

Summary
Appropriate growth and synaptic integration of GABAergic inhibitory interneurons are essential for functional neural circuits in the brain. Here, we demonstrate that disruption of primary cilia function following the selective loss of ciliary GTPase Arl13b in interneurons impairs interneuronal morphology and synaptic connectivity, leading to altered excitatory/inhibitory activity (E/I) balance. The altered morphology and connectivity of cilia mutant interneurons and the functional deficits are rescued by either chemogenetic activation of ciliary GPCR signaling or the selective induction of Sstr3, a ciliary-GPCR, in Arl13b deficient cilia. Our results thus define a specific requirement for primary cilia-mediated GPCR signaling in interneuronal connectivity and inhibitory circuit formation.
Keywords: Primary cilia, interneurons, circuitry, GPCR signaling, Joubert syndrome related disorders, autism spectrum disorders
eTOC Blurb
Growth and connectivity of interneurons are fundamental to the formation of functional neural circuitry. Guo et al. show that primary cilia signaling, via Arl13b-GPCR pathway, promotes interneuronal connectivity and inhibitory circuit formation. Primary cilia signaling is a non-synaptic signaling mechanism through which environmental signals can shape and refine interneuronal networks.
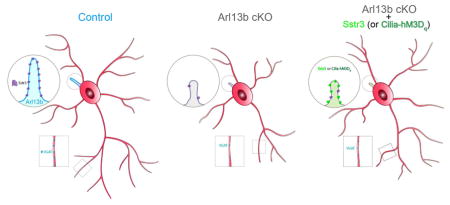
Introduction
Appropriate connectivity and integration of GABAergic interneurons (INs) are essential for the formation of functional neuronal circuitry in the brain. Disrupted IN circuit formation, homeostasis and the resultant excitatory/inhibitory (E/I) imbalance is a convergent mechanism underlying neurodevelopmental disorders such as autism spectrum disorders (ASD), schizophrenia, and intellectual disabilities (Bourgeron, 2009; Kepecs and Fishell, 2014; Marín, 2012). INs, originating from the ventral telencephalon, migrate widely in different trajectories to populate distinct brain regions and integrate into local neural circuits (Guo and Anton, 2014; Marín, 2013; Touzot et al., 2016). Extensive efforts have been made toward understanding the fate specification and migration of INs (Kepecs and Fishell, 2014; Marín, 2013). However, the developmental mechanisms that regulate the assembly of INs into functional circuits are not well understood. Local environment, through synaptic or non-synaptic mechanisms are thought to exert profound influence over the formation and architecture of IN circuits (Betley et al., 2009; Close et al., 2012b; De Marco García et al., 2011; De Marco García et al., 2015; Kepecs and Fishell, 2014; Pieraut et al., 2014). Here, we tested whether primary cilium, an essential sensor of environmental cues and a signal transduction hub, plays a determinant role in the environmental modulation of IN morphology and connectivity necessary for circuit assembly (Guemez-Gamboa et al., 2014).
Primary cilia contain signal transduction components necessary to transmit efficient intracellular responses to extracellular stimuli. The critical role of primary cilia in brain development and function is evident in human ciliopathies (Guemez-Gamboa et al., 2014; Guo et al., 2015), where disrupted cilia function lead to brain malformations and behavioral deficits. Many candidate genes for neurodevelopmental disorders such as ASD, schizophrenia, and intellectual disabilities affect cilia function (Hildebrandt et al., 2011; Lee and Gleeson, 2011; Louvi and Grove, 2011; Marley and Von Zastrow, 2012). Therapeutic drugs such as lithium used in the treatment of neurobehavioral disorders can modulate cilia mediated chemosensory functions in neurons(Miyoshi et al., 2009). Further, neuronal cilia are enriched in signaling machinery such as neurotransmitter receptors, GPCRs, ion channels, and Ca2+ and thus have been hypothesized to non-synaptically modulate neuronal function (Green and Mykytyn, 2014). The association of cilia dysfunction with neurodevelopmental disorders and the specialized signaling abilities of cilia raise the intriguing possibility that impaired cilia function during brain development and the resultant changes in neural circuitry may contribute to the emergence of intellectual disabilities and enhance the sensitivity for neuropsychiatric disorders (NPDs) such as ASD.
We therefore examined how disrupted primary cilia signaling affects the formation and function of striatal interneuronal circuitry, dysfunction of which is widely implicated in the pathophysiology of a spectrum of NPDs (Clark et al., 2009; Shepherd, 2013). The striatum integrates glutamatergic inputs from the cortex and thalamus and send GABAergic, inhibitory projections to neurons in the basal ganglia (Evans et al., 2012; Gittis and Kreitzer, 2012). To assess the specific requirement of interneuronal primary cilia signaling in the construction of inhibitory circuit, we conditionally disrupted primary cilia function in the striatal INs originating from the medial ganglionic eminence (MGE). Arl13b, a small GTPase of the Arf/Arl family, is specifically localized to cilia and is required for cilium’s ability to function as a signaling hub (Cantagrel et al., 2008b; Caspary et al., 2007; Guo et al., 2015; Higginbotham et al., 2012b; Higginbotham et al., 2013). Deletion of Arl13b impairs primary ciliary signal transduction, without physically ablating ciliary structure, and thus provides a unique model for examining the specific role of primary cilia signaling in interneuronal morphology, connectivity, and synaptic integration. Our results demonstrate that the disruption of cilia function perturbs interneuronal circuit development. Connectivity of cilia mutant striatal interneurons (INs), ciliary Ca2+ signaling, and ciliary localization of GPCRs such as Sstr3 are altered. Notably, deficits in the morphology and connectivity of cilia mutant INs are rescued by the cilia-selective induction of Sstr3 or chemogenetic, DREADD mediated activation of GPCR signaling in Arl13b deficient cilia. Together, these studies define a hitherto unknown role of primary cilia-GPCR signaling in striatal IN connectivity.
Results
Primary ciliary signaling is required for interneuronal morphology
To assess the functional significance of primary cilia in the emergence of IN morphology and connectivity, we selectively inactivated Arl13b in the developing INs. Nkx2.1-Cre line, which expresses Cre in medial ganglionic eminence (MGE) and preoptic area, was crossed with a floxed allele of Arl13b (Xu et al., 2008). MGE gives rise to the majority of GABAergic Parvalbumin+ (PV+) and somatostatin+ (SST+) INs in the striatum. Nkx2.1Cre thus enables specific interneuron targeting and does not affect striatal medium spiny neuron (MSN) population. A Cre reporter Lox-STOP-Lox-Ai3 allele was incorporated to label INs with YFP. Arl13b is absent in the primary cilia of Cre+/YFP+ INs (Figure S1A–D) in Nkx2.1Cre; Arl13bLox/lox; Ai3 (Nkx-Arl13b; Ai3) mice. Deletion of Arl13b in MGE did not adversely affect the production or survival of INs (Data not shown).
We first assessed the consequences of Arl13b deletion on interneuronal morphological development. At P30, when the morphological maturation of INs is largely complete (Chattopadhyaya et al., 2004), immunolabeling of the PV+ and SST+ INs revealed significantly reduced dendritic and axonal complexity throughout the striatum (Figure 1A–D). Similar changes were also evident in PV+ INs in cortex and hippocampus (Figure S2A–D). To further quantify the changes in interneuronal dendritic and axonal processes, newborn Nkx-Arl13b; Ai3 mice and control littermates were injected with Cre inducible AAV2-CAG-FLEX-tdTomato virus to sparsely label Cre+ INs. Reconstruction of labeled INs at P30 revealed significantly reduced axonal length, axonal branching, as well as dendritic complexity in mutant PV+ (Figure 1E, F, I) and SST+ INs (Figure 1G, H, J). Together, these results suggest that deletion of Arl13b in interneuronal cilia leads to striatal IN morphological defects.
Figure 1. Deletion of Arl13b in interneurons results in morphological defects.

(A–B) Striatal PV+ interneurons were labeled with anti-PV antibodies in Nkx2.1Cre; Arl13blox/+ (A) and Nkx2.1Cre; Arl13blox/lox (B) brains. (C, D) Striatal SST+ INs were labeled with anti-SST antibody in Nkx2.1Cre; Arl13blox/+ (C) and Nkx2.1 Cre; Arl13blox/lox (D) brains. (E–H) Representative images of PV+ (E, F) or SST+ INs (G, H) interneurons from AAV2-FLEX-tdTomato injected Nkx2.1Cre; Arl13blox/+ (E, G) and Nkx2.1Cre; Arl13blox/lox (F, H) brains. Insets (E–H) show co-labeling of tdTom+ neurons with PV (E, F) and SST (G, H) antibodies. (I–J) Quantification of morphological defects of PV+ (I) and SST+ (J) INs in Nkx2.1Cre; Arl13blox/lox brains [P30]. Data shown are mean ± SEM. *P<0.05 (Student’s t-test;). (K–L) Representative images of tdTom+ INs from ParvCre; Arl13blox/+; Ai9 (K) and ParvCre; Arl13blox/lox; Ai9 (L) brains [P60]. Neurons were co-labeled with anti-NeuN antibodies. Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[PV+ axonal length] = 5.32281E-06, p[PV+ axonal node] = 0.0005, p[PV+ dendritic length] = 0.0001, p[PV+ dendritic node] = 0.0003, p[SST+ axonal length] = 7.92531E-06, p[SST+ axonal node] = 0.0002, p[SST+ dendritic length] = 0.002, p[SST+ dendritic node] = 0.0003). 42 cells from 4 different brains were analyzed per group. Scale bar, 25μm (A–D); 50μm (E–H); 20μm (K, L). See also Figure S1, Figure S2, and Figure S3.
Nkx2.1-Cre is active in INs from the time of birth. To examine if primary ciliary signaling regulates interneuron morphological development, independently of their potential function in early IN development, we generated Parv-Cre; Arl13bLox/lox; Ai9 (Parv-Arl13b; Ai9) mice, in which Cre is expressed in PV+ INs from around postnatal week two, after the completion of IN generation and placement (Korotkova et al., 2010; Dehorter et al, 2015) (Figure S3A–B). At P30, cortical interneuron morphology was not affected in Parv-Arl13b cortex (Higginbotham et al, 2012). However, by P60 in Parv-Arl13b; Ai9 mice, a significant reduction in PV+ interneuronal process complexity in the striatum (Figure 1K, L), cortex, and hippocampus (Figure S3C–F) was evident without associated changes in the PV+ cell density. PV+ neurite density (tdTom+) was reduced by 45.5 ± 2.2% in Parv-Arl13b; Ai9 mice compared to controls (Figure 1K, L). Together, these analyses of Arl13b deletion in INs at different developmental stages indicate a specific requirement for primary ciliary signaling in the morphological development of striatal INs.
Primary ciliary signaling is required for IN synaptic connection
The morphological defects observed in Arl13b deficient PV+ and SST+ INs prompted us to examine their synaptic connections. Since PV+ INs preferentially form perisomatic synapses, we examined the density of YFP+ boutons of PV+ INs around the soma of NeuN+ medium spiny neurons (MSNs) in control and Arl13b deficient striatum. Compared to controls, PV+ perisomatic boutons were significantly reduced in both density and size in Nkx-Arl13b; Ai3 INs (Figure 2A–D). Similarly, the density and size of tdTomato+ or VGAT+ perisomatic boutons were also reduced in Parv-Arl13b; Ai9 (Figure 2E–L) striatum. We also observed a similar reduction in the average density of perisomatic boutons in the cortex of Nkx-Arl13b (Figure S2E–H) and Parv-Arl13b (Figure S3G–J) mice compared to controls. Further, to analyze synaptic boutons of single IN axons at high resolution, we imaged virally (AAV2-CAG-FLEX-tdTomato) labeled PV+ and SST+ INs in control and Nkx-Arl13b mice. Arl13b deficient axons of INs displayed reduced synaptic bouton density and size (Figure 2M–T). A similar reduction in the average density of VGAT+ presynaptic boutons along the axons of Arl13b deficient INs is also evident in vitro (Figure S4). Together, these results demonstrate that ciliary Arl13b deletion disrupts synaptic connectivity of INs.
Figure 2. Changes in synaptic connectivity of Arl13b deficient interneurons.

(A, B) Striatal neurons were labeled with anti-PV, anti-GFP, and anti-NeuN antibodies in Nkx2.1Cre; Arl13blox/+; Ai3 (A) and Nkx2.1Cre; Arl13blox/lox; Ai3 (B) brains. (A′, B′) PV+ or YFP+ perisomatic synaptic boutons (arrowheads) in control (A′, A″) and in Nkx2.1Cre; Arl13blox/lox (B′, B″) brains. Striatal medium spiny neurons (blue) are NeuN positive. (C, D) Quantification of PV+ perisomatic bouton density and size in control and Nkx2.1Cre; Arl13blox/lox; Ai3 brains [P30]. Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[C] = 0.0001, p[D] = 0.0001). (E–F) Representative images of striatal medium spiny neurons co-labeled with anti-NeuN antibodies in ParvCre; Arl13blox/+; Ai9 (E) and ParvCre; Arl13blox/lox; Ai9 (F) brains [P60]. Arrowheads indicate tdTom+ perisomatic boutons. (G, H) Representative images of striatal medium spiny neurons co-labeled with anti-VGAT antibodies and DAPI in ParvCre; Arl13blox/+; Ai9 (G) and ParvCre; Arl13blox/lox; Ai9 (H) brains. Arrowheads (G–H) indicate VGAT+ perisomatic boutons. (I–L) Quantification of PV+ perisomatic bouton density and size in control and ParvCre; Arl13blox/lox; Ai9 brains. Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[I] = 0.0001, p[J] = 0.0004, p[K] = 0.0003, p[L] = 0.0003). (M–P) tdTomato labeled PV+ (M, N) and SST+ (O, P) INs from AAV2-FLEX-tdTomato injected Nkx2.1Cre; Arl13blox/+ (M, O) and Nkx2.1Cre; Arl13blox/lox (N, P) brains show synaptic boutons (arrowheads) along single axons. (Q–T) Quantification of the synaptic bouton density and size of PV+ (Q, R) and SST+ (S, T) INs in Nkx2.1Cre; Arl13blox/lox brains. Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[Q] = 0.003, p[R] = 0.0004, p[S] = 0.0001, p[T] = 0.001). 42 cells from 4 different brains were analyzed per group. Scale bar, 8.5μm (A, B); 4.3μm (A′, A″, B′, B″); 2.5μm (E–H); 2μm (M–P). See also Figure S2 and Figure S3.
Functional impact of impaired primary cilia signaling in interneurons
To assess the electrophysiological consequences of impaired primary cilia signaling and IN morphology on interneuronal synaptic activity, we performed patch-clamp recordings in the dorsal striatum of brain slices from control and Nkx-Arl13b; Ai9 mice. Miniature inhibitory and excitatory postsynaptic currents (mIPSCs and mEPSCs) were sampled from MSNs. Compared to control littermates, recordings from MSNs of Nkx-Arl13b; Ai9 mice revealed a decrease in mIPSC frequency in cilia mutant mice (Figure 3A, B). In contrast, no changes in mIPSC amplitude (Figure 3C), mEPSC frequency (Figure 3D, E), or mEPSC amplitude (Figure 3D, F) were found in MSNs of Nkx-Arl13b; Ai9 mice. The specific decrease in the inhibitory synaptic input onto projecting MSNs of cilia mutant mice, leads to excitatory/inhibitory synaptic imbalance (Figure 3G). Thus, consistent with the cellular characterization of IN synaptic connectivity, these electrophysiological data confirm a functional deficit in the synaptic activities of cilia mutant INs.
Figure 3. The functional impact of impaired primary cilia signaling in interneurons.

(A) Representative patch-clamp electrophysiological recordings showing mIPSCs in striatal MSNs of Nkx2.1Cre; Arl13blox/+ (control) and Nkx2.1Cre; Arl13blox/lox (IN cKO) mice. (B, C) Quantification of mIPSC frequency (B) and amplitude (C). *P<0.05 (Student’s t-test, mIPSC frequency: t(15) = 3.167, p = 0.0066; control, n=6; IN cKO, n =11). (D) Representative recordings showing mEPSCs in striatal MSNs of Nkx2.1Cre; Arl13blox/+ and Nkx2.1Cre; Arl13blox/lox brains. (E, F) Quantification of mEPSC frequency (E) and amplitude (F). Data shown are mean ± SEM. (Student’s t-test; mIPSC amplitude, mEPSC frequency, mEPSC amplitude: t-values <1). (G) Overall excitatory/inhibitory ratio in striatal MSNs of Nkx2.1Cre; Arl13blox/+ and Nkx2.1Cre; Arl13blox/lox brains (Student’s t-test: t(14)=2.277; p = 0.0390; control, n=6; IN cKO, n=10).
Primary ciliary GPCR signaling activity is required for interneuron connectivity
To define the ciliary signaling mechanisms underlying the disrupted connectivity of INs, we analyzed changes in primary cilia signaling characteristics in Arl13b deficient INs. Calcium signaling is a hallmark of ciliary signaling activity (Belgacem and Borodinsky, 2011; DeCaen et al., 2013; Su et al., 2013). We therefore examined cilia-specific calcium dynamics in Arl13b deficient INs using a genetically encoded single fluorescence calcium indicator (GECO1.0) targeted selectively to primary cilia (Cilia-GECO1.0) (Su et al., 2013). ATP is known to trigger an increase in ciliary calcium level that normally proceeds through primary cilia from the base to tip (Su et al., 2013). ATP stimulation of Nkx-Arl13b INs showed a 63 ± 8.2% reduction in Ca2+ elevation compared to controls (Figure 4A–C).
Figure 4. Defective calcium signaling in Arl13b deficient primary cilia.

(A–B) Time-lapse imaging of interneurons from Nkx2.1Cre; Arl13blox/+ (A) and Nkx2.1Cre; Arl13blox/lox (B) striatum expressing Cilia-G-GECO1.0. were treated with 10μM ATP. Insets (A–B) show changes in fluorescence intensity in pseudocolor (Red [High], Blue [Low]). (C) Quantification of changes in the fluorescence intensity of Cilia-G-GECO1.0 in control and mutant cilia. Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[−ATP] = 0.002, p[+ATP] = 0.0004). 15 neurons from 3 independent experiments were analyzed per group. (D–G) Time-lapse imaging of wild type (D, E) and Arl13b null (F, G) MEF cells expressing Cilia-R-GECO1.0 and Cilia-GFP treated with 10μM ATP. Arrowhead and arrow (D, F) indicate cilia tip and base, respectively. Insets (D, F) show fluorescence intensity in pseudocolor. (H, I) RFP fluorescence/GFP fluorescence ratio at cilia base (H) and tip (I) before and after administration of ATP in wild type (n=9) and Arl13b null cells (n=11). Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p = 0.002). (J, K) Changes in calcium indicator (GECO-RFP)/GFP fluorescence intensity at cilia base (Arrow [D, K]; J) and tip (Arrowhead [D, F]; K) before (−ATP, T=0) and after ATP (+ATP) in wild type (n=9) and Arl13b null cilia (n=11). Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[J] = 0.0006, p[K] = 0.0002). (L–M) Time-lapse imaging of spontaneous calcium dynamics in interneuronal primary cilia from Nkx2.1Cre; Arl13blox/+; Ai9 (L) and Nkx2.1Cre; Arl13blox/lox; Ai9 (M) striatum. Arrowheads and arrow point to dynamic calcium hot spots in control cilia. Such hot spots are comparatively rare in Arl13b deficient cilia (N). Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p[N] = 0.0005). Scale bar, 2μm (A, B, L, M); 1.4μm (D–G). See also Figure S4.
In wt mouse embryonic fibroblasts (MEFs), Cilia-GECO1.0 displayed a uniform activity along the whole primary cilia (Figure 4D–E). However, Ca2+ activity in mutant cilia is preferentially detected in the basal area (Figure 4F, arrow) with weaker activity throughout the rest of the cilium (Figure 4F), suggesting a deficiency in ciliary Ca2+ signal transduction. Upon ATP stimulation, Ca2+ spike in wt cells appeared first at base and subsequently traveled towards the tip of the cilium (Figure 4D). In contrast, mutant cilia showed a 55±4.7% and 77±3.6% reduction in Ca2+ elevation at cilia base and tip, respectively (Figure 4D, F, H–K).
To further characterize the ciliary, calcium dynamics of INs, we electroporated Cilia-GECO1.0 plasmids into the GE of control and Nkx-Arl13b; Ai9 embryos (E15.5) and imaged interneuronal ciliary calcium dynamics in brain slices prepared at P0. Primary cilia in control neurons display Ca2+ “hot spots” that show changes in intensity (Figure 4L, arrowheads and arrow). These Ca2+ “hot spots” periodically emerge, travel, and disappear within control primary cilia, indicating dynamic signal transduction activities. In contrast, Arl13b deficient cilia are characterized by the lack of such dynamic Ca2+ intensity changes or “hot spot” activity (Figure 4M, N). Together, these observations suggest that Arl13b deletion lead to primary cilia signal transduction defects in INs.
Calcium dynamics in cilia is a hallmark of ciliary G-protein-coupled receptor (GPCR) signaling activity (Cantagrel et al., 2008b; Caspary et al., 2007). Considering the pivotal role of GPCR signaling in the formation, function, and plasticity of neural circuits, we first asked if the ciliary expression of somatostatin receptor 3 (Sstr3), a GPCR known to be highly enriched throughout neuronal cilia and crucial for striatal functions (Green et al., 2012), is disrupted in Arl13b deficient INs. Sstr3 is aberrantly distributed in Nkx-Arl13b cilia and compared to control, only (11±9%) express it normally. Expression of wild type Arl13b, but not non ciliary Arl13b (Arl13bV358) in Nkx-Arl13b INs rescues this defect (Arl13b: 82±12%, Arl13bV358: 15±12%). Arl13b deficiency driven changes in ciliary targeting of GPCRs such as Sstr3 may underlie cilia signaling defects. We therefore asked if expression of Sstr3 in Arl13b- deficient interneuron cilia could compensate for the ciliary signal transduction deficiency and ameliorate the IN connectivity defects. We generated Nkx2.1-Cre; Arl13bLox/lox; Ai9; Sstr3-GFP (O’Connor et al., 2013) (Nkx-Arl13b-Sstr3) mice to test this We observed that in Nkx-Arl13b-Sstr3 mice, in which Sstr3 expression is induced in Arl13b deficient interneurons (Figure 5A–E), Sstr3-GFP localizes to cilia and was not detected in non ciliary compartments of the INs. Arl13b deficient cilia are likely to have retained elements of mechanisms needed for ciliary localization of GPCR receptors (e.g., Tubby and Tulp3; Badgandi et al., 2016), albeit at disrupted levels. Perhaps this is what enables the localization of over expressed Sstr3-GFP to Arl13 deficient cilia in Nkx-Arl13b-Sstr3 mice. We examined IN morphology and synaptic connectivity in Nkx-Arl13b-Sstr3 mice as before and found that Sstr3 expressing Arl13b deficient INs exhibited similar morphology as controls (Figure 5A–C). Further, both in vivo and in dissociated INs, we observed a similar rescue of the synaptic bouton density and size following Sstr3 expression (Figure 5F–O; Figure S5). Importantly, patch-clamp recordings confirmed a rescue of the mIPSC frequency of MSNs and E/I imbalance in Nkx-Arl13b-Sstr3 mice (Figure 5P–V). Further, expression of a non functional Sstr3 point mutant (D124E; Green et al., 2016) that disrupts ligand binding, but localizes to cilia, fails to rescue Arl13b deficient phenotype (Figure S5D). Thus, the expression of Sstr3, a ciliary-GPCR, in Arl13b deficient INs helped rescue the defective IN morphology and synaptic connectivity.
Figure 5. Primary cilia-driven Sstr3 signaling regulates interneuron morphology and connectivity.

(A–C) Striatal tdTom+ INs in Nkx2.1Cre; Arl13blox/+; Ai9 (A), Nkx2.1Cre; Arl13blox/lox; Ai9 (B), and Nkx2.1Cre; Arl13blox/lox; Ai9; Sstr3-GFP (C) brains [P30]. (A′–C′) Higher-magnification images of tdTom+ striatal INs from outlined area in panels A–C. (D) Induced Sstr3-GFP expression in primary cilia of Nkx2.1Cre; Arl13blox/lox; Sstr3-GFP; Ai9 INs. (E) Altered Sstr3 localization in Arl13b deficient IN primary cilia and re-expression after induction of Sstr3-GFP. Co-labeling of primary cilia with anti-ACIII and anti-Sstr3 antibodies in Nkx2.1Cre; Arl13blox/+; Ai9, Nkx2.1Cre; Arl13blox/lox; Ai9 and Nkx2.1Cre; Arl13blox/lox; Ai9+; Sstr3-GFP INs. (F–K) Perisomatic boutons (arrowheads) labeled with tdTom (F–H) and anti-VGAT antibodies (I–K) in Nkx2.1Cre; Arl13blox/+; Ai9 (F, I), Nkx2.1Cre; Arl13blox/lox; Ai9 (G, J) and Nkx2.1Cre; Arl13blox/lox; Sstr3-GFP; Ai9 (H, K) brains [P30]. (L–M) Quantification of tdTom+ perisomatic bouton density (L) and size (M). Data shown are mean ± SEM. *P<0.05 (One-way ANOVA: F2,42 [L] = 23.0; p = 5.53616E-07; post-hoc p[L, lox/+ vs. lox/lox] = 6.08809E-08, post-hoc p[L, lox/lox vs. lox/lox-Sstr3] = 3.26839E-05; F2,42 [M] = 6.54; p = 0.004; post-hoc p[M, lox/+ vs. lox/lox] = 3.64284E-05, post-hoc p[M, lox/lox vs. lox/lox-Sstr3] = 0.008). n = 15 cells from 3 different brains per group. (N–O) Quantification of VGAT+ perisomatic bouton density (N) and size (O). Data shown are mean ± SEM. *P<0.05 (One-way ANOVA: F2,42 [N] = 13.35; p = 5.6345E-05; post-hoc p[N, lox/+ vs. lox/lox] = 5.21332E-06, post-hoc p[N, lox/lox vs. lox/lox-Sstr3] = 0.004; F2,42 [O] = 17.68; p = 6.04031E-06; post-hoc p[O, lox/+ vs. lox/lox] = 4.23172E-06, post-hoc p[O, lox/lox vs. lox/lox-Sstr3] = 0.0009). n = 15 cells from 3 different brains per group. (P–U) Patch-clamp electrophysiological recordings of striatal MSNs from Nkx2.1Cre; Arl13blox/+; Ai9 (control, n=6), Nkx2.1Cre; Arl13blox/lox; Ai9 (mutant, n=10) and Nkx2.1Cre; Arl13blox/lox; Sstr3-GFP; Ai9 (rescue, n=4) brains. (P, S) Representative patch-clamp electrophysiological recordings from MSNs showing mIPSCs (P) and mEPSCs (S). (Q, R) Quantification of mIPSC frequency (Q) and amplitude (R). *P<0.05 (One-way ANOVA, mIPSC frequency: F2,20 = 7.7; p = 0.003; post-hoc p[ctrl vs. IN cKO] < 0.01, post-hoc p[Rescue vs. IN cKO] < 0.05; control, n = 6; IN cKO, n = 11; rescue, n = 6). (T, U) Quantification of mEPSC frequency (T) and amplitude (U). (V) Rescue of overall excitatory/inhibitory ratio in striatal MSNs of Nkx2.1Cre; Arl13blox/lox; Sstr3-GFP; Ai9 mice. Data shown are mean ± SEM. *P<0.05 (One-way ANOVA: F2,19 = 4.8; p = 0.021; post-hoc p[ctrl vs. IN cKO] < 0.05, p[Rescue vs. IN cKO] < 0.05; control, n = 6; IN cKO, n = 10; rescue, n = 6). Scale bar, 37.6μm (A–C); 18μm (A′–C′); 6.2μm (D); 1.3μm (E); 5μm (F–K). See also Figure S5.
To further examine the function of cilia specific GPCR signaling in striatal interneuronal circuit development, we used a chemogenetic approach to gain precise spatio-temporal control of GPCR signaling in primary cilia and evaluated if activation of ciliary GPCR signaling rescues IN circuit disruption in cilia mutants. This approach entails IN cilia-specific expression of mutant GPCRs (Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) that have been evolved to be activated by the pharmacologically inert small molecule clozapine N-oxide (CNO) (Armbruster et al., 2007; Dong et al., 2010). hM3Dq DREADD couples to Gq and can induce Ca2+ activation when activated by CNO.
We generated cilia-targeted hM3Dq DREADD (Cilia-hM3Dq) and found that Cilia-hM3Dq displayed highly enriched ciliary expression in INs and MEFs (Figure 6A; Figure S6). INs were then co-transfected with cilia- targeted Cilia-hM3Dq and cilia calcium indicator Cilia-RFP-GECO1.0. Upon 10μM CNO treatment, control cilia co-expressing Cilia-hM3Dq and Cilia-RFP-GECO1.0 showed a 74 ± 12% increase of relative RFP intensity (Figure 6B–C). Thus cilia targeted DREADD allow us to manipulate cilia-specific GPCR and related Ca2+ signaling.
Figure 6. DREADD mediated chemogenetic activation of primary cilia-GPCR signaling rescues morphological and synaptic defects in Arl13b deficient interneurons.

(A) Expression of Cilia-hM3Dq in the primary cilia of tdTom+ control interneurons. (B) Changes in ciliary Ca2+ dynamics upon CNO (10μM) exposure in control INs expressing Cilia-hM3Dq (green) and Cilia-R-GECO1.0 (red). Arrowhead indicates cilia tip from where changes in fluorescence intensity were measured. Insets (B) show changes of fluorescence intensity in pseudocolor (Red [High], Blue [Low]). Time elapsed is indicated in seconds. (C) Quantification of changes in cilia-GECO1.0 intensity upon CNO treatment. Data shown are mean ± SEM. *P<0.05 (Student’s t-test, p = 0.0006). 15 cells from 3 independent experiments were analyzed per group. (D–F) Activation of ciliary GPCR signaling rescued Arl13b deficient phenotype. Anti-VGAT antibody labeled tdTom+ INs from Nkx2.1Cre; Arl13blox/+; Ai9 (D), and Nkx2.1Cre; Arl13blox/lox; Ai9 mice expressing Cilia-hM3Dq (E, F). INs in D–E and F were treated with DMSO and CNO, respectively. Insets (E, F) show high-magnification images of Cilia-hM3Dq expression (arrowhead). (D′–F′) High-magnification images of axonal segments in D–F showing VGAT+ synaptic boutons (arrowheads). (G, H) Quantification of VGAT+ synaptic bouton density (I) and size (J) in tdTom+ interneurons. Data shown are mean ± SEM. *P<0.05 (One-way ANOVA: F2,33[G] = 15.92; p[G] = 1.44379E-05; post-hoc p[G][lox/+ vs. lox/lox (DMSO) = 5.46811E-06, p[G][lox/lox (CNO) vs. lox/lox (DMSO) = 0.0002; F2,33[H] = 13.07; p[H] = 6.59073E-05; post-hoc p[H][lox/+ vs. lox/lox (DMSO) = 8.88938E-05, p[H][lox/lox (CNO) vs. lox/lox (DMSO) = 0.001). 12 cells from 3 independent experiments were analyzed per group. Scale bar, 5.4μm (A); 2μm (B); 40μm (D–F); 7.8μm (D′–F′). See also Figure S6.
To evaluate if modulation of ciliary GPCR signaling can rescue IN circuit disruption in cilia mutants, we transfected INs from Nkx-Arl13b; Ai9 brains with Cilia-hM3Dq. INs were then maintained in media supplemented with CNO (10μM) or DMSO for 2 weeks. We hypothesized that activation of ciliary-GPCR signaling in the mutant INs may rescue the mutant morphology and connectivity phenotype. Consistent with our hypothesis, in Arl13b deficient INs, activation of ciliary-GPCR signaling leads to a rescue of morphology and synaptic connections (Figure 6D–H).
Human mutations in ARL13B disrupt interneuron connectivity
To date, four different mutations in ARL13B have been identified in Joubert syndrome patients (Figure 7A) (Cantagrel et al., 2008b; Thomas et al., 2015). To test whether the expression of the mutant alleles of human ARL13B affects interneuron connectivity, we generated Cre- inducible, human wild type ARL13B and human mutant AAV constructs (R79Q, W82X, Y86C, R200C) and expressed them in INs dissociated from Nkx-Arl13b brains. This approach enables us to selectively express human wild type or mutant ARL13B in Arl13b null INs and query the effects of human ARL13B mutations on IN morphology and connectivity. While wild-type human ARL13B rescued the morphological and synaptic defects in the Arl13b deficient INs, JSRD causing variants R79Q, W82X, Y86C failed to do so (Figure 7 B–G, J–M). Expression of JSRD causing variant R200C partially rescued the morphological, but not the synaptic connection defects in Arl13b deficient INs (Figure 7H, J–M). Together, these results demonstrate that expression of disease causing alleles of human ARL13B in INs disrupt primary ciliary signal transduction and thus interneuronal connectivity.
Figure 7. The effect of Jourbert-Sydrome linked human ARL13B alleles in Arl13b deficient interneurons.

(A) Schematic of human ARL13B mutations and mouse non-cilia targeted Arl13b. (B–I) Dissociated INs from Nkx2.1Cre; Arl13blox/+; Ai9 (B) or Nkx2.1Cre; Arl13blox/lox; Ai9 (C–I) mice were transfected with AAV-mCherry (B, C), AAV-mCherry-ARL13B (D), R79Q (E), W82X (F), Y86C (G), R200C (H), and mV358A (I). Inset (I) shows non-ciliary expression of Arl13bV358A. (B′–I′) Labeling with anti-VGAT antibodies show inhibitory presynaptic boutons (arrowhead) along mCherry+ IN axons (B–I). (J, K) Quantification of total dendritic (J) and axonal length (K). (L, M) Quantification of VGAT+ bouton density (L) and size (M) along axons. Data shown are mean ± SEM. 12 cells from 3 independent experiments were analyzed per group. *P<0.05 (One-way ANOVA: F7,88 = 13.27[J], 17.61[K], 25.75[L], 23.52[M]; p = 1.43E-11[J], 2.11E-14[K], 8.13E-19[L], 1.06E-17[M]; post-hoc p[lox/+ vs. lox/lox = 0.001[J], 7.50E-08[K], 2.34E-06[L], 0.0003[M]; post-hoc p[lox/+ vs. lox/lox-ARL13B = 0.06[J], 0.1[K], 0.08[L], 0.09[M]; post-hoc p[lox/+ vs. lox/lox-R79Q] = 9.38E-06 [J], 6.50E-05[K], 6.44E-06[L], 0.0003[M]; post-hoc p[lox/+ vs. lox/lox-W82X] = 0.009 [J], 3.51E-08[K], 1.15E-05[L], 2.14E-06[M]; post-hoc p[lox/+ vs. lox/lox-Y86C] = 0.002 [J], 6.42E-06[K], 7.67E-08[L], 9.10E-06[M]; post-hoc p[lox/+ vs. lox/lox-R200C] = 0.46[J], 0.06[K], 0.002[L], 0.004[M]; post-hoc p[lox/+ vs. lox/lox-V358A] = 0.0001[J], 6.91E-05[K], 5.2771E-08[L], 1.63E-05[M]. Scale bar, 40μm (B–I); 10μm (B′–I′).
We further investigated whether the requirement for Arl13b in interneuron development is specifically due to its ciliary expression and activity by using a nonciliary form of Arl13b, Arl13bV358A. A point mutation (V-A) at residue 358 abolishes the cilia localization of Arl13b, but retains its normal GTPase activity (Higginbotham et al., 2012). Similar to the human mutations, Arl13bV358A did not rescue the developmental defects in Arl13b deficient INs (Figure 7I, J–M), suggesting that primary ciliary localization and activity of Arl13b is required for its role in the regulation of interneuronal morphology and connectivity.
Discussion
Our results show that primary cilia signaling is essential for the connectivity of striatal interneurons and disruption of this process can lead to circuit malformation in the striatum, a known contributor to the emergence of neuropsychiatric disorders. Further, these circuit malformations can be ameliorated by the ciliary activation of GPCRs in cilia mutant INs.
Earlier studies have demonstrated that primary cilia and Arl13b activities also modulate interneuronal migration in embryonic cerebral cortex (Baudoin et al., 2012; Higginbotham et al., 2012). The current analysis of postnatal interneuronal development in striatum indicate that the ciliary effect on interneuronal morphology and connectivity is distinct from the embryonic effect and likely not a consequence of it. The density of interneurons in postnatal striatum was not affected in Nkx-Arl13 mice (control: 17.6±2.4/10Kμm2, Nkx-Arl13: 18.8±1.6/10Kμm2) and, importantly, post migratory, interneuron- specific inactivation of Arl13b in Parv-Arl13 mice and disrupted growth and connectivity of isolated Arl13b- deficient interneurons in vitro clearly demonstrate the selective effect of Arl13b in post migratory interneuronal morphology and connectivity (Figures 1, 2, and S4). Although how primary cilia modulate distinct aspects of neuronal development (i.e., neurogenesis, migration, growth, and connectivity) remains to be fully deciphered (Guo et al., 2015; Harwell et al., 2012), many known signaling pathways that regulate interneuronal migration (e.g., Erb4/Neuregulin-1; Flames et al., 2004), also exert migration- independent effects on interneuron circuitry and function (Fazzari et al., 2010).
The connectivity and synaptic integration of GABAergic interneurons are influenced by local environment (Chattopadhyaya, 2011; Chattopadhyaya et al., 2004; Chattopadhyaya et al., 2007; Maroof et al., 2013; Pieraut et al., 2014), modulated by experience and activity (Chattopadhyaya et al., 2004; Chattopadhyaya et al., 2007; Close et al., 2012a; Close et al., 2012b; De Marco García et al., 2011; Denaxa et al., 2012; Donato et al., 2013; Kinney et al., 2006; Pieraut et al., 2014). Primary cilium is a sensory organelle of environmental stimuli. Our results demonstrate that disruption of primary ciliary signaling in PV+ and SST+ striatal INs results in deficits in their morphology, connectivity, and function. These observations reveal a non-synaptic signaling mechanism through which environmental signals can shape and refine neuronal networks. We also observed similar defects in cilia mutant INs in cerebral cortex and hippocampus, indicating a general requirement for the primary cilia mediated signaling in the development of interneuronal morphology and connectivity.
Imbalance in E/I ratio caused by disrupted GABAergic interneuron network homeostasis has been proposed as a major converging mechanism of NPDs (Consortium et al., 2013; Consortium et al., 2015; Lee and Gleeson, 2011; Parikshak et al., 2013; Voineagu et al., 2011). The prolonged postnatal maturation and sensitivity to environmental modulation of the GABAergic networks may facilitate this common biological vulnerability. Our identification of a primary cilia-based mechanism in interneuron functional connectivity illustrates how local environment can impact circuit assembly and function through atypical avenues. A spectrum of behavioral characteristics of NPDs have been associated with human ciliopathies (Alvarez Retuerto et al., 2008; Amann-Zalcenstein et al., 2006; Lee and Gleeson, 2011; Marley and Von Zastrow, 2012; Hildebrandt et al., 2011). Many candidate genes for ASD, schizophrenia, and intellectual disabilities exert potent effects on cilia function (Hildebrandt et al., 2011; Marley et al., 2013; Marley and Von Zastrow, 2012). While increasing number of investigations have focused on causative, individual genetic variants of NPDs, the identification of common biological nodes of vulnerabilities that underlie the pathophysiology of NPDs is equally critical for the development of effective therapeutic interventions for NPDs. The association of cilia dysfunction with NPDs (Green and Mykytyn, 2014; Guo et al., 2015; Lee and Gleeson, 2011) raises the intriguing possibility that primary cilium, unique in its ability to function as an integrative sensor and conveyor of critical environmental signals during circuit formation, represents one such node of vulnerability.
How does primary cilia signaling participate in the environmental modulation of interneuron connectivity? Further, how can primary cilium, a few μm long, small organelle localized at cell soma, exert a physiological effect on the whole neuron? Our findings on interneuron ciliary Ca2+ dynamics and ciliary-GPCR signaling shed some light on these questions. Recent studies using genetically encoded, cilia targeted calcium indicators revealed that primary cilium is a specialized calcium signaling hub that holds a much higher resting, as well as a wider dynamic range of Ca2+ concentration than the cytoplasm (DeCaen et al., 2013; Delling et al., 2013; Su et al., 2013). Primary cilia are known to utilize calcium as a second messenger for triggering signaling cascades (Green and Mykytyn, 2014). We adapted this Ca2+ indicator tool to provide a glimpse into the dynamic calcium signaling within interneuronal cilia. The ciliary calcium dynamics in INs is likely a reflection of the signaling activities within cilia as they probe and respond to the surrounding environment. The loss of this signaling dynamics in mutants is indicative of the changes in signaling emanating from Arl13b deficient cilia.
Primary cilium is emerging as a unique cellular compartment for enriched G-protein-coupled receptor (GPCR) signaling in many cell types (Adamantidis et al., 2005; Berbari et al., 2008; Brailov et al., 2000; Brear et al., 2014; Händel et al., 1999; Hwang and Mukhopadhyay, 2015; Loktev and Jackson, 2013; Omori et al., 2015; Soetedjo et al., 2013). Studies in olfactory epithelium and photoreceptors have demonstrated that activation of ciliary GPCRs is capable of trigging signaling cascades that lead to calcium influx and ultimately the depolarization of the cell (Green and Mykytyn, 2014). Genetic and pharmacological approaches have shown that function of ciliary-GPCRs such as Sstr3, 5-HT6, MCHR1 impact cognitive functions including learning and memory, anxiety and depression, seizure activity, and feeding behaviors. Deficits in these functions are commonly associated with human ciliopathies (Adamantidis et al., 2005; Einstein et al., 2010; Qiu et al., 2008; Svenningsson et al., 2007; Hildebrandt et al., 2011; Lee and Gleeson, 2011). In this study, using chemogenetic activation of cilia-targeted DREADDs (hM3Dq) and induced ciliary expression of Sstr3, we show that ciliary-GPCR signaling is a key regulator of interneuronal connectivity. Shh signaling appears not to affect IN connectivity (Xu et al., 2005). Sstr3 ligand somatostatin is abundantly expressed in the brain. However, the dynamics of somatostatin and other GPCR ligands’ access to cilia during IN connectivity remains to be elucidated. Additionally, Sstr3 can form heterodimers with other Sstrs, as well as with other GPCRs and receptor tyrosine kinases to generate synergistic, antagonistic or completely unique signals due to changes in G protein coupling (Einstein et al., 2010; Green et al., 2012; Viollet et al., 2008). Recent studies also indicate that normal ciliary signaling involves ectocytosis of activated GPCRs, such as Sstr3, from ciliary tips (Nager et al., 2016). The nature of such interactions in IN cilia is yet to be fully understood. Further, Sstr3 is likely to be one of the many GPCRs contributing to IN development. The expression and function of other cilia localized GPCRs (e.g., D1R, D2R, D5R, NPY2R, NPY5R, PGR15L, Gpr161, GPR19/83/88, GAL2R, GAL3R, HTR6, KISS1R, MCHR1, NPFFR1, P2RY1, PRHLR, and QRFPR) in INs remains to be characterized (Badgandi et al., 2017; Loktev and Jackoson, 2013). The lack of rescue with D124E-Sstr3 confirms the importance of cilary Sstr3 activity in INs, but other non- ciliary roles of Sstr3 in these neurons cannot be ruled out. Functional evaluation of non-cilia targeted Sstr3 in Arl13b deficient INs will help clarify this further. Nevertheless, our findings provide an example of the impact of ciliary-GPCR signaling on inhibitory network construction and function.
Four distinct mutations (p. R79Q, p. W82X, Y86C, p. R200C) in highly conserved domains of ARL13B have been identified in JSRD patients: the R79Q substitution interferes with GTP binding, W82X introduces a stop codon in exon 3, Y86C is a missense mutation in switch II region, and R200C is a missense mutation within the C terminal, coiled-coiled domain (Cantagrel et al., 2008a; Cantagrel et al., 2008b; Miertzschke et al., 2014; Thomas et al., 2015). Three of the mutations, R79Q, W82X, and Y86C are clustered in the GTP-binding domain and are known to destabilize the structure or functional integrity of the GTP-binding domain. Our results show that the expression of R79Q, W82X, and Y86C fail to rescue the morphological and synaptic connectivity defects of Arl13b deficient INs, suggesting that these disease causing alleles may disrupt cilia functions during IN development and connectivity. Interestingly, R200C, the mutation in the coiled-coiled domain, partially rescued IN morphology, but did not affect synaptic connectivity. R200C mutation, which is known to destabilize the GTP-binding domain structure, may have disrupted Arl13b activity and there might be a differential requirement for distinct functional domains of Arl13b between IN morphological vs. synaptic connection development. Further, different ARL13B mutations may also affect the axoneme structure of cilia, in addition to disrupting downstream signaling partners such as INPP5E (Caspary et al., 2007; Cevik et al., 2010; Humbert et al., 2012) necessary for ciliary GPCR targeting. In the absence of Arl13b, the B-tubules of the 9 microtubule doublets in the axoneme are often open and not attached to the A-tubule of the doublets (Caspary et al., 2007). Anterograde intraflagellar transport trains, containing ciliary components, move along the B-tubules (Stepanek and Pigino, 2016). Disruption of this process, together with the loss of downstream effectors such as INPP5E necessary for GPCR localization, are likely to have contributed to the disrupted Sstr3 receptor localization and signaling defects in Arl13 deficient cilia. In addition to Arl13b, Tubby and Tulp3 can also regulate Sstr3 localization (Badgandi et al., 2016). Presence of some of these proteins, even at disrupted levels, in Arl13b deficient cilia, may have enabled localization of over expressed Sstr3-GFP to Arl13 deficient cilia in Nkx-Arl13b-Sstr3 mice. Creation of Arl13b structure-function models in which ciliary structure defects and signaling receptor transport/localization defects can be clearly separated will be highly useful in fully understanding the functions of Arl13b. A structurally ‘normal’ Arl13b cilia mutant model that lacks a normal repertoire of GPCR signaling receptors will help understand the relative contributions of Arl13b to cilia structure and signaling.
In the future, it will be important to examine the possible participation of ciliary signaling in the conveyance of experience-driven environmental regulation of interneuronal connectivity and plasticity during critical period (Baraban et al., 2009; Chattopadhyaya et al., 2004; Southwell et al., 2010). Recent studies identified IGF1, a ligand for Igf1R receptors in cilia (Higginbotham et al., 2013; Lehtinen et al., 2011), as a sensory experience regulated gene necessary to sculpt the synaptic connections of vasoactive intestinal peptide (VIP) interneurons (Mardinly et al., 2016). It is also possible that ciliary signaling is continuously required in mature neurons for the modulation and maintenance of neuronal network homeostasis. Further, the source and the identity of the various environmental cues that trigger primary cilia signaling in INs and whether such signaling enables INs to function cooperatively (Karnani et al., 2016) are yet to be fully characterized. The exact downstream signaling cascades of ciliary signaling, including ciliary-GPCR signaling, and their precise cellular and physiological effect on neuronal function during cognitive activities such as learning and memory also remain to be elucidated. Future efforts aimed at answering these questions will further our understanding of the newly identified role of primary cilia in neuronal circuit formation and function.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were cared for according to the guidelines approved by the University of North Carolina. Light/dark cycle in the vivarium is 7/7 hours. Animals were housed in groups of 3 adults per cage. Arl13b null mice (Arl13bhnn/hnn) were generated as described in Caspary et al., 2007 (Caspary et al., 2007). Arl13b was conditionally inactivated in interneurons by crossing Arl13bLox/Lox mice, in which Arl13b exon 2 is flanked by LoxP sites (Su et al., 2012), with either Nkx2.1-Cre (Xu et al., 2008), Ai3 (Stachniak et al., 2014) mice or Parv-Cre (Hippenmeyer et al., 2005), Ai9 (Stachniak et al., 2014) mice. The Sstr3 expression was induced in Arl13b deleted interneurons by crossing Arl13blox/lox, Nkx2.1-Cre mice with Cre inducible Sstr3GFP (O’Connor et al., 2013), Ai9 mice (Stachniak et al., 2014). Littermate Arl13blox/+, Cre+ mice served as controls. Both males and females were used in all of the experiments.
METHOD DETAILS
Cilia targeted DREADD, calcium indicators, Sstr3 mutant, and human ARL13B variants
The cilia targeted DREADD (Cilia-hM3Dq) was generated by ligating a cilia-targeting-sequence [CLVCCWFKKSKTRKIKP; (Follit et al., 2010)] to the C-terminal of hM3D-mCitrine in a modified pcDNA3.1 vector containing CAG promoter (gift from Dr. Bryan Roth, University of North Carolina). The cilia targeted calcium indicator and GFP [Cilia-GECO1.0 and Cilia-GFP (Su et al., 2013)] are gifts from Dr. Takanari Inoue (John Hopkins University). Myc tagged D124E Sstr3 mutant in pcDNA3.1 vector is a gift from Dr. Kirk Mykytyn (Ohio State University, Green et al., 2016). The Cre-inducible adeno-associated virus (AAV) vectors expressing ARL13B variants and mCherry were generated using a pAAV-DIO-EF1a shuttle vector as described in Cardin et al., 2010 (Cardin et al., 2010). Each ARL13B variant (ARL13B, R79A, W82X, Y86C, R200C, mV358A; Higginbotham et al., 2012b) and mCherry were inserted into AAV-DIO-EF1a vector under the control of loxP sites.
In utero electroporation and ciliary calcium imaging
Lateral ventricles of embryonic day 14.5 control and littermate Nkx2.1-Arl13b mouse embryos were electroporated as described previously (Guo et al., 2015; Higginbotham et al., 2012b). Briefly, 1–2 μl of Cilia-GECO1.0 plasmid DNA (2 μg/μl) were injected into the lateral ventricles of E16.5 brains and electroporated using 5 pulses at 30 V for 50ms at 950ms intervals through the uterine wall using a BTX ElectroSquarePorator (ECM 830). Embryos were then allowed to develop in vivo. Cortices were removed from the electroporated pups at P0, coronally sectioned (250 μm) in a vibratome (Leica VT 1000S), mounted on nucleopore membrane filters, placed in glass-bottom FluoroDish chambers (World Precision Instruments, Inc.) and maintained in neuralbasal/B27/L-Glutamine/GlutaMaxTM-I medium at 37°C/5% CO2. Striatal interneuronal (Ai9+) primary cilia (GECO1.0+) were repeatedly imaged at 20 seconds intervals using a Zeiss780 live cell confocal laser scanning system. Changes in ciliary Ca2+ indicator intensity were quantified and analyzed using Zeiss LSM Image Browser or ImageJ software (Higginbotham et al., 2012a; Yokota et al., 2007). Plasmids used for in utero electroporation were prepared using the EndoFree Plasmid kit (Qiagen).
AAV injections
AAV viruses (AAV2-FLEX-tdTomato) were injected unilaterally into the striatum of control and Nkx2.1-Arl13b mice at P1. Briefly, animals were cold anesthetized and 100nl of the viruses (1012 GC/ml) were injected over a 1-min period using a Hamilton microliter syringe (32 gauge needle) attached to an injection apparatus (World Precision Instruments). The needle was held in place for 1 min after each injection before slow retraction. Neuronal morphology and synaptic connections were analyzed at P30.
Cell lines and transfection
Mouse embryonic fibroblast (MEF) cells were derived from wild type and Arl13bhnn/hnn E11.5 embryos as previously described (Higginbotham et al., 2012). Mouse kidney inner medullary collecting duct (IMCD3, CRL-2123) epithelial cells and MEF cells were grown in DMEM/F12 media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) at 37°C/5% CO2. Cells were allowed to reach 80% confluency and ciliogenesis was induced by serum starvation for 24 hours. Cells were then transfected using Invitrogen Lipofectamine 2000 reagent according to manufacture’s instructions. Sex of the cell lines are unknown.
Primary interneuron culture and transfection
Medial ganglionic eminence from E15.5 Arl13blox/+; Nkx2.1Cre; Ai9 or Arl13blox/lox; Nkx2.1Cre; Ai9 embryos were dissociated and plated (20,000 cells/cm2) on laminin/poly-L-lysine coated glass covers slips in Neurobasal-A/5% B27/1% GlutaMax/1% P/S media. Transfection of interneurons was performed using Invitrogen Lipofectamine 2000 according to manufacture’s instructions. Some of the cultures were supplemented with SAG (200 nM; Calbiochem) or recombinant mouse Shh N-terminus (10μg/ml; R&D Systems). Sex of the embryos were not determined.
Ca2+ imaging in vitro
Interneurons or MEFs were transfected with Cilia-GECO1.0, and or Cilia-DREADDs and imaged at an acquisition rate of 0.5 Hz using a Zeiss 780 live cell confocal microscope. Cells were maintained in DMEM (Gibco) containing 0.9 mM Ca2+. The effects of ATP or CNO were assessed by recording for 3 min before and after addition. GECO1.0 fluorescence values were normalized against background and to the average baseline values measured prior to ATP or CNO addition. Image analysis was performed using Zeiss LSM Image Browser or ImageJ software.
Immunohistochemistry
Mouse brains were fixed with 4% paraformaldehyde, embedded in 3% agarose and sectioned (50 μm thick) with a Vibratome (VT1000S; Leica Microsystems). Neuronal cultures were fixed in 4% paraformaldehyde for 30 minutes. Sections and cells were blocked with PBS/10% goat serum/0.2% Triton X-100 for 1 h, and incubated in primary antibodies overnight at 4 °C or 3 hours at room temperature. The primary antibodies used were anti-GFP (chicken, 1:1000; Abcam, ab13970), anti-RFP (rabbit, 1:500; Rockland, cat. nr. 600-401-379), anti-Caspase 3 (rabbit, 1:1000; Millipore, ab3623), anti-PH3 (rabbit polyclonal, 1:1000; Millipore, cat. nr. 07-424), anti-Arl13b (mouse, 1:500; NeuroMabs, cat. nr. 75-287), anti-Sstr3 (goat, 1:100; Santa Cruz, sc-11617), anti-5ht6 (rabbit, 1:1000, Abcam, ab103016), anti-MCHR1 (goat, 1:100; Santa Cruz, sc-5534), anti-ACIII (rabbit polyclonal, 1:100; Santa Cruz, sc-56855), anti-PV (rabbit, 1:1000, Millipore, AB15736), anti-PV (mouse, 1:1000; Millipore, MAB1572), anti-SST (rat, 1:500, Millipore, MAB354), anti-Calretinin (rabbit, 1:500; Abcam, ab702), anti-VGAT (rabbit, 1:1000; Synaptic System, cat. nr. 131003), anti-NeuN (mouse, 1:1000, Millipore, MAB377). After three washes in 1×PBS, sections and cells were incubated with secondary antibodies (1:1,000) at room temperature for 2 h, washed and mounted with Citifluor anti-fading solution (Agar). AlexaFluor 488, 647 or Cy3-conjugated (Invitrogen and Jackson ImmunoResearch) secondary antibodies were used. Nuclei were counterstained with DAPI (Sigma).
Patch Clamp Electrophysiology
Mice were anesthetized with pentobarbital and transcardial perfusions were performed using an ice-cold sucrose cutting solution containing the following (in mM): 225 sucrose, 119 NaCl, 1.0 NaH2P04, 4.9 MgCl2, 0.1 CaCl2, 26.2 NaHCO3, 1.25 glucose, 305 mOsm. Brains were then removed and submerged in the cutting solution, while coronal sections 300μm thick were taken using a vibrating blade (Leica, VT 1200). Slices were then placed in warm aCSF (32°C) containing the following (in mM): 119 NaCl, 2.5 KCl, 1.0 NaH2P04, 1.3 MgCl, 2.5 CaCl2, 26.2 NaHCO3, 15 glucose, 305 mOsm. After one hour of recovery, slices were perfused with warm aCSF (32°C) containing 1 μM tetrodotoxin (Sigma-Aldrich). Neurons were visualized using differential interference contrast through an upright 40x water-immersion objective mounted on an upright microscope (Olympus BX51WI). Fluorescent imaging using a mercury lamp (Olympus U-RFL-T) was used to identify striatal tdTomato+ interneurons. TdTomato negative medium spiny neurons were identified by their characteristic late spiking firing pattern. Voltage-clamp recordings were obtained using glass electrodes (3–5 MΩ) back-filled with cesium methylsulfonate internal solution containing of the following (in mM): 117 Cs methanesulfonic acid, 20 HEPES, 2.8 NaCl, 5 TEA, 2 ATP, 0.2 GTP, pH 7.35, mOsm 280. mEPSCs were obtained by holding neurons at -70mV for 5 minutes, whereas mIPSCs were obtained by holding neurons at +40mV for 5 minutes. Data acquisition occurred at 1 kHz sampling rate through a MultiClamp 700B amplifier connected to a Digidata 1440A digitizer (Molecular Devices). Additional details on patch-clamp recordings are available in Otis et al., 2014. Data were analyzed using a template analysis (Clampfit 10.3). Excitatory/inhibitory ratio was calculated as follows: (mEPSC frequency x mEPSC amplitude)/(mIPSC frequency x mIPSC amplitude).
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad or Excel was used for data analysis. Two-tailed Student’s t test and ANOVA test were performed using GraphPad. Data were collected and processed blindly. All experiments were independently repeated for at least 3 or more times. All data are expressed as means ± standard error of the mean (SEM). Statistical details are included in figure legends.
Supplementary Material
Highlights.
Primary cilia signaling regulates interneuronal morphology and synaptic connectivity
Ciliary GPCR signaling is critical for emergence of interneuronal connectivity.
Cilia signaling is a non-synaptic mechanism to sculpt interneuronal circuits.
Disrupted cilia signaling may underlie circuit malformations in ciliopathies
Acknowledgments
This research was supported by NIH grants NS 090029 and MH060929 to E.S.A and by the confocal imaging core of an NINDS institutional center core grant (5P30NS045892). We thank Dr. B. Roth (UNC) and Dr. B. Yoder (The Hepatorenal Fibrocystic Disease Core Center, UAB [DK074038]) for assistance with reagents and helpful comments. The authors declare no competing financial interests.
Footnotes
Author contributions
J.G., H.H., J.O., A.A, K.M., T.C, G.S., and E.A. designed the experiments and supervised the project. J.G., J.O., and H.H. conducted the experiments and analyzed the data. J.G., H.H., J.O., G.S., and E.A. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamantidis A, Thomas E, Foidart A, Tyhon A, Coumans B, Minet A, Tirelli E, Seutin V, Grisar T, Lakaye B. Disrupting the melanin-concentrating hormone receptor 1 in mice leads to cognitive deficits and alterations of NMDA receptor function. The European journal of neuroscience. 2005;21:2837–2844. doi: 10.1111/j.1460-9568.2005.04100.x. [DOI] [PubMed] [Google Scholar]
- Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, Geschwind DH. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Human Molecular Genetics. 2008;17:3887–3896. doi: 10.1093/hmg/ddn291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann-Zalcenstein D, Avidan N, Kanyas K, Ebstein RP, Kohn Y, Hamdan A, Ben-Asher E, Karni O, Mujaheed M, Segman RH, et al. AHI1, a pivotal neurodevelopmental gene, and C6orf217 are associated with susceptibility to schizophrenia. European journal of human genetics: EJHG. 2006;14:1111–1119. doi: 10.1038/sj.ejhg.5201675. [DOI] [PubMed] [Google Scholar]
- Armbruster BA, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Southwell DG, Estrada RC, Jones DL, Sebe JY, Alfaro-Cervello C, García-Verdugo JM, Rubenstein JLR, Alvarez-Buylla A. Reduction of seizures by transplantation of cortical GABAergic interneuron precursors into Kv1.1 mutant mice. Proc Natl Acad Sci U S A. 2009;106:15472–15477. doi: 10.1073/pnas.0900141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgandi HB, Hwang SH, Shimada IS, Loriot E, Mukhopadhyay S. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. Journal of Cell Biology. 2017;216:743–760. doi: 10.1083/jcb.201607095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudoin JP, Viou L, Launay PS, Luccardini C, Espeso Gil S, Kiyasova V, Irinopoulou T, Alvarez C, Rio JP, Boudier T, Lechaire JP, Kessaris N, Spassky N, Métin C. Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron. 2012;76:1108–22. doi: 10.1016/j.neuron.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Belgacem YH, Borodinsky LN. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proceedings of the National Academy of Sciences. 2011;108:4482–4487. doi: 10.1073/pnas.1018217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Johnson AD, Lewis JS, Askwith CC, Mykytyn K. Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Molecular Biology of the Cell. 2008;19:1540–1547. doi: 10.1091/mbc.E07-09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley JN, Wright CVE, Kawaguchi Y, Erdélyi F, Szabó G, Jessell TM, Kaltschmidt JA. Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell. 2009;139:161–174. doi: 10.1016/j.cell.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T. A synaptic trek to autism. Current opinion in neurobiology. 2009;19:231–234. doi: 10.1016/j.conb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Vergé D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain research. 2000;872:271–275. doi: 10.1016/s0006-8993(00)02519-1. [DOI] [PubMed] [Google Scholar]
- Brear AG, Yoon J, Wojtyniak M, Sengupta P. Diverse cell type-specific mechanisms localize G protein-coupled receptors to Caenorhabditis elegans sensory cilia. Genetics. 2014;197:667–684. doi: 10.1534/genetics.114.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Atti-Bitach T, Holden KR, Dobyns WB, et al. Mutations in the Cilia Gene ARL13B Lead to the Classical Form of Joubert Syndrome. The American Journal of Human Genetics. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Developmental Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Cevik S, Hori Y, Kaplan OI, Kida K, Toivenon T, Foley-Fisher C, Cottell D, Katada T, Kontani K, Blacque OE. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J Cell Biol. 2010;188:953–969. doi: 10.1083/jcb.200908133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B. Molecular mechanisms underlying activity-dependent GABAergic synapse development and plasticity and its implications for neurodevelopmental disorders. Neural plasticity. 2011;2011:734231. doi: 10.1155/2011/734231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark L, Chamberlain SR, Sahakian BJ. Neurocognitive mechanisms in depression: implications for treatment. Annual review of neuroscience. 2009;32:57–74. doi: 10.1146/annurev.neuro.31.060407.125618. [DOI] [PubMed] [Google Scholar]
- Close J, Xu H, De Marco García N, Batista-Brito R, Rossignol E, Rudy B, Fishell G. Satb1 is an activity-modulated transcription factor required for the terminal differentiation and connectivity of medial ganglionic eminence-derived cortical interneurons. J Neurosci. 2012;32:17690–17705. doi: 10.1523/JNEUROSCI.3583-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium C.-D.G.o.t.P.G. Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI, Ripke S, Santangelo S, Sullivan PF. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, N.a.P.A.S.o.t.P.G, (IIBDGC), I.I.B.D.G.C., and IIBDGC, I.I.B.D.G.C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nature neuroscience. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marco García NV, Karayannis T, Fishell G. Neuronal activity is required for the development of specific cortical interneuron subtypes. Nature. 2011;472:351–355. doi: 10.1038/nature09865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marco García NV, Priya R, Tuncdemir SN, Fishell G, Karayannis T. Sensory inputs control the integration of neurogliaform interneurons into cortical circuits. Nature neuroscience. 2015;18:393–401. doi: 10.1038/nn.3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Delling M, Vien TN, Clapham DE. Direct recording and molecular identification of the calcium channel of primary cilia. Nature. 2013;504:315–318. doi: 10.1038/nature12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehorter N, Ciceri G, Bartolini G, Lim L, del Pino I, Marín O. Tuning of fast-spiking interneuron properties by an activity-dependent transcriptional switch. Science. 2015;349:1216–20. doi: 10.1126/science.aab3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504:311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaxa M, Kalaitzidou M, Garefalaki A, Achimastou A, Lasrado R, Maes T, Pachnis V. Maturation-promoting activity of SATB1 in MGE-derived cortical interneurons. Cell Reports. 2012;2:1351–1362. doi: 10.1016/j.celrep.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Dong S, Allen JA, Farrell M, Roth BL. A chemical-genetic approach for precise spatio-temporal control of cellular signaling. Molecular Biosystems. 2010;6:1376–1380. doi: 10.1039/c002568m. [DOI] [PubMed] [Google Scholar]
- Einstein EB, Patterson CA, Hon BJ, Regan KA, Reddi J, Melnikoff DE, Mateer MJ, Schulz S, Johnson BN, Tallent MK. Somatostatin signaling in neuronal cilia is critical for object recognition memory. J Neurosci. 2010;30:4306–4314. doi: 10.1523/JNEUROSCI.5295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AE, Kelly CM, Precious SV, Rosser AE. Molecular regulation of striatal development: a review. Anatomy Research International. 2012;2012:106529. doi: 10.1155/2012/106529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzari P, Paternain AV, Valiente M, Pla R, Luján R, Lloyd K, Lerma J, Marín O, Rico B. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature. 2010;464:1376–80. doi: 10.1038/nature08928. [DOI] [PubMed] [Google Scholar]
- Flames N, Long JE, Garratt AN, Fischer TM, Gassmann M, Birchmeier C, Lai C, Rubenstein JL, Marín O. Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron. 2004;44:251–61. doi: 10.1016/j.neuron.2004.09.028. [DOI] [PubMed] [Google Scholar]
- Follit JA, Li L, Vucica Y, Pazour GJ. The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. The Journal of cell biology. 2010;188:21–28. doi: 10.1083/jcb.200910096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Kreitzer AC. Striatal microcircuitry and movement disorders. Trends in Neurosciences. 2012;35:557–564. doi: 10.1016/j.tins.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Gu C, Mykytyn K. Heteromerization of ciliary G protein-coupled receptors in the mouse brain. PloS one. 2012;7:e46304. doi: 10.1371/journal.pone.0046304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Mykytyn K. Neuronal primary cilia: an underappreciated signaling and sensory organelle in the brain. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2014;39:244–245. doi: 10.1038/npp.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JA, Schmid CL, Bley E, Monsma PC, Browon A, Bohn LM, Mykytyn K. Recruitment of b-arrestin into neuronal cilia modulates somatostatin receptor subtype 3 ciliary localization. Mol Cell Biol. 2016;36:223–235. doi: 10.1128/MCB.00765-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guemez-Gamboa A, Coufal NG, Gleeson JG. Primary Cilia in the Developing and Mature Brain. Neuron. 2014;82:511–521. doi: 10.1016/j.neuron.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Anton ES. Decision making during interneuron migration in the developing cerebral cortex. Trends in cell biology. 2014 doi: 10.1016/j.tcb.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Higginbotham H, Li J, Nichols J, Hirt J, Ghukasyan V, Anton ES. Developmental disruptions underlying brain abnormalities in ciliopathies. Nature Communications. 2015;6:7857. doi: 10.1038/ncomms8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Händel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, Höllt V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89:909–926. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- Harwell CC, Parker PRL, Gee SM, Okada A, McConnell SK, Kreitzer AC, Kriegstein AR. Sonic hedgehog expression in corticofugal projection neurons directs cortical microcircuit formation. Neuron. 2012;73:1116–1126. doi: 10.1016/j.neuron.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Eom TY, Mariani LE, Bachleda A, Hirt J, Gukassyan V, Cusack CL, Lai C, Caspary T, Anton ES. Arl13b in Primary Cilia Regulates the Migration and Placement of Interneurons in the Developing Cerebral Cortex. Developmental Cell. 2012;23:925–938. doi: 10.1016/j.devcel.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Guo J, Yokota Y, Umberger NL, Su CY, Li J, Verma N, Hirt J, Ghukasyan V, Caspary T, et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nature neuroscience. 2013;16:1000–1007. doi: 10.1038/nn.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. The New England journal of medicine. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, Arber S. A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLOS Biology. 2005;3:e159. doi: 10.1371/journal.pbio.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert MC, Weihbrecht K, Searby CC, Li Y, Pope RM, Sheffield VC, Seo S. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A. 2012;109:19691–19696. doi: 10.1073/pnas.1210916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SH, Mukhopadhyay S. G-protein-coupled receptors and localized signaling in the primary cilium during ventral neural tube patterning. Birth defects research Part A, Clinical and molecular teratology. 2015;103:12–19. doi: 10.1002/bdra.23267. [DOI] [PubMed] [Google Scholar]
- Karnani MM, Jackson J, Ayzenshtat I, Tucciarone J, Manoocheri K, Snider WG, Yuste R. Cooperative Subnetworks of Molecularly Similar Interneurons in Mouse Neocortex. Neuron. 2016;90:86–100. doi: 10.1016/j.neuron.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Current Opinion in Neurology. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 2011;69:893–905. doi: 10.1016/j.neuron.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktev AV, Jackson PK. Neuropeptide Y Family Receptors Traffic via the Bardet-Biedl Syndrome Pathway to Signal in Neuronal Primary Cilia. Cell Reports. 2013;5:1316–1329. doi: 10.1016/j.celrep.2013.11.011. [DOI] [PubMed] [Google Scholar]
- Louvi A, Grove EA. Cilia in the CNS: the quiet organelle claims center stage. Neuron. 2011;69:1046–1060. doi: 10.1016/j.neuron.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly AR, Spiegel I, Patrizi A, Centofante E, Bazinet JE, Tzeng CP, Mandel-Brehm C, Harmin DA, Adesnik H, Fagiolini M, et al. Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature. 2016;531:371–375. doi: 10.1038/nature17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O. Interneuron dysfunction in psychiatric disorders. Nature Reviews Neuroscience. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- Marín O. Cellular and molecular mechanisms controlling the migration of neocortical interneurons. The European Journal of Neuroscience. 2013;38:2019–2029. doi: 10.1111/ejn.12225. [DOI] [PubMed] [Google Scholar]
- Marley A, Choy RWY, von Zastrow M. GPR88 reveals a discrete function of primary cilia as selective insulators of GPCR cross-talk. PloS one. 2013;8:e70857. doi: 10.1371/journal.pone.0070857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley A, Von Zastrow M. A simple cell-based assay reveals that diverse neuropsychiatric risk genes converge on primary cilia. PLoS ONE. 2012;7:e46647. doi: 10.1371/journal.pone.0046647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell stem cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miertzschke M, Koerner C, Spoerner M, Wittinghofer A. Structural insights into the small G-protein Arl13B and implications for Joubert syndrome. The Biochemical journal. 2014;457:301–311. doi: 10.1042/BJ20131097. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Kasahara K, Miyazaki I, Asanuma M. Lithium treatment elongates primary cilia in the mouse brain and in cultured cells. Biochemical and biophysical research communications. 2009;388:757–762. doi: 10.1016/j.bbrc.2009.08.099. [DOI] [PubMed] [Google Scholar]
- Nager AR, Goldstein JS, Herranz-Pe’rez V, Portran D, Ye F, Garcia-Verdugo JM, Nachury MV. An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell. 2017;168:252–263. doi: 10.1016/j.cell.2016.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor AK, Malarkey EB, Berbari NF, Croyle MJ, Haycraft CJ, Bell PD, Hohenstein P, Kesterson RA, Yoder BK. An inducible CiliaGFP mouse model for in vivo visualization and analysis of cilia in live tissue. Cilia. 2013;2:8. doi: 10.1186/2046-2530-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Chaya T, Yoshida S, Irie S, Tsujii T, Furukawa T. Identification of G Protein-Coupled Receptors (GPCRs) in primary cilia and their possible involvement in body weight control. PloS one. 2015;10:e0128422. doi: 10.1371/journal.pone.0128422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis JM, Fitzgerald MK, Mueller D. Infralimbic BDNF/TrkB enhancement of GluN2B currents facilitates extinction of a cocaine-conditioned place preference. J Neurosci. 2014;34:6057–6064. doi: 10.1523/JNEUROSCI.4980-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieraut S, Gounko N, Sando R, Dang W, Rebboah E, Panda S, Madisen L, Zeng H, Maximov A. Experience-dependent remodeling of basket cell networks in the dentate gyrus. Neuron. 2014;84:107–122. doi: 10.1016/j.neuron.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Zeyda T, Johnson B, Hochgeschwender U, deLecea L, Tallent MK. Somatostatin receptor subtype4 couples to the M-current to regulate seizures. J Neurosci. 2008;28:3567–3576. doi: 10.1523/JNEUROSCI.4679-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GMG. Corticostriatal connectivity and its role in disease. Nature reviews Neuroscience. 2013;14:278–291. doi: 10.1038/nrn3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetedjo L, Glover DvA, Jin H. Targeting of vasoactive intestinal peptide receptor 2, VPAC2, a secretin family G-protein coupled receptor, to primary cilia. Biology open. 2013;2:686–694. doi: 10.1242/bio.20134747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Southwell DG, Froemke RC, Alvarez-Buylla A, Stryker MP, Gandhi SP. Cortical plasticity induced by inhibitory neuron transplantation. Science (New York, NY) 2010;327:1145–1148. doi: 10.1126/science.1183962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachniak TJ, Ghosh A, Sternson SM. Chemogenetic synaptic silencing of neural circuits localizes a hypothalamus→midbrain pathway for feeding behavior. Neuron. 2014;82:797–808. doi: 10.1016/j.neuron.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanek L, Pigino G. Microtubule doublets are double-track railways for intraflagellar transport trains. Science. 2016;352:721–724. doi: 10.1126/science.aaf4594. [DOI] [PubMed] [Google Scholar]
- Su CY, Bay SN, Mariani LE, Hillman MJ, Caspary T. Temporal deletion of Arl13b reveals that a mispatterned neural tube corrects cell fate over time. Development (Cambridge, England) 2012;139:4062–4071. doi: 10.1242/dev.082321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Phua SC, Derose R, Chiba S, Narita K, Kalugin PN, Katada T, Kontani K, Takeda S, Inoue T. Genetically encoded calcium indicator illuminates calcium dynamics in primary cilia. Nature methods. 2013;10:1105. doi: 10.1038/nmeth.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Qi H, Carruthers R, Witkin JM, Nomikos GG, Greengard P. Biochemical and behavioral evidence for antidepressant-like effects of 5-HT6 receptor stimulation. J Neurosci. 2007;27:4201–4209. doi: 10.1523/JNEUROSCI.3110-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Cantagrel V, Mariani L, Serre V, Lee JE, Elkhartoufi N, de Lonlay P, Desguerre I, Munnich A, Boddaert N, et al. Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. European journal of human genetics: EJHG. 2015;23:621–627. doi: 10.1038/ejhg.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzot A, Ruiz-Reig N, Vitalis T, Studer M. Molecular control of two novel migratory paths for CGE-derived interneurons in the developing mouse brain. Development. 2016;143:1753–1765. doi: 10.1242/dev.131102. [DOI] [PubMed] [Google Scholar]
- Viollet C, Lepousez G, Loudes C, Videau C, Simon A, Epelbaum J. Somatostatinergic systems in brain: networks and functions. Molecular and cellular endocrinology. 2008;286:75–87. doi: 10.1016/j.mce.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Tam M, Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. The Journal of Comparative Neurology. 2008;506:16–29. doi: 10.1002/cne.21529. [DOI] [PubMed] [Google Scholar]
- Xu Q, Wonders CP, Anderson SA. Sonic hedgehog maintains the identity of cortical interneuron progenitors in the ventral telencephalon Nkx2.1-lineage cells in the mouse telencephalon. Development. 2005;132:4987–4998. doi: 10.1242/dev.02090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.