

Abstract
Rationale:
Axenfeld–Rieger syndrome (ARS) is a rare autosomal dominant disorder with ocular anterior segment dysgenesis and systemic anomalies.
Patient concerns:
A 28-year-old Chinese Han female was referred to Beijing Tongren Eye Center for progressive decrease of the visual acuity on her right eye in the past month.
Diagnoses:
The patient was diagnosed as ARS with retinal detachment based on series of ophthalmic examinations performed.
Interventions:
A pars plana vitrectomy was performed to manage the retinal detachment.
Outcomes:
Her best-corrected visual acuity was slightly improved after surgery.
Lessons:
ARS is a developmental defect of ocular anterior segment with various clinical manifestations which might cause misdiagnosis.
Keywords: anterior segment dysgenesis, autosomal dominant, Axenfeld–Rieger syndrome, embryotoxon, retinal detachment
1. Introduction
Axenfeld–Rieger syndrome (ARS) is a rare autosomal dominant disorder with an incidence of 1:200,000 worldwide. It has both systemic and ocular anterior segment dysgenesis.[1] The ocular manifestations include featured posterior embryotoxon, changes in iris and anterior angle. Developmental anomalies of anterior angle cause increase of outflow resistance and ocular hypertension in nearly 50% of the cases.[1] The characteristic craniofacial signs like maxillary hypoplasia, dental features including hypodontia, oligodontia and microdontia, and umbilical anomalies were also reported in ARS patients.[2] It has been widely accepted that mutations in genes forkhead box C1 (FOXC1, chromosomes 6p25) and pituitary homeobox 2 (PITX2, chromosomes 4q25) encoding transcription factors may lead to ARS.[3] Another locus chromosome 13q14 was also related to ARS but its function has not been identified yet.[4] Till now, there have been only a very few cases reported the clinical features of ARS in Chinese Han population.[5–7] In the present case, we aimed to report a Chinese female ARS patient with retinal detachment. A brief review of literature on this disease will also be provided.
2. Case presentation
A 28-year-old Chinese Han female (weight: 53 kg, height: 169 cm) was referred to Beijing Tongren Eye Center on December 7, 2016. Her visual acuity of right eye progressively decreased in the past month. She did not receive any medications that were harmful to eyes before the reduction of visual acuity. The present study was approved by the Institutional Review Board for the Protection of Human Subjects of Beijing Tongren Hospital and adhered to the tenets of the Declaration of Helsinki. An informed consent was obtained from the patients.
A comprehensive ophthalmic examination was performed. The best-corrected visual acuity (BCVA) was hand motion (HM)/20 cm (OD) and 0.50 (OS) (LogMAR chart), while the intraocular pressure (IOP) was 10 mm Hg (OD) and 20 mm Hg (OS) (Goldmann applanation tonometry, Suzhou City, Jiangsu Province, China), respectively. The slit-lamp microscope examination of right anterior segment showed corneal edema with characteristic corneal posterior embryotoxon (Fig. 1A). The diameters of bilateral cornea were within the normal range (Fig. 1). The corneal endothelium count of both eyes by corneal specular microscopy (Robo SP-8000, Konan Medical, Nishinomiya, Japan) indicated almost no normal-shaped corneal endothelial cells existed (Fig. 2). The iris changes included stromal hypoplasia with irregular-shaped pupils (Fig. 1). Gonioscopy suggested an iridocorneal adhesion of the anterior angle on her right eye at all 4 quadrants. The retinal photography of the both eyes was available as corneal edema and cataract (Fig. 1). The B scan indicated retinal detachment on her right eye.
Figure 1.
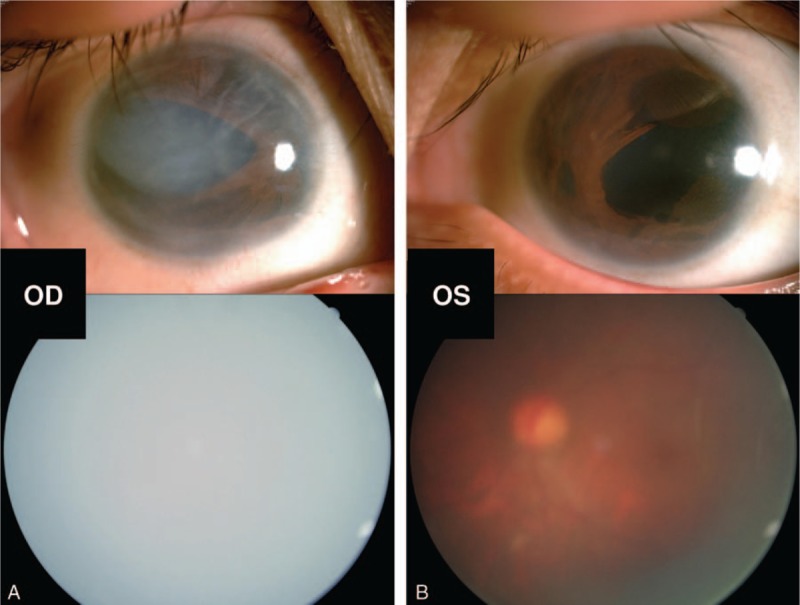
Ocular traits of the recruited patient. Biomicroscopic photograph of the anterior segment showed that corneal edema with characteristic corneal posterior embryotoxon and iris changes of stromal hypoplasia with irregular-shaped pupils of her right eye (A upper). The traits of the left anterior segment included stromal hypoplasia and irregular-shaped pupil (B upper). The fundus photography of bilateral eyes was not clear because of corneal edema and cataract (A, B lower).
Figure 2.
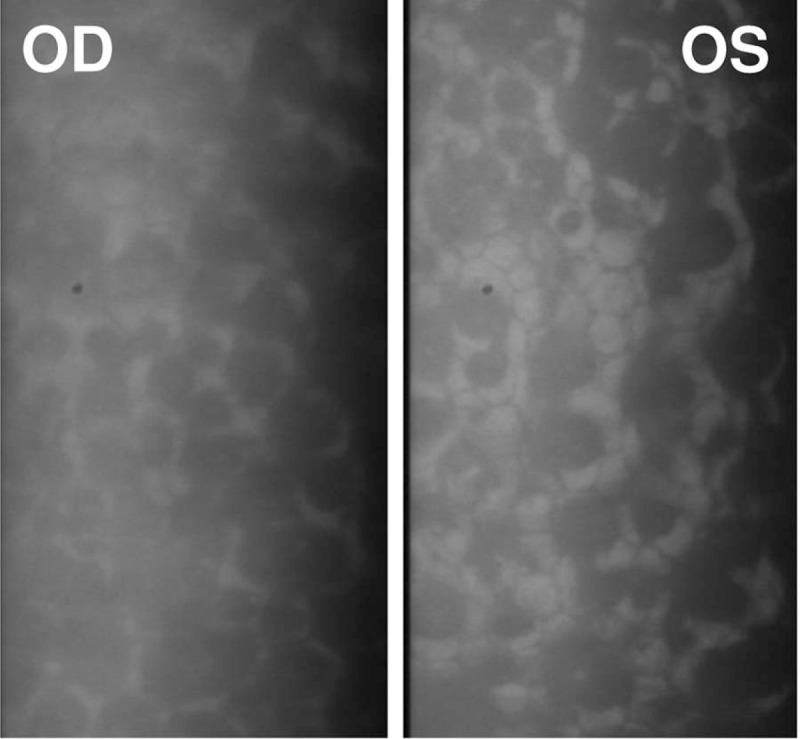
Corneal specular microscopy of bilateral eyes performed in the patient. There were almost no normal-shaped corneal endothelium present and the corneal endothelium count of both eyes was 0 cells/mm2.
The patient had a history of dental abnormalities of hypodontia of the maxillary anterior teeth in both primary and permanent dentition. She received orthodontic treatment at 15-year-old (Fig. 3B). We identified craniofacial anomalies, including midface hypoplasia, hypertelorism, and telecanthus (Fig. 3A). Other systemic examinations showed redundant periumbilical skin (Fig. 3C). Diagnosis of ARS was made based on characteristic ocular and systemic signs above. The patient received a pars plana vitrectomy for the retinal detachment on her right eye in Beijing Tongren Eye Center on February 6, 2017. One week later, she had BCVA recovered to HM/50 cm and IOP lowered to 12 mm Hg. Therefore, further treatment for IOP was not necessary for her.
Figure 3.
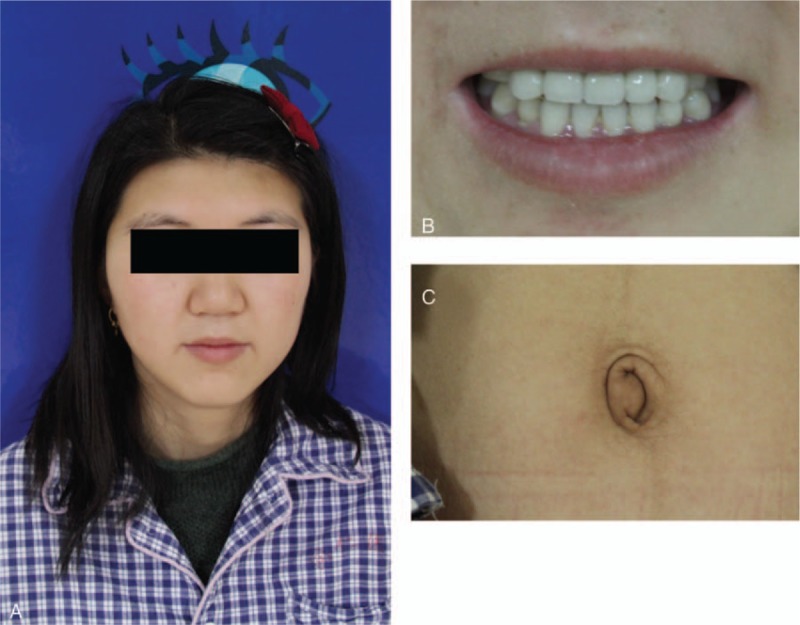
The systemic traits of the reported patient. There were midface hypoplasia, hypertelorism, and telecanthus present in this patient (A). She had a history of dental abnormalities of hypodontia of the maxillary anterior teeth in both primary and permanent dentition and received orthodontic treatment at 15-year-old (B). Other physical examinations showed redundant periumbilical skin (C).
We also studied her family history. Her father had glaucoma on the both eyes and retinal detachment on his right eye. There was no anterior segment anomaly present recorded, and no anomalies in the periumbilical skin, either. However, he had hypodontia of the maxillary anterior teeth in both primary and permanent dentition, which was similar to his elder daughter. This patient also had a younger sister of 5-year-old, who also suffered from blurred vision and had developmental anomalies in maxillary anterior teeth.
3. Discussion
ARS was first described by German ophthalmologist Theodor Axenfeld in 1920 as featured posterior embryotoxon and prominent iris strands extending from the peripheral iris to this line. In 1934, Rieger described a case with changes in the iris like stromal hypoplasia, polycoria, and corectopia in addition to the same features described by Axenfeld.[8] ARS covers a spectrum of inherited disorders and traditionally was classified into 3 subcategories: Axenfeld anomaly refers to the condition with a prominent, anteriorly displaced Schwalbe line named posterior embryotoxon, and prominent iris strands extending from the peripheral iris to this line; Rieger anomaly refers to the condition with central iris changes like stromal hypoplasia and irregular-shaped pupils along with features mentioned in Axenfeld anomaly; Reiger syndrome refers to the condition with Rieger anomaly and other systemic features.[9] The diagnosis of the patient here should be Reiger syndrome since she presented ocular anomalies of Rieger anomaly together with systemic anomalies. However, we could easily found that most of the ARS cases are sharing an overlap of features within this spectrum so that the delineation of each of these is not clear. Thus, the term ARS has been used since 1985,[1] and Ozeki et al[10] reported that Axenfeld anomaly accounted for 71%, Rieger anomaly accounted for 10%, while Reiger syndrome covered 19% of all ARS cases.
Besides of typical manifestations, other unusual ocular anomalies have also been reported. Espana et al[11] and Parikh et al[12] reported 2 cases with unusual presentation of detached Schwalbe line suspended in anterior chambers, respectively. The anterior segment structure Schwalbe line has its genesis from neural crest cells, the impaired development of which is related to the pathogenesis of ARS[1] may explain such clinical manifestation. Hypoplasia of extraocular muscles derived from mesodermal complex also appeared in ARS. Bhate and Martin[13] reported a 6-year-old boy with hypoplasia of right inferior rectus muscle presenting exotropia; while Park et al[14] reported another 4-year-old girl with more posteriorly insertion of superior oblique presenting exotropia and dissociated vertical deviation (DVD). Other posterior segment abnormality also appeared in ARS case reports. In 1989, Spallone reported 3 cases of ARS in 1 family, and among them 2 patients suffered from retinal detachment due to proliferative vitreoretinopathy.[15] More recently, another ARS family was presented by Kelberman et al.[16] In their study, the most severely affected individual received vitreoretinal surgery also because of retinal detachment due to persistent hyperplastic primary vitreous. Interestingly, this patient in our study also presented retinal detachment because of proliferative vitreoretinopathy. Thus, she received vitreoretinal surgery, and the postoperative BCVA was improved.
In addition to atypical ocular features that showed in ARS patients, atypical systemic anomalies were also reported. Titheradge et al[17] found 3 cases with mental deficiencies. The authors implicated the adjacent neuronally expressed genes to PITX2 as potentially causal. Dr Ferrer reported a 7-year-old ARS patient that showed mild mitral insufficiency, which processed to moderate mitral regurgitation.[18] Cardiac anomalies were also found by Gripp et al in 2013.[19] This 21 months old patient received valve replacement surgery because of heart failure. Echocardiogram of the patient showed dysplastic arcade mitral, mildly hypoplastic left ventricular outflow tract and aortic arch. However, with carefully systemic evaluation and consultation with other specialties, female patient did not have mental deficiencies and problems within cardiac system. Other underappreciated feature like thyreoid dysfunction was also exhibited in Reiger syndrome.[20]
Mutations in the FOXC1[21] and PITX2[22] genes were associated with ARS in an autosomal dominant inherited manner and accounted for approximate 40% to 70% of all cases.[23] These 2 genes are closely related to the development of ocular anterior segment. FOXC1 belongs to the forkhead family of transcription factors, the biological function of which is to regulate migration and differentiation of mesenchymal cells in the embryogenesis.[24]FOXC1 mutations include missense mutations in forkhead domain, frameshift and nonsense mutations throughout the gene, deletion of the whole gene, and duplications.[6,25–27] In ARS, mutations in FOXC1 are related to ocular, heart, and hearing defects.[28] PITX2 belongs to the bicoid-class of homeodomain protein family and is able to regulate the development of ocular anterior segment and several extraocular tissues like heart.[29] Typically, mutations in PITX2 that are associated with ARS include splice-site mutations, deletions of coding exons, and chromosomal translocations.[7,25,30,31] Using microarray analysis, Titheradge et al[17] found novel overlapping microdeletions with PITX2 in 4 unrelated ARS patients. Mutations in PIXT2 appear to be related with ocular, dental, and umbilical anomalies in ARS.[28]
In addition to FOXC1 and PITX2 genes, mutations in the CYP1B1 gene (mapped to cytochrome P4501B1) was first reported to lead to the occurrence of ARS in 2006,[32] which presented clinical manifestations belonged to Reiger syndrome.[33,34] CYP1B1 belongs to cytochrome P450 super family of drug metabolizing enzymes and may play roles in the metabolism of substrates essential for ocular development.[35] More recently, another novel heterozygous PRDM5 gene (mapped to chromosome 4q26) missense variant has been identified using whole exome sequencing.[36] PRDM5 protein belongs to PRDM proteins family and is crucial for the development and maintenance of extracellular matrix (ECM), which may explain its involvement in the development of ARS.[37]
Since around 50% of ARS patients will develop glaucoma, conventional glaucoma surgeries like trabeculectomy and trabeculotomy are still required.[5,12] With a 20-year follow-up, Mandal and Pehere[38] confirmed the safety and effectiveness of the combination of trabeculotomy and trabeculectomy for ARS children with early-onset of glaucoma. Mitomycin C (MMC) or the newly developed Ologen Collagen matrix reducing subconjunctival fibrosis may effectively lower IOP in long-term.[39,40] As to the maxillary hypoplasia and dental anomalies of ARS, specialized oral and maxillofacial surgery, special dental care, and application of orthodontic unit may be helpful.[41] In the present case report, the patient underwent orthodontic surgery at 15-year-old (Fig. 3B).
There are some limitations in the present case:
-
(1)
Only 1 ARS patient was presented because it is a kind of rare autosomal dominant disorder in the ophthalmic clinic. We are not able to reach any convincing conclusions about the clinical features and prognosis of this disease in Chinese Han population. More subjects should be recruited in the future.
-
(2)
We did not know the entire family history of the patient and did not obtain the ocular and physical examinations from her father and younger sister, and we did not perform gene analysis for this family.
-
(3)
Although the patient received orthodontic treatment at 15-year-old, the radiological observation should be performed so that we could learn more about the hypodontia of the maxillary anterior teeth.
Acknowledgments
The authors thank the enrolled patient for her cooperation. The authors also thank Dr Ying Zhou from the Department of Stomatology, Jiaxing Traditional Chinese Medicine Hospital Affiliated to Zhejiang Chinese Medical University for the assistance in the dental assessment of the patient and Dr Yalong Dang from Department of Ophthalmology, University of Pittsburgh School of Medicine, Pittsburgh, United States for polishing the language for us.
Footnotes
Abbreviations: ARS = Axenfeld–Rieger syndrome, BCVA= best-corrected visual acuity, FOXC1 = forkhead box C1, IOP = intraocular pressure, PITX2 = pituitary homeobox 2.
The authors have no funding and conflicts of interest to disclose.
References
- [1].Shields MB, Buckley E, Klintworth GK, et al. Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Surv Ophthalmol 1985;29:387–409. [DOI] [PubMed] [Google Scholar]
- [2].Childers NK, Wright JT. Dental and craniofacial anomalies of Axenfeld-Rieger syndrome. J Oral Pathol 1986;15:534–9. [DOI] [PubMed] [Google Scholar]
- [3].Tumer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet 2009;17:1527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Phillips JC, del Bono EA, Haines JL, et al. A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet 1996;59:613–9. [PMC free article] [PubMed] [Google Scholar]
- [5].Ma J, Zhong Y, Zhao C, et al. Axenfeld-Rieger syndrome in monozygotic twins. J Glaucoma 2011;20:584–6. [DOI] [PubMed] [Google Scholar]
- [6].Du RF, Huang H, Fan LL, et al. A novel mutation of FOXC1 (R127L) in an Axenfeld-Rieger Syndrome family with glaucoma and multiple congenital heart diseases. Ophthalmic Genet 2016;37:111–5. [DOI] [PubMed] [Google Scholar]
- [7].Yin HF, Fang XY, Jin CF, et al. Identification of a novel frameshift mutation in PITX2 gene in a Chinese family with Axenfeld-Rieger syndrome. J Zhejiang Univ Sci B 2014;15:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Idrees F, Vaideanu D, Fraser SG, et al. A review of anterior segment dysgeneses. Surv Ophthalmol 2006;51:213–31. [DOI] [PubMed] [Google Scholar]
- [9].Rao A, Padhy D, Sarangi S, et al. Unclassified Axenfeld-Rieger Syndrome: a case series and review of literature. Semin Ophthalmol 2016;1–8. [DOI] [PubMed] [Google Scholar]
- [10].Ozeki H, Shirai S, Ikeda K, et al. Anomalies associated with Axenfeld-Rieger syndrome. Graefes Arch Clin Exp Ophthalmol 1999;237:730–4. [DOI] [PubMed] [Google Scholar]
- [11].Espana EM, Mora R, Liebmann J, et al. Bilateral prominent schwalbe ring in the anterior chamber in a patient with axenfeld-rieger syndrome and megalocornea. Cornea 2007;26:379–81. [DOI] [PubMed] [Google Scholar]
- [12].Parikh RS, Parikh SR, Debashish B, et al. Unusual presentation in Axenfeld-Rieger syndrome. Indian J Ophthalmol 2011;59:312–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bhate M, Martin FJ. Unilateral inferior rectus hypoplasia in a child with Axenfeld-Rieger syndrome. J AAPOS 2012;16:304–6. [DOI] [PubMed] [Google Scholar]
- [14].Park SW, Kim HG, Heo H, et al. Anomalous scleral insertion of superior oblique in Axenfeld-Rieger syndrome. Korean J Ophthalmol 2009;23:62–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Spallone A. Retinal detachment in Axenfeld-Rieger syndrome. Br J Ophthalmol 1989;73:559–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kelberman D, Islam L, Holder SE, et al. Digenic inheritance of mutations in FOXC1 and PITX2: correlating transcription factor function and Axenfeld-Rieger disease severity. Hum Mutat 2011;32:1144–52. [DOI] [PubMed] [Google Scholar]
- [17].Titheradge H, Togneri F, McMullan D, et al. Axenfeld-Rieger syndrome: further clinical and array delineation of four unrelated patients with a 4q25 microdeletion. Am J Med Genet Part A 2014;164A:1695–701. [DOI] [PubMed] [Google Scholar]
- [18].Sanchez Ferrer F, Grima Murcia MD. Progressive moderate mitral regurgitation in a children with Axenfeld-Rieger syndrome. The importance of cardiologic follow up. Arch Argent Pediatr 2016;114:e417–20. [DOI] [PubMed] [Google Scholar]
- [19].Gripp KW, Hopkins E, Jenny K, et al. Cardiac anomalies in Axenfeld-Rieger syndrome due to a novel FOXC1 mutation. Am J Med Genet Part A 2013;161A:114–9. [DOI] [PubMed] [Google Scholar]
- [20].Ornek N, Ogurel R, Ornek K. Congenital hypothyroidism in Rieger Syndrome. Ophthalmic Genet 2016;37:86–8. [DOI] [PubMed] [Google Scholar]
- [21].Mirzayans F, Gould DB, Heon E, et al. Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. Eur J Hum Genet 2000;8:71–4. [DOI] [PubMed] [Google Scholar]
- [22].Vaux C, Sheffield L, Keith CG, et al. Evidence that Rieger syndrome maps to 4q25 or 4q27. J Med Genet 1992;29:256–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol 2000;130:107–15. [DOI] [PubMed] [Google Scholar]
- [24].Pierrou S, Hellqvist M, Samuelsson L, et al. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J 1994;13:5002–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Reis LM, Tyler RC, Volkmann Kloss BA, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet 2012;20:1224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Micheal S, Siddiqui SN, Zafar SN, et al. A novel homozygous mutation in FOXC1 causes Axenfeld Rieger Syndrome with congenital glaucoma. PloS One 2016;11:e0160016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim GN, Ki CS, Seo SW, et al. A novel forkhead box C1 gene mutation in a Korean family with Axenfeld-Rieger syndrome. Mol Vis 2013;19:935–43. [PMC free article] [PubMed] [Google Scholar]
- [28].D’Haene B, Meire F, Claerhout I, et al. Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Invest Ophthalmol Vis Sci 2011;52:324–33. [DOI] [PubMed] [Google Scholar]
- [29].Gage PJ, Camper SA. Pituitary homeobox 2, a novel member of the bicoid-related family of homeobox genes, is a potential regulator of anterior structure formation. Hum Mol Genet 1997;6:457–64. [DOI] [PubMed] [Google Scholar]
- [30].Seifi M, Footz T, Taylor SA, et al. Novel PITX2 gene mutations in patients with Axenfeld-Rieger syndrome. Acta Ophthalmol 2016;94:e571–9. [DOI] [PubMed] [Google Scholar]
- [31].Yun JW, Cho HK, Oh SY, et al. Novel c.300_301delinsT mutation in PITX2 in a Korean family with Axenfeld-Rieger syndrome. Ann Lab Med 2013;33:360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chavarria-Soley G, Michels-Rautenstrauss K, Pasutto F, et al. Primary congenital glaucoma and Rieger's anomaly: extended haplotypes reveal founder effects for eight distinct CYP1B1 mutations. Mol Vis 2006;12:523–31. [PubMed] [Google Scholar]
- [33].Tanwar M, Dada T, Dada R. Axenfeld-Rieger Syndrome associated with congenital glaucoma and cytochrome P4501B1 gene mutations. Case Rep Med 2010;2010: Article ID 212656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Milla E, Mane B, Duch S, et al. Survey of familial glaucoma shows a high incidence of cytochrome P450, family 1, subfamily B, polypeptide 1 (CYP1B1) mutations in non-consanguineous congenital forms in a Spanish population. Mol Vis 2013;19:1707–22. [PMC free article] [PubMed] [Google Scholar]
- [35].Stoilov I, Jansson I, Sarfarazi M, et al. Roles of cytochrome p450 in development. Drug Metab Drug Interact 2001;18:33–55. [DOI] [PubMed] [Google Scholar]
- [36].Micheal S, Siddiqui SN, Zafar SN, et al. Whole exome sequencing identifies a heterozygous missense variant in the PRDM5 gene in a family with Axenfeld-Rieger syndrome. Neurogenetics 2016;17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fog CK, Galli GG, Lund AH. PRDM proteins: important players in differentiation and disease. Bioessays 2012;34:50–60. [DOI] [PubMed] [Google Scholar]
- [38].Mandal AK, Pehere N. Early-onset glaucoma in Axenfeld-Rieger anomaly: long-term surgical results and visual outcome. Eye (Lond) 2016;30:936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Radhakrishnan OK, Pahuja K, Patel K, et al. OLOGEN((R)) implant in the management of glaucoma in an unusual case of Axenfeld-Rieger syndrome. Oman J Ophthalmol 2014;7:90–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mandal AK, Prasad K, Naduvilath TJ. Surgical results and complications of mitomycin C-augmented trabeculectomy in refractory developmental glaucoma. Ophthalmic Surg Lasers 1999;30:473–80. [PubMed] [Google Scholar]
- [41].Bender CA, Koudstaal MJ, van Elswijk JF, et al. Two cases of axenfeld-rieger syndrome, report of the complex pathology and treatment. Cleft Palate Craniofac J 2014;51:354–60. [DOI] [PubMed] [Google Scholar]