

ABSTRACT
Intestinal permeation enhancers (PEs) are agents aimed to improve oral delivery of therapeutic drugs with poor bioavailability. The main permeability barrier for oral delivery is the intestinal epithelium, and PEs act to increase the paracellular and/or transcellular passage of drugs. Transcellular passage can be achieved by cell membrane permeabilization and/or by endocytic uptake and subsequent transcytosis. One broad class of PEs is surfactants which act by inserting into the cell membrane, thereby perturbing its integrity, but little is known about how the dynamics of the membrane are affected. In the present work, the interaction of the surfactants lauroyl-L-carnitine, 1-decanoyl-rac-glycerol, and nonaethylene glycol monododecyl ether with the intestinal epithelium was studied in organ cultured pig jejunal mucosal explants. As expected, at 2 mM, these agents rapidly permeabilized the enterocytes for the fluorescent polar tracer lucifer yellow, but surprisingly, they all also blocked both constitutive -and receptor-mediated pathways of endocytosis from the brush border, indicating a complete arrest of apical membrane trafficking. At the ultrastructural level, the PEs caused longitudinal fusion of brush border microvilli. Such a membrane fusogenic activity could also explain the observed formation of vesicle-like structures and large vacuoles along the lateral cell membranes of the enterocytes induced by the PEs. We conclude that the surfactant action of the PEs selected in this study not only permeabilized the enterocytes, but profoundly changed the dynamic properties of their constituent cell membranes.
KEYWORDS: 1-decanoyl-racglycerol, brush border, enterocyte, intestinal permeation enhancers, lauroyl-L-carnitine, nonaethylene glycol monododecyl ether, small intestine
Introduction
Limited oral bioavailability of several drugs, such as peptides, other types of macromolecules or hydrophilic small molecules, together with a desire to offer “needle-free” administration, has long spurred a quest for development of non-injection formulations of drugs with poor membrane permeability.1 Within this context, interest has focused on intestinal permeation enhancers (PEs), also commonly termed “absorption enhancers," which are agents aimed to improve oral delivery of therapeutic drugs.2–5 Here, the main obstacle to overcome is the intestinal mucosal barrier.6–8 Under normal physiologic conditions, this is a selective barrier upheld to allow uptake of dietary nutrients, vitamins and minerals while simultaneously protecting the organism from invasion or intoxication by luminal pathogens. Limited diffusion through the luminal “unstirred” mucus layer may present the initial barrier for drugs, but the primary obstacle to overcome is the poor transepithelial passage.2,3 Generally, some PEs therefore act selectively to increase the paracellular permeability by opening of tight junctions between adjacent epithelial cells, whereas others may act by increasing transcellular pathways. Either mode of action typically involves functional interactions with the enterocyte cell membrane. The majority of PEs acting to increase transcellular permeability are broadly defined chemically as surfactants; i. e. amphiphilic, surface active agents with a capacity to adsorb to the enterocyte cell membrane. By a detergent-like action, they insert into the outer leaflet of the bilayer and subsequently reach the inner leaflet by a flip-flop mechanism. This incorporation of PEs typically renders the cell membrane more fluid and causes it to expand, leading to a loss of integrity and an increase in permeability.2,9 At higher concentrations, PEs may cause solubilization of membrane constituents, and ultimatively, buckling and lysis of the cell membrane itself.2
Numerous studies have been performed over the years on PEs in search of candidates suitable for use in oral drug delivery,1–3 but with the focus having foremost been concentrated on their permeabilizing properties, little is yet known about how these agents affect the dynamic properties of their primary target, the intestinal brush border. Despite the lipid raft organization of this membrane10 and the rigid microvillus structure defining its shape,11,12 the enterocyte brush border is a highly dynamic cell membrane. Thus, uptake studies in organ cultured mucosal explants, using both lipophilic -and polar tracers, have revealed a widespread constitutive apical endocytosis.13,14 Furthermore, a second, receptor-mediated and clathrin-dependent pathway is inducible, for instance by enterotoxins.15 In the present work, we investigated the interaction of 3 commonly used surfactant PEs, lauroyl-L-carnitine (LC), 1-decanoyl-rac-glycerol (DG), and nonaethylene glycol monododecyl ether (C12E9), with enterocyte cell membranes, using a mucosal organ culture system. Of these, LC is a main PE in Peptelligence™, which has been licensed for clinical development and successfully reached a phase III trial of oral calcitonin.16 Predictably, all 3 PEs permeabilized enterocytes for the fluorescent polar tracer lucifer yellow (LY), but surprisingly, they also blocked both constitutive and receptor-mediated pathways of endocytosis from the brush border. At the ultrastructural level, the PEs caused longitudinal fusion of brush border microvilli. In addition, they caused formation of vesicle-like structures and large vacuoles along the lateral sides of the enterocytes. Overall, these findings indicate that the surfactant action of the PEs studied not only permeabilized the enterocytes, but profoundly changed the dynamic properties of their cell membranes.
Materials and methods
Materials
Lauroyl-L-carnitine (L-carnitine dodecanoyl ester), 1-decanoyl-rac-glycerol (monocaprin), nonaethylene glycol monododecyl ether (C12E9), heat-labile enterotoxin, B subunit, from Escherichia coli (LTB), and fluorescein 5(6)-isothiocyanate (FITC) were supplied by Sigma-Aldrich (www.sigmaaldrich.com), lucifer yellow CH (ammonium salt), Alexa 488 hydrazide, Alexa 594-conjugated secondary antibodies for immunofluorescence microscopy, ProLong antifade reagent with DAPI, Alexa 488-conjugated phalloidin, and a monoclonal antibody to Na+/K+-ATPase (α-chain) from Thermo Scientific (www.thermodanmark.dk). A rabbit antibody to pig intestinal aminopeptidase N was described previously.17
Animals
Animal experimentation in Denmark is subject to ethical evaluation by the Ministry of Justice's Council for Animal Experimentation. Animal experimentation included in this work was performed only by licensed staff at the Department of Experimental Medicine, the Panum Institute, University of Copenhagen, and was covered by license 2012–15–2934–00077 issued to the Dept. of Experimental Medicine.
Segments of porcine jejunum, taken about 2 m from the pylorus of overnight fasted, postweaned animals, were surgically removed from the anaesthetized animals. After excision of the tissue, the animals were killed by an injection with pentobarbital/lidocaine (1 mg/kg bodyweight). As this study contained no further animal experimentation, no specific approval by an ethics committee was required.
Organ culture of pig jejunal mucosa
Organ culture of intestinal tissue was performed essentially as described previously.18 Briefly, after removal from the animals, jejunal segments (∼20 cm) were quickly cut open longitudinally and placed in ice-cold RPMI medium. Mucosal tissue specimens weighing ∼0.1 g were excised with a scalpel and transferred to metal grids in organ culture dishes to which 1 ml of RPMI medium was added. The dishes were placed in an incubator kept at 37 oC and subjected to 15 min of preincubation without any additions to the medium. The permeation enhancers (PEs) lauroyl-L-carnitine (LC), 1-decanoyl-rac-glycerol (DG), and nonaethylene glycol monododecyl ether (C12E9) were dissolved in 10 mM Na2HPO4, 0.9% NaCl, pH 7.0 (PBS) and added to the culture medium at a final concentration of 2 mM. The following microscopy markers for cellular uptake were added together with the PEs: Lucifer yellow (0.5 mg/ml), Alexa 488 hydrazide (10 µg/ml), and FITC-LTB (10 µg/ml).
After addition of the above reagents, the explants were cultured in the presence of the probes for 1 h at 37 oC, and after culture they were carefully rinsed in fresh medium and immersed in a fixative overnight at 4 oC.
Preparation of FITC-conjugated LTB
FITC-LTB was prepared for use in fluorescence microscopy as follows: 0.5 mg of LTB and 0.25 mg of FITC were dissolved in 1 ml 50 mM sodium borate, pH 8.5. After incubation in the dark for 1 h at room temperature with magnetic stirring, unconjugated dye was removed by extensive dialysis against PBS. The FITC-LTB was finally aliquoted and stored in the dark at -20 oC until use.
Fluorescence microscopy
Explants were fixed at 4oC for 2 h or overnight in 4% paraformaldehyde in 0.1 M sodium phosphate, pH 7.2 (buffer A). Following fixation and a quick rinse in buffer A, the explants were immersed overnight in 25% sucrose in buffer A before mounting in Tissue-Tek and sectioning (∼7 µm thick sections cut parallel to the crypt-villus axis) at -19 oC in a Leica CM1850 cryostat. For immunolabeling, the sections were incubated for 1 h at room temperature with antibodies to aminopeptidase N (diluted 1:500) or Na+/K+-ATPase (diluted 1:100) in 50 mM Tris-HCl, 150 mM NaCl, 0.5% ovalbumin, 0.1% gelatin, 0.2% teleostan gelatin, 0.05% Tween 20, 0.05% Triton X-100, pH 7.2 (buffer B). Incubation with Alexa 488/594-conjugated secondary antibodies (diluted 1:200) was performed for 1 h at room temperature in buffer B. For staining of the actin cytoskeleton, sections were incubated for 1 h with Phalloidin in buffer B (0.4 U/ml).
Controls with omission of primary antibodies were routinely included in the labeling experiments.
Hematoxylin-eosin staining of fixed tissue sections was performed according to a standard protocol.
All sections were finally mounted in antifade mounting medium with DAPI and examined in a Leica DM 4000B microscope fitted with a Leica DFC495 digital camera. Images were obtained using Leica HCX PL Fluotar objectives with the following magnification/numerical aperture: 20x/0.40, 40x/0.65, 63x/0.90, and 100x/1.30. The following filter cubes were used: I3 (band pass filter, excitation 450–490 nm), TX2 (band pass filter, excitation 560/40 nm), and A4 (band pass filter, excitation 360/40 nm).
Electron microscopy
Cultured explants for electron microscopy were fixed in 3% glutaraldehyde (v/v), 2% (w/v) paraformaldehyde in buffer A overnight at 4 oC. After a rinse in buffer A, the explants were post-fixed in 1% osmium tetroxide in buffer A for 1 h at 4 oC, dehydrated in acetone and finally embedded in Epon. Ultrathin sections were cut in a Pharmacia LKB Ultrotome III, stained with 1% (w/v) uranyl acetate in H2O and lead citrate, and finally examined in a Zeiss EM 900 electron microscope equipped with a Mega View II camera.
Preparation of microvillus membrane vesicles and treatment with PEs
Pig jejunal mucosa was fractionated by the divalent cation precipitation method.19 Briefly, the mucosa was scraped from the gut and homogenized in 10 volumes of 2 mM Tris-HCl, 50 mM mannitol, pH 7.1, containing 10 μg/ml aprotinin and leupeptin. After centrifugation at 500 g, 5 min, MgCl2 was added to the supernatant (final concentration: 10 mM), and after 10 min on ice, the preparation was centrifuged at 1.500 g, 10 min. The Mg2+- precipitated pellet (containing intracellular- and basolateral membranes) was discarded, and the resulting supernatant centrifuged at 48.000 g, 30 min, to yield a pellet of microvillus membrane vesicles that were stored at -20 oC until use.
For subsequent treatment with PEs, microvillus vesicles (∼1 mg/ml) were resuspended in 25 mM HEPES-HCl, 150 mM NaCl, pH 7.1, and samples of 75 µl were mixed with 25 µl of LC, DG or C12E9 at a final concentration range of 0–5 mM. After 10 min incubation at 37 oC and rapid cooling on ice, the vesicle suspensions were centrifuged at 25.000 x g, 10 min, to yield a membrane pellet and a supernatant of solubilized protein. Both fractions were collected and subjected to SDS/PAGE in 10% gels as described by Laemmli.20 After electrophoresis and electrotransfer onto Immobilon membranes, protein was visualized by staining with Coomassie brilliant blue.
Results
LC, DG and C12E9 as surfactants and PEs in organ culture of mucosal explants
LC, DG and C12E9 (Fig. 1) are all compounds capable of exerting surfactant action on cell membranes, with LC and C12E9 belonging to the broad chemical class of soluble PEs, while DG is classified as an insoluble surfactant.2 Thus, they share a common ability to adsorb to, and insert into, the outer leaflet of cell membranes, and, at higher concentrations, even to solubilize both lipids –and proteins from the bilayer. Owing to differences in their chemistry, they may be expected to perturb the integrity of the lipid bilayer to a varying extent and thus to affect the membrane dynamics and overall cellular permeability differently.
Figure 1.
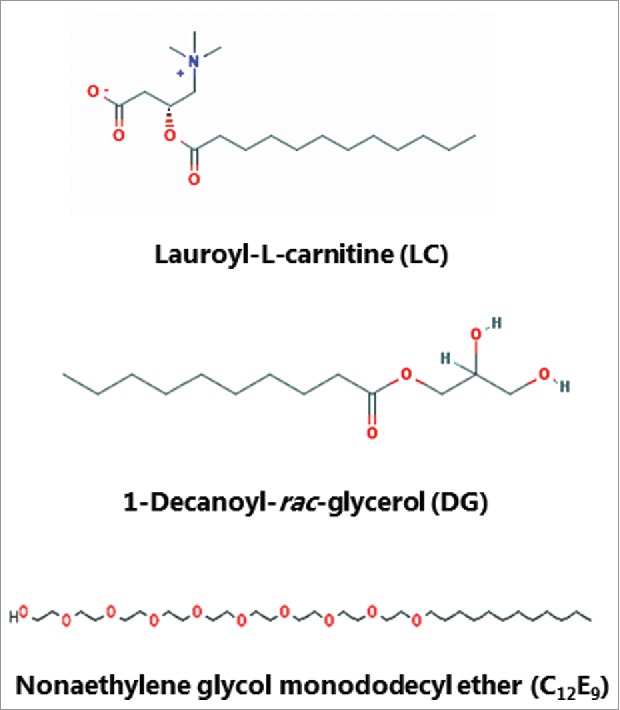
The chemical structure of lauroyl-L-carnitine (LC), 1-decanoyl-rac-glycerol (DG) and nonaethylene glycol monododecyl ether (C12E9). (The images were downloaded from the PubChem Open Chemistry Data Base (https://pubchem.ncbi.nlm.nih.gov/).)
Figure 2 shows the surfactant action of the PEs, as measured by their concentration-dependent ability to solubilize brush border proteins, including the major digestive hydrolases, from outside-out microvillus membrane vesicles. In this experiment solubilization was shown as transfer of proteins from the membrane-bound pellet fraction (P) to the supernatant fraction (S) following centrifugation and SDS/PAGE, and in the absence of any of the PEs little or no protein was released from the P to the S fraction. LC and C12E9 exhibited roughly similar profiles with only moderate solubilization at 0.5 mM, gradually increasing to almost complete release of all major components at 5 mM from the P to the S fraction. In contrast, DG only weakly solubilized the brush border proteins even at 5 mM. In subsequent studies with mucosal explants, a fixed concentration of 2 mM was chosen for all 3 PEs as a balanced compromise between tissue damage and enhancer effect. In contrast to the brush border membrane of intact mucosal tissue, microvillus vesicles are not protected by mucus and should therefore be expected to be more susceptible to surfactant action of PEs at low concentrations.
Figure 2.
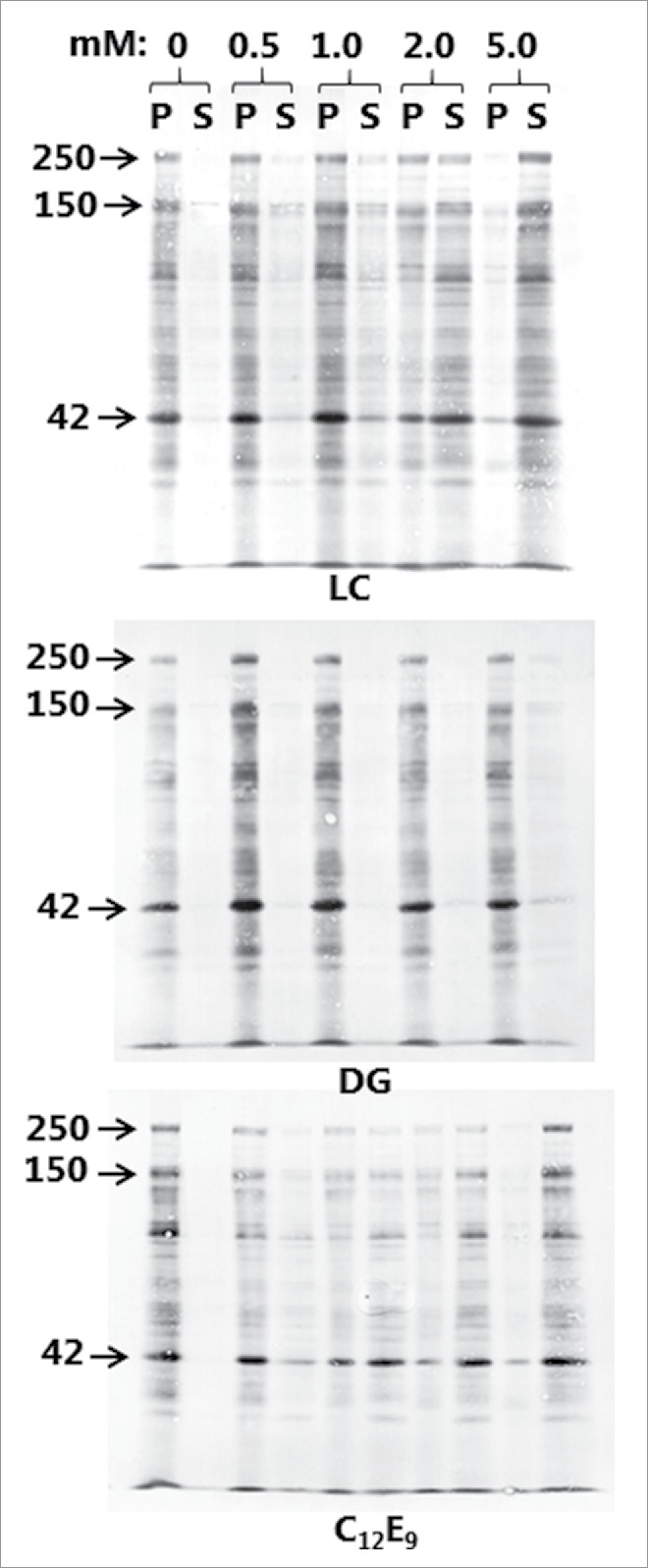
Solubilization of microvillus vesicles by LC, DG, and C12E9. Microvillus membranes were prepared as described in Methods and treated with PEs at a range of concentrations as indicated. After treatment, the pellet (P) -and supernatant (S) fractions following a centrifugation were subjected to SDS/PAGE. Arrows indicate the molecular mass-values of the brush border enzymes sucrose-isomaltase (250 kDa), aminopeptidase N (150 kDa) and the cytoskeleton protein actin (42 kDa).
Organ culture of intestinal mucosal explants is an in vivo-like model suitable for studies on short-term effects of compounds interacting with the mucosal epithelium,18,21,22 and we have previously used it to investigate the action of different types of enterotoxins, including cholera toxin B,15 S. aureus enterotoxins A- and B23 and LTB from E. coli,22 as well as that of another type of PE, glycol chitosan.24 As shown in Fig. 3, LC, DG and C12E9 all caused foci of denudation to form at the tip of villi, whereas the epithelium appeared to be intact along the sides of the villi. A similar denuding effect of other PEs on the intestinal epithelium has previously been reported by others and is rapidly repaired by villus contraction and resealing of the epithelium under normal in vivo conditions.2,25,26 Thus, the organ culture system used here faithfully mimicked the known effect of the PEs on the overall morphology of the epithelium. In all the results presented below we focused on how the PEs affected the parts of the absorptive epithelium where no gross effect on tissue morphology was apparent, i. e. along the sides of the villi rather than at the extrusion zone at the tip.
Figure 3.
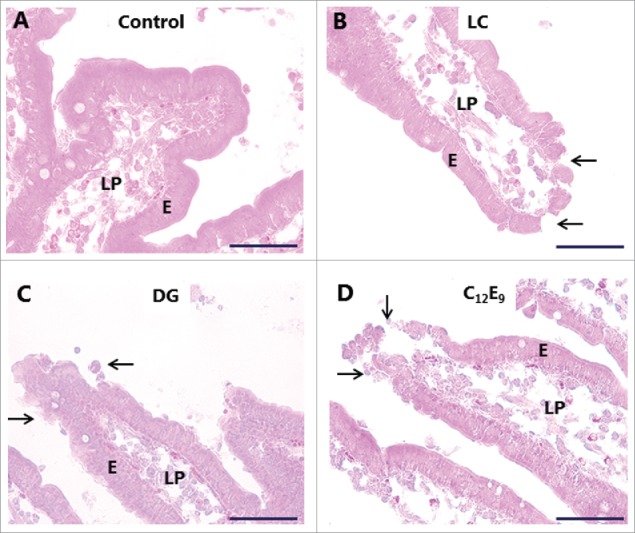
Hematoxylin-eosin stained sections showing villi of mucosal explants cultured for 1 h in the absence (A) or presence of 2 mM of LC (B), DG (C) or C12E9 (D), as described in Methods. All 3 PEs caused denudation at foci near the villus tips (arrows), whereas the epithelium along the sides of the villi generally remained intact. Enterocytes (E) and lamina propria (LP) are indicated. The images shown of each situation are representative of at least 5 images. Bars: 50 µm.
PEs induce permeabilization of enterocytes and inhibit endocytic uptake
LY is a small (molecular mass 444) polar tracer normally non-permeable to cell membranes.27 It has previously been used in an enhancer study to assess paracellular permeability in Caco-2 monolayers,28 and we have used it in the mucosal organ culture system for studying epithelial cell integrity during fat absorption.29 In control explants cultured in the presence of LY for 1 h, uptake into distinct subapical punctae just below the brush border of enterocytes could be seen (Fig. 4A), as previously observed.29 These punctae are early endosome antigen-1 (EEA-1)-positive and thus represent early endosomes in the terminal web region (hence termed “TWEEs” for “terminal web early endosomes”), and they have previously been visualized by use of lipophilic markers of endocytosis.13,14 Otherwise, the cytoplasm of the enterocytes was largely devoid of staining, indicating that the integrity of cell membranes is generally preserved during culture. Labeling of the intercellular space along the lateral sides of the epithelial cells and accumulation in areas of the lamina propria implies that the tight junctions between adjacent cells are permeable to LY. As seen in Fig. 4B-D, both LC, DG and C12E9 caused an uptake of LY into the cytoplasm and most nuclei of the enterocytes, indicating a widespread cell membrane permeabilization. A conspicuous cell-to-cell variation in labeling intensity was evident, resulting in a mosaic appearance of the epithelium. Subapical LY-positive punctae were not readily detectable, suggestive of an impaired endocytosis, but alternatively, a punctate labeling may have been obscured by a high background of diffuse cytoplasmic staining. Therefore, to explore this issue further, another polar tracer of low molecular mass (570), Alexa 488 hydrazide, was used. Like LY, Alexa 488 hydrazide was previously reported to be taken up into TWEEs in enterocytes,30 and as shown in Fig. 5A, it appeared in the control as a string of subapical punctae in the enterocytes similar to LY (Fig. 4A). In addition, little or no Alexa 488 hydrazide was seen laterally along the enterocytes, suggesting that the tight junctions are less permeable to this tracer. Unlike LY, Alexa 488 hydrazide was not staining the cytoplasm to any great extent in the presence of PEs, implying that the cell membranes did not become leaky to this tracer (Fig. 5D). Remarkably, in the presence of DG, little or no uptake of Alexa 488 hydrazide into TWEEs was detected, indicating that the apical constitutive pinocytotic uptake of small molecules is blocked by DG Similar results were obtained for LG and C12E9 (data not shown).
Figure 4.
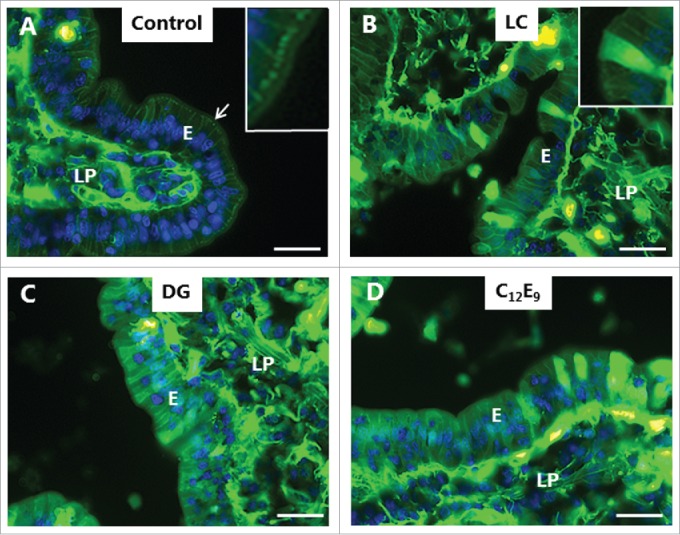
PE-induced permeabilization of enterocytes revealed by LY. Mucosal explants cultured for 1 h in the absence (A) or presence of 2 mM of LC (B), DG (C) or C12E9 (D), and in the presence of 0.5 mg/ml of the fluorescent polar tracer LY. All images shown were captured at identical settings of the microscope. A: A narrow line of punctate labeling of LY is seen just below the enterocyte brush border (arrow and enlarged insert), but diffuse cytoplasmic staining of the cytosol is weak or absent. LY is also seen in the paracellular space between enterocytes and, most strongly, in areas of the lamina propria. B-D: LC, DG, and C12E9 all caused a diffuse cytoplasmic staining of the enterocytes in a mosaic pattern, but subapical punctae were not detected. The images shown of each situation are representative of at least 5 images. Bars: 20 µm.
Figure 5.
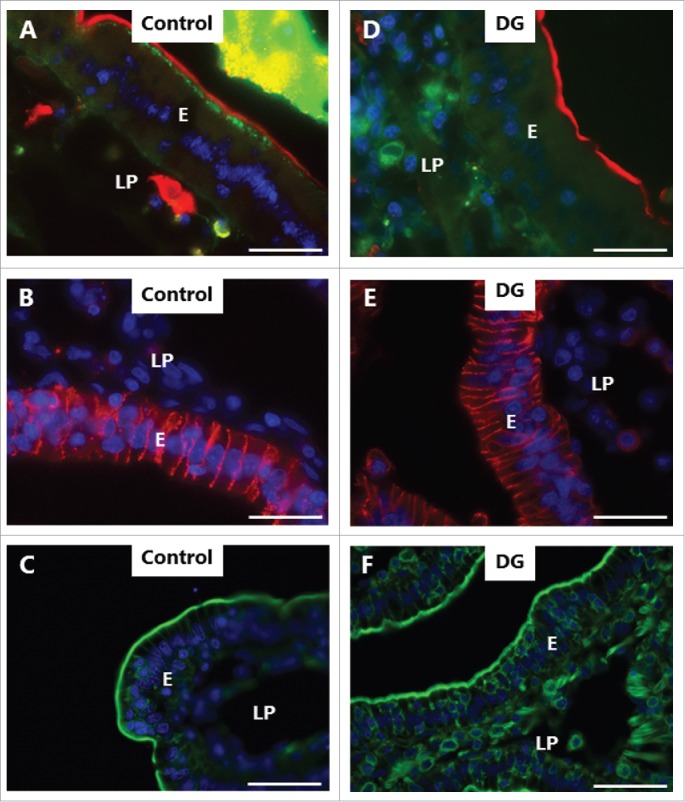
PE-induced inhibition of constitutive endocytosis revealed by Alexa 488 hydrazide. Mucosal explants cultured for 1 h in the absence (A-C) or presence (E-F) of 2 mM DG and in the presence of 10 µg/ml of the fluorescent polar tracer Alexa 488 hydrazide (green color in A, D). Sections were immunolabeled for aminopeptidase N (red color in A, D), Na+/K+-ATPase (red color in B, E), or with Alexa 488-conjugated phalloidin (green color in C, F). All images shown were captured at pairwise identical settings of the microscope. A: The fluorescent polar tracer Alexa 488 hydrazide is taken up into distinct subapical punctae like LY (Fig. 4), but is not visible in the paracellular space and only weakly stains the lamina propria. D: No Alexa 488 hydrazide accumulated in subapical punctae, and diffuse cytoplasmic staining of the enterocytes was moderate in comparison with LY (Fig. 4). Neither the brush border marker aminopeptidase N, the basolateral marker Na+/K+-ATPase, nor the cortical cytoskeleton marker phalloidin were visibly affected by DG. The images shown of each situation are representative of at least 5 images. Bars: 20 µm.
Figure 5 also shows the localization of aminopeptidase N (apical membrane marker), Na+/K+-ATPase (basolateral membrane marker), and phalloidin (actin cytoskeleton marker) in controls and DG-treated explants. The distribution of none of these markers was visibly perturbed by DG, suggesting that the overall cell membrane polarity and gross cortical cytoskeletal architecture of the enterocytes were not compromised by DG. Similar results were obtained for LG and C12E9 (data not shown).
In addition to the constitutive pinocytosis of small molecules visualized by LY and Alexa 588 hydrazide, the enterocyte brush border possesses another uptake mechanism, receptor-mediated endocytosis. Bacterial toxins belonging to the heat-labile enterotoxin family of AB5 multimer proteins, such as cholera toxin B and E. coli LTB,31,32 exploit this pathway for intoxication. Glycolipids in the brush border, notably ganglioside GM1, act as cell surface receptors for LTB,33 and as shown in Fig. 6A, FITC-LTB efficiently labeled the villus surface. In addition to the surface labeling, subapical punctae were also seen, indicative of an endocytotic uptake. By comparison, LC, DG and C12E9 all caused a marked reduction in LTB binding to the villus surface, and few, if any, distinct subapical punctae were detectable (Fig. 6 B-D). In case of C12E9, a more diffuse apical staining was seen in some enterocytes, suggestive of an entry into the cytoplasm (Fig. 6D).
Figure 6.
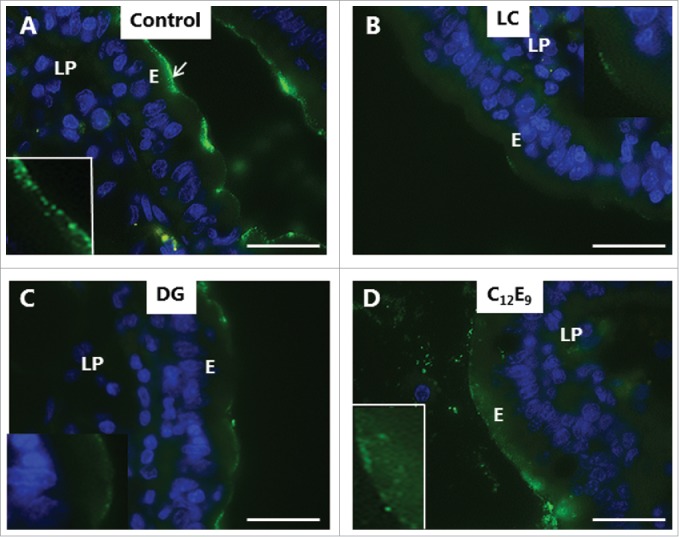
Inhibition of enterotoxin uptake by PEs. Mucosal explants cultured for 1 h in the presence of 10 µg/ml FITC-LTB and in the absence (A) or presence of 2 mM of LC (B), DG (C) or C12E9 (D). A: Punctate labeling with FITC-LTB is seen at, and below, the apical surface of the enterocytes. B-D: Reduced labeling at the apical surface and only few subapical punctae. In the presence of C12E9, a diffuse staining of the apical cytoplasm was seen. (Inserts in A and D show enlarged image details.) The images shown of each situation are representative of at least 5 images. Bars: 20 µm.
Collectively, the results presented above indicate a common functional mechanism of interaction between the 3 PEs studied and the mucosal epithelium. Firstly, they all permeabilized the cell membranes of the enterocytes, rendering them leaky to a compound like LY. Secondly, they blocked both the constitutive -and the ligand-induced endocytotic uptake from the brush border.
PEs functionally perturb enterocyte cell membranes
Electron microscopy was used for examining in greater structural detail the epithelial changes caused by the PEs, and Fig. 7A, C shows that the overall microvillus length was not markedly affected by LC, and that tight junctions between adjacent cells likewise appeared normal. However, microvesiculation of entire, single microvilli was occasionally seen, indicating a breakdown of the longitudinal actin cytoskeleton. In cross sections (Fig. 7B, D-F), a highly regular hexagonal array of brush border microvilli was seen in control explants, but remarkably, LC and DG disrupted this organization by causing lateral fusion of adjacent microvilli. Thus, pair-wise fused microvilli were frequently observed, and occasionally structures consisting of 3 fused microvilli were also detected. The 2 central bundles of the actin cytoskeleton in the microvillus dimers apparently remained intact, resulting in oblong oval microvilli. Finally, Fig. 7F depicts pairs of microvilli connected by a “stalk," suggestively in the early stage of complete fusion. However, in enterocytes closer to the denudation zone at the tip of villi, apical membrane buckling- and blebbing occurred (Fig. 8A, B). Here, electron micrographs revealed a more extensive deterioration of the brush border with widespread fragmentation and disappearance of microvilli. The formation of blebs seemed to be a process resulting from available surplus apical membrane following the collapse of the microvillus architecture and blocked endocytosis (Fig. 8C, D).
Figure 7.
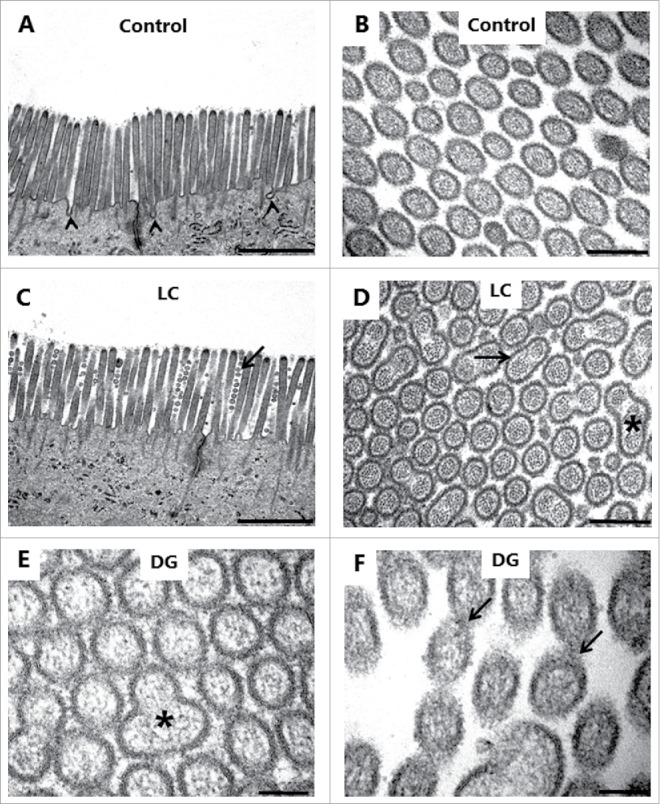
PE action on the enterocyte brush border. Electron micrographs of the microvillus membrane from mucosal explants cultured for 1 h in the absence (A, B) or presence of 2 mM LC (C, D) or 2 mM DG (E, F). A, C: Longitudinal sections of microvilli. Only in the control were membrane invaginations frequently seen between adjacent microvilli (arrowheads), implying an ongoing endocytosis. LC induced sporadic microvesiculation of microvilli (arrow). B, D-E: Cross sections of microvilli. LC and DG disrupted the hexagonal order by inducing widespread pair-wise fusion of microvilli (arrow), and occasionally 3 fused microvilli of various shapes were also seen (asterisks). F: Cross section of microvilli at high magnification. Arrows indicate pairs of microvilli connected by a “stalk," possibly in the early stage of fusion. The images shown of each situation are representative of at least 5 images. Bars: 1 µm (A, C); 0.2 µm (B, D); 0.1 µm (E, F).
Figure 8.
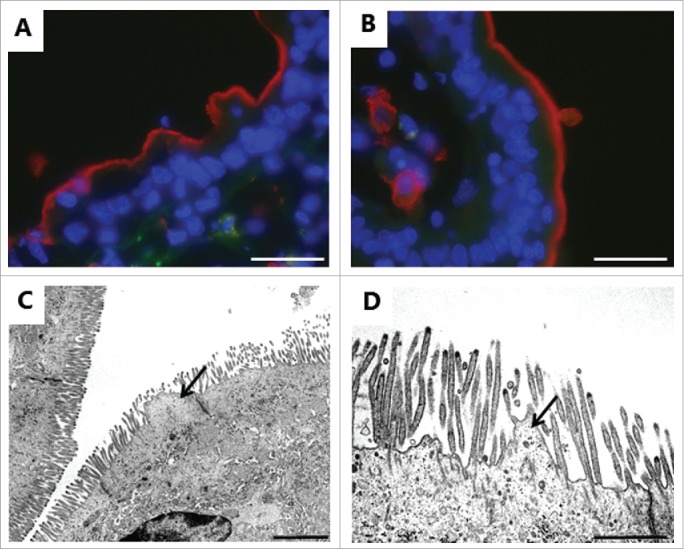
Deterioration of the brush border near a zone of denudation induced by 2 mM LC. A, B: Immunofluorescent images of sections labeled for the apical membrane marker aminopeptidase N as in Fig. 5, showing buckling (A) and blebbing (B) of the brush border. C, D: Electron micrographs showing microvillus deterioration and formation of blebs by surplus membrane (arrows). The images shown of each situation are representative of at least 5 images. Bars: 20 µm (A, B); 2 µm (C); 1 µm (D).
As shown in Fig. 9, also the lateral parts of the enterocyte cell membrane were affected by LC. In controls, this membrane is seen as a distinct, meandering border with only a narrow intercellular space between adjacent cells (Fig. 9A). LC caused an extensive vacuolization in the lateral region of the cells together with the appearance of numerous cytoplasm-filled, vesicle-like structures. Although the vacuoles were lighter in color than the cytoplasm, they seemed to contain material of cellular origin (Fig. 9B-D).
Figure 9.
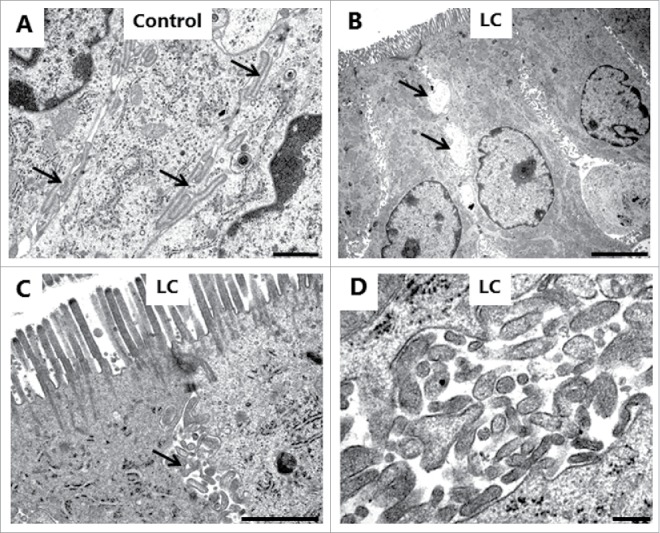
LC action on the enterocyte lateral cell membranes. Electron micrographs of the lateral cell membranes from mucosal explants cultured for 1 h in the absence (A) or presence of 2 mM LC (B-D). A: Highly meandering lateral cell membranes between adjacent cells (arrows). B: Formation of vacuoles (arrows) along the intercellular space. C: Formation of vesicle-like structures filled with cytoplasmic material (arrows). D: Lateral vesicles shown in higher magnification. The images shown of each situation are representative of at least 5 images. Bars: 1 µm (A, C); 5 µm (B); 0.2 µm (D).
In summary, PEs perturbed both the apical –and lateral aspects of the enterocyte cell membrane. The formation of fused microvilli directly indicates that they render the cell membrane spontaneously fusogenic. Conceivably, a similarly induced fusogenic activity might also underlie the formation of vacuoles and vesicle-like structures along the lateral surfaces, either by causing fusion of meandering parts of membranes belonging to the same cell or by fusion of membrane parts of adjacent cells. In contrast, no gross changes in the morphology of the tight junctions were revealed by electron microscopy despite the fact that long chain acylcarnitines are known to act as tight junction openers.34 Most likely, this reflects that modulation of tight junction permeability can be achieved without causing a full disintegration of the junctional complex.
Discussion
Acyl carnitines, acyl glycerols, and ethoxylates are all examples of surfactant types of agents that have been commonly explored as candidate PEs in commercial formulations for oral drug delivery.2 Understandably, most studies performed on these agents so far have focused on various parameters reflecting their ability to increase epithelial permeability, i. e. typically determination of transepithelial electrical resistance (TEER) and transport rates of soluble markers across monolayers of epithelial cells, such as Caco-2 cells.34–37 The organ culture system used in the present work is not suitable for quantitative measurements of this type, but instead allows visualization of the impact of PEs on the brush border membrane of authentic mucosal tissue. Here, our results show that surfactant action on cell membranes exerts a wider biologic effect than simply increasing paracellular –or transcellular permeability. One essential feature of cell membrane functioning is the membrane-confined trafficking connecting the cell surface with the intracellular cellular compartments, and the results obtained revealed that LC, DG, and C12E9 all blocked both the constitutive –and receptor-mediated endocytosis from the brush border. This observation indicates that irrespective of the particular surfactant chemical structure, incorporation into the bilayer is sufficient to perturb the ability of the membrane to engage in any type of endocytosis. Surfactants are generally thought to fluidize the membrane and cause swelling, ultimately leading to loss of integrity and permeabilization.2 It is now also evident that such changes in bilayer organization must be equally prohibitive of the processes involved in the formation of a membrane invagination and its progression to an endocytotic vesicle. Likewise, the formation of the peculiar-looking apical fused microvilli, as well as lateral vesicle-like structures and vacuoles, most likely can be ascribed to a common surfactant-induced membrane fusogenic activity. To occur, a membrane fusion process must typically be initiated by the formation of a focal hemifusion connection, commonly referred to as a “stalk," which subsequently leads to a larger fusion pore.38,39 However, spontaneous membrane fusion in living cells would be highly detrimental and is normally prevented by repulsive forces between the opposing bilayers, resulting from electrostatic repulsion of equally charged surfaces and from hydration, together with the lateral tension of the bilayer interface. When required to occur under physiologic processes, such as exocytosis, a specialized fusion machinery, including SNARE proteins, acts to overcome this energy barrier.40 Spontaneous fusion events in artificial model membranes can be induced, for instance by Ca2+ and polyethylene glycol,41,42 but to our knowledge fusion of microvilli has not previously been reported as a consequence of PE action. Notably, however, the same striking membrane defect was previously observed in myosin VI (myo6) knockout mice.43 Myo6 is a cytoskeleton protein localized primarily in the inter-microvillus/terminal web region of enterocytes, and its absence causes disorganization of the brush border morphology as well as reduced endocytotic uptake of luminal lactoferrin. Although seemingly unrelated phenomena, this observation also implies a functional relationship between the membrane fusion defect and an impaired endocytotic membrane trafficking.
In conclusion, concurrently with the continued pharmacological interest in surfactants and their use in formulations of oral drugs,2 there is also a widespread safety concern regarding this use.3,26 The main unsettled issues here are questions related to the effectiveness of epithelial repair mechanisms following damage caused by PEs, and fears that PEs may also inadvertently enable absorption of luminal pathogens (the so-called bystander argument). In this context, the novel effects on cell membrane functioning described in the present work add to the complexity of what is currently known about surfactant PE action in the gut and ought to be taken into consideration when assessing this type of agents as candidate PEs. The mucosal explant culture system used here may prove a valuable tool for future studies addressing these problems.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
Karina Rasmussen and Lotte Niels-Christiansen are thanked for excellent technical assistance.
Funding
The study was supported by grants from Brødrene Hartmanns Fond (A26557) and Læge Sofus Carl Emil Friis og hustru Olga Doris Friis legat.
References
- [1].Moroz E, Matoori S, and Leroux JC. Oral delivery of macromolecular drugs: where we are after almost 100years of attempts. Adv Drug Deliv Rev. 2016;101:108-21. doi: 10.1016/j.addr.2016.01.010. PMID:26826437 [DOI] [PubMed] [Google Scholar]
- [2].Maher S, Mrsny RJ, and Brayden DJ. Intestinal permeation enhancers for oral peptide delivery. Adv Drug Deliv Rev. 2016;106(Pt B):277-319. doi: 10.1016/j.addr.2016.06.005; PMID:27320643 [DOI] [PubMed] [Google Scholar]
- [3].Aungst BJ. Absorption enhancers: applications and advances. AAPS.J. 2012;14(1):10-18. doi: 10.1208/s12248-011-9307-4. PMID:22105442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fasinu P, Pillay V, Ndesendo VM, du Toit LC, Choonara YE. Diverse approaches for the enhancement of oral drug bioavailability. Biopharm Drug Dispos. 2011;32(4):185-209. doi: 10.1002/bdd.750. PMID:21480294 [DOI] [PubMed] [Google Scholar]
- [5].Yewale C, Patil S, Kolate A, Kore G, Misra A. Oral absorption promoters: opportunities, issues, and challenges. Crit Rev Ther Drug Carrier Syst. 2015;32(5):363-87. doi: 10.1615/CritRevTherDrugCarrierSyst.2015011865. PMID:26559432 [DOI] [PubMed] [Google Scholar]
- [6].Konig J, Wells J, Cani PD, García-Ródenas CL, MacDonald T, Mercenier A, Whyte J, Troost F, Brummer RJ. Human intestinal barrier function in health and disease. Clin Transl Gastroenterol. 2016;7(10):e196; doi: 10.1038/ctg.2016.54. PMID:27763627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Blikslager AT, Moeser AJ, Gookin JL, Jones SL, Odle J. Restoration of barrier function in injured intestinal mucosa. Physiol Rev. 2007;87(2):545-64. doi: 10.1152/physrev.00012.2006. PMID:17429041 [DOI] [PubMed] [Google Scholar]
- [8].France MM and Turner JR. The mucosal barrier at a glance. J Cell Sci. 2017;130(2):307-14. doi: 10.1242/jcs.193482. PMID:28062847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lichtenberg D, Robson RJ, and Dennis EA. Solubilization of phospholipids by detergents. Structural and kinetic aspects. Biochim Biophys Acta. 1983;737(2):285-304. doi: 10.1016/0304-4157(83)90004-7. PMID:6342675 [DOI] [PubMed] [Google Scholar]
- [10].Danielsen EM and Hansen GH. Lipid raft organization and function in brush borders of epithelial cells. Mol Membr Biol. 2006;23(1):71-79. doi: 10.1080/09687860500445604. PMID:16611582 [DOI] [PubMed] [Google Scholar]
- [11].Delacour D, Salomon J, Robine S, Louvard D. Plasticity of the brush border - the Yin and Yang of intestinal homeostasis. Nat Rev Gastroenterol Hepatol. 2016;13(3):161-74. doi: 10.1038/nrgastro.2016.5. PMID:26837713 [DOI] [PubMed] [Google Scholar]
- [12].Crawley SW, Mooseker MS, and Tyska MJ. Shaping the intestinal brush border. J Cell Biol. 2014;207(4):441-451. doi: 10.1083/jcb.201407015. PMID:25422372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hansen GH, Rasmussen K, Niels-Christiansen LL, Danielsen EM. Endocytic trafficking from the small intestinal brush border probed with FM dye. Am J Physiol Gastrointest Liver Physiol. 2009;297(4):G708-15. doi: 10.1152/ajpgi.00192.2009. PMID:19679822 [DOI] [PubMed] [Google Scholar]
- [14].Danielsen EM and Hansen GH. Generation of stable lipid raft microdomains in the enterocyte brush border by selective endocytic removal of non-raft membrane. PLoS One. 2013;8(10):e76661; doi: 10.1371/journal.pone.0076661. PMID:24124585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hansen GH, Dalskov SM, Rasmussen CR, Immerdal L, Niels-Christiansen LL, Danielsen EM. Cholera toxin entry into pig enterocytes occurs via a lipid raft- and clathrin-dependent mechanism. Biochemistry. 2005;44(3):873-82. doi: 10.1021/bi047959+. PMID:15654743 [DOI] [PubMed] [Google Scholar]
- [16].Binkley N, Bolognese M, Sidorowicz-Bialynicka A, Vally T, Trout R, Miller C, Buben CE, Gilligan JP, Krause DS, Oral Calcitonin in Postmenopausal Osteoporosis (ORACAL) Investigators . A phase 3 trial of the efficacy and safety of oral recombinant calcitonin: the Oral Calcitonin in Postmenopausal Osteoporosis (ORACAL) Trial. J Bone Miner Res. 2012;27(8):1821-9. doi: 10.1002/jbmr.1602. PMID:22437792 [DOI] [PubMed] [Google Scholar]
- [17].Hansen GH, Sjöström H, Norén O, Dabelsteen E. Immunomicroscopic localization of aminopeptidase N in the pig enterocyte. Implications for the route of intracellular transport. Eur J Cell Biol. 1987;43(2):253-9. PMID:2885197 [PubMed] [Google Scholar]
- [18].Danielsen EM, Sjöström H, Norén O, Bro B, Dabelsteen E. Biosynthesis of intestinal microvillar proteins. Characterization of intestinal explants in organ culture and evidence for the existence of pro-forms of the microvillar enzymes. Biochem J. 1982;202(3):647-54. doi: 10.1042/bj2020647. PMID:7092836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Booth AG and Kenny AJ. A rapid method for the preparation of microvilli from rabbit kidney. Biochem J. 1974;142(3):575-81. doi: 10.1042/bj1420575. PMID:4464842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(259):680-5. doi: 10.1038/227680a0. PMID:5432063 [DOI] [PubMed] [Google Scholar]
- [21].Kagnoff MF, Donaldson RM Jr., and Trier JS. Organ culture of rabbit small intestine: Prolonged in vitro steady state protein synthesis and secretion and secretory IgA secretion. Gastroenterology. 1972;63(4):541-51. PMID:4627771 [PubMed] [Google Scholar]
- [22].Lorenzen US, Hansen GH, and Danielsen EM. Organ culture as a model system for studies on enterotoxin interactions with the intestinal epithelium. Methods Mol Biol. 2016;1396:159-166. doi: 10.1007/978-1-4939-3344-0_14. PMID:26676046 [DOI] [PubMed] [Google Scholar]
- [23].Danielsen EM, Hansen GH, and Karlsdottir E. Staphylococcus aureus enterotoxins A- and B: Binding to the enterocyte brush border and uptake by perturbation of the apical endocytic membrane traffic. Histochem Cell Biol. 2013;139(4):513-24. doi: 10.1007/s00418-012-1055-8. PMID:23180309 [DOI] [PubMed] [Google Scholar]
- [24].Danielsen ET and Danielsen EM. Glycol chitosan: A stabilizer of lipid rafts in the intestinal brush border. Biochim Biophys Acta. 2017;1859(3):360-7. doi: 10.1016/j.bbamem.2016.12.017. PMID:28034633 [DOI] [PubMed] [Google Scholar]
- [25].Moore R, Carlson S, and Madara JL. Rapid barrier restitution in an in vitro model of intestinal epithelial injury. Lab Invest. 1989;60(2):237-44. PMID:2915518 [PubMed] [Google Scholar]
- [26].McCartney F, Gleeson JP, and Brayden DJ. Safety concerns over the use of intestinal permeation enhancers: a mini-review. Tissue Barriers. 2016;4(2):e1176822; doi: 10.1080/21688370.2016.1176822. PMID:27358756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hanani M. Lucifer yellow - an angel rather than the devil. J Cell Mol Med. 2012;16(1):22-31. doi: 10.1111/j.1582-4934.2011.01378.x. PMID:21740513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gopalakrishnan S, Pandey N, Tamiz AP, Vere J, Carrasco R, Somerville R, Tripathi A, Ginski M, Paterson BM, Alkan SS. Mechanism of action of ZOT-derived peptide AT-1002, a tight junction regulator and absorption enhancer. Int J Pharm. 2009;365(1-2):121-30. doi: 10.1016/j.ijpharm.2008.08.047. PMID:18832018 [DOI] [PubMed] [Google Scholar]
- [29].Danielsen EM, Hansen GH, Rasmussen K, Niels-Christiansen LL. Permeabilization of enterocytes induced by absorption of dietary fat. Mol Membr Biol. 2013;30(3):261-72. doi: 10.3109/09687688.2013.780642. PMID:23527550 [DOI] [PubMed] [Google Scholar]
- [30].Danielsen EM and Hansen GH. Small molecule pinocytosis and clathrin-dependent endocytosis at the intestinal brush border: two separate pathways into the enterocyte. Biochim Biophys Acta. 2016;1858(2):233-243. doi: 10.1016/j.bbamem.2015.11.022. PMID:26615917 [DOI] [PubMed] [Google Scholar]
- [31].Moss J and Vaughan M. Mechanism of action of choleragen and E. Coli heat-labile enterotoxin: activation of adenylate cyclase by ADP-ribosylation. Mol Cell Biochem. 1981;37(2):75-90. doi: 10.1007/BF02354931. PMID:6268961 [DOI] [PubMed] [Google Scholar]
- [32].Sixma TK, Kalk KH, van Zanten BA, Dauter Z, Kingma J, Witholt B, Hol WG. Refined structure of escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J Mol Biol. 1993;230(3):890-918. doi: 10.1006/jmbi.1993.1209. PMID:8478941 [DOI] [PubMed] [Google Scholar]
- [33].Fukuta S, Magnani JL, Twiddy EM, Holmes RK, Ginsburg V. Comparison of the carbohydrate-binding specificities of cholera toxin and escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect Immun.. 1988;56(7):1748-53. PMID:3290106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hochman JH, Fix JA, and LeCluyse EL. In vitro and in vivo analysis of the mechanism of absorption enha.ncement by palmitoylcarnitine. J Pharmacol Exp Ther. 1994;269(2):813-22. PMID:8182550 [PubMed] [Google Scholar]
- [35].Shima M, Kimura Y, Adachi S, Matsuno R. The relationship between transport-enhancement effects and cell viability by capric acid sodium salt, monocaprin, and dicaproin. Biosci Biotechnol Biochem. 1998;62(1):83-86. doi: 10.1271/bbb.62.83. PMID:27393356 [DOI] [PubMed] [Google Scholar]
- [36].Sha X et al.. Effect of self-microemulsifying drug delivery systems containing labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477-86. doi: 10.1016/j.ejps.2005.01.001. PMID:15784337 [DOI] [PubMed] [Google Scholar]
- [37].Tomita M, Doi N, and Hayashi M. Effects of acylcarnitines on efflux transporting system in Caco-2 cell monolayers. Eur J Drug Metab Pharmacokinet. 2010;35(1-2):1-7. doi: 10.1007/s13318-010-0001-1. PMID:21495260 [DOI] [PubMed] [Google Scholar]
- [38].Kozlovsky Y, Chernomordik LV, and Kozlov MM. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys J. 2002;83(5):2634-51. doi: 10.1016/S0006-3495(02)75274-0. PMID:12414697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kozlovsky Y and Kozlov MM. Stalk model of membrane fusion: solution of energy crisis. Biophys J. 2002;82(2):882-95. doi: 10.1016/S0006-3495(02)75450-7. PMID:11806930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Han J, Pluhackova K, and Bockmann RA. The multifaceted role of SNARE proteins in membrane fusion. Front Physiol. 2017;8:5; doi: 10.3389/fphys.2017.00005. PMID:28163686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pannuzzo M, De Jong DH, Raudino A, Marrink SJ. Simulation of polyethylene glycol and calcium-mediated membrane fusion. J Chem Phys. 2014;140(12):124905; doi: 10.1063/1.4869176. PMID:24697479 [DOI] [PubMed] [Google Scholar]
- [42].Lentz BR. PEG as a tool to gain insight into membrane fusion. Eur Biophys J. 2007;36(4-5):315-26. doi: 10.1007/s00249-006-0097-z. PMID:17039359 [DOI] [PubMed] [Google Scholar]
- [43].Hegan PS, Giral H, Levi M, Mooseker MS. Myosin VI is required for maintenance of brush border structure, composition, and membrane trafficking functions in the intestinal epithelial cell. Cytoskeleton (Hoboken.). 2012;69(4):235-51. doi: 10.1002/cm.21018. PMID:22328452 [DOI] [PMC free article] [PubMed] [Google Scholar]