

Abstract
Methylation of CpG motifs in DNA is involved in the control of gene expression and in several other epigenic effects. It suppresses also the immuno-stimulation properties of bacterial or viral DNAs that contain CpGs. However, effects of methylation on the DNA structure and dynamics are not clear. Here we carried out a 10 ns MD simulation, confronted to an NMR analysis, of a hexadecanucleotide with the cAMP responsive element (CRE) DNA methylated at its center: d(GAGATGAmCGTCATCTC)2 (CREmet). Methylation does not introduce significant structure modification but reduces the dynamics. Molecular mechanics and generalized Born solvation energy calculations showed that the stiffness of CREmet arises from both a restriction of the conformational space by the bulky methyl groups and a folding of DNA around the hydrophobic methyls. The latter effect is favored when the GpA steps belonging to the TGA binding half-sites adopt the BII conformation. The inability of the methylated DNAs to interact with their protein partners—either transcription factors for gene regulation or a Toll-like receptor for immunostimulation—could result from both the obstacle created by methyls, preventing crucial interactions, and the loss of DNA flexibility, reducing its adaptability. Results are discussed in the light of NMR and crystallographic data.
INTRODUCTION
Methylation of CpG cytosines in DNA is thought to be an epigenetic event that plays a crucial role in the control of gene expression in mammalian cells. It is essential for embryonic development, inactivation of specific genes on the X chromosome and parental imprinting (1). It also plays a role in chromatin structure and function (2). Moreover, the silencing of many genes involved in tumorigenesis by DNA methylation suggests that this event is important in cancer development (3,4). Actually, DNA methylation is a potent suppressor of gene activity, especially when it occurs in the promoter region of genes (3) and one possible mechanism for gene repression is the binding inhibition of transcription factors to their specific sites (5). For instance, the binding of the transcription factors ATF/CREB to the cAMP DNA responsive element [CRE: d(TGACGTCA)2] is inhibited by the methylation of the CpG motif located at the center of CRE between the binding half-sites (6). Inhibition occurs despite the fact that the modified cytosines in the CpG motif are not directly contacted by the proteins (6).
Another striking point is the immunostimulatory properties exhibited by DNA fragments containing CpG motifs with two purines before and two pyrimidines after (7), as it is the case for the CRE consensus sequence. Methylation of CpG suppresses these properties, likely through inhibition of the oligonucleotide binding to a Toll-like receptor, newly discovered, that triggers the immune response (8). However, the structural reasons for the loss of activity are not known.
X-ray and NMR studies, including the present NMR analysis, of singly CpG-methylated DNA indicate that the structure of oligomers is not significantly altered by methylation (9–12). However, a recent solid-state deuterium NMR investigation of the EcoRI restriction binding site has revealed a significant modification of the DNA dynamics at the methylation site (13).
The present MD simulation and NMR study were performed mainly in order to understand the fine structural and dynamic effects of methylation on the recognition properties of CRE. We used a palindromic 16mer duplex d(GAGATGAmCGTCATCTC)2, termed CREmet, that contains the C5-cytosine methylated CRE consensus sequence d(TGAmCGTCA)2 at its center. At least two reasons motivated the choice of this hexadecaoligonucleotide: it is the minimal size to obtain stable interactions of CRE with the bZIP transcription factors (14), and the refined NMR and MD structures of the unmethylated 16mer, CREnat, are available (15,16).
Globally, results from the 10 ns MD simulations of CREnat and CREmet revealed significantly different dynamics, which were not amenable to the NMR analysis. The most important feature is that CpG methylation reduces the DNA flexibility, especially regarding the phosphodiester backbone at the TGA binding half-sites flanking CpG. Molecular mechanics and generalized Born solvation energy calculations revealed that the relative stiffness of methylated CRE is caused by both a reduction of the accessible conformational space due to the steric hindrance of the bulky methyl groups, and to a kind of folding of the DNA molecule around the hydrophobic methyl groups. The ‘folding’ of CREmet is generated by the blocking in the BII conformation of the GpA phosphate at the TGA half-sites, while native CRE undergoes spontaneous BI/BII transitions. This mechanism appears to compensate for a degradation of the solvation energy due to the addition of methyl groups.
The binding inhibition of the specific protein factors to methylated CRE could be explained by two concurring structural effects: (i) the obstacle created by the methyl groups, which sterically hinder the specific contacts between the conserved residues Arg301/Arg301′ and the CpG motif (17), and (ii) the loss of flexibility induced by CpG methylation that makes the adaptation of CREmet to protein partners difficult.
Although this work deals with the methylation effects on the functionality of CRE as a response element, present results could be used to explain the antagonizing effects of methylation on the immunostimulatory properties of CpG-containing DNA fragments.
MATERIALS AND METHODS
Molecular dynamics
MD simulation was carried out under NPT conditions (303 K, 1 atm) with the suite of programs AMBER 4.1 (18) and the modified version of the Cornell et al. force field (parm95), parm98 (19,20), identically to MD simulation of unmethylated CRE (16). Water was simulated with the TIP3P model (21). All bond lengths involving hydrogen atoms were restrained using the SHAKE algorithm (tolerance = 0.0005) (22), and a time step of 2 fs was used. Electrostatic interactions were calculated using the Particle Mesh Ewald summation method, with a 10–6 Ewald convergence tolerance (23,24). Methylated cytosines were added to the residue library of AMBER. The charges of the methylated cytosines were parameterized consistently to the Cornell et al. (19) force field (Dennis Sprous, TRIPOS, personal communication), i.e. by ab initio HF/6–31G** calculations followed by RESP fitting (18,19). The charges of the methylated and unmethylated cytosines are reported in Table 1. Periodic boundary conditions were used with a rectangular unit cell. The initial box was truncated to achieve a minimum distance of ∼11 Å beyond all DNA atoms in all directions, resulting in a box size of 49 × 49 × 87 Å and 4750 water molecules. Thirty neutralizing Na cations were placed using a simple energy minimization algorithm (Leap module of AMBER 5.0). The starting DNA conformation was the AMBER-generated canonical B-DNA double helix. The protocol followed for the minimization, heating, equilibration and production phases of the MD simulation was identical to the one used for CREnat, and has been previously described in detail (16). In summary, after 2250 cycles of energy minimization, the system was heated to 303 K over 10 ps. During each of these phases, harmonic constraints were imposed on the atomic positions of the oligomer and of the sodium counterions. After an NVE equilibration period of 5 ps at 303 K, the simulation was performed under NPT conditions (303 K, 1 atm) using coupling constants of 0.2 ps. The restraints were relaxed over a period of 25 ps until a free system was achieved, and a free equilibration was carried out for 5 ps. Then a Maxwell distribution of the velocities was assigned to the system and followed by 10 ps of free equilibration. This procedure was repeated before the beginning of the production phase and after 50 and 100 ps of production. During the 10 ns production phase, the translations and rotations of the DNA oligomer were removed every 100 steps of MD. The calculations were performed on a Silicon Graphics Origin200 workstation.
Table 1. Values of the atomic charges of the unmethylated (19) and methylated (Dennis Sprous, personal communication) cytosines used in the MD simulations of native and methylated CRE, respectively.
| Atom |
Unmethylated cytosine |
Methylated cytosine |
| N1 |
–0.0339 |
0.0325 |
| C6 |
–0.0183 |
–0.2117 |
| H6 |
0.2293 |
0.2334 |
| C5 |
–0.5222 |
–0.1611 |
| H5 |
0.1863 |
– |
| C7 |
– |
–0.0262 |
| H71 |
– |
0.0231 |
| H71 |
– |
0.0231 |
| H73 |
– |
0.0231 |
| C4 |
0.8439 |
0.7629 |
| N4 |
–0.9773 |
–0.9888 |
| H41 |
0.4314 |
0.4314 |
| H42 |
0.4314 |
0.4314 |
| N3 |
–0.7748 |
–0.7863 |
| C2 |
0.7959 |
0.8049 |
| O2 | –0.6548 | –0.6548 |
Structural analysis
We used the following numbering for the DNA sequence: (5′-G1A2G3A4T5G6A7mC8G9T10C11A12T13C14T15C16:G17A18G19A20T21G22A23mC24G25T26C27A28T29C30T31C32-3′). CURVES v5.2 was used to analyze the DNA structures stored every 1 ps of MD (25). This work refers to the local parameters output by CURVES. To avoid end effects, only the 14 central base pairs were taken into account for the analysis. For the MD simulation (10 ns of production), each parameter was measured 10 000 times.
MD results for CREmet are presented in comparison with the MD structure of CREnat (16). They are discussed relative to the experimental NMR data on CREmet and to a variety of published experimental data regarding the impact of C5-cytosine methylation on DNA structure. The 3.0 Å X-ray structure of unmethylated CRE bound to CREB (17) (PDB ID 1DH3) is used to propose possible explanations for the experimentally observed abolishment of specific factor binding to methylated CRE (6).
Calculation of internal and solvation energies
The energies of the molecules were estimated using the MM_GBSA approach implemented in AMBER 6.0. Internal energy was calculated with the ANAL program (18), and represents the sum of bond, angle, dihedral, van der Waals and electrostatic contributions of the in vacuo DNA–DNA interactions. An infinite cutoff was used for all interactions, and the dielectric was set to 1.0. The solvation energy was calculated with the GB and the MSMS programs, respectively for the electrostatic and the hydrophobic contributions. The hydrophobic contribution is determined with solvent-accessible surface area (SASA)-dependent term (26): Enonpolar = γ*SASA, where γ = 0.0072 kcal/(mol.Å2), taking a probe radius of 1.4 Å for the computation of the surface area. The electrostatic contribution is estimated with a modified generalized Born solvation model, which has been parameterized to be consistent with the Cornell et al. (19) AMBER atomic charges (27). The dielectric was set to 1.0 and 80.0 inside and outside the molecule, respectively. For each molecule, the energies were computed every ps of MD, and a statistical analysis gave the averaged values and the standard deviations.
NMR measurements
CREmet was synthesized by the solid-phase method, purified by reversed-phase HPLC, dialyzed against water and lyophilized. Oligonucleotide concentrations were determined by UV light absorption measurements at 260 nm using an extinction coefficient value of 7800 M–1cm–1base–1. The duplex was dissolved at 1.7 mM concentration in a phosphate buffer containing 0.2 mM EDTA at pH 7, ionic strength I = 0.1. NMR spectra were acquired at 500 MHz on an AMX 500 Bruker spectrometer and processed using an X32-BRUKER station or on a Silicon Graphics INDIGO R4000 work station with the 2.35 Felix Software (Biosym Technologies). NMR data collection and processing was made following the same protocol as that used for CREnat (15). In order to assign all non-exchangeable protons (except H5′1 and H5′2), COSY, TOCSY and NOESY spectra were measured in 2H2O; exchangeable protons in the amino groups of the cytosines were assigned according to NOESY-jr spectra recorded in H2O. All phosphorus signals were assigned according to hetero-TOCSY and hetero-COSY. All NMR experiments were conducted at 30°C.
RESULTS
Structure and dynamics of methylated versus native CRE
A MD trajectory of 10 ns was simulated with the methylated 16mer duplex d(GAGATGAmCGTCATCTC)2, CREmet. The atom coordinates were saved and analyzed every 1 ps, and the results take into account the trajectory after 500 ps from its starting point (i.e. data collected through 9.5 ns). Results were compared to our previous MD simulation on the native sequence d(GAGATGACGTCATCTC)2, CREnat (16). The latter has shown that a trajectory longer than 5 ns is necessary to get a convergence of the global DNA structural parameters (bending) for a 16mer duplex, and that 10 ns are sufficient to render the sequence dependency of the phosphodiester backbone conformation, the most flexible part of a DNA duplex.
We found that the average structure of CRE is only slightly modified by methylation. The root mean square deviation (r.m.s.d.) between the averaged MD structures of CREmet and CREnat for the coordinates of heavy atoms in the 14 central residues of the DNA (excluding the methyl groups of methylated cytosines) was only 0.2 Å, and the r.m.s.d. was >0.2 Å for the central CpG dinucleotide.
A careful examination of the inter-base pair, pair axis, base–base and sugar structural parameters showed that the values of CREnat (16) and CREmet averaged over the MD trajectories (0.5–10 ns) were not significantly different. For each parameter, the difference was less than half the standard deviation (SD). The most noticeable, although very slight, divergences were found for the twist angles at the TpG/CpA and GpA/TpC steps in the TGA:TCA half-sites, with averaged values of 32° and 36°, respectively, for CREnat, and of 29° and 38°, respectively, for CREmet. A slight but noticeable divergence was also found for the averaged shifts at the ApC/GpT steps situated between the TGA:TCA half-sites and the central CpG dinucleotide, with values of 0.3 and 0.6 Å for CREnat and CREmet, respectively.
Larger differences between CREmet and CREnat were reflected by the parameters’ SD, related to the oligomer’s local flexibilities. Globally, SD values were inferior in CREmet proving that methylation decreases the flexibility, especially in the central consensus sequence (TGACGTCA)2 (Fig. 1). In particular, the SD of twists at GpA and ApC in the consensus octamer were 7° and 6°, respectively, for CREnat, and 5° at both steps, for CREmet. The SD of the propeller twist at the central C:G base pairs was also significantly lower in CREmet (8°) compared to CREnat (9°).
Figure 1.

Standard deviations of the inter-base pair twist and shift parameters of CREnat (solid line) and CREmet (dashed line) as a function of the dinucleotide step. Angles are indicated in ° and distances in Å. Values are averaged over the two symmetrical half-sites.
Regarding the conformation of the phosphodiester backbone, the patterns of BI/BII (BI, ɛ in trans and ζ in gauche; BII, ɛ in gauche and ζ in trans) percentages reflect a sequence dependence that is moderately affected by the methylation (Fig. 2A). Curiously, no significant change was observed at the methylation site, CpG, while the BII percentage increased from 72 to 91% at the GpA step incorporated in the TGA binding half-site. The high BII percentage noted at the GpA step belonging to the half-site does not have its counterpart at the other GpA step flanked by the ApG and ApT steps. The BII percentage at the adjacent TpG and ApC steps also changes upon methylation. Globally, methylation seems to lower the flexibility of phosphate groups in the TGAC half-site, and a direct correlation was noted between the BI/BII transitions at GpA and its neighboring TpG and ApC steps, these passing from BI to BII only when GpA stays in BI. The BII populations at the TpG and ApC steps seem then under the control of the GpA step. A plot of the BII population at GpA as a function of the MD simulation length (Fig. 2B) showed that the difference between the CREnat and CREmet trajectories converges after 5 ns, and could not be due to a problem of MD simulation length resulting in a poor sampling of the backbone conformations. Actually, one observed many attempts of the GpA phosphate in CREmet to acceed the BI state, but they were unsuccessful and the phosphate returned to the BII state (Fig. 2C). The latter plot can be compared to that of CREnat given in Figure 2D.
Figure 2.

(A) MD percentage population of the BII state in CREnat (gray bars) and in CREmet (black bars) as a function of the dinucleotide step. (B) Percentage population of the BII state as a function of the simulation length, for CREnat (solid line) and CREmet (dashed line). (C) and (D) Difference between the ɛ and ζ dihedral angles at the GpA step in a TGAC half-site of CREmet (C) and of CREnat (D), as a function of the simulation time.
In summary, methylation alters the dynamics of CRE in rigidifying the DNA, especially the phosphodiester backbone as far as the BI/BII transitions are concerned. Rather surprisingly, effects are less pronounced at the methylated CpG step than at the neighboring steps. Moreover, effects are larger on the 5′ residues than on the 3′ residues and are even relayed to a step further in the 5′ direction, suggesting steric conflicts between the methylated cytosines and the 5′ residues in the major groove. In the next sections we will examine (i) whether any correlation exists between the observed BI/BII transitions at GpA and the other modified structural parameters, and (ii) whether the structural impact of CpG methylation is due to intrinsic DNA mechanical properties or rather to an indirect effect via the solvent.
Correlation between BI/BII transitions and helical parameters
Theoretical studies have shown that BI/BII transitions are associated with a change of some helical parameters like, for instance, the twist, the roll and the Xdisp (28,29). Such correlations were also observed in MD trajectories of CREnat and CREmet for BI/BII transitions and variations of twist, slide and shift parameters at the GpA steps. The BII to BI transition at GpA paralleled the decrease of the twist at GpA/TpC and the increase of the twist at TpG/CpA, the decrease of the slide at GpA/TpC and the decrease of the absolute values of the shifts at GpA/TpC and ApC/GpT (data not shown). The correlation with the twist at ApC/GpT and with the propeller twist at C:G was less clear, although CREnat and CREmet displayed a different behavior (Fig. 3). The transition from BII to BI resulted in a slight increase and decrease of the twist at ApC/GpT in CREnat and CREmet, respectively (Fig. 3A). It had no noticeable impact on the propeller at C:G in CREnat while it slightly lowered the propeller in CREmet (Fig. 3B). The ranges of values for the ApC twist and the C:G propeller for the CREmet BI state appeared as sub-domains of their corresponding ranges for the CREnat BI state: when CREmet was in BI, only twist values <35° and propeller twist values <25° were populated.
Figure 3.

(A) Relationship between the twist at ApC/GpT and the value of ɛ–ζ at GpA in the TGAC half-sites, in CREmet (left) and in CREnat (right). (B) Relationship between the propeller twist at the C:G base pairs and the value of ɛ–ζ at GpA in the TGAC half-sites, in CREmet (left) and in CREnat (right). Values are extracted every ps of the MD trajectories.
Therefore, one can deduce a correlation for the difference between the helical parameter values, and especially between the SD, and the difference of BI/BII populations at the GpA/TpC steps of CREnat and CREmet. The weaker flexibility of the CREmet helix is related to the higher rigidity of its backbone, that is to a lower rate of BI/BII transitions. Furthermore, in the BI state, CREnat appears more flexible than CREmet.
Curvature and flexibility
The BI occupation rate of GpA in each TGA:TCA half-site, was ∼1/3 for CREnat and only ∼1/10 for CREmet. Defining the BIIBII, BIBII and BIBI global DNA conformations as the DNA states with both GpA phosphates in BII, one in BI and one in BII, and both in BI, respectively, percentages of the BIIBII, BIBII and BIBI sub-states were found equal to 83, 16 and 1%, respectively, for CREmet, and 49, 48 and 3%, respectively, for CREnat (Fig. 4). Thus, CREmet displays a clear preference for a global BIIBII conformation, while CREnat appears much more flexible, adopting more freely the BI and BII conformations.
Figure 4.

Relationship between ɛ–ζ at the G6pA7 step and ɛ–ζ at the symmetrical G22pA23 step, in CREnat (left) and in CREmet (right). Values are extracted every ps of the MD trajectories.
Regarding the helix curvature in CREnat and CREmet, the averaged values and SD of rolls and tilts were very similar for their central octamers. The slight positive rolls at CpG (5.7 ± 7.3° and 7.0 ± 6.9° in CREnat and CREmet, respectively) and the tilts at GpA/TpC (3.8 ± 4.6° and 4.0 ± 4.4° in CREnat and CREmet, respectively) generated an averaged global curvature in both helices toward the major groove (∼7–8°) (16). But CREmet was slightly less flexible than CREnat also with regard to the DNA curvature (SD of rolls and tilts were smaller in CREmet at all dinucleotides).
Internal and solvation energies
The fact that CpG methylation exerts a global effect on the dynamics of the DNA helix is reflected by several correlated alterations, although their precise origin(s) cannot be detected. To search the involved mechanism, an energetic study was performed on the BI and BII sub-conformations of CREnat and CREmet. The internal and solvation energies were calculated using the MM_GBSA routine of the AMBER package.
The local energetic contribution of the methylated cytosines was calculated in order to identify the interactions that would strongly disfavor the BI state in CREmet. The van der Waals and electrostatic energies for the interactions of methylated cytosines with adjacent nucleotides were found to be similar in the BI and BII states. The ‘BI-phobicity’ of CREmet did not seem to result from a damage in the stacking energy of the central C:G base pairs. The more restricted ranges of values for the twist at ApC/GpT and the propeller twist at C:G in CREmet than in CREnat suggested that the methyl groups interfere with the helix rearrangements during BI/BII transitions (the r.m.s.d. between the averaged BI and BII sub-states were 0.9 Å for CREmet and 0.6 Å for CREnat). Reduction of the conformational space in the BI state of CREmet allows avoidance of a steric clash, but maintains a good local energetic contribution. The diminution of the BI occupation rate in CREmet seems to partly result from a loss of freedom in the BI sub-conformation, signifying that impact of methylation on the double helix is at least partly entropic.
The internal and hydration energies of native and methylated 16mers were calculated for the three global sub-states BIIBII, BIBII and BIBI. The averaged and SD values of the electrostatic, van der Waals and covalent (bond + angle + dihedral) internal energies, and of the electrostatic GB and non-polar solvation energies, are reported in Table 2, together with the values of the SASAs. In both CREnat and CREmet, the transitions from BIIBII to BIBII, i.e. the transitions of a single GpA phosphate, increase the DNA internal energy, essentially through an electrostatic contribution, and are then unfavorable. Conversely, they decrease the solvation energy, essentially due to the electrostatic GB contribution, and appear favorable. Globally, the total energy change (internal + solvation) is positive for CREmet and CREnat. The transitions from BIIBII to BIBI, i.e. transitions of the two GpA phosphates, decrease the internal energy in both CREnat and CREmet, again essentially due to the electrostatic contribution, and are then favorable. The solvation energy is increased, essentially due to the electrostatic GB contribution, and appears thus unfavorable. Here again, the total energy change is positive and similar for CREmet and CREnat.
Table 2. Averaged values (± SD) of the internal electrostatic, van der Waals and covalent energies, of the solvation energy calculated with the modified generalized Born (GB) model, and of the SASA, for CREnat and CREmet, as a function of the BIBI, BIBII and BIIBII sub-conformations.
| Conformation at GpA |
CREnat |
CREmet |
| Electrostatic energy (kcal/mol) | ||
| BIBI | 2580.08 (78.15) | 2677.18 (70.56) |
| BIBII | 2609.88 (67.23) | 2750.93 (59.73) |
| BIIBII | 2598.73 (64.26) | 2743.78 (64.40) |
| van der Waals energy (kcal/mol) | ||
| BIBI | –289.94 (11.53) | –289.47 (10.21) |
| BIBII | –289.79 (11.63) | –292.35 (11.25) |
| BIIBII | –289.44 (11.32) | –292.99 (11.26) |
| Covalent energy (kcal/mol) | ||
| BIBI | 1422.79 (20.14) | 1423.10 (21.35) |
| BIBII | 1421.43 (20.67) | 1424.05 (20.98) |
| BIIBII | 1420.76 (20.75) | 1422.64 (20.73) |
| Total internal energy (kcal/mol) | ||
| BIBI | 3712.93 (77.30) | 3810.81 (69.09) |
| BIBII | 3741.52 (66.51) | 3882.63 (60.58) |
| BIIBII | 3730.05 (62.52) | 3873.43 (63.10) |
| GB solvation energy (kcal/mol) | ||
| BIBI | –9898.31 (75.98) | –9837.37 (70.86) |
| BIBII | –9934.43 (67.17) | –9917.68 (60.59) |
| BIIBII | –9925.49 (63.05) | –9910.45 (63.86) |
| SASA (Å2) | ||
| BIBI | 6150.00 (48.76) | 6213.97 (47.83) |
| BIBII | 6120.46 (48.07) | 6158.79 (41.88) |
| BIIBII | 6115.44 (46.92) | 6139.17 (43.62) |
Our energetic data do not actually account for any difference in the BI/BII dynamics between CREnat and CREmet, as these present the same total transition energies. A major reason could be the approximations used in the MM_GBSA method, especially for estimation of the solvation energy (implicit water, and non-inclusion of counter-ions screening), which prevents the detection of fine structural alterations. Another reason would be the role of entropic effects (see above). Whatever is the exact origin of the effects, the GBSA results on the solvation energy appear very interesting. First, Esolvation is worse for CREmet than for CREnat, for all the DNA sub-states (Table 2), probably due to the hydrophobicity of the two additional methyl groups. Secondly, the difference between the averaged solvation energies of CREnat and CREmet appears strongly reduced upon the passage from BIBI to BIIBII (from 61 to 15 kcal/mol). The passage is correlated with a decrease from 64 to 24 Å2 of the difference between the solvent accessible surface areas of CREnat and CREmet (Fig. 5). The unfavorable impact of the apolar methyl groups on the solvation energy is probably for a large part compensated by the BI to BII transition of the 5′ GpA step. This could also explain why CREmet spends more time in the BIIBII sub-state compared to CREnat. Snapshots from the MD trajectory of CREmet in the BIBI (t = 4301 ps) and in the BIIBII (t = 4440 ps) sub-conformations (Fig. 6) reveal that in the BII sub-state (i) the GpA phosphates are closer to one another in the major groove, and (ii) the methyl groups are buried in the bottom of the groove. The latter effect recalls the burying of hydrophobic residues that follows protein folding, and a detailed study of the water molecules network and rearrangement during the BI/BII transition in CREnat and CREmet would be of great interest. Recent MD simulation and spectroscopic studies have actually revealed the migration of water molecules during such DNA transitions (30,31).
Figure 5.

Averaged values (± SD) of the SASA (top) and of the solvation energy calculated with the modified generalized Born (GB) model (bottom), for CREnat (solid line) and CREmet (dashed line), as a function of the BIBI, BIBII and BIIBII DNA sub-conformations.
Figure 6.
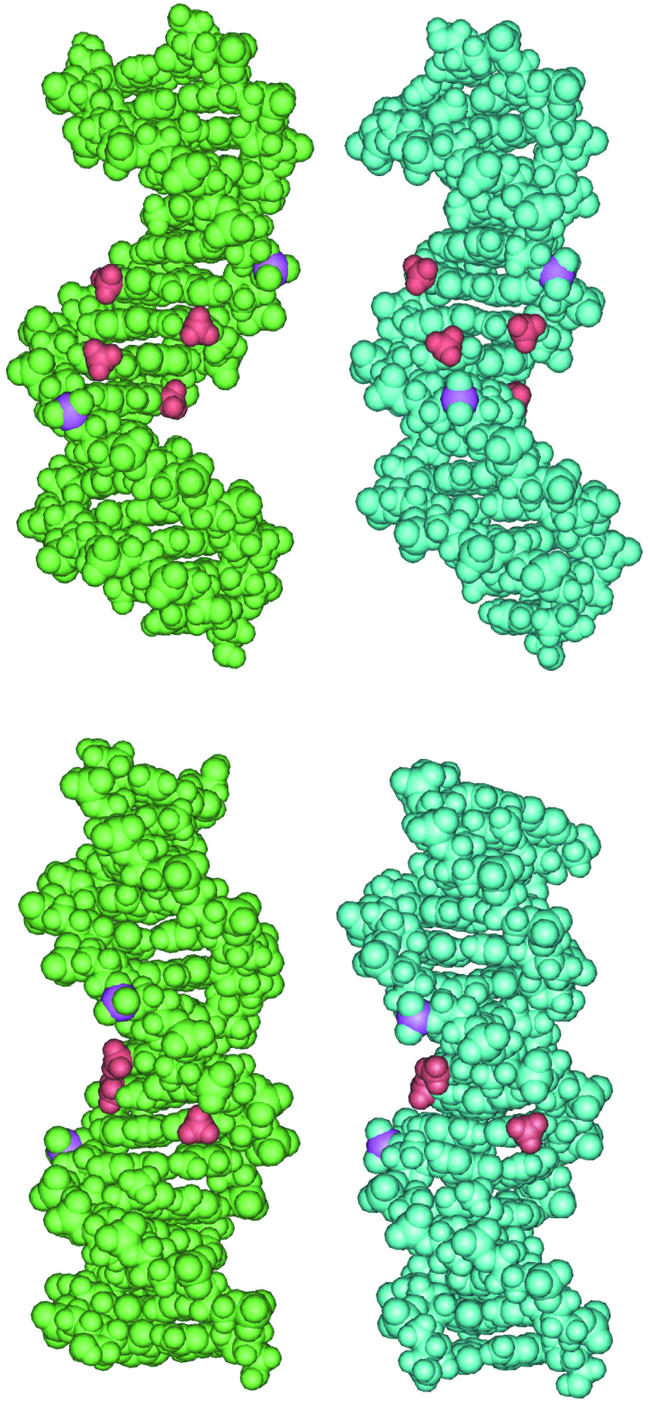
Snapshots from the MD trajectory of CREmet for the BIBI (t = 4301 ps) (left) and the BIIBII (t = 4440 ps) (right) sub-conformations. The phosphorus atoms at the GpA steps are pink, and the methyl groups at AmCGT in the center of CREmet are red. Front (top) and profile (bottom) views of the major groove.
The supposed folding of CRE in response to methylation seems to depend on the DNA sequence and to concern only residues located before the methylation site. For instance, methylation reinforces the preference of the 5′ GpA step for the BII state but does not affect the 3′ GpT step. Single-site methylation would then induce very local DNA foldings.
Impact of CpG methylation on the structure and dynamics of CRE: MD versus NMR
The NMR results on CREnat have already been reported (15). Those on CREmet have been obtained in the frame of this study. Unfortunately, a comparison of the NMR refined structures of these molecules is prevented by the strong impact of the refinement software on the final structures (16). The major drawbacks of the software are the high negative rolls favored at the flexible pyrimidine–purine steps CpG and TpG/CpA and the bending of CRE toward the minor groove, contrasting with most of the experimental data. However, incorporation of the inter-nucleotide distances H3′–H6/8 seems to prevent bias on the roll values (32). The CREmet structure treated in this way displays more usual roll values, that is less negative, compared with the former roll values of CREnat (–6 and –4° versus –19 and –12° at CpG and TpG/CpA, respectively).
Sugar puckers of CREmet were determined using nOe data and J couplings similarly to CREnat (33). The agreement between the MD and NMR results is illustrated by the MD averaged phase values staying within the NMR experimental uncertainties. Moreover, the similarities between MD values of CREnat and CREmet are reproduced by NMR (Fig. 7A) (16). When chemical shifts were compared, the differences between CREnat and CREmet were generally very small (<0.2 p.p.m.). Regarding the bases, the major differences (between CREnat and CREmet) concerned H6 of C8, H8 of G9 and CH3 of T10 (+0.16, +0.08 and +0.05 p.p.m., respectively) which are protons located close to the CpG methyl groups in the major groove. Effects might result either from a change in the electronic environment, or from weak structural rearrangements caused by the bulky methyl groups. The differences in chemical shifts are smaller for sugars, with the largest values found for H2′2 of A7 (–0.06 p.p.m.), H2′ of C8 (–0.05 p.p.m.) and H1′ of G6 (–0.04 p.p.m.). In the central GAC segment, similar observations were made for the phosphate chemical shifts: –0.08, +0.06 and –0.07 p.p.m. at G6, A7 and C8, respectively (Table 3). The weakness of the effects suggests that CpG methylation produces only a slight modification of the averaged structure of CRE, and/or a change of the solvent organization around GAC, as suggested by MD.
Figure 7.

(A) Sugar phase angles as a function of the nucleotide in CREmet: MD values averaged over 9.5 ns (bold line) and NMR data with error bars (thin lines). (B) Percentage population of the BII state as a function of the dinucleotide step, in CREmet (black bars) and in CRE (gray bars), calculated from the 31P chemical shift measurements using the Roongta–Gorenstein relationship (34,35).
Table 3. Differences between the phosphorus chemical shifts in CREnat and CREmet (CREnat–CREmet).
| GpA |
ApG |
GpA |
ApT |
TpG |
GpA |
ApC |
CpG |
GpT |
TpC |
CpA |
ApT |
TpC |
CpT |
TpC |
| –0.04 | 0.00 | –0.01 | 0.00 | 0.01 | –0.08 | 0.06 | –0.07 | 0.02 | 0.04 | 0.00 | –0.01 | 0.02 | 0.01 | 0.03 |
The phosphorus chemical shifts may help to assess the dynamics of the phosphodiester backbone as they provide the relative populations of the phosphate BI and BII states from the Roongta–Gorenstein relationship (34,35). Results for CREmet were similar to those obtained for CREnat (16). There was also a good global qualitative agreement between MD and NMR, with the GpA, CpG and CpA steps displaying the most populated BII states in both cases (>30%) (Figs 2A and 7B). Quantitatively, the BII percentage averaged over the whole CREmet sequence equalled 20% in NMR and 21% in MD. However, the variations of the BII rate as a function of the sequence showed significant divergence: the sequence dependence of the BII percentage was found to be higher with MD calculations than with NMR measurements, with spreads of 0–90% and 5–40%, respectively. In particular, the very high levels of the BII population rates and the difference between CREnat and CREmet obtained by MD for the GpA/TpC steps (∼70 and ∼90%, respectively) were not reproduced by NMR (∼30% for CREnat and CREmet). Neither did NMR predict the blocking of TpG and ApC in BI.
DISCUSSION
Here we showed using MD simulation that the averaged conformation of the CRE double helix is only weakly affected by CpG methylation. In contrast, there was a significant alteration of the DNA dynamics by methylation. CREmet appears less flexible than CREnat, in terms (i) of BI/BII transitions at the TpG, GpA and ApC steps incorporated in the TGAC binding half-sites, and (ii) of a correlated reduction of the SDs of several helical parameters, that is the twist at GpA and ApC and the propeller twist at C:G. These modifications of the DNA dynamics likely result from two concurring effects: the bulkiness of the methyl groups responsible for the reduction of the accessible conformational space, and a degradation of the solvation energy due to the hydrophobicity of the methyl groups compensated by a kind of DNA folding around the methyl groups (by maintaining the 5′ GpA phosphates in the BII sub-state).
Below we discuss the divergence between the MD and the NMR results. The results were compared to experimental data provided by crystallographic and NMR studies on various methylated DNAs, and inhibition by methylation of the CRE recognition by specific bZIP transcription factors was observed.
MD versus NMR
The particular behavior of the half-site GpA step showed by MD simulation could be an artifact due to a bias of the force field since BI/BII transitions are known to depend on both the stacking energy between the adjacent bases (29) and the parameterization of the sugar dihedral angles (20). This cannot be the case since the other GpA step of CRE displays only 30% of BII conformation (Fig. 2A). Moreover, a 7 ns MD simulation of the pseudo-palindromic TPA responsive element sequence (TRE), d(GAGATGACTCATCTC)2, which differs from CRE by the absence of a single C:G base pair at the center, provides a BII value of 50% for the half-site GpA in the first strand and 35% in the second strand (S.Derreumaux and S.Fermandjian, unpublished results). The electrostatic parameterization of the methyl groups, possibly with underestimated polarities, could be another problem of the force field. Proton charges in methyl groups are +0.077 for thymines and only +0.023 for methylated cytosines. Thus, the aptitude of the added methyl groups to form C-H…O hydrogen bonds with water molecules (36,37) would be underestimated, and the hydrophobic influence of methyl groups on DNA solvation and structure overestimated. Crystallographic studies of the hydration of methyl groups actually lead to diverging results. While some of them stipulate a significant hydration of methyl groups (38), others argue that methyl groups create hydrophobic pockets in the major groove and prevent the approach of solvent molecules (39,40).
Another question arising from MD simulation concerns the relevance of the Roongta–Gorenstein relationship that correlates the phosphorus chemical shift and the BI/BII percentages, where pure BI corresponds to –4.6 p.p.m. and pure BII to –3.0 p.p.m. chemical shifts. This has been established from experimental data almost exclusively for phosphates preferentially in BI (34,35). According to current MD results, the use of the Roongta–Gorenstein relationship requires caution. It consists of a linear approximation of a function that could be non-linear, and the chemical shifts depend not only on the ɛ and ζ dihedral angles describing the BI/BII conformations but also on several other structural parameters (34). It is also noticeable that BII conformations are often found by X-ray crystallography (see the statistical analysis in 30) while NMR has never predicted a BII preferential state (>50%) at ambient temperature. Most of chemical shifts predict at most a BI/BII equilibrium, where the BI state always predominates (34,35,41,42). Strongly downfield-shifted phosphates (around –3 p.p.m.) are found either for highly distorted DNA structures, as those found for mismatches (34,43), or for DNAs under elevated temperatures (>50°C) (34) and damaged by UV radiations (cis–syn cyclobutane thymine dimer) (44). Moreover, the 31P chemical shifts reported in two of these studies were –4 p.p.m. for a pure BI state, and –2 and –2.5 p.p.m. for a pure BII state (43,44), which do not correspond to the scale of chemical shift values proposed by Roongta et al. (34). Thus, MD simulation could provide values helpful for the establishment of a more complex relationship between the phosphorus chemical shifts and the BI/BII structures.
Impact of cytosine methylation on DNA structure and dynamics
Numerous experimental studies (NMR, radiocrystallography, gels) show that the effect of cytosine methylation on the DNA structure depends on the DNA sequence and the density of methylation sites. Single-site methylation generates very fine structural modifications in DNA fragments of at least 8 bp (9,11–13,42), and the effects depend on the sequence context (12,42). Some bending effect has been observed by gel studies, but again, this depends on the sequence (45,46). A comparison of the 1.75 Å crystal structure of d(CCAGGCmCTGG)2 with its unmethylated counterpart indicates that methylation does not cause visible steric conflicts but results in a slight structural rearrangement with the methylated base pairs pushed into the minor groove and making the major groove deeper (9). This rearrangement is interpreted by the authors as a strategy to reduce solvent exposure of the cytosine apolar methyl groups. Our theoretical investigation of CREmet agrees with all the above experimental findings and also with a recent solid-state deuterium NMR analysis of d(CGCGAATTmCGCG)2 versus d(CGCGAATTCGCG)2, which concluded that there was a significant reduction in the phosphodiester backbone mobility at the level of the methylated cytosine (13).
The higher the number of methylation sites, the larger is the impact on the DNA structure, but here again the alteration seems related to solvation effects. Methylation of cytosines in the alternate sequences poly(dG–dC)·poly(dG–dC) allows the formation of Z-DNA at weaker ionic strength than is needed for the unmethylated sequences (47). X-ray structures of the methylated and unmethylated versions of d(CGCGCG)2 suggests that the stabilization of Z-DNA relative to B-DNA is partly due to the fact that the methyl groups are less exposed to the solvent in the Z form than in the B form (39). Methylation of two cytosines in the decamer d(GmCGmCGCGCGC)2 generates an A form duplex that appears bent and folded around dehydrated methyl groups, with a small major groove (40). A recent crystal structure of d(GGCGmCC)2 reveals a new DNA structure, called E-DNA, with a deep major groove and a shortened distance between the phosphates in the modified base pairs (48). Finally, an X-ray study of d(GmCGmCGC)2 and d(GmCCGGC)2, and of their unmethylated counterparts reveals that methylation significantly increases the ability of these sequences to crystallize as an A-DNA, probably because it lowers their intrinsic solubility (49).
The above experimental data support our MD simulation results on the impact of cytosine methylation on the local or the global structure and the dynamics of CRE. Our results provide the first picture of an overall and detailed change of structure, of dynamics and of solvation produced by the methylation of a CpG step within a 16mer DNA. We learn also about the mechanisms by which methylation leads to structural rearrangements as a function of the DNA sequence.
Impact of CpG methylation on CRE-specific recognition by bZIP proteins
CpG methylation abolishes the specific recognition of CRE by the bZIP transcription factors (CREB/ATF) and thereby the transcriptional activity, both in vitro and in vivo (6). However, the cytosine of CpG is not directly contacted by the CREB/ATF proteins (17), so that the binding inhibition produced by methylation could result from structural or thermodynamic effects. Actually, if CpG is artificially methylated in the recently reported CRE–CREB crystal complex (17), the added methyl groups interfere with the site-specific interactions between the conserved arginine 301 (Arg243 in GCN4) and the guanine of CpG (Fig. 8A). These site-specific interactions have been shown to be necessary for the recognition of CRE by CREB (50,51). Yet in the CRE–CREB complex, the Arg301–CpG contacts are not symmetric: the arginine of one monomer, called Arg301′, is hydrogen bonded like both arginines in the CRE–GCN4 complex (17) to the N7 atom of the guanine of CpG and to the phosphate group of the adjacent ApC step on one DNA strand; the arginine of the other monomer, termed Arg301, hydrogen bonds to both the O6 and the N7 atoms of the guanine of the CpG on the other strand, but does not contact the backbone. Addition of one methyl group creates an obstacle to the interaction of Arg301′ with DNA, while the interaction of the other arginine, Arg301, appears less disturbed. Moreover, one of the two water molecules bridging Arg301 to CRE appears to clash (Fig. 8B) (17).
Figure 8.
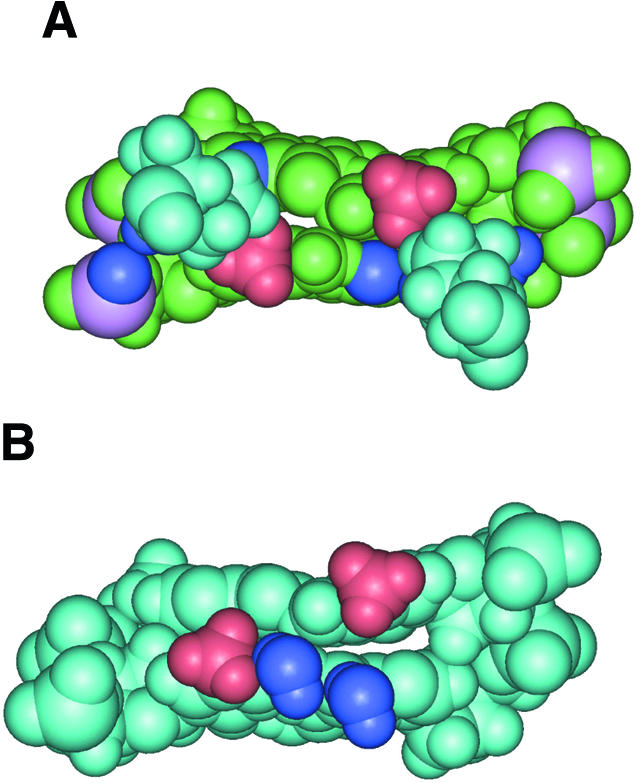
(A) Interactions between the methyl groups of artificially methylated central cytosines of CRE (red) and the conserved arginines 301/301′ (pale blue) in the crystal complex of native CRE with CREB. The atoms in dark blue are those that show direct contact with the Arg301/301′: O6 and N7 of the central guanines, and a phosphate oxygen at ApC (17). The phosphorus atoms are pink. (B) Interactions between the methyl groups of artificially methylated central cytosines of CRE (red) and the water molecules linking arginine 301 and CRE (dark blue) in the crystal complex of native CRE with CREB (17). The arginines have been omitted for clarity.
Thus, the introduction of methyl groups on CpG either prevents the site-specific interactions of the crucial arginines 301 and 301′ (enthalpic effect) or significantly reduces the conformational space of the CREB–CpG contacts (entropic effect). On the other hand, CREmet appears less flexible than CREnat and the energetic cost needed to form a functional complex would be higher for CREmet than for CREnat. In particular, methylation almost blocks the GpA step of the binding half-sites in the BII conformation, while the adjacent TpG and CpA steps are in the BI conformation. In contrast, in the CREB–CRE complex GpA is in the BI state and in one DNA strand TpG is in BII (Fig. 9A) (17). Superimposition of the MD averaged BIIBII sub-conformation of CREmet (occupancy rate >80%) with the structure of CRE in the complex (that is the heavy atoms of the 8mer consensus sequence) shows that the distances between the NH3+ group of lysines (Lys305/Lys305′) and the contacted PO4– groups at GpA increase from 3.8 and 4.5 Å in CRE–CREB to 4.6 and 5.5 Å in <CREmet_BIIBII>–CREB (Fig. 9B). The Lys305–GpA interactions are fundamental as mutagenesis studies have shown that Lys305–DNA contacts are responsible for a great part for the specific recognition of CRE by CREB proteins (52,53). Then, the higher rigidity of the CREmet backbone as compared to CREnat would require additional work by the protein to fit the DNA structure.
Figure 9.

(A) Difference between the ɛ and ζ dihedral angles as a function of the dinucleotide step: averaged MD values ± SD for CREnat (blue line) and CREmet (red line), and values found for CREnat in the crystal complex with CREB in the first (solid green line) and the second DNA strands (dashed green line) (17). (B) Superposition of the MD averaged BIIBII sub-conformation of CREmet (dark blue) with the structure of CRE in the complex with CREB (pale blue). The lysines 305 interacting with the backbone at the GpA steps are shown in pink. (C) Superposition of the MD averaged BIIBII sub-conformation of CREmet (dark blue) with the structure of CRE in the complex with CREB (pale blue). The lysines 304 interacting with the hydrated magnesium ion (black) are gray, and the methyl groups of the central cytosines of CREmet are red.
The <CREmet_BIIBII>–CREB complex further shows a close contact between the methyl groups and the hexahydrated magnesium ion located in a small room just in between the bZIP fork and the DNA (Fig. 9C), while no clash is visible in the crystal structure of CRE artificially methylated in the complex. Direct implication of Mg2+ in the binding of CREB with CRE is a new finding. As the Mg2+ ions strongly enhance the binding efficiency of CREB for CRE (17), this suggests that the inhibiting effect of methylation is at least partly due to the fact that the BIIBII conformation of CRE prevents the binding of CREB to Mg2+.
As a whole, the binding inhibition of a specific factor to methylated CRE could be explained by concurring effects: a steric clash of methyl groups with Arg301/Arg301′ and with a linking water molecule, and an unfavorable BIIBII conformation of CREmet. It can also be suggested that the hydrophobic context created by two additional methyl groups is unfavorable for the interaction with the charged arginines and with the water linking molecules, and with the magnesium ion. To prove these hypotheses, MD simulations of both CREmet–CREB and CREnat–CREB complexes would be necessary.
Impact of methylation on the immunostimulatory CpG motif
Finally, accumulating evidence has revealed the potential of CpG-containing DNAs as adjuvants for vaccination strategies for cancer, allergy and infectious diseases. In bacterial DNA, the CpGs are predominantly unmethylated whereas vertebrate DNA presents a low frequency of CpGs and ∼75% of these are methylated (54). Such differences seem sufficient to generate a broad spectrum of host immune responses (55–60). Curiously, the CRE sequence that is commonly found in mammalian DNAs displays a CpG step flanked by two 5′ purines and two 3′ pyrimidines, which represents the best immunostimulatory sequence. Similarly to CRE, all the CpG containing immuno-stimulatory oligonucleotides are deactivated by methylation. Here again the bulky methyl groups could create an obstacle to the interactions with protein partners and a loss of flexibility required for the adjustment of molecules.
CONCLUSION
The present comparison of CREnat and CREmet showed that the oligonucleotides have very similar averaged structures, but significantly different dynamics as proved by the standard deviations of the helical structural parameters and the frequency of backbone BI/BII transitions. Molecular mechanics and generalized Born solvation energy calculations suggested that the relative stiffness of CREmet is due to both a reduction of the accessible conformational space caused by steric hindrance of the bulky methyl groups, and to a kind of DNA folding around the methyl groups favored by the BII conformation of the half-site GpA phosphate group just before the methylation site. This seems to occur in response to a degradation of the solvation energy provoked by the added methyl groups.
Resolution of the NMR refined structures of CREnat and CREmet was not sufficient to allow a rigorous comparison of these structures with the MD structures. We thus resorted to the NMR data. Both the sugar puckers determined by NMR and the base and sugar proton chemical shifts indicated that the averaged structures of CREnat and CREmet were very similar, in agreement with the calculated averaged structures. However, our NMR values did not account for any change in the dynamics due to methylation, as they correspond to time-averaged measurements. Particularly, the phosphorus chemical shifts did not reflect the change of BI/BII populations predicted by MD simulation.
Comparison of our MD results with reported experimental data confirms that the extent of the DNA structural modifications due to methylation depends on the DNA sequence and the number of methylation sites, and seems often related to an attempt to bury the methyl groups inside the groove of the double helix. The MD approach further allows study of the dynamic impact of methylation, which seems crucial.
Abolishment of the recognition of methylated CRE by specific protein factors could be due to at least two combined effects: (i) the CpG methyl groups which sterically hinder the specific contacts of the conserved residues Arg301/Arg301′ with CpG, and (ii) a loss of DNA flexibility as shown by the GpA steps of the binding half-sites frozen in the BII conformation, making the fitting of methylated CRE to its protein partner difficult.
On the other hand, the results presented here shed new light on how methylation inhibits the immunostimulatory activity of CpG-containing oligonucleotides (58). Strong immuno-stimulatory oligonucleotides contain sequences similar to CRE, that is two purines before and two pyrimidines after a CpG step. Reference to inhibition produced by methylation in the CRE transcription factor system suggests that immuno-stimulation deactivation results from the inability of methylated oligonucleotides to efficiently interact with their Toll-like receptors in the cell (8).
Finally, a large number of cellular processes involve specific protein–nucleic acid interactions. Some of these interactions are physiologically regulated by post-translational modifications including DNA methylation. But there are also undesirable events such as punctual mutations that can alter the quality of interactions. Generally, conformational effects are small but biological consequences significant as shown by the two examples given above. Here we showed that MD simulation is an excellent tool for detection of small structure or dynamic modifications in DNA, these being not amenable to classical NMR analysis.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dennis Sprous for kindly giving the charge values for the methylated cytosine, and Miguel Elizondo Riojas for his precious help regarding the use of the generalized Born model in AMBER. This work was supported by Fondation pour la Recherche Médicale.
References
- 1.Robertson K.D. and Jones,P.A. (2000) DNA methylation: past, present and future directions. Carcinogenesis, 21, 461–467. [DOI] [PubMed] [Google Scholar]
- 2.Leonhardt H., Rahn,H.-P. and Cardoso,M.C. (1999) Functional links between nuclear structure, gene expression, DNA replication and methylation. Crit. Rev. Eukaryot. Gene Expr., 9, 345–351. [DOI] [PubMed] [Google Scholar]
- 3.Jones P.A. and Laird,P.W. (1999) Cancer epigenetics comes of age. Nat. Genet., 21, 163–167. [DOI] [PubMed] [Google Scholar]
- 4.Momparler R.L. and Bovenzi,V. (2000) DNA methylation and cancer. J. Cell Physiol., 183, 145–154. [DOI] [PubMed] [Google Scholar]
- 5.Tate P.H. and Bird,A.P. (1993) Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev., 3, 226–231. [DOI] [PubMed] [Google Scholar]
- 6.Iguchi-Arriga S.M.M. and Schaffner,W. (1989) CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev., 3, 612–619. [DOI] [PubMed] [Google Scholar]
- 7.Krieg A.M. (2000) The role of CpG motifs in innate immunity. Curr. Opin. Immunol., 12, 35–43. [DOI] [PubMed] [Google Scholar]
- 8.Hemmi H., Takeuchi,O., Kawai,T., Kaisho,T., Sato,S., Sanjo,H., Matsumoto,M., Hoshino,K., Wagner,W., Takeda,K. and Akira,S. (2000) A Toll-like receptor recognizes bacterial DNA. Nature, 408, 740–745. [DOI] [PubMed] [Google Scholar]
- 9.Heinemann U. and Hahn,M. (1992) C-C-A-G-G-C-m5C-T-G-G helical fine structure, hydration and comparison with C-C-A-G-G-C-C-T-G-G. J. Biol. Chem., 267, 7332–7341. [PubMed] [Google Scholar]
- 10.Lefebvre A., Mauffret,O., El Antri,S., Monnot,M., Lescot,E. and Fermandjian,S. (1995) Sequence dependent effects of CpG cytosine methylation. Eur. J. Biochem., 229, 445–454. [DOI] [PubMed] [Google Scholar]
- 11.Mayer-Jung C., Moras,D. and Timsit,Y. (1997) Effect of cytosine methylation on DNA–DNA recognition at CpG steps. J. Mol. Biol., 270, 328–335. [DOI] [PubMed] [Google Scholar]
- 12.Marcourt L., Cordier,C., Couesnon,T. and Dodin,G. (1999) Impact of C5-cytosine methylation on the solution structure of d(GAAAACGTTTTC)2. An NMR and molecular modelling investigation. Eur. J. Biochem., 265, 1032–1042. [DOI] [PubMed] [Google Scholar]
- 13.Geahigan K.B., Meints,A.G., Hatcher,M.E., Oban,J. and Drobny,G.P. (2000) The dynamic impact of CpG methylation in DNA. Biochemistry, 39, 4939–4946. [DOI] [PubMed] [Google Scholar]
- 14.Talanian R.V., McKnight,J.C., Rutkowski,R. and Kim,P.S. (1992) Minimum length of a sequence-specific DNA binding peptide. Biochemistry, 31, 6871–6875. [DOI] [PubMed] [Google Scholar]
- 15.Chaoui M., Derreumaux,S., Mauffret,O. and Fermandjian,S. (1999) An intrinsic curvature towards the minor groove in the cAMP-responsive element DNA found by combined NMR and molecular modelling studies. Eur. J. Biochem., 259, 877–886. [DOI] [PubMed] [Google Scholar]
- 16.Derreumaux S. and Fermandjian,S. (2000) Bending and adaptability to proteins of the cAMP DNA-responsive element: molecular dynamics contrasted with NMR. Biophys. J., 79, 656–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schumacher M.A., Goodman,R.H. and Brennan,R.G. (2000) The structure of a CREB bZIP somatostatin CRE complex reveals the basis for selective dimerization and divalent cation-enhanced DNA binding. J. Biol. Chem., 275, 35242–35247. [DOI] [PubMed] [Google Scholar]
- 18.Pearlman D.A., Case,D.A., Caldwell,J.W., Ross,W.S., Cheatham,T.E., DeBolt,S., Ferguson,D., Seibel,G. and Kollman,P. (1995) AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comp. Phys. Comm., 91, 1–41. [Google Scholar]
- 19.Cornell W.D., Cieplak,P., Bayly,C.I., Gould,I.R., Merz,K.M., Ferguson,D.M., Spellmeyer,D.C., Fox,T., Caldwell,J.W. and Kollman,P.A. (1995) A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc., 117, 5179–5197. [Google Scholar]
- 20.Cheatham T.E.,III, Cieplak,P. and Kollman,P.A. (1999) A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn., 16, 845–862. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen W.L., Chandrasekhar,J. and Madura,J.D. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys., 79, 926–935. [Google Scholar]
- 22.Ryckaert J.-P., Ciccotti,G. and Berendsen,H.J.C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys., 23, 327–341. [Google Scholar]
- 23.Darden T., York,D. and Pedersen,L. (1993) Particle mesh Ewal: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys., 98, 10089–10092. [Google Scholar]
- 24.Essmann U., Perera,L., Berkowitz,M.L., Darden,T.A., Lee,H. and Pedersen,L.G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys., 103, 8577–8593. [Google Scholar]
- 25.Lavery R. and Sklenar,H. (1988) The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J. Biomol. Struct. Dyn., 6, 63–91. [DOI] [PubMed] [Google Scholar]
- 26.Sitkoff D., Sharp,K.A. and Honig,B. (1994) Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem., 98, 1978–1988. [Google Scholar]
- 27.Jayaram B., Sprous,D. and Beveridge,D.L. (1998) Solvation free energy of biomacromolecules: parameters for a modified generalized Born model consistent with the AMBER force field. J. Phys. Chem. B., 102, 9571–9576. [Google Scholar]
- 28.Hartmann B., Piazzola,D. and Lavery,R. (1993) BI-BII transitions in B-DNA. Nucleic Acids Res., 21, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertrand H.-O., Ha-Duong,T., Fermandjian,S. and Hartmann,B. (1998) Flexibility of the B-DNA backbone: effects of local and neighbouring sequences on pyrimidine–purine steps. Nucleic Acids Res., 26, 1261–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winger R.H., Liedl,K.R., Rüdisser,S., Pichler,A., Hallbrucker,A. and Mayer,E. (1998) B-DNA’s BI→BII conformer substate dynamics is coupled with water migration. J. Phys. Chem. B, 102, 8934–8940. [Google Scholar]
- 31.Pichler A., Rüdisser,S., Mitterböck,M., Huber,C.G., Winger,R.H., Liedl,K.R., Hallbrucker,A. and Mayer,E. (1999) Unexpected BII conformer substate population in unoriented hydrated films of the d(CGCGAATTCGCG)2 dodecamer and native B-DNA from salmon testes. Biophys. J., 77, 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefebvre A., Fermandjian,S. and Hartmann,B. (1997) Sensitivity of NMR internucleotide distances to B-DNA conformation: underlying mechanics. Nucleic Acids Res., 25, 3855–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaoui M., Mauffret,O., Lefebvre,A., Lescot,E., Tevanian,G. and Fermandjian,S. (1997) A new method to determine sugar conformation from joint use of 2D and 3D NMR data. J. Biomol. NMR, 10, 187–192. [Google Scholar]
- 34.Roongta V.A., Jones,C.R. and Gorenstein,D.G. (1990) Effect of distorsions in the deoxyribose phosphate backbone conformation of duplex oligodeoxyribonucleotide dodecamers containing GT, GG, GA, AC and GU base-pair mismatches on 31P NMR spectra. Biochemistry, 29, 5245–5258. [DOI] [PubMed] [Google Scholar]
- 35.Gorenstein D.G. (1994) Conformation and dynamics of DNA and protein-DNA complexes by 31P NMR. Chem. Rev., 94, 1315–1338. [Google Scholar]
- 36.Wahl M.C. and Sundaralingam,M. (1997) C-H···O hydrogen bonding in biology. Trends Biochem. Sci., 22, 97–102. [DOI] [PubMed] [Google Scholar]
- 37.Gu Y., Tapas,K. and Scheiner,S. (1999) Fundamental properties of the CH···O interaction; is it a true hydrogen bond? J. Am. Chem. Soc., 121, 9411–9422. [Google Scholar]
- 38.Mayer-Jung C., Moras,D. and Timsit,Y. (1998) Hydration and recognition of methylated CpG steps in DNA. EMBO J., 17, 2709–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fujii S., Wang,A.H.-J., van der Marel,G., van Boom,J.H. and Rich,A. (1982) Molecular structure of (m5dC-dG)3: the role of the methyl group on 5-methyl cytosine in stabilizing Z-DNA. Nucleic Acids Res., 10, 7879–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tippin D.B., Ramakrishnan,B. and Sundaralingam,M. (1997) Methylation of the Z-DNA decamer d(GC)5 potentiates the formation of A-DNA: crystal structure of d(Gm5CGm5CGCGCGC). J. Mol. Biol., 270, 247–258. [DOI] [PubMed] [Google Scholar]
- 41.El antri S., Bittoun,P., Mauffret,O., Monnot,M., Convert,O., Lescot,E. and Fermandjian,S. (1993). Effect of distortions in the phosphate backbone conformation of six related octanucleotide duplexes on CD and 31P NMR spectra. Biochemistry, 32, 7079–7088. [DOI] [PubMed] [Google Scholar]
- 42.Lefebvre A., Mauffret,O., Lescot,E., Hartmann,B. and Fermandjian,S. (1996) Solution structure of the CpG containing d(CTTCGAAG)2 oligonucleotide: NMR data and energy calculations are compatible with a BI/BII equilibrium at CpG. Biochemistry, 35, 12560–12569. [DOI] [PubMed] [Google Scholar]
- 43.Chou S.-H., Cheng,J.-W. and Reid,B.R. (1992) Solution structure of [d(ATGAGCGAATA°]2. Adjacent G:A mismatches stabilized by cross-strand base-stacking and BII phosphate groups. J. Mol. Biol., 228, 138–155. [DOI] [PubMed] [Google Scholar]
- 44.McAteer K., Jing,Y., Kao,J., Taylor,J.-S. and Kennedy,M.A. (1998) Solution-state structure of a DNA dodecamer duplex containing a cis–syn thymine cyclobutane dimer, the major UV photoproduct of DNA. J. Mol. Biol., 282, 1013–1032. [DOI] [PubMed] [Google Scholar]
- 45.Diekmann S. (1987) Temperature and salt dependence of the gel migration anomaly of curved DNA fragments. Nucleic Acids Res., 15, 247–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hodges-Garcia Y. and Hagerman,P. (1992) Cytosine methylation can induce local distorsions in the structure of duplex DNA. Biochemistry, 31, 7595–7599. [DOI] [PubMed] [Google Scholar]
- 47.Behe M. and Felsenfeld,G. (1981) Effects of methylation on a synthetic polynucleotide: the B-Z transition in poly(dG-m5dC) poly(dG-m5dC). Proc. Natl Acad. Sci. USA, 78, 1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vargason J.M., Eichman,B.F. and Shing Ho,P. (2000) The extended and eccentric E-DNA structure induced by cytosine methylation or bromination. Nat. Struct. Biol., 7, 758–761. [DOI] [PubMed] [Google Scholar]
- 49.Mooers B.H.M., Schroth,G.P., Baxter,W.W. and Shing Ho,P. (1995) Alternating and non-alternating dG-dC hexanucleotides crystallize as canonical A-DNA. J. Mol. Biol., 249, 772–784. [DOI] [PubMed] [Google Scholar]
- 50.Walton K.M., Rehfuss,R.P., Chrivia,J.C., Lochner,J.E. and Goodman,R.H. (1992) A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol. Endocrinol., 6, 647–655. [DOI] [PubMed] [Google Scholar]
- 51.Fujii Y., Shimizu,T., Toda,T., Yanagida,M. and Hakoshima,T. (2000) Structural basis for the diversity of DNA recognition by bZIP transcription factors. Nat. Struct. Biol., 7, 889–893. [DOI] [PubMed] [Google Scholar]
- 52.Kim J., Tzamairas,D., Ellenberg,T., Harrison,S.C. and Struhl,K. (1993) Adaptability at the protein–DNA interface is an important aspect of sequence recognition by bZIP proteins. Proc. Natl Acad. Sci. USA, 90, 4513–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J. and Struhl,K. (1995) Determinants of half-site spacing preferences that distinguish AP-1 and ATF/CREB bZIP domains. Nucleic Acids Res., 23, 2531–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bird A.P. (1993) Functions for DNA methylation in vertebrates. Cold Spring Harb. Symp. Quant. Biol., 58, 281–285. [DOI] [PubMed] [Google Scholar]
- 55.Ballas Z.K., Rasmussen,W.L. and Krieg,A.M. (1996) Induction of NK activity in murine and human cells by CpG motifs in oligodeoxynucleotides and bacterial DNA. J. Immunol ., 157, 1840–1845. [PubMed] [Google Scholar]
- 56.Cowdery J.S., Chace,J.H., Yi,A.K. and Krieg,A.M. (1996) Bacterial DNA induces NK cells to produce IFN-γin vivo and increases the toxicity of lipopolysaccharides. J. Immunol., 156, 4570–4575. [PubMed] [Google Scholar]
- 57.Klinman D.M., Yi,A.K., Beaucage,S.L., Conover,J. and Krieg,A.M. (1996) CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12 and interferon γ. Proc. Natl Acad. Sci. USA, 93, 2879–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krieg A.M., Yi,A.K., Matson,S., Waldschmidt,T.J., Bishop,G.A., Teasdale,R., Koretzky,G.A. and Klinman,D.M. (1995) CpG motifs in bacterial DNA trigger direct B-cell activation. Nature, 374, 546–549. [DOI] [PubMed] [Google Scholar]
- 59.Sester D.P., Stacey,K.J., Sweet,M.J., Beasley,S.J., Cronau,S.L. and Hume,D.A. (1999) The actions of bacterial DNA on murine macrophages. J. Leukoc. Biol., 66, 542–548. [DOI] [PubMed] [Google Scholar]
- 60.Sparwasser T., Miethke,T., Lipford,G., Erdmann,A., Hacker,H., Heeg,K. and Wagner,H. (1997) Macrophages sense pathogens via DNA motifs: Induction of tumor necrosis factor-α-mediated shock. Eur. J. Immunol., 27, 1671–1679. [DOI] [PubMed] [Google Scholar]