


Abstract
Whereas in Escherichia coli DNA mismatch repair is directed to the newly synthesized strand due to its transient lack of adenine methylation, the molecular determinants of strand discrimination in eukaryotes are presently unknown. In mammalian cells, cytosine methylation within CpG sites may represent an analogous and mechanistically plausible means of targeting mismatch correction. Using HeLa nuclear extracts, we conducted a systematic analysis in vitro to determine whether cytosine methylation participates in human DNA mismatch repair. We prepared a set of A·C heteroduplex molecules that were either unmethylated, hemimethylated or fully methylated at CpG sequences and found that the methylation status persisted under the assay conditions. However, no effect on either the time course or the magnitude of mismatch repair events was evident; only strand discontinuities contributed to strand bias. By western analysis we demonstrated that the HeLa extract contained MED1 protein, which interacts with MLH1 and binds to CpG-methylated DNA; supplementation with purified MED1 protein was without effect. In summary, human DNA mismatch repair operates independently of CpG methylation status, and we found no evidence supporting a role for CpG hemimethylation as a strand discrimination signal.
INTRODUCTION
Biosynthetic errors that arise during DNA replication are normally corrected by the broadly conserved DNA mismatch repair (MR) pathway (1,2). The strand discrimination capability for mismatch correction is presumed to rely on targeting a mismatch-dependent exonucleolytic processing event to the newly synthesized strand. In the model organism Escherichia coli, adenine hemimethylation within GATC sequences provides the information that allows for strand discrimination. Following replication, the DNA strands are transiently hemimethylated, with the methyl groups residing in the parental strand. Proximal to a DNA mismatch, the MutHLS repair pathway [methyl-directed mismatch repair (MMR)] activates the latent endonuclease activity of MutH, which structurally resembles a family of restriction endonucleases (3). MutH introduces a mismatch-promoted nick in the unmethylated DNA strand, which marks an initiation point for exonucleolytic processing that removes a ‘long patch’ tract, minimally encompassing the mismatch and nick in the unmethylated partner within hemimethylated DNA.
How strand discrimination occurs in eukaryotes, or in any organism except for E.coli and related Gram-negative prokaryotes, has not been established. Postulated mechanisms include the following. (i) Covalent strand marking such as hemimethylation at CpG sites (4–6), analogous to the pathway in E.coli. (ii) Utilization of strand discontinuities such as nicks, which target repair in vitro (7) but are likely to be much more common in lagging than leading strand biosynthesis. Single strand nicks can support both human MR and the exonucleolytic removal of mismatches in the absence of synthesis in vitro (8), suggesting that human MR is not limited to correcting mismatches that arise during replication. (iii) The presence of a MutS homolog with an inherent endonuclease activity (9), putatively eliminating the need for both MutL and MutH homologs. Such a mechanism has been proposed for prokaryotes lacking an obvious MutL homolog, and for meiotic events where the strand signal available following replication might be absent. (iv) A direct association of human MR proteins with the replication machinery via the processivity factor PCNA (10–12). Such an association is proposed to allow access for the human MR components to the DNA terminus where synthesis occurs. In this paradigm, DNA ends serve as markers to reveal the strand harboring biosynthetic errors. Consistent with this model, a direct interaction between PCNA and mismatch recognition proteins has been demonstrated via the interaction of a specific protein motif within the MSH3 or MSH6 polypeptides (12); strong support for such a model has been established in Saccharomyces cerevisiae (13,14). PCNA has also been shown to be a necessary component of mismatch repair in vitro in a step prior to strand excision (10,11).
The human MED1 protein (6), also called MBD4 (15), was recently isolated as a protein that interacts with human MLH1 by two-hybrid analysis and by immunoprecipitation studies. Because MED1 is capable of binding CpG-methylated DNA and nicking supercoiled plasmid substrates (6), it makes an attractive candidate for an activity capable of discriminating DNA strands following replication. Not only is MED1 placed at the scene of mismatch correction by virtue of its interaction with MLH1, this interaction may have functional consequences. In fact, transfection of a construct lacking the MeCpG binding domain into MR-proficient human cell lines introduces microsatellite instability (6). The observation that MED1 functions as a glycosylase at G·T, G·U or certain modified base sites (15–17) suggests a role for MED1 in maintaining genomic integrity independent of MR, but it does not rule out the possibility that MED1 contributes a role in DNA strand discrimination via its interaction with MLH1.
An independent assessment supporting a role for CpG methylation in mismatch correction has been reported (4,5). Based on experiments in monkey CV1 cells infected with mismatched simian virus 40 (SV40) DNA molecules, it has been suggested that cytosine hemimethylation may be a determinant of strand discrimination. Indeed, following DNA replication, mammalian DNA possesses a transient, strand-specific CpG hemimethylation in the parental strand. Maintenance cytosine methyltransferases then restore full methylation to hemimethylated CpG sites (18). In this model, a transitory lack of cytosine methylation on the daughter strand would provide the strand discrimination signal. However, the validity of this study has been questioned since a direct involvement of the mismatch repair pathway in this system could not be demonstrated (7). More recently, Petranovic et al. (19) assessed the ability of internal cytosine methylation at CCMeGG sites to influence the outcome of mismatch correction events in Xenopus laevis egg extracts, but the tested repair events were found to be independent of such methylation.
In this work, we conducted a systematic biochemical analysis to determine whether CpG hemimethylation is competent to serve as a strand discrimination signal in vitro using nuclear extracts prepared from HeLa cells. We were unable to find evidence that CpG hemimethylation altered the outcome of authentic MR events we monitored, or that it contributed a MR function in heteroduplexes lacking a known strand signal. In addition, neither hemimethylation nor full methylation at CpG sequences was found to have an effect on the magnitude or rate of mismatch correction in vitro. While this data may not be taken as proof that CpG hemimethylation does not serve as a strand discrimination signal in human cells, it is consistent with the hypothesis that the MR pathway recognizes and processes DNA mismatches independent of the CpG methylation status.
MATERIALS AND METHODS
DNA modifying enzymes
Most of the restriction enzymes and DNA methylases (HhaI, HpaII and the SssI CpG methylase) used in this work were purchased from New England Biolabs. The Bsp106I used in substrate construction was obtained from Stratagene, while the ATP-dependent Exonuclease V used to degrade linear DNA molecules in the substrate preparation was obtained from Amersham.
Extracts and proteins
Nuclear extracts were prepared from HeLa cells purchased from the National Cell Culture Center (Minneapolis, MN) using a published protocol (20). Expression and purification of the histidine-tagged human MED1 protein has been described (6).
Preparation of DNA substrates
A protocol for generating heteroduplexes using f1 phage DNAs has been described (21), which was modified in this work for simplified smaller-scale substrate construction. All buffer conditions were identical to the published protocol unless noted. As a brief example, linearized duplex phage DNA (f1MR1) was denatured and reannealed with a 5-fold excess of its circular single-stranded phage partner (f1MR3 in this case) to make a single A·C mismatch within a 6.4 kb molecule. In order to prepare a substrate with a nick 125 bp removed from the mismatch (reading 5′→3′ along the shorter distance from nick to mismatch), the double-stranded form was linearized with Sau96I. When the nick was placed 3100 bp from the mismatch, the f1MR1 was first digested with Bsp106I, generating a substrate where the distance from nick to mismatch was roughly equivalent in either direction (Fig. 1).
Figure 1.

Construction of CpG hemimethylated heteroduplexes. A·C substrate molecules were constructed based on a modification of the method of Su et al. (21). The double-stranded (replicative) form of f1MR1 phage DNA (1) was linearized with Bsp106I and fully methylated with the SssI CpG methylase to give (2). A small, representative fraction of the 272 CpG methylation sites is presented to mark the methylated strands. The strands were denatured in the presence of an excess of circular single-stranded f1MR3 phage DNA (3), identical in sequence except for a single base replacement at the mismatch site. Annealing yields an A·C mismatch with a remote nick at the Bsp106I site (4). Ligation in the presence of ethidium bromide yields the covalently closed and positively supercoiled derivative (5). Molecules linearized at the Sau96I site instead of the Bsp106I site were also prepared using this approach.
In this work, the excess single strands from the annealing reaction were scavenged by passage over a small column of benzoylated napthoylated DEAE (BND) cellulose (Sigma). For a 1 ml reaction containing 100 µg of double-stranded DNA and 500 µg of single-stranded viral partner, the denaturation–reannealing reaction was first dialyzed for 4 h against 10 mM Tris–HCl (pH 8.0) and 1 mM EDTA (TE buffer) containing 1 M NaCl. The solution was then passed by gravity flow through a 200 µl bed of BND cellulose packed into a 5 cm glass pipette to remove single-stranded DNA. A small plug of silanized glass wool was used to retain the resin. The combined fractions containing double-stranded DNA were dialyzed for 4 h against TE and treated with 0.25 U of ATP-dependent Exonuclease V (Amersham) per microgram of double-stranded DNA using the buffer conditions recommended by the supplier, except that 5 mM MgCl2 was used instead of 30 mM over a time course of 1 h. The digestion was terminated by bringing the reaction to 10 mM Na–EDTA (pH 8.0), and the short nucleotide products and nucleotide monophosphates were removed by ultrafiltration through a Centricon 100 concentrator (Microcon). The concentrated sample (∼50 µl) was diluted to 500 µl with TE, reconcentrated and the process repeated to yield a final volume of ∼100 µl. This protocol yielded ∼10 µg of heteroduplex DNA.
Restrictions on the choice of heteroduplexes
An A·C mismatch was chosen over G·T for the experiments to determine whether hemimethylation is capable of directing human MR. Both mismatches arise frequently during replication and are efficiently repaired. However, G·T can also result from spontaneous deamination of 5-methylcytosine. We sought to avoid a lesion (G·T) that might be recognized and processed by either the human MED1 or thymine DNA glycosylase activity (15,16,22), to avoid confusion about the origin of mismatch processing. This choice also allows each strand to be scored independently for mismatch correction, based on sensitivity to one of two overlapping restriction enzymes (HindIII or XhoI; 21). Because XhoI is sensitive to full or hemimethylation at CpG sequences, the digestion-based assay is only applicable when the hemimethylated strand is excised as part of the repair process, restoring sensitivity to XhoI. The dinucleotide insertion mismatch /CA\ was chosen to determine whether complete CpG methylation affected MR, because the restriction enzymes used to score for repair in each strand (BglI or XcmI; 23) are insensitive to the CpG methylation status. The 3′-/CA\ substrate was prepared from the 5′-/CA\ substrate as previously described by ligation of the original nick and re-introduction of a nick in the opposite strand using the f1 phage gene II protein (23).
Methylation of substrate DNAs
In order to fully methylate the f1-based homo- or heteroduplex 6.4 kb DNA molecules with SssI methylase at CpG sequences, the protocol of the supplier (NEB) was slightly modified. As an example, 1 µg of heteroduplex DNA was incubated in a volume of 20 µl of NEB buffer 2 containing 4 U of SssI methylase and 32 µM S-adenosyl-l-methionine. After a 2 h incubation, the molecules were apparently completely protected from digestion with a large excess (15 U/100 ng DNA) of either HinPII (at GCGC sites) or HpaII (at CCGG sites) restriction enzymes. A control incubation with unmethylated DNA yielded apparently complete digestion under the same conditions (data not shown for HpaII).
Hemimethylated heteroduplex DNA substrates
In order to prepare A·C-containing substrate molecules where one strand was selectively methylated, the linearized double-stranded DNA partner was first fully methylated with SssI CpG methylase. When the fully methylated duplex was denatured and re-annealed in the presence of its unmethylated circular, single-stranded partner, a hemimethylated heteroduplex is produced. For each of the hemimethylated DNA substrates reported in this work, the corresponding unmethylated A·C substrate was given an identical treatment, including a mock methylation reaction that omitted only the SssI methylase. Because CpG methylation blocks the enzymatic activity of the Bsp106I enzyme used to linearize a subset of the molecules, digestion in all cases was performed prior to methylation.
Covalently closed A·C heteroduplexes
The starting material used to prepare the supercoiled A·C substrates, either hemimethylated or lacking CpG methylation, were the heteroduplexes described above with a nick at the Bsp106I site (+3100 bp from the mismatch). The nicked substrates (25 µg in a 600 µl reaction) were treated with T4 ligase (50 U/µg DNA) in the presence of 1 pmol ethidium bromide/ng f1MR1 DNA (21). The positively supercoiled product was separated from the open circular form by ultracentrifugation at 60 000 r.p.m. for 15 h in a Beckman VTi 65 rotor. Prior to centrifugation, the samples were diluted to the 2 ml centrifuge tube volume so that the final solution contained 50% CsCl and 15 µM ethidium bromide. After centrifugation, the ethidium bromide was removed by butanol extraction and the CsCl removed by dialysis against three 1 l exchanges of TE.
MR assays
Mismatch correction was assayed based on the sensitivity of repaired DNA to one of two overlapping restriction enzymes as previously described (20,21,23). An aliquot of 100 ng (24 fmol) of heteroduplex DNA and 50 µg of HeLa nuclear protein were used in each assay. However, because Bsp106I is sensitive to CpG methylation, this enzyme could not be used to linearize the recovered substrate molecules following extract treatment. Instead, 1 U of AlwNI was used for each assay. As previously described for an A·C heteroduplex, HindIII was used to score for conversion to AT (potential MMR), while XhoI identifies molecules converted to GC (nick-directed MR). Both enzymes were used at 1 U/100 ng DNA, at which concentration neither cuts the mismatch in the absence of correction (controls not shown). The buffer NEB2 from New England Biolabs was used for all assays, supplemented with 100 µg/ml BSA and 100 µg/ml RNase A in all cases, and with 20 mM NaCl when HindIII was used to identify repaired molecules. This suppresses the ability of HindIII to cut an A·C mismatch under the conditions described above; homoduplex control DNA is fully digested under these conditions. In the case of the two /CA\ insertion mismatches, BglI or XcmI represent the two enzymes that identify MR (23). The XcmI-based assays were performed in NEB2 buffer, while the BglI digests were performed in a unique buffer (45 mM Tris–HCl pH 6.8 containing 100 mM NaCl and 10 mM MgCl2). Both assays employed 1 U of enzyme per 100 ng DNA.
Purification of MED1 and western analysis
A histidine-tagged version of MED1 was expressed in E.coli and purified as previously described (6). Western analysis was conducted using enhanced chemiluminescence (Amersham), as previously described (6). The antibody used was a polyclonal mouse serum raised against recombinant MED1 protein (A.Bellacosa, unpublished material).
RESULTS
Construction of CpG hemimethylated heteroduplex substrates
Our strategy for preparing hemimethylated molecules is presented in Figure 1. It is based on the method of Su et al. (21) and Parsons et al. (23), and yields mismatch-containing substrates of 6.4 kb with a single A·C mismatch. Circular phage double-stranded DNA (f1MR1) was linearized with either Bsp106I at a site 3100 bp removed from the base that will participate in the mismatch (shown in Fig. 1) or with Sau96I at a site 125 bp removed (not shown). In this work, the linear heteroduplex molecules were then methylated with the SssI CpG methylase (see Materials and Methods). The strands were denatured by briefly raising the pH, then allowed to re-anneal with a 5-fold molar excess of a circular, single-stranded phage molecule that differs by a single base to generate an A·C mismatch. The product heteroduplex is methylated only on the strand derived from the double-stranded partner, and it contains a single, defined nick site 3100 bp away from the mismatch in the case of the Bsp106I cut (Fig. 1), or 125 bp removed from the mismatch when Sau96I was used to linearize the molecule (not shown). Ligation of the nick in the presence of ethidium bromide affords the corresponding covalently closed supercoiled heteroduplex, which was isolated by ultracentrifugation in a CsCl gradient. In each case, the corresponding unmethylated heteroduplex was prepared using the same reagents carried through the protocol in an identical fashion, except that SssI methylase was omitted from the incubation reaction where the DNA was mock methylated.
Methylation status of the substrates
The SssI methylase activity is distributive and highly efficient under the conditions used (Materials and Methods), and the enzyme is presumed to methylate each of the 270 CpG sites present. The extent of the methylation reaction was judged by the apparently complete protection from HinP1I and HpaII digestion (data not shown for HpaII digestion), for which 20 GCpGC (HinP1I) and 13 CCpGG (HpaII) sites are present, respectively. We infer that CpG sites found in sequence contexts other than those tested are predominately methylated, although most other sites could not be assayed. For reference, Figure 2 shows a linear map of sites within 85 bp of the A·C mismatch in each direction, showing the 14 CpG sites and two HinPI sites present within this region. This map is used to show that a spectrum of CpG sites are available proximal to the mismatch, including two sites for which methylation can be verified.
Figure 2.

CpG sites located within f1MR1. The site of the A·C mismatch is shown in double-stranded DNA, and the scale below is in base pairs. CpG hemimethylation sites are marked with vertical lines above the scale. The asterisks indicate GCGC sites protected from digestion with HinP1I, one of the enzymes used to detect CpG methylation. Note that nick-directed MR is predicted to excise the hemimethylation sites spanning the nick and mismatch, as well as sites proximal to but beyond the mismatch (8). The two possible repair outcomes are shown below the arrow and characterized to the left.
Predicted outcomes if CpG methylation directs mismatch correction
If CpG methylation were able to direct MR, in a fashion analogous to the dam-directed system in E.coli, the unmethylated strand is predicted to be targeted for excision (1). In this scenario, an endonuclease would be expected to contribute a single-strand nicking activity that serves as a prerequisite to excision of the long-patch repair tract. Such a strand break would then serve as the discriminatory signal for removal of the mismatched base in the unmethylated strand. If CpG methyl-directed MR were occurring under our assay conditions, an increase in the frequency of removal of the mismatched base present in the unmethylated strand would be expected (A·C→AT). If the single nick introduced during the synthesis of the A·C heteroduplex is accessed to distinguish the strands, then the hemimethylated strand spanning the nick and mismatch is predicted to be excised and replaced (A·C→GC). In the case of the molecule with a single nick at the Bsp106I site, the substrate has hundreds of hemimethylated CpG sites that lie physically closer to the mismatch than the nick site (Fig. 1). In effect, a competition would be established between the frequent hemimethylated CpG sites and the single remote nick as strand discrimination signals in a relaxed, circular substrate.
Nick-directed repair operates independently of CpG hemimethylation
The substrates we used (21) were designed to allow restriction enzyme analysis of strand-specific A·C correction to either GC (partial removal of the methylated strand, based on XhoI sensitivity) or AT (repair of the unmethylated strand, based on HindIII sensitivity). As shown in Figure 3, the extent of repair on each strand was evaluated between matched substrates, which were either hemimethylated or unmethylated at CpG sites. When the heteroduplexes have a strand-specific nick 3100 bp removed from the mismatch (or 3340 bp along the longer tract), correction of the mismatch in either strand is relatively poor (Fig. 3A). Of the 24 fmol substrate introduced into the repair assay, 4.2 fmol of repair represents 17% of the total molecules, suggesting that nick-directed MR is relatively inefficient when the nick is so far removed. Most importantly, the extent of repair in either strand is indistinguishable when comparing the hemimethylated and unmethylated heteroduplexes, independent of which strand is examined. If hemimethylation contributed to MR under the assay conditions, repair is predicted to be biased toward the unmethylated (continuous) strand in the hemimethylated substrates. Within the error of the experiment, there is no evidence for CpG methyl-directed MR under the assay conditions that support nick-directed MR (Fig. 3A).
Figure 3.

The extent of MR on relaxed circular molecules is independent of CpG hemimethylation. (A) Correction of the A·C heteroduplex to either AT or GC was scored by restriction analysis (21). The relaxed heteroduplex substrate (24 fmol) contained a site-specific nick 3100 bp removed from the mismatch (Bsp106I site; see Fig. 1). When comparing unmethylated (open bars) with CpG hemimethylated substrate (shaded bars), no significant difference was observed in the magnitude of either repair event. If CpG hemimethylation were to direct repair, an increase in A·C to AT, or removal of the base in the unmethylated partner strand, would be expected. (B) The same comparison between unmethylated and hemimethylated substrate was made in a substrate that contained a proximal repair-directing nick 125 bp removed from the mismatch. The magnitude of nick-directed repair (A·C to GC) was substantially increased in comparison with the substrate containing a remote nick (+3100 bp), but CpG hemimethylation had no significant effect. Four assays were averaged, and the error bars reflect one standard deviation from the mean.
When assaying MR in vitro using human nuclear extracts, the nick is traditionally positioned substantially closer to the mismatch (not closer than 125 bp; 20). Under these conditions, the presence of the nick strongly biases repair to the nicked strand over the continuous strand. The effect of CpG hemimethylation on such a substrate was therefore tested in a molecule where the separation between nick and mismatch was 125 bp. As expected, the proximal nick is a more effective strand discrimination signal. As shown in Figure 3B, ∼40% of the A·C mismatches were corrected to GC in the absence of CpG hemimethylation. In this case, mismatch correction was highly biased towards the nicked strand (in excess of 5:1). The presence of CpG hemimethylation did yield a slight increase in the magnitude of nick-directed MR, although this result was neither statistically significant nor consistent with methyl-directed MR.
At a minimum, these experiments demonstrate that frequent CpG hemimethylation cannot compete with a single strand break as a strand discrimination signal, even if the nick is situated kilobases from the mismatch. Hemimethylation had no effect on the repair of either strand in either substrate under the assay conditions tested. Incubation for an additional 3 h beyond the standard time course of the assay, monitored in 1 h intervals, yielded no change in the result (data not shown). At these later time points, ∼50% of the heteroduplex molecules remain unrepaired, suggesting that they are not substrates for a putative methyl CpG-directed MR.
Hemimethylation in covalently closed heteroduplexes
The heteroduplexes containing a site-specific nick, even placed kilobases from the mismatch, represent relaxed DNA as they enter the assay. However, an activity presumed to nick DNA at CpG hemimethylated sites might require the torsional stress of supercoiling, released upon nicking, to drive the reaction. Alternatively, one might imagine a hierarchy of repair-directing signals, where strand breaks are more effective than hemimethylation, yet either can serve as a signal. We therefore ligated the nicked substrate molecules in the presence of ethidium bromide to yield positively supercoiled molecules (Fig. 4). Although ethidium bromide unwinds the DNA and yields positive supercoils upon ligation, such molecules serve as effective substrates for the MutH endonuclease in E.coli during MMR in vitro when all of the reaction components are present (24,25). Similar supercoiling activates f1 phage molecules for nicking by the f1 gene II protein, which contributes a single strand break at a precise location (23–25). In these examples, both of which involve introduction of a site- and strand-specific nick, relaxed template molecules either function less efficiently or do not function at all.
Figure 4.
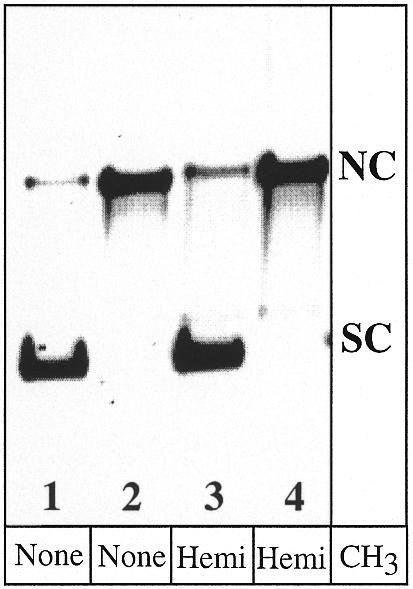
Isolation of positively supercoiled (SC), unmethylated or hemimethylated DNA heteroduplexes. Lanes 1 and 2 represent A·C heteroduplexes unmethylated at CpG sequences, while lanes 3 and 4 are hemimethylated. Lanes 1 and 3 show the supercoiled product used in the repair assays (see Materials and Methods), with ∼10% contaminating nicked circular species (NC). Lanes 2 and 4 show nicked circular DNA substrate prior to ligation.
MR assays using covalently closed and supercoiled heteroduplexes, either hemimethylated or unmethylated at CpG sites, were then tested as templates for mismatch correction in vitro. As shown in Figure 5, conversion of the A·C mismatch to either GC or AT occurs with poor and equivalent efficiency, independent of the presence of CpG hemimethylation. Roughly 5% (1 of 24 fmol) of the A·C substrate molecules were repaired with each strand bias (AT versus GC). If hemimethylation were to direct MR, a bias in the correction of A·C to AT would be expected; these data therefore do not support a role for hemimethylation at CpG sites in directing repair on covalently closed substrates. As for the low levels of mismatch correction, it is difficult to determine whether this represents a nick-independent repair event. The presence of ∼10% randomly nicked substrate molecules, which unavoidably arise during recovery or storage of the supercoiled molecules, could also account for the observed repair. It should be noted that Holmes et al. (20) observed similar low levels of mismatch correction without strand bias in heteroduplexes that were putatively covalently closed.
Figure 5.

The extent of MR on covalently closed and supercoiled molecules. The magnitude of mismatch conversion in each DNA strand was assayed on unmethylated and CpG hemimethylated A·C substrates. In the absence of a site-specific nick, ∼5% of the heteroduplexes were converted to each possible product, independent of the methylation status. The data represent three independent assays, and the error bars reflect one standard deviation from the mean. If CpG hemimethylation were to direct repair, an increase in the frequency of A·C repaired to AT would be expected.
Alternative explanations for the failure to identify MeCpG-directed MR
Even though our HeLa nuclear extracts failed to access the available hemimethylation at CpG sites to discriminate between the strands, several factors that might mask such repair were examined. First, despite the fact that each of the components required for nick-directed MR are present, it is possible that an activity essential for CpG methyl-directed MR is missing from the HeLa extracts. For example, the MED1 DNA repair protein has recently been identified as an interactor of MLH1 that can bind to hemimethylated and fully methylated DNA, and is therefore a plausible candidate. Although MED1 appears to function mainly as a thymine and uracil glycosylase specific for G·T and G·U mismatches, it might also affect strand discrimination via its interaction with MLH1 and binding to MeCpG sequences (6,15,16). Alternatively, the MED1 protein might be present, but in reduced amounts or in an inactive state. Finally, the extracts might contain a maintenance methylase that rapidly converts the hemimethylated sites to full methylation at a rate that precluded access to these sites as a strand signal. Each of these possibilities is addressed in turn below.
We first asked whether MED1 is present in the HeLa nuclear extracts used for our experiments. By western analysis (Fig. 6), the amount of nuclear extract used in a MR assay (50 µg) was found to contain ∼50 ng of MED1 protein. For reference, extracts prepared in this way contain ∼100 ng of MutSα (MSH2/MSH6) (26). Originally isolated by virtue of its interaction with MLH1, MED1 therefore appears to be present in a stoichiometry roughly equivalent in magnitude to that of the MR proteins in our nuclear extracts. The concentration of MED1 in a 15 µl assay is consequently ∼40 nM (based on monomeric protein), which is an 8-fold excess over the concentration (5 nM) that supports glycosylase assays in vitro (16). This amount of MED1 also represents a 25-fold molar excess over the heteroduplex substrate (24 fmol) in each assay. While the MED1 in the nuclear extract was not purified to homogeneity and assayed, there is no reason to expect a priori that the protein is not active, or that it is present in an amount insufficient to support strand discrimination.
Figure 6.

Detection of MED1 protein in HeLa extracts by western analysis. A set of known amounts of purified, recombinant MED1 protein was used to estimate that the HeLa extracts used for MR assays contained ∼1 ng of MED1 protein in each 1 µg of total extract protein.
In spite of the fact that MED1 was present in assay system, we sought to determine whether supplementing the extract with purified, active MED1 protein (4,13) could alter the outcome of MR assays on hemimethylated DNA templates that were either relaxed (nicked) or covalently closed. The relaxed substrate contained a nick 3100 bp away from the mismatch at the Bsp106I site, which offers hundreds of hemimethylated CpG sites closer to the mismatch than the single nick. MED1 (either 40 or 400 ng) was added to HeLa nuclear extract (50 µg) prior to initiating MR assays, but this protein neither inhibited nick-directed MR, nor stimulated repair in the unmethylated strand (data not shown). This amount of MED1 protein (∼0.5 to ∼5 pmol) represents a 20–200-fold excess of protein monomer over DNA heteroduplex used in each assay (24 fmol).
The failure of our HeLa nuclear extracts to access CpG hemimethylation as a strand discrimination signal could also be explained by the presence of a potent DNA methyltransferase activity in the extract. In this model, the hemimethylated CpG sites would become fully methylated over a time course that preceded mismatch correction, thus removing the information needed for strand distinction. To test this possibility, unmethylated, CpG hemimethylated and fully methylated DNA heteroduplexes were incubated with HeLa nuclear extract under MR assay conditions. The recovered DNA molecules were then assayed for the gain or loss of CpG methylation, using a large excess of HinP1I restriction enzyme (Fig. 7). Using a ratio of 15 U of HinP1I per 100 ng DNA, the enzyme digests frequently within hemimethylated DNA, providing a means to assess the methylation status at these specific sites. In addition, 1 U of AlwNI was used in each assay to linearize all the DNA molecules, since incubation with HeLa nuclear extract results in the formation of large catenated networks that can be released upon linearization (27,28).
Figure 7.
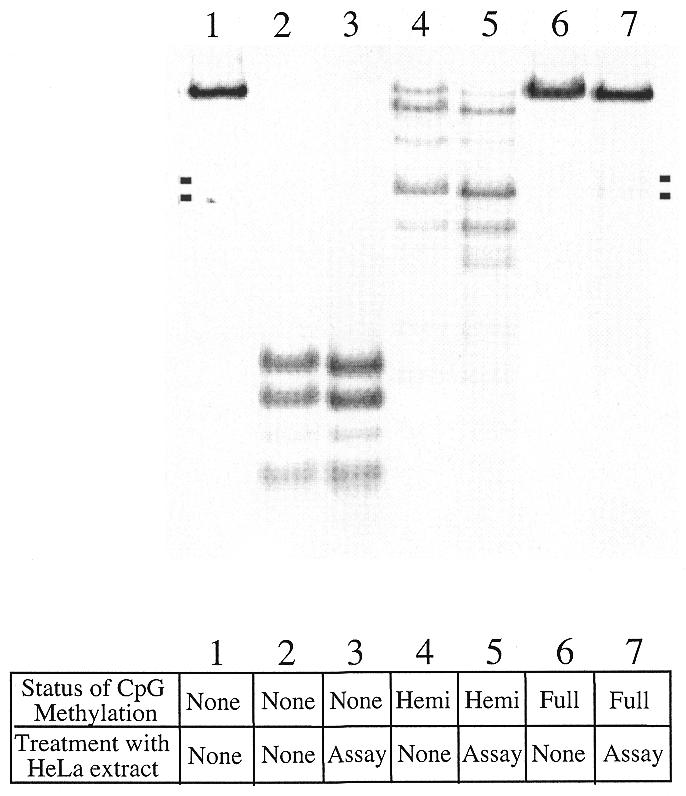
MR-competent HeLa extracts do not support either de novo or maintenance CpG methylation, nor demethylation, under assay conditions. An A·C heteroduplex of 6.4 kb, which was unmethylated (lanes 1–3), hemimethylated (lanes 4 and 5) or fully methylated at CpG sites (lanes 6 and 7) was incubated under assay conditions in the presence or absence of HeLa nuclear extract and then treated with an excess of HinP1I restriction enzyme (15 U/100 ng DNA, except in lane 1). HinP1I digests to apparent completion in unmethylated DNA, at a set of sensitive sites in hemimethylated DNA, and is largely unable to digest in fully methylated DNA. Pairwise comparisons are made between substrate that was treated with HeLa extract under assay conditions (lanes 3, 5 and 7) and substrate that was incubated under parallel conditions in the absence of HeLa extract (lanes 2, 4 and 6). Lane 1 represents linearized, unmethylated substrate. Under the assay conditions, there is no evidence for de novo methylation (compare lanes 3 and 2), maintenance methylation at CpG hemimethylated sites (compare lanes 5 and 4), or for removal of CpG methylation except for a trace level of a site-specific loss associated with mismatch excision (compare lanes 7 and 6). The two small marks on each side of the gel represent the sizes of two bands that would result if the DNA were cut at or near the mismatch site.
We found no evidence for significant de novo or maintenance methylation under the assay conditions, or for substrate demethylation, as judged by the comparable restriction sensitivity of the HeLa nuclear extract-treated and untreated DNA substrate molecules. Unmethylated DNA molecules (Fig. 7, lanes 2 and 3) show no evidence of protection from HinP1I digestion when tested at CCGG sites, either before or after exposure to HeLa nuclear extract under MR assay conditions. Most important to this work, we found no evidence for conversion of hemimethylated to fully methylated molecules. Hemimethylated molecules with a nick 125 bp removed from the mismatch, when recovered from HeLa extract, demonstrate a nearly identical pattern of incomplete protection from HinP1I digestion when compared with substrates not exposed to HeLa extract (Fig. 7, lanes 4 and 5). The appearance of a slight increase in two DNA fragments following HeLa treatment (marked by small, solid bars on each side of Fig. 7) apparently indicates that exonucleolytic removal of the mismatch also removed a hemimethylated and protected HinP1I site proximal to the mismatch in a fraction of the molecules (see Fig. 2). When fully methylated heteroduplexes were assayed, parallel results were obtained; treatment with HeLa nuclear extract resulted in the loss of a small fraction of the HinP1I-protected sites, but the methylation status was largely independent of HeLa exposure. This indicates that the extract is not removing tracts of the substrate DNA concomitant with MR, except for the tract required for mismatch excision.
Nick-directed MR is independent of CpG methylation in both strands
In order to test whether complete CpG methylation affected nick-directed MR, we prepared two fully methylated heteroduplexes by incubation with SssI methylase (see Materials and Methods). Both contained a /CA\ dinucleotide insertion within the same sequence context (23), but they differed in the relative positioning of the mismatch and repair-directing nick. The first substrate (5′-/CA\) presents a nick 125 bp removed from the mismatch so that exonucleolytic digestion proceeds 5′→3′ from the nick towards the mismatch. The second substrate has roughly the same separation (131 bp), but exonucleolytic processing occurs 3′→5′ towards the mismatch (3′-/CA\). Because the human MR pathway is bidirectional (8), capable of accessing a strand break in either strand or orientation with respect to the mismatch, the presence of CpG methylation could affect mismatch correction selectively, such as impeding or enhancing repair only when the repair-directing nick is positioned in a specific orientation. The A·C substrate described above could not be used in this case, since exonucleolytic removal of the mismatch leaves CpG hemimethylation at the XhoI site when A·C conversion to GC is assessed. This modification interferes with the ability to quantitate mismatch correction using XhoI (data not shown).
Figure 8 shows the extent of nick-directed mismatch correction on both the nicked and continuous DNA strand in a standard assay, where both strands are fully methylated at CpG sites. The data set on the left describes the substrate containing a strand break with a 5′ polarity (reading 5′→3′ from nick to mismatch), while the right panel describes substrate with a 3′ polarity. Unmethylated substrates are presented as open bars; fully methylated substrates at CpG sites are given as filled bars. Because the experiments represent the average of two assays, due to substrate limitation, error bars are not presented. For both substrates, the magnitude of nick-directed mismatch correction was very slightly reduced between unmethylated and fully methylated DNA. This diminution does not appear to be statistically significant, although this judgement is based only on two independent assays. Similarly, the low levels of mismatch correction in the putatively continuous strand as a template are also within the error of the assay data, although slightly increased in the fully methylated substrate. It is possible that repeating this experiment a large number of times might yield a very minor difference in strand bias for these specific substrates that is attributable to CpG methylation. Nonetheless, it is clear from this experiment that CpG methylation, when present on both DNA strands, had a very minor effect at best on the strand specificity and magnitude of any of the four repair events tested.
Figure 8.

Nick-directed MR is effectively independent of double-stranded CpG methylation. Two dinucleotide insertion mismatches were assayed, both of which contained an /CA\ insertion. The two substrates represent each possible orientation of the nick with respect to the mismatch [reading 5′→3′ (5′-/CA\) versus 3′→5′ (3′-/CA\) from nick to mismatch]. The extent of mismatch correction in each strand was determined in paired substrates where one heteroduplex was unmethylated (open bars), the other fully methylated at CpG sites (shaded bars). For each substrate, ∼50% (12 fmol) of the heteroduplexes were corrected based on removing the heterology in the nicked strand. No evidence for a significant effect of CpG methylation on the magnitude of mismatch correction in either strand was observed.
DISCUSSION
Based on results obtained in monkey CV1 cells infected with mismatched SV40 DNA molecules, it has been suggested that cytosine hemimethylation at CpG sites may be a determinant of strand discrimination in mammalian MR (6). In this model, cytosine methylation would be functionally equivalent to adenine methylation in E.coli: the transitory lack of this epigenetic modification would mark the newly synthesized strand for incision by MMR. However, the validity of this study has been questioned, since the nature of this system (infection with SV40 DNA and recovery of ‘repaired’ infectious viral DNA) precludes the demonstration of a direct involvement of MR (7). Furthermore, it has been argued that interpretation of the results in this study was complicated by the use of an infectious mixture of SV40 DNA molecules harboring two different mismatches (2).
Our goal in this paper was to determine, using a defined biochemical assay system, whether CpG methylation can provide a functional means of discriminating DNA strands following replication in human cells. This assumes that DNA synthesis incorporates unmethylated cytosine into the growing DNA strands and that mismatch correction competes successfully with maintenance methylation following replication. We approached this by preparing mismatch-containing circular DNA molecules where one strand is fully methylated at CpG sites and asking whether such hemimethylation is competent to drive mismatch correction in vitro, using nuclear extracts prepared from HeLa cells. We found no biochemical evidence to support such a model, but it must be clear that we do not take our data to represent proof that CpG methylation cannot serve in this capacity. What is clear is that human MR operates independently of hemimethylation or complete methylation at CpG sequences in vitro as it presumably occurs in vivo.
Recently, the identification of the MED1 protein as an interactor of MLH1 that binds to CpG-methylated DNA revealed biochemical clues to a possible role of cytosine methylation in DNA repair processes in humans (6). As judged by coimmunoprecipitation experiments, MED1 and MLH1 form a complex in vivo in human cells and the interaction seems to have functional consequences. Indeed, transfection into microsatellite-stable SW480 tumor cell line of a deletion mutant lacking the MeCpG-binding domain but maintaining the region of interaction with MLH1 is associated with microsatellite instability of a β-galactosidase reporter gene (6). The most plausible interpretation of this finding is that the association with microsatellite instability, rather than reflecting a direct role of MED1 in long-patch MMR, is more likely due to the MED1 deletion mutant sequestering MLH1 in mislocalized, non-functional complexes (17). In fact, the assignment of specific G·T glycosylase activity to this protein, along with its ability to initiate repair of bases not found in native DNA (15,16,29), gives this protein a role outside mismatch correction. The presence of the MeCpG binding domain has been proposed to target MED1 to regions rich in cytosine methylation, sites where deamination of such methylated bases yields a substrate for the enzyme.
Further evidence that the physiological role of MED1 may lie outside mismatch correction comes from the observation that the alterations of the MED1 gene in primary tumors are frameshift mutations that target two coding polyadenine tracts, likely as a result of a generalized microsatellite instability (30,31). The tumors were all defective in either MSH2 or MLH1, based on an immunohistochemistry survey, and MED1 mutations appeared more commonly in tumors of later clinical stages (30,31). These data support the contention that MED1 mutations arose late in tumor progression as a result of the mutator phenotype conferred by the loss of a critical MR protein, such as MSH2 or MLH1. If a required enzymatic contribution of MED1 were strand discrimination for human MR, its contribution to mutagenesis would be predicted to be epistatic with respect to essential MR proteins such as MSH2 or MLH1. Taken together, these data are consistent with a role for MED1 independent of methyl-directed MR, as our data indicate, but still do not provide a conclusive exclusion from MR.
Implications for the capacity of MR to operate independently of the CpG methylation status
In addition to the possibility that covalent marking of cytosine by methylation could provide a strand discrimination signal, CpG methylation changes the physical properties of DNA. For example, CpG methylation increases the helical pitch of DNA (32), and such methylation invites binding of other proteins to the modified DNA (33). Critical to this work is the observation that mismatch recognition was apparently independent of the CpG methylation status based on the outcome of an in vitro assay. This is most notable for the A·C substrate, where a CpG site is positioned 1 bp from the mismatch. While we did not carry out a survey of the effect of CpG methylation on mismatch recognition, or its ability to affect access to the nick that directs repair in the standard assay, it is clear that CpG hemimethylation did not alter the rate-limiting step in mismatch correction in vitro. Nonetheless, the possibility remains that the CpG hemimethylation presumed to exist after DNA replication has the potential to either positively or negatively affect the likelihood that a specific mismatch might be recognized or processed, or that specific individual steps in the overall MR pathway might be affected. The rate of exonucleolytic removal of the mismatch might be altered, for example, but the individual mechanistic step would need to be isolated to make such a determination. The data presented in this work suggest that the rate-limiting step in MR, when assayed in vitro, is apparently unaffected by cytosine methylation or hemimethylation.
In this work, we failed to observe a role for CpG hemimethylation as a strand discrimination signal in vitro. We attempted to rule out testable explanations that might also account for this finding. Nonetheless, relatively unlikely scenarios are possible that are more difficult to address. It is still possible that CpG methylation can direct human MR, but only when it occurs in a specific sequence context not found within the phage substrate we used. Counter to this argument is the fact that a latent endonuclease activated by mismatch recognition is likely to encounter any common sequence involving CpG in the 6.4 kb phage DNA substrate used. As an example, if a recognition sequence involving four bases is proposed with a central CpG sequence (i.e. XCpGY), the phage-based substrate we used would present all 16 possibilities, with frequencies ranging from 10 TCpGA to 24 TCpGC sites. One might also imagine a requirement for methylation within CpG-rich sequences. The f1MR1 molecules contain 15 CGCG sequences, but no longer CG repeats. Alternatively, despite the fact that the HeLa extracts used are competent in nick-directed MR, it is possible that one or more components are lost in the extract preparation that are essential for methylcytosine-directed MR. This seems unlikely in light of the fact that the nuclear extracts support nick-directed repair, and that one of the methylated CpG binding proteins, MED1, is well represented in these extracts. Finally, one might imagine that methyl-directed human MR only functions in a particular temporal context, perhaps only processing lesions that were generated by replication. In this scenario, hemimethylation might fail as a strand signal in our in vitro repair assay because the mismatches and hemimethylation sites are introduced into the extract, not generated by a replication event. While these represent untested possibilities, we do not consider them likely.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Dr Keith Iams for providing thoughtful comments on the manuscript, and to recognize the excellent cell culture service provided by the National Cell Culture Center (Minneapolis, MN). In addition, we would like to thank the National Institutes of Health for supporting this research.
References
- 1.Modrich P. (1991) Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet., 25, 229–253. [DOI] [PubMed] [Google Scholar]
- 2.Jiricny J. (1998) Replication errors: cha(lle)nging the genome. EMBO J., 17, 6427–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ban C. and Yang,W. (1998) Structural basis for MutH activation in E. coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J., 17, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hare J.T. and Taylor,J.H. (1985) One role for DNA methylation in vertebrate cells is strand discrimination in mismatch repair. Proc. Natl Acad. Sci. USA, 82, 7350–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hare J.T. and Taylor,J.H. (1989) Methylation in eucaryotes influences the repair of G/T and A/C DNA basepair mismatches. Cell Biophys., 15, 29–40. [DOI] [PubMed] [Google Scholar]
- 6.Bellacosa A., Cicchillitti,L., Schepis,F., Riccio,A., Yeung,A.T., Matsumoto,Y., Golemis,E.A., Genuardi,M. and Neri,G. (1999) MED1, a novel human methyl-CpG binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc. Natl Acad. Sci. USA, 96, 3969–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 8.Fang W.H. and Modrich,P. (1993) Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem., 268, 11838–11844. [PubMed] [Google Scholar]
- 9.Malik H.S. and Henikoff,S. (2000) Dual recognition-incision enzymes might be involved in mismatch repair and meiosis. Trends Biochem. Sci., 25, 414–418. [DOI] [PubMed] [Google Scholar]
- 10.Umar A., Buermeyer,A.B., Simon,J.A., Thomas,D.C., Clark,A.B., Liskay,R.M. and Kunkel,T.A. (1996) Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell, 87, 65–73. [DOI] [PubMed] [Google Scholar]
- 11.Gu L., Hong,Y., McCulloch,S., Watanabe,H. and Li,G.M. (1998) ATP dependent interaction of human mismatch repair proteins and dual role of PCNA in mismatch repair. Nucleic Acids Res., 26, 1173–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark A.B., Valle,F., Drotschmann,K., Gary,R.K. and Kunkel,T.A. (2000) Functional interaction of PCNA with MSH2-MSH6 and MSH2-MSH3 complexes. J. Biol. Chem., 275, 36498–36501. [DOI] [PubMed] [Google Scholar]
- 13.Chen C., Merrill,B.J., Lau,P.J., Holm,C. and Kolodner,R.D. (1999) Saccharomyces cerevisiae pol30 (proliferating cell nuclear antigen) mutations impair replication fidelity and mismatch repair. Mol. Cell. Biol., 19, 7801–7815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson R.E., Kovvali,G.K., Guzder,S.N., Amin,N.S., Holm,C., Habraken,Y., Sung,P., Prakash,L. and Prakash,S. (1996) Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. J. Biol. Chem., 271, 27987–27990. [DOI] [PubMed] [Google Scholar]
- 15.Hendrich B., Hardeland,U., Ng,H.H., Jiricny,J. and Bird,A. (1999) The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- 16.Petronzelli F., Riccio,A., Markham,G.D., Seeholzer,S.H., Stoerker,J., Genuardi,M., Yeung,A.T., Matsumoto,Y. and Bellacosa,A. (2000) Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J. Biol. Chem., 275, 32422–32429. [DOI] [PubMed] [Google Scholar]
- 17.Bellacosa A. (2001) Role of the MED1 (MBD4) gene in DNA repair and human cancer. J. Cell Physiol., in press. [DOI] [PubMed] [Google Scholar]
- 18.Wigler M., Levy,D. and Perucho,M. (1981) The somatic replication of DNA methylation. Cell, 24, 33–40. [DOI] [PubMed] [Google Scholar]
- 19.Petranovic M., Vlahovic,K., Zahradka,D., Dzidic,S. and Radman,M. (2000) Mismatch repair in Xenopus egg extracts is not strand-directed by DNA methylation. Neoplasma, 47, 375–381. [PubMed] [Google Scholar]
- 20.Holmes J.,Jr, Clark,S. and Modrich,P. (1990) Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc. Natl Acad. Sci. USA, 87, 5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su S.S., Lahue,R.S., Au,K.G. and Modrich,P. (1988) Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem., 263, 6829–6835. [PubMed] [Google Scholar]
- 22.Scharer O.D., Kawate,T., Gallinari,P., Jiricny,J. and Verdine,G.L. (1997) Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc. Natl Acad. Sci. USA, 94, 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons R., Li,G.M., Longley,M.J., Fang,W.H., Papadopoulos,N., Jen,J., de la Chapelle,A., Kinzler,K.W., Vogelstein,B. and Modrich,P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell, 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 24.Lahue R.S. and Modrich,P. (1988) Methyl-directed DNA mismatch repair in Escherichia coli. Mutat. Res., 198, 37–43. [DOI] [PubMed] [Google Scholar]
- 25.Lahue R.S., Au,K.G. and Modrich,P. (1989) DNA mismatch correction in a defined system. Science, 245, 160–164. [DOI] [PubMed] [Google Scholar]
- 26.Drummond J.T., Li,G.M., Longley,M.J. and Modrich,P. (1995) Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 27.Holden J.A. and Low,R.L. (1985) Characterization of a potent catenation activity of HeLa cell nuclei. J. Biol. Chem., 260, 14491–14497. [PubMed] [Google Scholar]
- 28.Larson E.D. and Drummond,J.T. (2001) Human mismatch repair and G/T mismatch binding by hMutSαin vitro is inhibited by adriamycin, actinomycin D and nogalamycin. J. Biol. Chem., 276, 9775–9783. [DOI] [PubMed] [Google Scholar]
- 29.Petronzelli F., Riccio,A., Markham,G.D., Seeholzer,S.H., Genuardi,M., Karbowski,M., Yeung,A.T., Matsumoto,Y. and Bellacosa,A. (2000) Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J. Cell Physiol., 185, 473–480. [DOI] [PubMed] [Google Scholar]
- 30.Bader S., Walker,M., Hendrich,B., Bird,A., Bird,C., Hooper,M. and Wyllie,A. (1999) Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene, 18, 8044–8047. [DOI] [PubMed] [Google Scholar]
- 31.Riccio A., Aaltonen,L.A., Godwin,A.K., Loukola,A., Percesepe,A., Salovaara,R., Masciullo,V., Genuardi,M., Paravatou-Petsotas,M., Bassi,D.E., Ruggeri,B.A., Klein Szanto,A.J., Testa,J.R., Neri,G. and Bellacosa,A. (1999) The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet., 23, 266–268. [DOI] [PubMed] [Google Scholar]
- 32.Gruenbaum Y., Cedar,H. and Razin,A. (1982) Substrate and sequence specificity of a eukaryotic DNA methylase. Nature, 295, 620–622. [DOI] [PubMed] [Google Scholar]
- 33.Hendrich B., Abbott,C., McQueen,H., Chambers,D., Cross,S. and Bird,A. (1999) Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm. Genome, 10, 906–912. [DOI] [PubMed] [Google Scholar]