

Abstract
The discovery of epidermal growth factor receptor (EGFR) mutations and subsequent demonstration of the efficacy of genotype-directed therapies with EGFR tyrosine kinase inhibitors (TKIs) marked the advent of the era of precision medicine for non-small-cell lung cancer (NSCLC). First- and second-generation EGFR TKIs, including erlotinib, gefitinib and afatinib, have consistently shown superior efficacy and better toxicity compared with first-line platinum-based chemotherapy and currently represent the standard of care for EGFR-mutated advanced NSCLC patients. However, tumors invariably develop acquired resistance to EGFR TKIs, thereby limiting the long-term efficacy of these agents. The T790M mutation in exon 20 of the EGFR gene has been identified as the most common mechanism of acquired resistance. Osimertinib is a third-generation TKI designed to target both EGFR TKI-sensitizing mutations and T790M, while sparing wild-type EGFR. Based on its pronounced clinical activity and good safety profile demonstrated in early Phase I and II trials, osimertinib received first approval in 2015 by the US FDA and in early 2016 by European Medicines Agency for the treatment of EGFR T790M mutation-positive NSCLC patients in progression after EGFR TKI therapy. Recent results from the Phase III AURA3 trial demonstrated the superiority of osimertinib over standard platinum-based doublet chemotherapy for treatment of patients with advanced EGFR T790M mutation-positive NSCLC with disease progression following first-line EGFR TKI therapy, thus definitively establishing this third-generation TKI as the standard of care in this setting. Herein, we review preclinical findings and clinical data from Phase I–III trials of osimertinib, including its efficacy in patients with central nervous system metastases. We further discuss currently available methods used to analyze T790M mutation status and the main mechanisms of resistance to osimertinib. Finally, we provide an outlook on ongoing trials with osimertinib and novel therapeutic combinations that might continue to improve the clinical outcome of EGFR-mutated NSCLC patients.
Keywords: osimertinib, T790M mutation, epidermal growth factor receptor, tyrosine kinase inhibitors
Introduction
Lung cancer is the leading cause of cancer mortality, representing more than one-quarter of all cancer deaths in 2016.1 About 85% of lung cancers are non-small-cell lung cancer (NSCLC), comprising adenocarcinoma, squamous cell carcinoma, and large-cell lung cancer, which are generally diagnosed as locally advanced or metastatic disease. For patients with advanced NSCLC, platinum-based chemotherapy has historically been the cornerstone of systemic treatment, although it offers modest improvement in overall survival (OS).2 The discovery of oncogenic drivers, including epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) rearrangements, mainly found in adenocarcinoma, and the development of specific molecularly targeted agents, has radically changed the therapeutic landscape for advanced NSCLC patients.3,4 The remarkable efficacy of treatment with EGFR tyrosine kinase inhibitors (TKIs), including gefitinib, erlotinib and afatinib, has been largely established in several prospective trials, resulting in higher objective response rate (ORR), longer progression-free survival (PFS) and better tolerability and quality of life (QoL), compared with standard platinum-based chemotherapy in EGFR-mutated NSCLC patients.5–12 Similarly, the first-in-class ALK TKI, crizotinib, showed superior efficacy than chemotherapy in NSCLC patients with ALK rearrangements.13 These targeted agents currently represent the standard, first-line treatment of such molecularly defined patients with advanced NSCLC.14 The combination of erlotinib and bevacizumab represents another first-line treatment option for patients with NSCLC harboring activating EGFR mutations.14–16 After progression to crizotinib, ALK-rearranged NSCLC patients can benefit from treatment with next-generation ALK inhibitors, including ceritinib, alectinib and brigatinib, which have been approved in this setting.17–19 Recently, for the relatively small subgroup of NSCLC patients harboring the BRAF V600E mutation, dabrafenib and trametinib combination has emerged as a novel, effective therapeutic option.20 In the past few years, immunotherapy has further expanded the therapeutic array for NSCLC patients and different immune checkpoint inhibitors targeting the PD-1/PD-L1 pathway, including nivolumab, pembrolizumab and atezolizumab, are currently approved in the second-line setting.21,22 The anti-PD-1 monoclonal antibody, pembrolizumab, has recently received approval also in the first-line setting, for those patients whose tumors have high PD-L1 expression (defined as PD-L1 expression on at least 50% of tumor cells), because of demonstration of longer PFS and OS, with fewer adverse events, than platinum-based chemotherapy.23
Novel promising targeted agents, including those directed against mesenchymal-epithelial transition (MET) and rearranged during transfection (RET), and different immunotherapies are under investigation and will probably further improve lung cancer management over the next few years.24
Targeting EGFR in NSCLC
The epidermal growth factor receptor (ErbB1 or human epidermal growth factor receptor 1 [HER1]) is a transmembrane receptor with tyrosine kinase activity involved in the regulation of cell proliferation, survival, differentiation and other crucial processes through activation of multiple downstream signaling cascades, including PI3K/AKT, RAS/RAF/mitogen-activated protein kinase (MAPK) and STAT pathways.25,26 EGFR pathway is often deregulated in a variety of tumor types, including NSCLC, through different molecular mechanisms involving gene encoding for EGFR, such as mutations or amplifications, altered expression of the EGFR protein, or its cognate ligands, EGF, or transforming growth factor-α, establishing autocrine loops that hyperactivate the receptor. Indeed, in squamous cell lung cancer, which is a particularly aggressive disease, EGFR gene amplification occurs in 7%–10% of cases and EGFR protein overexpression is most common in squamous- compared with the nonsquamous NSCLC.27,28 Somatic activating mutations of EGFR are instead predominant in adenocarcinoma.29 Activating EGFR mutations were first identified in NSCLC in 2004 and characterized as oncogenic mutations conferring high sensitivity to targeted inhibition by EGFR TKIs.30–32 EGFR gene mutations cluster in the region encoding for adenosine triphosphate (ATP)-binding pocket of the kinase domain (exons 18–21) and induce constitutive activation of the receptor and downstream pro-survival pathways and, consequently, confer oncogenic properties to cells which become dependent on EGFR for their survival.33 The most common, “classic” mutations are the in-frame exon 19 deletion and the exon 21 point mutation, resulting in a substitution of arginine for leucine at position 858 (L858%), accounting for about 90% of all mutations. Other relatively rare, sensitizing EGFR mutations have been described, such as G719X and L861Q mutations. The prevalence of EGFR mutations is higher in never-smokers, females and patients of East Asian ethnicity. Through a systematic review and meta-analysis, including 456 studies, Zhang et al have found that the overall pooled prevalence for EGFR mutations in patients with NSCLC is 32.3%, ranging from 38.4% in China to 14.1% in Europe.34 The first-generation, reversible EGFR TKIs, gefitinib and erlotinib, are orally bioavailable synthetic anilinoquinazolines designed to compete for ATP binding to the catalytic site of the receptor, switch off prosurvival signals, and cause tumor cell death, whereas the second-generation inhibitors, including afatinib and dacomitinib, are irreversible inhibitors with greater affinity for the EGFR kinase domain and can also inhibit and block signaling from other members of the ErbB family, thus providing enhanced EGFR blockade. The Iressa Pan-Asia Study, including advanced NSCLC patients with clinical characteristics predictive of response to TKIs (East Asian patients, adenocarcinoma, never- or mild-smokers), was the first study demonstrating the superiority of gefitinib compared with chemotherapy in patients with EGFR mutations, thus leading to its approval in this setting.5 Indeed, within this subgroup of patients, gefitinib was associated with a significantly longer PFS and ORR compared with chemotherapy. Subsequent multiple, randomized Phase III trials, exclusively enrolling patients with EGFR-mutated NSCLC, explored the use of gefitinib, erlotinib or afatinib as first-line treatment in comparison with standard platinum-based chemotherapy. In all studies, EGFR TKIs improved ORRs, PFS and QoL over chemotherapy.6–12,35 However, none of the above studies have shown any benefit in OS for any of the EGFR TKI over platinum-based chemotherapy, likely due to treatment crossover effects at progression. Only a pooled OS analysis of patients included in 2 Phase III trials, LUX-Lung 3 and LUX-Lung 6, showed a signifi-cant OS benefit in patients with exon 19 deletions treated with afatinib, compared with chemotherapy. The same OS difference was not seen in patients with L858R mutations.36 These positive results have led to the approval of gefitinib, erlotinib and afatinib, as upfront therapies of advanced NSCLC patients with EGFR mutations. These drugs have dramatically improved the therapeutic management of NSCLC patients and have also impacted on the diagnostic process, since tumor molecular profiling has been rapidly incorporated over the past few years into clinical routine practice to guide treatment selection.14,37
Mechanisms of resistance to first-line EGFR TKI therapy
Unfortunately, despite marked and often durable responses to EGFR TKIs, the vast majority of patients will develop progressive disease, generally within 1 year of treatment, due to development of acquired resistance that ultimately limits the long-term efficacy of these agents. A variety of mechanisms responsible for acquired resistance have been identified and can be broadly categorized into the following types: secondary mutations within the EGFR kinase domain, activation of alternative signaling pathways, namely bypass tracks, and phenotypic changes (including small-cell lung cancer transformation and epithelial to mesenchymal transition).38 The most common and best characterized mechanism of acquired resistance is represented by a C to T change at nucleotide 2369 in exon 20, which results in the substitution of the non-polar hydrophobic and larger amino acid methionine for the hydrophilic threonine residue within the ATP-binding cleft of the kinase domain.39–42 Other mechanisms include the activation of parallel signaling pathways, the so-called bypass tracks, leading to EGFR-independent growth, including amplification, overexpression and autocrine loops involving MET, HER2, AXL, insulin-like growth factor (IGF-1R), fibroblast growth factor receptor, and ephrin type-A receptor 2 or molecular alterations of key downstream molecules of the PI3K/AKT/mammalian target of rapamycin (mTOR) or the RAS/RAF/MAPK pathways.38,42–44 A crucial role in acquired resistance has also been demonstrated for other key mediators downstream of EGFR, including nuclear factor-κB, the proapoptotic protein BIM (BCL2-like 11) and STAT3.45–47
In addition, epithelial–mesenchymal transition and small-cell lung cancer transformation have been also correlated with resistance to EGFR TKIs.38,43
Until recently, no standard treatment options after failure of EGFR TKI therapy were available. Because of possible coexistence of different subclonal populations of tumor cells in EGFR TKI-resistant cancers, some evidence suggests that for those patients experiencing central nervous system (CNS) progressive disease or oligo-progressive disease, local therapy (e.g., surgery, radiotherapy, or both) to the site of progression to target the TKI-resistant subclones might be appropriate, with continuation of treatment with the same initial TKI.38,48–50
Systemic treatment is generally represented by platinum-based doublet or single-agent chemotherapies, with modest benefit in terms of PFS. A Phase III randomized trial failed to demonstrate the superiority of gefitinib plus cisplatin and pemetrexed compared with chemotherapy and placebo after progression on first-line gefitinib.51 In addition, the combination of gefitinib and chemotherapy had a detrimental effect on OS, mainly driven by T790M-positive mutation status detected by circulating tumor DNA (ctDNA) genotyping.52 Alternative treatment strategies, including investigational drugs or novel combinations, have been explored in clinical trials, but none of these has been approved as standard treatment in this setting.38
Therapeutic strategies targeting EGFR T790M
The EGFR T790M occurs in about 50%–60% of EGFR TKI-resistant tumors.39–42 This gatekeeper mutation, and consequent amino acid change, interferes with reversible EGFR TKIs binding through steric hindrance and is analogous to L1196M in ALK and T315I in the BCR-ABL fusion gene conferring resistance to corresponding TKIs. The presence of T790M can also restore the affinity of mutant receptor for ATP, thus reducing the potency of competitive inhibitors.53 The identification of T790M prompted the development of different therapeutic strategies to overcome resistance. Second-generation EGFR TKIs, including afatinib, dacomitinib and neratinib, are irreversible, non-selective EGFR inhibitors showing greater potency compared with first-generation TKIs in inhibiting T790M kinase activity in preclinical studies.54–56
However, clinical data suggested a rather limited activity of these drugs in the setting of acquired resistance to first-generation TKIs.38,57–60 These results have been largely attributed to the fact that the high rate of toxicities, mainly represented by skin and gastrointestinal adverse events (AEs), due to non-selective inhibition of wild-type EGFR, limited the ability to administer in vivo doses sufficient to effectively inhibit the T790M mutation. Based on promising preclinical data, the combination of afatinib and cetuximab was tested in Phase Ib study in heavily pretreated patients with EGFR-mutated NSCLC and in those resistant to gefitinib or erlotinib; the results showed significant clinical activity, with ORR of 29% and median PFS of 4.7 months.61 Results were comparable in both subgroups of patients with T790M-positive and T790M-negative tumors. However, a high incidence of grade ≥3 AEs were observed (mainly skin and gastrointestinal toxic effects), with 13% of patients discontinuing study drugs due to treatment-related AEs.61 To overcome pharmacodynamic shortcomings of second-generation EGFR TKIs, third-generation, mutant EGFR-selective TKIs have been designed, with preferential activity toward sensitizing mutations, as well as T790M resistance mutation over the wild-type receptor, thus likely reducing on-target toxicities. The first-described compound directed at EGFR T790M was WZ4002, identified by screening an irreversible kinase inhibitor library for drugs that bound EGFR T790M. The drug binds irreversibly to mutant EGFR at the C797 residue and demonstrated 100-fold less potency against WT EGFR and 30–100 fold more potency against EGFR T790M.62,63 While WZ4002 has not progressed to clinical development, other drugs with similar characteristics were developed and taken into early phase clinical trials. Among these, CO-1686 (rociletinib) and AZD9291 (osimertinib) were the first to demonstrate pronounced clinical activity as single agent in EGFR-mutated NSCLC patients with acquired resistance to erlotinib, gefitinib or afatinib.64–67 However, while clinical development of rociletinib was interrupted due to some concerns regarding its benefit–risk profile,22,68 osimertinib development proceeded and it was granted US Food and Drug Administration (FDA) approval in November 2015 for the treatment of patients with metastatic EGFR T790M mutation-positive NSCLC who have progressed on or after EGFR TKI therapy.69
Osimertinib
Design and mechanism of action
Osimertinib (TAGRISSO™, AZD9291; AstraZeneca) is a mono-anilino-pyrimidine compound that irreversibly and selectively targets EGFR TKI-sensitizing- and T790M resistance-mutant forms of EGFR, while sparing wild-type EGFR (Figure 1).65 Osimertinib binds to the EGFR kinase by targeting the cysteine-797 residue in the ATP-binding site via covalent bond formation. While WZ4002 and rociletinib share a number of common structural features, osimertinib has a distinct, unique chemical structure. In EGFR recombinant enzyme assays, osimertinib had nearly 200 times greater potency against L858R/T790M than wild-type EGFR, thus confirming its selectivity for mutant EGFR.65 Osimertinib showed a wide therapeutic window against EGFR T790M-resistant mutations in an in vitro model established to assess the mutation specificity of different EGFR TKIs.70 In murine in vivo studies, the metabolism of osimertinib produced at least 2 metabolites, AZ5104 and AZ7550; of these 2 metabolites, only AZ7550 had comparable potency and selectivity to the original compound, while AZ5104 was more powerful against mutant and wild-type EGFR forms, at the expense of minor selectivity. When tested across multiple other kinases, osimertinib showed minimal off-target kinase activity. At 1 μM, the compound showed inhibition >60% of a limited number of additional kinases, including ErbB2/4, ACK1, ALK, BLK, BRK, MLK1 and MNK2, thereby supporting the overall selectivity of osimertinib. Interestingly, osimertinib did not show activity in vitro against the IGF-1R and insulin receptor, which also have a methionine gatekeeper in their kinase domains and this observation was confirmed in in vivo studies, thus suggesting a low risk of dose-limiting toxicities related to hyperglycemia, in contrast to rociletinib.65
Figure 1.

EGFR pathway and mechanism of action of osimertinib.
Abbreviation: EGFR, epidermal growth factor receptor; TGF, transforming growth factor.
Osimertinib showed similar potency to early generation reversible TKIs in inhibiting EGFR phosphorylation in EGFR cell lines harboring sensitizing EGFR mutations. However, unlike first-generation TKIs, it inhibited phosphorylation of EGFR in T790M mutant cell lines (H1975: L858R/T790M and PC-9VanR: ex19del/T790M) with higher potency (mean IC50 <15 nM) than wild-type EGFR (mean IC50: 480–1865 nM). In mutant EGFR cell lines, osimertinib also induced inhibition of downstream signaling substrates (pAKT, pERK) more potently compared with EGFR wild-type cell lines. In vitro studies on cell lines transfected with cDNAs encoding other rare EGFR mutations and HER2 mutations, which have been also correlated with EGFR TKIs acquired resistance, showed that AZ5104 had some activity against the EGFRvIII mutant, while osimertinib was effective in inhibiting the growth of cells harboring HER2 mutations (VC insertion at G776 in exon 20 and the most common exon 20 YVMA 776-779ins). Therefore, depending on the clinical exposures achieved, osimertinib and AZ5104 may also inhibit these targets.65
In mutant EGFR xenograft models, once daily dosing of osimertinib induced significant dose-dependent regression in both TKI-sensitizing and T790M-resistant EGFR-mutant disease models. Significant tumor shrinkage was observed at low doses. When osimertinib was administered as long-term daily oral dosing, complete and durable responses were observed in these EGFR-mutant xenografts, with no evidence of tumor progression out to 200 days of treatment and with good tolerability in the animals, showing <5% of starting body weight loss.65 Finally, osimertinib demonstrated its antitumor activity by inducing significant tumor shrinkage in in vivo transgenic mouse tumor models harboring EGFR L858R or L858R+T790M mutants. In these models, the pharmacodynamic properties of osimertinib to inhibit its target and downstream pathway were further confirmed.
Based on promising results from preclinical studies, in which osimertinib largely demonstrated its role as a mutant-selective EGFR inhibitor, with sustained antitumor activity, subsequent clinical trials were conducted to develop this drug and identify its best use in therapy.
Pharmacokinetics (PK)
Osimertinib demonstrated good bioavailability, was widely distributed in tissues and had moderate clearance resulting in a half-life of around 3 h after oral dosing in the mouse. Preliminary clinical PK analysis on a small number of NSCLC patients indicated that osimertinib and its active metabolites, AZ5104 and AZ7550, have a half-life of at least 50 h, longer than would be predicted from the preclinical data, which resulted in a desirable flat PK profile after multiple once daily dosing.65
Key secondary endpoint of the Phase I AURA study, enrolling EGFR-mutated NSCLC patients who had developed resistance to treatment with EGFR TKIs, included determination of the PK of osimertinib. Patients in the dose-escalation cohorts received a single dose of osimertinib (in capsule form, from 20 to 240 mg once daily) followed by a PK evaluation period; after 7 days, they received the same oral dose daily, with additional PK assessment after daily continuous dosing. PK analyses showed that osimertinib exposure, maximal plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC) increased in a dose-proportional manner across the range from 20 to 240 mg after single and multiple doses, thus exhibiting linear PK.67
The PK of osimertinib was extensively characterized following a single-dose administration in healthy volunteers and advanced NSCLC patients of the AURA trial following single- and multiple-dose administration. The PK profiles between patients and healthy volunteers appeared similar, although there was an apparent higher clearance in healthy volunteers. Absorption was slow, regardless of formulation (capsule, solution or tablet). The median time to maximum plasma concentration (Cmax) of osimertinib after single oral dosing was 6 h (range 3–24 h) across all doses. Dose-proportional increase in osimertinib exposure (20–240 mg) was observed after single and multiple dosing. The distribution of the drug was extensive and clearance low to moderate, resulting in a mean half-life of ~48 h. Osimertinib steady state was achieved by 15 days of once daily dosing, witĥ3- to 4-fold accumulation in exposure over single dose, resulting in a low Cmax to Cmin (minimal concentration) ratio (1.6), suggesting that concentrations are maintained throughout the dosing interval.71 The main metabolic pathways of osimertinib were oxidation (predominantly CYP3A) and dealkylation in vitro. Two circulating pharmacologically active metabolites, AZ5104 and AZ7550, were measured in plasma at ~10% of osimertinib at steady state. In healthy volunteers, food caused a small increase in osimertinib exposure compared with fasted administration, but this was not considered clinically significant. Osimertinib exposure appeared to be similar in both Asian and non-Asian patients.71 In the Phase II AURA 2 study,72 192 patients were included in the PK analyses, which further demonstrated a flat PK for osimertinib and confirmed previous results from Planchard et al.71
On the basis of population PK analyses, no clinically significant differences in the PK of osimertinib were observed based on age, sex, ethnicity, body weight, smoking status, mild or moderate or severe renal impairment, or mild or moderate hepatic impairment.73
Development of the drug
Phase I and II studies
The Phase I/II AURA trial was conducted to determine the safety and efficacy of osimertinib in patients with advanced NSCLC who had disease progression after previous treatment with EGFR-TKIs.67 In the study were included patients with known EGFR-sensitizing mutations or those who had prior clinical benefit from EGFR TKI therapy and had a documented disease progression while on treatment. Key secondary objectives included the determination of the maximum dose of the agent associated with an acceptable level of AEs, the PK and the antitumor activity. A total of 253 patients were included in the study: 31 in the dose-escalation cohorts and 222 in 5 dose-expansion cohorts. At data cut-off date (August 1, 2014), enrollment into additional cohorts, including patients who had not received prior treatment, and Phase II extension were ongoing.67 Most of the patients (62%) were Asian and women (62%) and 80% had received prior chemotherapy. In the dose-escalation part of the study in which patients received osimertinib at doses of 20–240 mg per dose, once daily, no dose-limiting toxicities occurred at any dose level and a maximum tolerated dose could not be defined. In the expansion cohorts, all patients underwent tumor biopsy for central assessment of EGFR T790M status and 138 (62%) were found positive for this mutation.
Of the 239 patients evaluable for response, 123 (51%) had a confirmed objective response, including partial response in 122 patients and 1 complete response, with a disease control rate (DCR) of 84%.67 The majority of patients (85%) showed a long-lasting response of ≥6 months. The median PFS was 8.2 months. In the subgroup of T790M-positive NSCLC patients evaluable for response (n=127), osimertinib showed high activity, with an ORR of 61%, a DCR of 95% and a median PFS of 9.6 months (95% CI 8.3 to not reached). In contrast, for the 61 evaluable patients with no detectable EGFR T790M, the ORR was 21% and PFS was 2.8 months (95% CI 2.1–4.3) (Table 1). The most frequent AEs were diarrhea (47% of patients), rash (40%) nausea and decreased appetite (21%). Among these, only the frequency of diarrhea and rash seemed to be dose dependent. Indeed, at doses of 160 and 240 mg, there was an increase in the incidence and severity of AEs, possibly explained by initial inhibition of non-mutant EGFR. Grade ≥3 AEs were observed in 32% of patients; however, the percentage of patients that needed to reduce the dose or discontinue the treatment was low. Treatment-related serious AEs were observed in 6% of patients. Six cases of potential pneumonitis-like events occurred, all leading to discontinuation of treatment, with resolution. However, pneumonia was reported as a fatal adverse event in 1 patient. Six patients had hyperglycemia and 11 a corrected QT interval prolongation, but none of these events led to drug dose reduction or discontinuation. Based on clinical activity and safety data, the dose of 80 mg once daily was selected for further clinical investigation.67
Table 1.
AURA clinical trials
| Trial | Phase | No. of patients | mPFS (months) | RR (%) | OS (months) |
|---|---|---|---|---|---|
| AURA | I/II | 253: - 31 (dose-escalation cohorts) - 222 (dose-expansion cohorts): - 138: T790M + - 62: T790M − - 22: T790M unknown |
- T790M + : 9.6 (95% CI 8.3–NR); - T790M − : 2.8 (95% CI 2.1–4.3) |
- T790M + : 61 (95% CI 52–70); - T790M − : 21 (95% CI 12–34) |
– |
| AURA2 | II | 210 | 9.9 (95% CI 8.5–12.3) | 70 (95% CI 64–77) | 1-year OS rate: 81% (95% CI 75–86) |
| AURA extension | II | 411 (T790M +) | 12.3 (95% CI 9.5–13.8) | 62 (95% CI 54–68) | 1-year OS rate: 79% (95% CI 72–84) |
| AURA3 | III | 416: - experimental arm (279) - chemotherapy arm (140) |
10.1 (95% CI 8.3–12.3) 4.4 (95% CI 4.2–5.6) HR 0.30; (95% CI 0.23–0.41; p<0.001) |
71 (95% CI 65–76] 31 (95% CI 24–40) Odds ratio 5.39; 95% CI 3.47–8.48; p<0.001 |
– |
Notes: ‘–’ indicates not reported.
Abbreviations: HR, hazard ratio; NR, not reached; OS, overall survival; PFS, progression-free survival; RR, response rate.
The encouraging efficacy and safety results from the Phase I study, led to AURA2, a multicenter, Phase II, single-arm study testing osimertinib at 80 mg orally daily in locally advanced or metastatic NSCLC patients with EGFR T790M-positive mutations, centrally confirmed by the Cobas® EGFR Mutation Test v2, who had progressed on prior EGFR TKI therapy.72 Patients could have received more additional lines of treatment, including platinum-based chemotherapy. Patients with asymptomatic, stable CNS metastases not requiring steroids could also be enrolled. A total of 210 patients were enrolled and treated with osimertinib. At data cutoff (November 1, 2015), with a median duration of treatment follow-up of 13 months, 58% of patients remained on treatment. Among the 199 evaluable patients, ORR as assessed by blinded independent central review (BICR), the primary endpoint, was 70%, including 3% of complete responses, with a DCR of 92%. The high proportion of objective responses was consistent across all subgroups, including patients with CNS metastases. Median duration of response (DoR) was 11.4 months and the median PFS was 9.9 months (95% CI 8.5–12.3) (Table 1). A higher number of objective responses, as well as a longer PFS, were noted in the group of patients with exon 19 deletions (ORR: 77%, 95% CI 69–84 and PFS: 10.9 months, 95% CI 9.5–13.6) than patients with L858R mutations (ORR: 59%, 95% CI 46–71 and PFS: 8.5 months, 95% CI 6.9–12.7). However, this difference was not significant and, as the authors underlined, could be in part explained by an apparent higher incidence of EGFR T790M in tumors with EGFR 19 deletions in the study population.72 One-year survival was 81%, while OS data were still immature. Osimertinib displayed a manageable toxicity profile. The most common possibly treatment-related grade 3 or 4 AEs were prolonged electrocardiogram QT (2%), neutropenia (1%) and thrombocytopenia (1%). Only 5% of patients discontinued osimertinib due to an AE. Among the reported deaths, the only death possibly related to treatment as reported by the investigator was due to interstitial lung disease (ILD). An improvement or stability of lung cancer symptoms and functioning domains from QoL was observed with osimertinib. For example, most patients reported a reduction in chest pain.72
The Phase II AURA extension study confirms the high activity of osimertinib at 80 mg, once-daily, in patients with EGFR-sensitizing and T790M-positive NSCLC progressing after EGFR TKI treatment.74
Preliminary results from the extension component of AURA had been previously presented in a preplanned pooled analysis with the Phase II AURA2 study.75 In this first analysis, including 411 EGFR T790M-positive patients, ORR was 66% and median PFS was 11.0 months (95% CI 9.6–12.4).75 In a recent publication that reported only data from the AURA extension, the ORR (by BICR) was 62% among 198 evaluable patients, with a DCR of 90% and median DoR of 15.2 months (Table 1).74 ORRs were high, ranging from 53% to 68%, across all predefined subgroups, including second/third line or more, Asian/non-Asian, male/female, age <65/≥65, exon 19 del/L858R, duration of most recent EGFR TKI: <6 months/≥6 months, CNS metastases: present/absent, never smoker/ever smoker, last EGFR TKI treatment: <30 days before osimertinib/≥30 days before osimertinib. Median PFS was 12.3 months (95% CI 9.5–13.8). Subset analysis revealed a nonsignificant trend toward longer median PFS in patients with exon 19 deletions (12.6 months) compared with L858R (9.6 months) and in Asian (12.6 months) versus non-Asian patients (9.7 months). Osimertinib showed substantial activity in patients with CNS metastases: 16 (64%) of 25 evaluable patients had an objective response, including 4 complete responses. Median PFS in patients with CNS metastases was encouraging, although it was shorter than in those without (7.1 vs 13.7 months, respectively). Median follow-up for OS was 13.8 months and 1-year survival rate was 79%. The safety profile was consistent with previous studies, with most AEs being mild and determining discontinuation of treatment only in 3% of patients. The most common possibly causally related AEs were diarrhea (43%; grade ≥3, <1%) and rash (40%; grade ≥3, <1%). ILD was reported in 8 patients (4%), including 3 fatal. QT prolongation was reported in 6 patients but only 3 were deemed possibly related to treatment. Patients had consistent and sustained improvements in key lung cancer symptoms, including dyspnea, cough, chest pain, and pain in arm or shoulder and in QoL compared with baseline.
Phase III studies
To confirm results of single-arm, Phase II studies, a randomized, Phase III trial (AURA3) was conducted to demonstrate the superiority of osimertinib over standard chemotherapy with platinum and pemetrexed for treatment of patients with EGFR-mutated and centrally confirmed T790M-positive advanced NSCLC with disease progression after first-line EGFR TKI therapy.76 A total of 419 patients were randomized in a 2:1 ratio to osimertinib (80 mg once daily) or pemetrexed plus carboplatin or cisplatin every 3 weeks, for up to 6 cycles, with maintenance pemetrexed allowed. The primary efficacy end point was PFS as determined by investigator assessments, according to response evaluation criteria in solid tumors (version 1.1). Secondary objectives included ORR by investigator, response duration, DCR, tumor shrinkage, OS, patient-reported outcomes (PROs) and safety. Predefined subgroup analyses included the assessment of PFS and response rate among patients for whom EGFR T790M status was determined by means of a plasma ctDNA test and in patients with CNS metastases. At data cut-off date (April 15, 2016), the mean duration of treatment was 8.6 months in the osimertinib group and 4.8 months in the platinum-pemetrexed group, and a total of 59% of patients in the osimertinib arm and 12% in the chemotherapy arm were still receiving treatment. In the platinum-pemetrexed group, 60% of patients at progression crossed over to receive osimertinib. After a median follow-up of 8.3 months, the study met its primary endpoint showing a significantly longer PFS for patients in the osimertinib arm compared with platinum chemotherapy arm (10.1 vs 4.4 months, HR: 0.30, 95% CI 0.23–0.41, p<0.001) (Table 1). The benefit was observed across all predefined subgroups, including patients with asymptomatic CNS metastases at baseline (PFS: 8.5 vs 4.2 months, HR 0.32, 95% CI 0.21–0.49) and patients positive for T790M on both tumor and plasma. Treatment with osimertinib was also associated with better ORR compared with chemotherapy (71% vs 31%, odds ratio: 5.39, 95% CI 3.47–8.48, p<0.001) (Table 1) in the overall population and in the subgroup of patients with T790M-positive status on both tumor and plasma samples. Responses were more durable with osimertinib (DoR: 9.7 vs 4.1 months). Despite the longer treatment duration with osimertinib, this was associated with lower incidence of grade ≥3 AEs (23%) compared with platinum-pemetrexed (47%) and lower discontinuation rate (7% vs 10%, respectively). The most common AEs with osimertinib were diarrhea, rash, dry skin and paronychia, while in the chemotherapy group, the AEs were nausea, decreased appetite, constipation and ane-mia. ILD-like AEs were reported in 10 patients (4%) in the osimertinib group (including 1 fatal) and in 1 patient (1%) in the platinum-pemetrexed group. Also, PROs were better in the osimertinib group than in the platinum-pemetrexed group.76
These results definitively established the role of osimertinib as a standard of care for patients who progress to first-line EGFR TKI and who harbor the T790M resistance mutation. Moreover, the findings of AURA3 support the feasibility of detecting the EGFR T790M resistance mutation from plasma ctDNA and their reliable predictive role.
Activity of osimertinib against CNS metastases
Brain metastases are a common problem in NSCLC patients and are associated with a significant symptomatic burden. Several reports have demonstrated a certain activity of early generation EGFR TKIs in EGFR-mutated NSCLC patients with brain and leptomeningeal metastases.77–80 It has also been demonstrated that a high-dose or pulsatile administration of high-dose EGFR TKIs may be able to improve their efficacy in these poor-prognosis patients.81–84 However, more than 30% of patients who progress during or after treatment with EGFR TKIs have brain and/or leptomeningeal metastases,85 which represent a clinical issue because of poor penetration of drugs, including targeter agents, through the blood–brain barrier (BBB).86
In preclinical studies, osimertinib demonstrated greater penetration of the BBB than gefitinib, afatinib or rociletinib and, in contrast to rociletinib, induced sustained tumor regression in an EGFR-mutated mouse brain metastases model at clinically relevant doses.87 By using positron emission tomography imaging, osimertinib displayed markedly greater and faster distribution into the cynomolgus monkey brain than rociletinib and gefitinib.87 A number of clinical reports, including subgroup analyses from the AURA trials, support these preclinical data by demonstrating relevant activity of osimertinib in EGFR mutation-positive patients with brain and/or leptomeningeal metastases.72,74,76,88 Moreover, a different CNS activity among third-generation EGFR TKIs has been reported in clinical setting.89 A Phase I trial (BLOOM; NCT02228369) is ongoing to assess the safety and preliminary activity of osimertinib (administered at 160 mg once daily) in patients with EGFR-mutated advanced NSCLC and leptomeningeal metastases, confirmed by positive cerebrospinal fluid (CSF). Responses are evaluated based on CSF cytology, intracranial imaging and neurological examination. Preliminary results on 21 patients from this trial have been recently reported.90 A confirmed intracranial response was observed in 7 patients: 2 patients had a confirmed CSF cytology clearance and 5 patients had neurological improvement. The safety profile of osimertinib was manageable.90
Overall, these results suggest that osimertinib can be an effective treatment option for EGFR-mutated patients with CNS disease, thus sparing patients from or delaying the use of radiotherapy, which, in the case of whole brain radiation (WBRT) can ultimately compromise patients’ cognitive function and QoL. Interestingly, due to the high risk of development of CNS metastases in EGFR-mutant NSCLC, an EGFR TKI specifically designed to penetrate the BBB and achieve high intracranial exposure, AZD3759, is currently being evaluated in an open-label, multicenter, Phase I study in patients with advanced EGFR mutation-positive NSCLC (NCT02228369).91
Cell-free ctDNA genotyping for EGFR T790M mutation
Genotyping post-progression tumor samples for EGFR T790M has become a crucial step for guiding treatment decisions and is recommended in current NSCLC guidelines.14,37 However, in clinical practice, rebiopsies of recurrent or metastatic tumors can often be difficult to obtain, due to the potential risks involved in invasive diagnostic procedures that can also be painful for patients. For the same reasons, it is rarely feasible to obtain serial biopsies from the same patient at different points during treatment. Moreover, tumor tissue from some biopsies is not sufficient for genetic analysis or, even when adequate, it only represents a single snapshot in time and is subject to selection bias resulting from tumor heterogeneity. Due to these limitations, a great number of studies in recent years have addressed the question of whether cell-free circulating DNA derived from tumors, also known as ctDNA, could be used as a surrogate liquid biopsy for noninvasive assessment of tumor genetic alterations, including EGFR TKI sensitizing- and T790M-resistant mutations.92 While tissue biopsies only describe genetic characteristics of the primary tumor, ctDNA is potentially able to provide a portrait of the whole tumor genome landscape of both primary and metastatic lesions. Serial analysis of ctDNA can track molecular dynamic evolution of the tumor during the course of treatment and identify the emergence of heterogeneous subclonal populations of tumor cells. A number of studies have demonstrated that the T790M mutation can be detected in plasma samples by highly sensitive genotyping assays, including droplet digital polymerase chain reaction (ddPCR) and the bead, emulsion, amplification and magnetics (BEAMing) technique.92–95
In a retrospective analysis of Phase I AURA, Oxnard et al reported a sensitivity of 70% of the plasma-based test for detecting T790M mutation. Remarkably, patients with T790M mutation detected in plasma ctDNA had comparable responses and PFS to osimertinib as those with positive tissue-based assay results.96 The predictive value of plasma ctDNA genotyping for T790M was prospectively confirmed in the Phase III AURA3 trial showing a longer PFS and higher ORR for osimertinib compared with platinum-pemetrexed chemotherapy (PFS: 8.2 vs 4.2 months, HR 0.42, 95% CI 0.29–0.61; ORR: 77% vs 39%, odds ratio, 4.96, 95% CI 2.49–10.15, p<0.001).76
However, due to the relatively high false negative rates with plasma T790M testing, the absence of this mutation in blood cannot exclude its presence in the tissue and, hence, its potential sensitivity to osimertinib. Indeed, in the analysis of AURA, tumour genotyping was positive for T790M in 45 out of 102 patients with T790M-negative plasma. This subgroup of patients had a good response to osimertinib with a median PFS of 16.2 months. For this reason, if plasma genotyping for T790M at disease progression is negative, a new tissue biopsy is highly recommended to correctly classify patients for treatment with osimertinib (Figure 2).
Figure 2.

ctDNA genotyping for EGFR T790M.
Abbreviations: EGFR, epidermal growth factor receptor; TKIs, tyrosine kinase inhibitors.
Remon et al further highlighted the predictive role of T790M mutation detected in plasma-based assay.97 In this study, the efficacy of osimertinib was evaluated in 48 EGFR-mutated, advanced NSCLC patients in progression after a prior EGFR TKI therapy, in whom T790M status was determined only in ctDNA by Inivata InVision™ assay (Inivata Ltd, Cambridge, UK), by using enhanced Tagged Amplicon-Sequencing (eTAm-Seq™ assay, Inivata Ltd). The ctDNA T790M mutation was found in 50% of patients and, among evaluable patients, osimertinib was associated with a partial response rate of 62.5% and a stable disease rate of 37.5%. In a small series of T790M-positive NSCLC who received osimertinib, no correlation was found between EGFR T790M mutation copy numbers evaluated by plasma-based assay (ddPCR) and response to osimertinib.98
Place in therapy
Osimertinib was the first third-generation EGFR TKI to receive accelerated approval by FDA in November 2015 for the treatment of patients with EGFR T790M-positive NSCLC whose disease had progressed on or after EGFR TKI therapy, and subsequent conditional approval by European Medicines Agency (EMA) in February 2016. In the AURA studies, EGFR T790M mutation status in tumor samples was assessed by using the Cobas EGFR Mutation Test v2 (Roche Molecular Systems, Pleasanton, CA, USA), which received initial FDA approval as companion diagnostic test to detect EGFR T790M mutant in tissue for osimertinib treatment. The approval of osimertinib by EMA already included the possibility of determining EGFR T790M mutation status using either a validated plasma-based or a tissue-based test. However, if the result of plasma detection in plasma is negative, due to the potential for false negative results, a tissue test is advisable wherever possible to determine EGFR T790M mutation (Figure 2). In the US, several months after the first approval of osimertinib, in September 2016, the FDA approved a label extension of the Cobas® EGFR Mutation Test v2 for use with plasma samples as a companion diagnostic for osimertinib.
Mechanisms of resistance to osimertinib
Similar to early generation EGFR TKIs, acquired resistance to osimertinib ultimately develops, thus limiting its long-term efficacy for EGFR T790M-mutated NSCLC patients. The mechanisms of resistance to third-generation EGFR TKIs, mainly osimertinib, are heterogeneous and can be divided into EGFR dependent and EGFR independent (Figure 3).99,100 The acquired mutation in C797S, the cysteine residue with which third-generation EGFR TKIs form a key covalent bond, has been reported as a potential mechanism of resistance to osimertinib, with its allelic context defining sensitivity to subsequent treatments.101,102 Serial ctDNA specimens collected from 15 osimertinib-treated subjects were analyzed by ddPCR. All were positive for the T790M mutation before treatment, but, upon developing osimertinib resistance, 6 cases acquired C797S resistance mutation in exon 20, 5 cases maintained T790M mutation but did not acquire the C797S mutation, and 4 cases lost T790M mutation despite the presence of the underlying EGFR activating mutation.101,102 The mechanism of resistance to third-generation EGFR TKIs was characterized in cell lines resistant to WZ4002, rociletinib and osimertinib. Three major drug-resistance-related EGFR mutations were identified: L718Q, L844V and C797S. All 3 mutations could cause resistance to WZ4002 and rociletinib, but only C797S mutation leads to osimertinib resistance.103 When C797S and T790M mutation is are in trans position, the cells can be sensitive to the combination of a first- and third-generation EGFR TKI, while when the 2 mutations are in cis position, the tumor cells cannot be suppressed by any EGFR TKI, alone or in combinations.104,105
Figure 3.
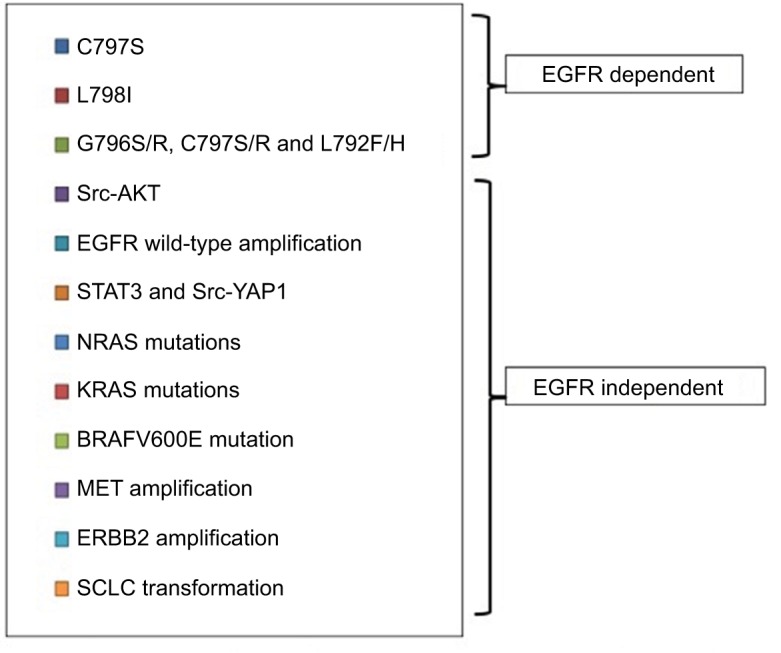
Mechanisms of resistance to third-generation EGFR TKI osimertinib.
Abbreviations: EGFR, epidermal growth factor receptor; TKIs, tyrosine kinase inhibitors.
The C797S mutation in the EGFR gene was found to confer resistance also to the third-generation EGFR TKI HM61713.102,106
Other EGFR mutations, including the L798I, can also confer resistance to osimertinib.107 Recently, in a patient with a L858R and T790M mutation progressing to osimertinib, a novel solvent-front mutation at Gly796(G796S/R) together with C797S/R and L792F/H, all in cis with T790M, emerged in plasma DNA.108
The Src-AKT pathway activation, as well as EGFR wild-type amplification, can contribute to acquired resistance to third-generation EGFR TKIs.109 EGFR wild-type allele amplification induces resistance to the mutation-selective EGFR inhibitors through EGFR ligand-induced activation.109
We have found that STAT3 and Src-YAP1 activation can be responsible for intrinsic resistance to first-, second- and third-generation EGFR TKIs.110 Considering that the EGFR ligand, amphiregulin, is a transcriptional target of YAP1,111 we can speculate a connection between YAP1 activation and the escape of tumor cells to third-generation EGFR TKIs through the activation of the wild-type EGFR.
Constitutive ERK activation through NRAS mutations and gain of copy number of wild-type NRAS or KRAS can be associated with resistance to osimertinib, as has been described in preclinical models.112 The concomitant inhibition of EGFR and ERK can prevent the emergence of resistance and be a better therapeutic approach than single EGFR inhibition.112,113 The BRAF V600E mutation was found in cells from malignant pleural effusion of a patient progressing to osimertinib. When these cells were cultured in vitro, they were significantly sensitive to the simultaneous blockade of EGFR and BRAF.114 According to case reports, bypass pathway activation via MET or ERBB2 amplification can be responsible for the resistance to third-generation EGFR TKIs.115,116 In a patient with high level of MET amplification, post-osimertinib treatment, single-agent crizotinib, was able to alleviate the symptoms of the disease.116 The amplification of MET, ERBB2 or KRAS G12S mutation was identified in tumor specimens from NSCLC patients who progressed with osimertinib or rociletinib treatment.100,117 Finally, transformation to SCLC has been described in 2 cases during osimertinib treatment, making the repetition of a biopsy a necessity at the time of resistance to third-generation EGFR TKIs, in order for an appropriate therapeutic decision to be made.118
Future perspectives: ongoing studies, novel agents and combination strategies
Osimertinib is currently being investigated in several trials as a single agent in different settings of disease and in novel, potentially synergistic, combinations with other targeted therapies or immunotherapies.
Updated efficacy results from 2 expansion cohorts, including patients with EGFR-mutated, advanced NSCLC who received 80 or 160 mg osimertinib as first-line treatment in the AURA study, demonstrated an impressive ORR of 77% (67% in the 80 mg- and 87% in the 160 mg cohort), with a DCR of 97% and a median DoR not reached. The median PFS was 19.3 months. The toxicity profile was manageable, with higher frequency of AEs and of dose reductions due to treatment-related AEs in the 160 mg cohort.119 The Phase III FLAURA (NCT02296125) trial is ongoing to compare the efficacy and safety of osimertinib with gefitinib or erlotinib in treatment-naïve EGFR-mutated NSCLC patients. A key secondary endpoint of the study is PFS in patients with T790M.
The presence of EGFR T790M in pretreatment tumors, which has been reported at different frequencies depending on the detection methods used,16,120–124 can further support the upfront use of osimertinib. In the EURTAC trial, pretreatment somatic EGFR T790M mutation was detected in about 65% of patients by using a highly sensitive method based on peptide nucleic acid clamping PCR and correlated with shorter PFS compared with patients without the resistant mutation.124 An ongoing study (AZENT; NCT02841579) has recently started to evaluate the safety and efficacy of osimertinib as first-line therapy for patients with concomitant sensitizing and T790M mutation.
Regarding the other settings of disease, an ongoing Phase III trial (AUDARA; NCT02511106), is evaluating osimer-tinib as adjuvant therapy compared with placebo in completed resected stage IB-IIIA, EGFR mutation-positive NSCLC.
Based on its favorable toxicity profile, osimertinib has been considered a particularly attractive candidate for combination therapeutic strategies. Preclinical studies also support a synergy of osimertinib with some targeted agents, for example, MEK inhibitors. The TATTON trial (NCT02143466) is a multi-arm, multicenter, Phase Ib trial, including a dose-escalation and a dose-expansion part, evaluating osimertinib in combination with the anti-PD-L1 monoclonal antibody, durvalumab (MEDI4736), or with the MET inhibitor, AZD6094, or with the MEK1/2 inhibitor, selumetinib, (AZD6244, ARRY-142886) in patients with advanced EGFR-mutated NSCLC. Inhibitors of the PD1/PD-L1 axis have been shown to be active in a broad range of malignancies.21,125 Durvalumab, which is currently approved for the treatment of locally advanced or metastatic urothelial carcinoma, has shown promising activity with durable responses as a single agent in pretreated patients with locally advanced or metastatic NSCLC, including patients with EGFR mutations, with greater activity in those patients with higher levels of PD-L1 expression (≥25%, assessed by the immunohistochemistry assay of Roche Ventana SP263, Roche Diagnostics, India-napolis, IN, USA).126 Updated results from the osimertinib and durvalumab combination arm presented at the European Lung Cancer Congress last year showed promising activity, but raised safety concerns due to a higher frequency of ILD events (38%, 13 of 34 patients, with 5 cases of grade 3/4) with the combination, compared with either single-agent treatment, especially in EGFR TKI treatment-naïve patients, although there was no apparent increase in ILD severity.127 Therefore, this combination arm was suspended.
Other possible approaches include combinations with antiangiogenic agents or anti-EGFR monoclonal antibodies. Indeed, an open-label, multicenter, Phase I study (NCT02789345) is ongoing to assess the safety and preliminary efficacy of ramucirumab or necitumumab in combination with osimertinib in patients with advanced EGFR T790M-positive NSCLC who have progressed after EGFR TKI therapy. An exploratory biomarker objective of this study includes the correlations between EGFR mutations in tissue and serial blood samples with clinical outcomes.128 In a recent appealing study, it was observed that acquired resistance mediated by T790M can occur, either by selective growth of preexisting clones positive for this mutation or by genetic evolution of initially T790M-negative drug-tolerant cells. In this last case, cells displayed a diminished apoptotic response to third-generation EGFR TKIs targeting T790M. Navitoclax (ABT-263), an inhibitor of BCL-2 family proteins, enhances the apoptotic response of these late resistant T790M cells and sensitivity to EGFR inhibition.129 The combination of navitoclax with the third-generation WZ4002 induced more apoptosis compared with WZ4002 alone in T790M patient-derived cell lines, in vitro-generated resistant T790M cell lines and in vivo xenograft tumors.129 A phase Ib trial is ongoing to evaluate the safety and tolerability of osimertinib and navitoclax combination in patients with EGFR-mutated NSCLC following resistance to prior EGFR TKI therapy (NCT02520778).
To overcome the C797S-mediated resistance, next-generation, selective inhibitors are needed. A novel allosteric, non-ATP competitive inhibitor (EAI045) of mutant EGFR has been recently identified.130,131 When combined with cetuximab, which can block EGFR dimerization by preventing EGF ligand binding, EAI045 markedly inhibited the proliferation of Ba/F3 cells bearing L858R/T790M mutation. These in vitro studies proved that EAI045 is active and selective for T790M-harboring EGFR mutants that are in a monomer state. In a mouse model of lung cancer harboring triple-mutant (L858R/T790M/C797S) EGFR, the combination of EAI045 and cetuximab was able to induce a remarkable tumor regression, thus suggesting this compound can overcome resistance from acquired T790M and C797S mutations. Notably, the next-generation ALK inhibitor brigatinib, with or without the combination of an anti-EGFR antibody, has recently demonstrated activity in preclinical models of triple-mutant tumors.132
Conclusion
Until recently, there were no standard therapeutic options in the setting of acquired resistance to initial EGFR TKIs and, outside clinical trials with investigational drugs, EGFR-mutated NSCLC patients generally received platinum-based or single-agent chemotherapy upon progression. Osimertinib has led to a paradigm shift in the management of lung cancer patients progressing to first- or second-generation TKIs, thus supporting the need in clinical practice for tumor genotyping at the time of disease progression on or after EGFR TKI therapy for biomarker evaluation of EGFR T790M. Osimertinib has also demonstrated activity in patients with CNS and leptomeningeal metastasis, which is particularly relevant, given the possibility to potentially spare these patients from radiotherapy or delay its use, including WBRT, that can ultimately compromise the cognitive function and QoL. Other novel, mutant-selective EGFR TKIs produced by different pharmaceutical companies, including EGF816, naquotinib (ASP8273), avitinib (AC0010) and PF-06747775, are in clinical development (Table 2) and have shown promising activity with manageable safety profile in early phase studies.133–139 More data from ongoing studies are eagerly awaited to confirm these preliminary results. Despite the evolving therapeutic landscape for EGFR-mutated NSCLC patients, there are still several issues to be addressed, such as the optimal therapeutic sequence of different generation EGFR TKIs. Osimertinib is being tested in the first-line setting, compared with standard EGFR TKI therapy, to see whether upfront use of an irreversible inhibitor could be a more effective strategy to improve clinical outcome and delay the resistance. Another possible strategy to prevent or delay the occurrence of resistance could be the upfront combination of EGFR inhibitors with other targeted agents. Indeed, the combination of erlotinib and bevacizumab results in a long PFS in EGFR-mutated NSCLC patients and, in the BELIEF trial, this combination was effective in presence of the T790M mutation, identified in 35% of patients, who attained a PFS of 16 months.16 The assessment of T790M mutation in ctDNA by using highly sensitive blood-based assays has already proven to be feasible in clinical practice and useful to identify those patients who are candidates for osimertinib. Moreover, a combined analysis of EGFR genotyping in matched blood and urine by a highly sensitive and quantitative assay using next-generation sequencing, as performed in the TIGER-X study of rociletinib, could further improve the chance of T790M mutation detection.140 Despite the high efficacy of osimertinib in T790M-mutant NSCLC, similar to other oncogene-addicted tumors, patients may ultimately progress due to acquisition of additional mechanism of resistance by tumor cells under the selective pressure of the EGFR TKI, including the C797S mutation. A novel EGFR inhibitor, EAI045, has shown to be able to overcome T790M and C797S resistance in in vitro and in vivo studies, although its clinical efficacy still needs to be investigated. Profiling of tumors at progression remains crucial in order to unravel the complex and heterogeneous resistance mechanisms to first-, second- and third-generation EGFR TKIs. The detection of new mechanisms of resistance to third-generation EGFR TKIs is feasible through ctDNA genotyping and suggests the need for new therapeutic approaches for these patients. Liquid biopsy, including not only ctDNA but also other tumor sources such as CTCs and exosomes, appears to be the most appealing strategy to study the dynamic evolution of oncogene-addicted tumor cells during treatment with different EGFR TKIs, thereby giving an early warning about possible recurrence and helping to establish novel, potentially targetable molecular alterations.
Table 2.
Third-generation EGFR TKIs in clinical development in NSCLC
| Agents | Binding mode | Selectivity | Status |
|---|---|---|---|
| Osimertinib (AZD9291) | Irreversible | EGFR TKI-sensitizing mutations and T790M | Approved, T790M + |
| Rociletinib (CO-1686) | Irreversible | EGFR TKI-sensitizing mutations and T790M | No longer in development |
| Olmutinib (BI1482694/HM61713) | Irreversible | EGFR TKI-sensitizing mutations and T790M | Approved (only South Korea), T790M + |
| ASP8273 | Irreversible | EGFR TKI-sensitizing mutations and T790M | Phase II, Phase III |
| Nazartinib (EGF816) | Irreversible | EGFR TKI-sensitizing mutations and T790M | Phase I/II, Phase II |
| PF-06747775 | Irreversible | EGFR TKI-sensitizing mutations and T790M | Phase I/II |
| Avitinib (AC0010) | Irreversible | EGFR TKI-sensitizing mutations and T790M | Phase I/II |
Abbreviations: EGFR, epidermal growth factor receptor; NSCLC, non-small-cell lung cancer; TKI, tyrosine kinase inhibitor.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Santarpia M, Karachaliou N, Rosell R. Beyond platinum treatment for NSCLC: what does the future hold? Expert Rev Anticancer Ther. 2017;17(4):293–295. doi: 10.1080/14737140.2017.1288103. [DOI] [PubMed] [Google Scholar]
- 3.Rosell R, Karachaliou N. Large-scale screening for somatic mutations in lung cancer. Lancet. 2016;387(10026):1354–1356. doi: 10.1016/S0140-6736(15)01125-3. [DOI] [PubMed] [Google Scholar]
- 4.Lazzari C, Spitaleri G, Catania C, et al. Targeting ALK in patients with advanced non small cell lung cancer: biology, diagnostic and therapeutic options. Crit Rev Oncol Hematol. 2014;89(3):358–365. doi: 10.1016/j.critrevonc.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 7.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11(2):121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;1(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 9.Han JY, Park K, Kim SW, et al. First-SIGNAL: first-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;30(10):1122–1128. doi: 10.1200/JCO.2011.36.8456. [DOI] [PubMed] [Google Scholar]
- 10.Inoue A, Kobayashi K, Maemondo M, et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin-paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002) Ann Oncol. 2013;24(1):54–59. doi: 10.1093/annonc/mds214. [DOI] [PubMed] [Google Scholar]
- 11.Sequist LV, Yang JCH, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 12.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 13.Blackhall F, Cappuzzo F. Crizotinib: from discovery to accelerated development to front-line treatment. Ann Oncol. 2016;27(Suppl 3):iii35–iii41. doi: 10.1093/annonc/mdw304. [DOI] [PubMed] [Google Scholar]
- 14.Novello S, Barlesi F, Califano R, Cufer T, et al. Metastatic non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(Suppl 5):v1–v27. doi: 10.1093/annonc/mdw326. [DOI] [PubMed] [Google Scholar]
- 15.Seto T, Kato T, Nishio M, et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol. 2014;15(11):1236–1244. doi: 10.1016/S1470-2045(14)70381-X. [DOI] [PubMed] [Google Scholar]
- 16.Rosell R, Dafni U, Felip E, et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): an international, multicentre, single-arm, phase 2 trial. Lancet Respir Med. 2017;5(5):435–444. doi: 10.1016/S2213-2600(17)30129-7. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, Savooji J, Liu D. Second- and third-generation ALK inhibitors for non-small cell lung cancer. J Hematol Oncol. 2016;9(1):19. doi: 10.1186/s13045-016-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santarpia M, Daffinà MG, D’Aveni A, et al. Spotlight on ceritinib in the treatment of ALK+ NSCLC: design, development and place in therapy. Drug Des Dev Ther. 2017;11:2047–2063. doi: 10.2147/DDDT.S113500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santarpia M, Altavilla G, Rosell R. Alectinib: a selective, next-generation ALK inhibitor for treatment of ALK-rearranged non-small-cell lung cancer. Expert Rev Respir Med. 2015;9(3):255–268. doi: 10.1586/17476348.2015.1009040. [DOI] [PubMed] [Google Scholar]
- 20.Planchard D, Besse B, Groen HJ, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–993. doi: 10.1016/S1470-2045(16)30146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santarpia M, Giovannetti E, Rolfo C, et al. Recent developments in the use of immunotherapy in non-small cell lung cancer. Expert Rev Respir Med. 2016;10(7):781–798. doi: 10.1080/17476348.2016.1182866. [DOI] [PubMed] [Google Scholar]
- 22.Karachaliou N, Sosa AE, Barron FB, et al. Pharmacological management of relapsed/refractory NSCLC with chemical drugs. Expert Opin Pharmacother. 2017;18(3):295–304. doi: 10.1080/14656566.2017.1285284. [DOI] [PubMed] [Google Scholar]
- 23.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 24.Dholaria B, Hammond W, Shreders A, Lou Y. Emerging therapeutic agents for lung cancer. J Hematol Oncol. 2016;9(1):138. doi: 10.1186/s13045-016-0365-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 26.Ciardiello F, Tortora G. EGFR antagonist in cancer treatment. N Engl J Med. 2008;358(11):1160–1174. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research N Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirsch FR, Varella-Garcia M, Bunn PA, Jr, et al. Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21(20):3798–3807. doi: 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 29.Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 31.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 32.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma SV, Bell DW, Settleman J, et al. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 34.Zhang YL, Yuan JQ, Wang KF, et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. 2016;7(48):78985–78993. doi: 10.18632/oncotarget.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenhalgh J, Dwan K, Boland A, et al. First-line treatment of advanced epidermal growth factor receptor (EGFR) mutation positive non-squamous non-small cell lung cancer. Cochrane Database Syst Rev. 2016;(5):CD010383. doi: 10.1002/14651858.CD010383.pub2. [DOI] [PubMed] [Google Scholar]
- 36.Yang JC, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141–151. doi: 10.1016/S1470-2045(14)71173-8. [DOI] [PubMed] [Google Scholar]
- 37.Ettinger DS, Wood DE, Aisner DL, et al. Non-small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15(4):504–535. doi: 10.6004/jnccn.2017.0050. [DOI] [PubMed] [Google Scholar]
- 38.Santarpia M, Gil N, Rosell R. Strategies to overcome resistance to tyrosine kinase inhibitors in non-small-cell lung cancer. Expert Rev Clin Pharmacol. 2015;8(4):461–477. doi: 10.1586/17512433.2015.1055252. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 40.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015;16(9):e447–e459. doi: 10.1016/S1470-2045(15)00246-6. [DOI] [PubMed] [Google Scholar]
- 44.Koch H, Busto ME, Kramer K, et al. Chemical proteomics uncovers EPHA2 as a mechanism of acquired resistance to small molecule EGFR kinase inhibition. J Proteome Res. 2015;14(6):2617–2625. doi: 10.1021/acs.jproteome.5b00161. [DOI] [PubMed] [Google Scholar]
- 45.Bivona TG, Hieronymus H, Parker J, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471(7339):523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karachaliou N, Codony-Servat J, Teixidó C, et al. BIM and mTOR expression levels predict outcome to erlotinib in EGFR-mutant non-small-cell lung cancer. Sci Rep. 2015;5:17499. doi: 10.1038/srep17499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HJ, Zhuang G, Cao Y, et al. Drug resistance via feedback activation of stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207–221. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 48.Gandara DR, Li T, Lara PN, et al. Acquired resistance to targeted therapies against oncogene-driven non-small-cell lung cancer: approach to subtyping progressive disease and clinical implications. Clin Lung Cancer. 2014;15(1):1–6. doi: 10.1016/j.cllc.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu HA, Sima CS, Huang J, et al. Local therapy with continued EGFR tyrosine kinase inhibitor therapy as a treatment strategy in EGFR-mutant advanced lung cancers that have developed acquired resistance to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2013;8(3):346–351. doi: 10.1097/JTO.0b013e31827e1f83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conforti F, Catania C, Toffalorio F, et al. EGFR tyrosine kinase inhibitors beyond focal progression obtain a prolonged disease control in patients with advanced adenocarcinoma of the lung. Lung Cancer. 2013;81(3):440–444. doi: 10.1016/j.lungcan.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 51.Soria JC, Wu YL, Nakagawa K, et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 2015;16(8):990–998. doi: 10.1016/S1470-2045(15)00121-7. [DOI] [PubMed] [Google Scholar]
- 52.Soria JC, Kim SW, Wu YL, et al. Gefitinib/chemotherapy vs chemotherapy in EGFR mutation-positive NSCLC after progression on 1st line gefitinib (IMPRESS study): final overall survival (OS) analysis. Ann Oncol. 2016;27(Suppl 6):vi416–vi454. [Google Scholar]
- 53.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105(6):2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 55.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kwak E. The role of irreversible HER family inhibition in the treatment of patients with non-small cell lung cancer. Oncologist. 2011;16(11):1498–1507. doi: 10.1634/theoncologist.2011-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 58.Katakami N, Atagi S, Goto K, et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol. 2013;31(27):3335–3341. doi: 10.1200/JCO.2012.45.0981. [DOI] [PubMed] [Google Scholar]
- 59.Ellis PM, Shepherd FA, Millward M, et al. Dacomitinib compared with placebo in pretreated patients with advanced or metastatic non-small-cell lung cancer (NCIC CTG BR.26): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2014;15(12):1379–1388. doi: 10.1016/S1470-2045(14)70472-3. [DOI] [PubMed] [Google Scholar]
- 60.Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(18):3076–3083. doi: 10.1200/JCO.2009.27.9414. [DOI] [PubMed] [Google Scholar]
- 61.Janjigian YY, Smit EF, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4(9):1036–1045. doi: 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou WJ, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riely GJ, Yu HA. EGFR: the paradigm of an oncogene-driven lung cancer. Clin Cancer Res. 2015;21(10):2221–2226. doi: 10.1158/1078-0432.CCR-14-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–1709. doi: 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]
- 67.Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 68.Van Der Steen N, Caparello C, Rolfo C, et al. New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong? Onco Targets Ther. 2016;9:6065–6074. doi: 10.2147/OTT.S97644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.US Food and Drug Administration FDA approves new pill to treat certain patients with non-small cell lung cancer. 2015. [Accessed May 15, 2017]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm472525.htm.
- 70.Hirano T, Yasuda H, Tani T, et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget. 2015;6(36):38789–38803. doi: 10.18632/oncotarget.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Planchard D, Brown KH, Kim D-W, et al. Osimertinib Western and Asian clinical pharmacokinetics in patients and healthy volunteers: implications for formulation, dose, and dosing frequency in pivotal clinical studies. Cancer Chemother Pharmacol. 2016;77(4):767–776. doi: 10.1007/s00280-016-2992-z. [DOI] [PubMed] [Google Scholar]
- 72.Goss G, Tsai CM, Shepherd FA, et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016;17(12):1643–1652. doi: 10.1016/S1470-2045(16)30508-3. [DOI] [PubMed] [Google Scholar]
- 73.TAGRISSO® (osimertinib) tablets, for oral use. US prescribing information. 2015. [Accessed May 15, 2017]. Available from: https://www.accessdata.fda.gov/drug-satfda_docs/label/2017/208065s006lbl.pdf.
- 74.Yang JC, Ahn MJ, Kim DW, et al. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA Study Phase II Extension Component. J Clin Oncol. 2017;35(12):1288–1296. doi: 10.1200/JCO.2016.70.3223. [DOI] [PubMed] [Google Scholar]
- 75.Yang J, Ramalingam SS, Jänne PA, et al. LBA2_PR: osimertinib (AZD9291) in pre-treated pts with T790M-positive advanced NSCLC: updated Phase 1 (P1) and pooled Phase 2 (P2) results. J Thorac Oncol. 2016;11(4):S152–S153. [Google Scholar]
- 76.Mok TS, Wu Y-L, Ahn M-J, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376(7):629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park SJ, Kim HT, Lee DH, et al. Efficacy of epidermal growth factor receptor tyrosine kinase inhibitors for brain metastasis in non-small cell lung cancer patients harboring either exon 19 or 21 mutation. Lung Cancer. 2012;77(3):556–560. doi: 10.1016/j.lungcan.2012.05.092. [DOI] [PubMed] [Google Scholar]
- 78.Hoffknecht P, Tufman A, Wehler T, et al. Efficacy of the irreversible ErbB family blocker afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)-pretreated nonsmall-cell lung cancer patients with brain metastases or leptomeningeal disease. J Thorac Oncol. 2015;10(1):156–163. doi: 10.1097/JTO.0000000000000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altavilla G, Arrigo C, Santarpia MC, et al. Erlotinib therapy in a patient with non-small-cell lung cancer and brain metastases. J Neurooncol. 2008;90(1):31–33. doi: 10.1007/s11060-008-9623-4. [DOI] [PubMed] [Google Scholar]
- 80.Proto C, Imbimbo M, Gallucci R, et al. Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of central nervous system metastases from non-small cell lung cancer: the present and the future. Transl Lung Cancer Res. 2016;5(6):563–578. doi: 10.21037/tlcr.2016.10.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clarke JL, Pao W, Wu N, et al. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. J Neurooncol. 2010;99(2):283–286. doi: 10.1007/s11060-010-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grommes C, Oxnard GR, Kris MG, et al. “Pulsatile” high-dose weekly erlotinib for CNS metastases from EGFR mutant non-small cell lung cancer. Neuro Oncol. 2011;13(12):1364–1369. doi: 10.1093/neuonc/nor121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Togashi Y, Masago K, Fukudo M, et al. Efficacy of increased-dose erlotinib for central nervous system metastases in non-small cell lung cancer patients with epidermal growth factor receptor mutation. Cancer Chemother Pharmacol. 2011;68(4):1089–1092. doi: 10.1007/s00280-011-1691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Milton DT, Azzoli GC, Heelan RT, et al. A phase I/II study of weekly high-dose erlotinib in previously treated patients with nonsmall cell lung cancer. Cancer. 2006;107(5):1034–1041. doi: 10.1002/cncr.22088. [DOI] [PubMed] [Google Scholar]
- 85.Heon S, Yeap BY, Britt GJ, et al. Development of central nervous system metastases in patients with advanced non-small cell lung cancer and somatic EGFR mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2010;16:5873–5882. doi: 10.1158/1078-0432.CCR-10-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zimmermann S, Dziadziuszko R, Peters S. Indications and limitations of chemotherapy and targeted agents in non-small cell lung cancer brain metastases. Cancer Treat Rev. 2014;40(6):716–722. doi: 10.1016/j.ctrv.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 87.Ballard P, Yates JW, Yang Z, et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res. 2016;22(20):5130–5140. doi: 10.1158/1078-0432.CCR-16-0399. [DOI] [PubMed] [Google Scholar]
- 88.Goss G, Tsai C, Shepherd F, et al. CNS response to osimertinib in patients with T790M-positive advanced NSCLC: pooled data from two phase II trials; IALSC Congress. 2016. MA16.11. [DOI] [PubMed] [Google Scholar]
- 89.Pareek V, Welch M, Ravera E, et al. Marked differences in CNS activity among EGFR inhibitors: case report and mini-review. J Thorac Oncol. 2016;11(11):e135–e139. doi: 10.1016/j.jtho.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 90.Yang JCH, Kim DW, Kim SW, et al. Osimertinib activity in patients (pts) with leptomeningeal (LM) disease from non-small cell lung cancer (NSCLC): updated results from BLOOM, a phase I study. J Clin Oncol. 2016;34(Suppl) abstr9002. [Google Scholar]
- 91.Ahn MJ, Kim DW, Kim TM, et al. Phase I study of AZD3759, a CNS penetrable EGFR inhibitor, for the treatment of non-small-cell lung cancer (NSCLC) with brain metastasis (BM) and leptomeningeal metastasis (LM) J Clin Oncol. 2016;34 abstr 9003. [Google Scholar]
- 92.Santarpia M, Karachaliou N, González-Cao M, et al. Feasibility of cell-free circulating tumor DNA testing for lung cancer. Biomark Med. 2016;10(4):417–430. doi: 10.2217/bmm.16.6. [DOI] [PubMed] [Google Scholar]
- 93.Taniguchi K, Uchida J, Nishino K, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011;17(24):7808–7815. doi: 10.1158/1078-0432.CCR-11-1712. [DOI] [PubMed] [Google Scholar]
- 94.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20(6):1698–1705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sundaresan TK, Sequist LV, Heymach JV, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin Cancer Res. 2016;22(5):1103–1110. doi: 10.1158/1078-0432.CCR-15-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non-small-cell lung cancer. J Clin Oncol. 2016;34(28):3375–3382. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Remon J, Caramella C, Jovelet C, et al. Osimertinib benefit in EGFR-mutant NSCLC patients with T790M-mutation detected by circulating tumour DNA. Ann Oncol. 2017;28(4):784–790. doi: 10.1093/annonc/mdx017. [DOI] [PubMed] [Google Scholar]
- 98.Buder A, Hochmair M, Holzer S, et al. Association between EGFR T790M mutation copy numbers in cell-free plasma DNA and response to osimertinib in advanced NSCLC. J Thor Oncol. 2017;12(1S):S1206. [Google Scholar]
- 99.Sullivan I, Planchard D. Osimertinib in the treatment of patients with epidermal growth factor receptor T790M mutation-positive metastatic non-small cell lung cancer: clinical trial evidence and experience. Ther Adv Respir Dis. 2016;10(6):549–565. doi: 10.1177/1753465816670498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med. 2016;10(4):383–388. doi: 10.1007/s11684-016-0488-1. [DOI] [PubMed] [Google Scholar]
- 101.Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol. 2016;9(1):59. doi: 10.1186/s13045-016-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ercan D, Choi HG, Yun CH, et al. EGFR mutations and resistance to irreversible pyrimidine- based EGFR inhibitors. Clin Cancer Res. 2015;21(17):3913–3923. doi: 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21(17):3924–3933. doi: 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rosell R, Karachaliou N. Lung cancer: using ctDNA to track EGFR and KRAS mutations in advanced-stage disease. Nat Rev Clin Oncol. 2016;13(7):401–402. doi: 10.1038/nrclinonc.2016.83. [DOI] [PubMed] [Google Scholar]
- 106.Song HN, Jung KS, Yoo KH, et al. Acquired C797S mutation upon treatment with a T790M-specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancer. J Thorac Oncol. 2016;11(4):e45–e47. doi: 10.1016/j.jtho.2015.12.093. [DOI] [PubMed] [Google Scholar]
- 107.Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ou SI, Cui J, Schrock AB, et al. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. 2017;108:228–231. doi: 10.1016/j.lungcan.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 109.Nukaga S, Yasuda H, Tsuchihara K, et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 2017;77(8):2078–2089. doi: 10.1158/0008-5472.CAN-16-2359. [DOI] [PubMed] [Google Scholar]
- 110.Chaib I, Karachaliou N, Pilotto S, et al. Co-activation of STAT3 and YES-associated protein 1 (YAP1) pathway in EGFR-mutant NSCLC. J Natl Cancer Inst. 2017;109(9) doi: 10.1093/jnci/djx014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang J, Ji JY, Yu M, et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. 2009;11(12):1444–1450. doi: 10.1038/ncb1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eberlein CA, Stetson D, Markovets AA, et al. Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75(12):2489–2500. doi: 10.1158/0008-5472.CAN-14-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tricker EM, Xu C, Uddin S, et al. Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. 2015;5(9):960–971. doi: 10.1158/2159-8290.CD-15-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ho CC, Liao WY, Lin CA, et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12(3):567–572. doi: 10.1016/j.jtho.2016.11.2231. [DOI] [PubMed] [Google Scholar]
- 115.Planchard D, Loriot Y, Andre F, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015;26(10):2073–2078. doi: 10.1093/annonc/mdv319. [DOI] [PubMed] [Google Scholar]
- 116.Ou SH, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. 2016;98:59–61. doi: 10.1016/j.lungcan.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 117.Ortiz-Cuaran S, Scheffler M, Plenker D, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22(19):4837–4847. doi: 10.1158/1078-0432.CCR-15-1915. [DOI] [PubMed] [Google Scholar]
- 118.Ham JS, Kim S, Kim HK, et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J Thorac Oncol. 2016;11(1):e1–e4. doi: 10.1016/j.jtho.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 119.Ramalingam S, Yang JC, Lee CK, et al. Osimertinib as first-line treatment for EGFR mutation-positive advanced NSCLC: updated efficacy and safety results from two Phase I expansion cohorts. J Thorac Oncol. 2016;11(4 Suppl):S152. [Google Scholar]
- 120.Denis MG, Vallée A, Théoleyre S. EGFR T790M resistance mutation in non small-cell lung carcinoma. Clin Chim Acta. 2015;444:81–85. doi: 10.1016/j.cca.2015.01.039. [DOI] [PubMed] [Google Scholar]
- 121.Su KY, Chen HY, Li KC, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol. 2012;30(4):433–440. doi: 10.1200/JCO.2011.38.3224. [DOI] [PubMed] [Google Scholar]
- 122.Yu HA, Arcila ME, Hellmann MD, et al. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann Oncol. 2014;25(2):423–428. doi: 10.1093/annonc/mdt573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee Y, Lee GK, Lee YS, et al. Clinical outcome according to the level of preexisting epidermal growth factor receptor T790M mutation in patients with lung cancer harboring sensitive epidermal growth factor receptor mutations. Cancer. 2014;120(14):2090–2098. doi: 10.1002/cncr.28711. [DOI] [PubMed] [Google Scholar]
- 124.Costa C, Molina MA, Drozdowskyj A, et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res. 2014;20(7):2001–2010. doi: 10.1158/1078-0432.CCR-13-2233. [DOI] [PubMed] [Google Scholar]
- 125.Diggs LP, Hsueh EC. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomarker Res. 2017;5(1):12. doi: 10.1186/s40364-017-0093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Garassino M, Cho B, Gray JE, et al. Durvalumab in ≥ 3rd-line EGFR mutant/ALK+, locally advanced or metastatic NSCLC: results from the phase 2 ATLANTIC study. Ann Oncol. 2017;28(Suppl 2):ii28–ii51. [Google Scholar]
- 127.Ahn MJ, Yang J, Yu H, et al. Osimertinib combined with durvalumab in EGFR-mutant non-small cell lung cancer: results from the TATTON phase Ib trial. J Thorac Oncol. 2016;11(4):S115. [Google Scholar]
- 128.Planchard D, Kris M, Besse B, et al. Phase 1 study of ramucirumab or necitumumab in combination with osimertinib (AZD9291) in advanced T790M-positive EGFR-mutant NSCLC. J Thor Oncol. 2017;12(1):S1071(P2.06–005). [Google Scholar]
- 129.Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–132. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017;385:51–54. doi: 10.1016/j.canlet.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 132.Uchibori K, Inase N, Araki M, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun. 2017;8:14768. doi: 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9(1):34. doi: 10.1186/s13045-016-0268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Barnes TA, O’Kane GM, Vincent MD, Leighl NB. Third-generation tyrosine kinase inhibitors targeting epidermal growth factor receptor mutations in non-small cell lung cancer. Front Oncol. 2017;7:113. doi: 10.3389/fonc.2017.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tan DSW, Yang JCH, Leighl NB, et al. Updated results of a phase 1 study of EGF816, a third-generation, mutant-selective EGFR tyrosine kinase inhibitor (TKI), in advanced non-small cell lung cancer (NSCLC) harboring T790M. J Clin Oncol. 2016;34(Suppl) abstr 9044. [Google Scholar]
- 136.Yu HA, Spira AI, Horn L, et al. Antitumor activity of ASP8273 300 mg in subjects with EGFR mutation-positive non-small cell lung cancer: interim results from an ongoing phase 1 study. J Clin Oncol. 2016;34 abstr 9050. [Google Scholar]
- 137.Xu X, Mao L, Xu W, et al. AC0010, an irreversible EGFR inhibitor selectively targeting mutated EGFR and overcoming T790M-induced resistance in animal models and lung cancer patients. Mol Cancer Ther. 2016;15(11):2586–2597. doi: 10.1158/1535-7163.MCT-16-0281. [DOI] [PubMed] [Google Scholar]
- 138.Husain H, Martins R, Goldberg S, et al. Phase 1 dose escalation of PF-06747775 (EGFR-T790M inhibitor) in patients with advanced EGFRm (Del 19 or L858R+/-T790M) NSCLC. J Thor Oncol. 2017;12(1):S1185. [Google Scholar]
- 139.Yu HA, Spira AI, Horn L, et al. Antitumor activity of ASP8273 300 mg in subjects with EGFR mutation-positive non-small cell lung cancer: interim results from an ongoing phase 1 study. J Clin Oncol. 2016;34(Suppl) abstr 9050. [Google Scholar]
- 140.Reckamp KL, Melnikova VO, Karlovich C, et al. A highly sensitive and quantitative test platform for detection of NSCLC EGFR mutations in urine and plasma. J Thorac Oncol. 2016;11(10):1690–1700. doi: 10.1016/j.jtho.2016.05.035. [DOI] [PubMed] [Google Scholar]