an ATPase-harboring ATP-binding cassette protein. Cardiac KATP channels are linked to cellular metabolism. Mutations in ABCC9 lead to the aberrant redistribution of the SUR2A protein conformation in the intrinsic ATP hydrolytic cycle, which translates into abnormal KATP channel phenotypes with compromised metabolic signal decoding. This dysfunction of the cardiac KATP channels has a proven correlation with DCM.[12] Gene KCND2 encodes the α-subunits of the voltage-gated potassium channel Kv4.2. Transgenic myocytes with dominant-negative N-terminal fragments of Kv4.2 demonstrated a reduction of transient outward K+ currents (Ito) and IK1 densities, and an increase in the duration of the action potential. T transgenic adult mice developed DCM with reduced peak aortic pressures, reduced peak systolic ventricular pressures, and increased LV end-diastolic pressures.[13]
The Kir2.x subfamilies (kir2.1, kir2.2, and kir2.3) regulate the inward rectifier current, Ik1, which affects the terminal phase of repolarization of the action potential and the stability of the resting membrane potential; and is involved in the arrhythmia that occurs in DCM patients.[14] The Ik1 current is conducted via ion channels, which consist of heteromeric assemblies of Kir2.1, Kir2.2, and Kir2.3 α subunits. Compared with the hearts of healthy controls obtained from organ donors, the gene and protein expression of Kir2.1 and Kir2.3 in the hearts of DCM patients who underwent heart transplants were upregulated; but those of Kir2.2 channels were downregulated.[15] In our present study, the identification of p.Glu334del of the KCNJ12 () gene by WES and the gene sequence annotation of samples taken from a family with DCM provided direct evidence that confirmed the relationship between FDCM and Kir2.2.
The activity of Kir channels depends critically on their conformation, which is modulated by phosphatidylinositol 4,5-bisphosphate (PIP2) and cholesterol. Kir2.2 channels are formed by the homotetrameric association of Kir2.2 subunits to form pore-conducting pathways, and do not contain tightly associated nonconducting auxiliary subunits.[16,17] The Kir channel is made of 2 transmembrane helices with cytoplasmic NH(2) and COOH termini, and an extracellular loop that folds back to form the pore-lining ion selectivity filter. PIP2 binds to and directly activates Kir2.2 with agonist-like properties. PIP2 binds at an interface between the transmembrane domain (TMD) and the cytoplasmic domain (CTD). The binding produces a large conformational change to initiate pore opening in Kir2.2.[18,19] CTDs of Kir channels modulate gating and inward rectification by structural changes. In the absence of PIP2, inward K+ current is occluded by ion-permeation pathways constructed by 4 cytoplasmic loops that form a girdle around the central pore (G-loop). The core structures of the CTD contain 3 β-sheets and 2 α-helices.[20] Mutations in these locations directly or indirectly affect channel activity by on-gating and particularly off-gating kinetics. The Kir2.2 (p.Glu334del) mutation is located in the third β sheet of the Kid2.2 CTD. We speculate that it influences the activity of the inward rectifier potassium channel by changing the core structure of the cytoplasmic pore, or by interacting with PIP2 or the modulator of cholesterol.
4.1. Limitations
There is an important limitation with the IV:1 absent. Although IK1 have been reported as correlated with DCM and arrhythmia, the gene KCNJ12 is the first identified. We hope to screen more DCM kindred to investigate the mutation of KCNJ12. We intend to further disclose the biological mechanism of KCNJ12 mutation in DCM at different levels.
5. Conclusions
Our study demonstrates that KCNJ12 gene plays an important role in the alteration of cardiac structure and conduction, and suggests that the KCNJ12 (p.Glu334del) may be a pathogenic mutation. In addition, we have observed that the patients who carry the KCNJ12 (p.Glu334del) have been characterized by DCM and arrhythmia. To our knowledge, this is the first report linking a mutation in the KCNJ12 gene to familial DCM.
In summary, our data provides direct evidence at a molecular level that in addition to IK1 variation in the action potential and resting membrane potential,[7] and Kir2.2 downregulation,[15] the KCNJ12 gene plays a role in DCM pathogenesis.
Acknowledgment
We would like to thank the members of the family with DCM that participated in this study.
Supplementary Material
Footnotes
Abbreviations: CTD = cytoplasmic domain, DCM = dilated cardiomyopathy, ICM = ischemic cardiomyopathy, TMD = transmembrane domain, WES = whole exome sequencing.
H-XY and KY contributed equally to this project.
This work was supported by the National Natural Science Foundation of China (81370340) and the Beijing Natural Science Foundation (7132103).
The authors report no conflicts of interest.
Data were submitted to the SRA database, available at: http://www.ncbi.nlm.nih.gov/gquery/?term=SRP066837. (Notes: Accession:SRX1457497 from II:2; Accession:SRX1457499 from II:5; Accession:SRX1457501 from III:1; Accession:SRX1457503 from III:3; Accession:SRX1457504 from III:5)
Supplemental Digital Content is available for this article.
References
- [1].Piran S, Liu P, Morales A, et al. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J Am Coll Cardiol 2012;60:283–9. [DOI] [PubMed] [Google Scholar]
- [2].Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010;31:2715–26. [DOI] [PubMed] [Google Scholar]
- [3].Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–47. [DOI] [PubMed] [Google Scholar]
- [4].McNally EM, Golbus JR, Puckelwartz MJ, et al. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest 2013;123:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang L, Hu A, Yuan H, et al. A missense mutation in the CHRM2 gene is associated with familial dilated cardiomyopathy. Circ Res 2008;102:1426–32. [DOI] [PubMed] [Google Scholar]
- [6].Dhamoon AS, Jalife J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005;2:316–24. [DOI] [PubMed] [Google Scholar]
- [7].Koumi S, Backer CL, Arentzen CE, et al. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patients with idiopathic dilated cardiomyopathy. Circulation 1995;92:164–74. [DOI] [PubMed] [Google Scholar]
- [8].Martin D, Abba MC, Molinolo AA, et al. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget 2014;5:8906–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bienengraeber M, Olson TM, Selivanov VA, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet 2004;36:382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wickenden AD, Lee P, Sah R, et al. Targeted expression of a dominant-negative K(v)4.2 K(+) channel subunit in the mouse heart. Circ Res 1999;85:1067–76. [DOI] [PubMed] [Google Scholar]
- [14].Cordeiro JM, Zeina T, Goodrow R, et al. Regional variation of the inwardly rectifying potassium current in the canine heart and the contributions to differences in action potential repolarization. J Mol Cell Cardiol 2015;84:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Szuts V, Ménesi D, Varga-Orvos Z, et al. Altered expression of genes for Kir ion channels in dilated cardiomyopathy. Can J Physiol Pharmacol 2013;91:648–56. [DOI] [PubMed] [Google Scholar]
- [16].Raab-Graham KF, Vandenberg CA. Tetrameric subunit structure of the native brain inwardly rectifying potassium channel Kir 2.2. J Biol Chem 1998;273:19699–707. [DOI] [PubMed] [Google Scholar]
- [17].Hugnot JP, Pedeutour F, Le Calvez C, et al. The human inward rectifying K+ channel Kir 2.2 (KCNJ12) gene: gene structure, assignment to chromosome 17p11.1, and identification of a simple tandem repeat polymorphism. Genomics 1997;39:113–6. [DOI] [PubMed] [Google Scholar]
- [18].Li J, Lü S, Liu Y, et al. Identification of the conformational transition pathway in PIP2 opening Kir channels. Sci Rep 2015;5:11289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 2011;477:495–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pegan S, Arrabit C, Zhou W, et al. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 2005;8:279–87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.